

Abstract
Recently we reported that rapid determination of enthalpy of binding can be achieved for a large number of congeneric agents or in combinatorial libraries, fairly efficiently. We show that using a Thermodynamic Craig plot can be very useful in dissecting the enthalpy and entropy contribution of different substituents on a common scaffold, in order to design potent, selective or pan-active compounds. In our implementation, the approach identified a critical Lys residue in the BIR3 domain of XIAP. We report for the first time that it is possible to target such residue covalently to attain potent and selective agents. Preliminary cellular studies in various models of leukemia, multiple myeloma and pancreatic cancers, suggest that the derived agents possess a potentially intriguing pattern of activity, especially for cell lines that are resistant to the pan-IAP antagonist and clinical candidate LCL161.
Keywords: Drug discovery, ΔH screening, ITC, PPIs, protein-protein interactions, XIAP, IAP, covalent inhibitors, lysine covalent, isothermal titration calorimetry
Graphical Abstract

Introduction
Apoptosis or programmed cell death is a natural cellular process designed to eliminate unwanted or damaged cells in the body. In healthy tissues, a well-regulated balance exists between pro- and anti-apoptotic proteins that work together to control the occurrence of this natural process. However, an imbalance in the expression of anti-apoptotic proteins can result in defective apoptosis. This phenomenon can in turn can lead to tumorigenesis with concomitant resistance of cancer cells to chemotherapy, radiotherapy, or even immunotherapy, given that these therapeutic strategies are aimed at inducing apoptosis. A common consequence of activating the pro-apoptotic cascade is the final activation of a class of cysteine proteases (caspases) that digest the cellular content. Critical regulators of apoptosis are the Inhibitors of Apoptosis Proteins (IAPs).1, 2 To date, eight members of this protein family have been identified and among these, the X-linked IAP (XIAP) has been shown to prevent apoptosis by directly binding to caspases. Structurally, XIAP contains three baculovirus IAP repeat (BIR) domains, and it has been shown that the third BIR domain (BIR3) potently binds to and inhibits caspase-9, while the second BIR domain (BIR2) and the linker between BIR1 and BIR2, potently inhibit the effector caspases: caspase-3 and caspase-7.3 In addition to XIAP, two other members of the family, namely cellular IAP1 (cIAP1) and cellular IAP2 (cIAP2), have been shown to interact with tumor necrosis factor receptor-associated factor 2 (TRAF2), and the resulting complex reportedly antagonizes the activation of caspase-8, hence, inhibiting TNF receptor-mediated apoptosis.4–7 Due to their ability to prevent caspase activation and inhibit apoptosis, it is not surprising that XIAP, cIAP1 and cIAP2 are overexpressed in many tumor cell lines and human tumor tissues, conferring a poor prognosis to anticancer treatments.8–12 These observations inspired a fervid drug hunt for possible effective inhibitors of these proteins.13–17 As mentioned above, apoptosis is a tightly regulated process, and in normal cells a natural IAP inhibitor, second mitochondria-derived activator of caspases (SMAC) has been identified. SMAC is a mitochondrial protein that when released into the cytosol following pro-apoptotic signals binds potently to both cIAP1/2 and XIAP, thus counteracting their anti-apoptotic activity.18–20 Proteolytic SMAC activation after mitochondrial release into the cytosol exposes an N-terminal tetrapeptide of sequence Ala-Val-Pro-Ile (AVPI) that mediates its interactions with XIAP, cIAP1 and cIAP2. In particular, dimeric SMAC binds to both the BIR2 and BIR3 domains of XIAP, hence, antagonizing the binding of XIAP to both caspase-9 and caspase-3/7.21–23 On the contrary, in cIAP1 and cIAP2, SMAC AVPI N-terminal peptide binds potently only to their BIR3 domain.5 On these premises, agents that could mimic SMAC AVPI peptide could serve as potential new therapeutic agents to restore apoptosis in tumors that are driven by XIAP and/or cIAP1/2 expression.24–45 Most SMAC mimetics reported to date are either pan-IAP antagonists, which means they potently inhibit XIAP, cIAP1 and cIAP2,23–26,28,30–43 while only few examples exist for compounds that are selective for cIAP1 or cIAP1/2.30 To date, several pan-IAP inhibitors have been shown to work as single agents in cellular and animal models6,19,26,36 and few have advanced7, 20, 27, 37 into clinical trials.4, 24, 26, 27, 42 Mechanistically, however, AVPI mimetics act quite distinctly depending on the cell lines and on the given agent’s relative affinity for XIAP versus cIAP1 or cIAP2, and the benefits of antagonizing one versus all members of the family remain to date an unsolved matter.30, 46 With the exception of a moderately selective XIAP BIR2 domain antagonist,47 no potent and selective agents have been reported that target XIAP alone. In our recent studies we reported that using a thermodynamic driven screening approach, small variations on the surface of BIR3 domains of XIAP, cIAP1, and cIAP2 could be potentially targeted to achieve such agents.48 Here we report detailed thermodynamic driven structure-activity relationship studies that led to innovative agents that target reversibly or covalently the BIR3 domain of XIAP. First, we demonstrated that careful considerations of enthalpy and entropy of binding can be used to derive potent and either pan-active or selective compounds. In addition, we demonstrated that selective covalent XIAP antagonists can be obtained by carefully targeting a unique Lys residue on its surface. Hence, not only the agents reported should help decipher the relative potential of XIAP versus cIAP1/2 as clinical targets, but open the way to the design of covalent inhibitors targeting binding site Lys residues. Finally, our studies demonstrated that a determination of thermodynamic parameters of binding can be successfully employed to analyze structure-activity relationships and to provide a general avenue to guide the lengthy and often unpredictable hit-to-lead optimization process.
Results
Thermodynamic driven design of novel IAP antagonists
Very recently we reported on a novel enthalpy-based screening strategy of focused combinatorial libraries aimed at identifying novel binding motifs targeting XIAP, cIAP1 or cIAP2.48 In that work, a tetrapeptide library of ~ 100,000 compounds of structures Ala-XXX, where X represented 46 natural and non-natural amino acids occupying the P2-P3-P4 positions of the SMAC-binding pocket on BIR3 (Figure 1a), revealed a novel consensus motif for XIAP BIR3 of sequence Ala-pTyr-Pro-(4F)Phe-NH2 or N-Me-Ala-(p-phosphonomethyl)Phe-Pro-Phe-NH2 (compound 1, Figure 1g). However, while this agent presented a markedly high enthalpy (ΔH) of binding for XIAP BIR3 of −12.2 kcal/mol (Figure 1g–i) compared to that for the reference peptide of −7.8 kcal/mol (Figure 1a–c; Table 1) or other pan-IAP inhibitors such as LCL161 (Figure 1d–f ΔH = −5.2 kcal/mol), AT-406, or GDC-0156 (ΔH = −6.5 kcal/mol and −5.2 kcal/mol, respectively; Supplementary Figure S3),26 it exhibited only a modest, yet encouraging, selectivity in inhibiting the BIR3 domain of XIAP compared to cIAP1/2 (Figure 1i).48 Hence, our working hypothesis was that selecting for ligands that displayed the largest ΔH of binding for the given target would also display the greatest selectivity.49 Therefore, based on the new identified consensus motif we sought to derive novel XIAP BIR3 targeting agents by iteratively synthesizing and testing a variety of phosphonomethyl or phosphate bioisosters at the P2 position of tetrapeptides of general sequence NMe-Ala-P2-Pro-Phe-NH2 (Table 1). Ranking the agents by enthalpy of binding to the BIR3 domain of XIAP revealed that compounds with a formal negative charge in P2, such as 4-sulfone-Phe (compound 2, Table 1) displayed the largest −ΔH for binding to the BIR3 domain of XIAP and, in agreement with our central hypothesis, these agents also displayed the largest selectivity especially for XIAP versus cIAP1, with IC50 values for compound 1 of 35 nM, 197.6 nM, for XIAP and cIAP1, respectively (Figure 1i), and IC50 values for compound 2 of 38.3 nM, 143.5 nM, for XIAP and cIAP1, respectively (Table 1). The reported IC50 values represented the ability of the agents to displace the binding of the given target from a biotinylated AVPI peptide in a Dissociation Enhanced Lanthanide Fluorescent Immunoassay (DELFIA) displacement assay platform, as we have recently described.48 To further assess if potency and selectivity could be further achieved also by varying the P4 position, we designed, synthesized and tested against the BIR3 domain of XIAP using an enthalpy screening approach a number of compounds with the general sequence NMe-Ala-Val-P3/P4 where P3/P4 represent Pro-Phe bioisosters (Table 1 and supplementary Figure S1). Out of 32 compounds synthesized and tested, we selected those compounds that displayed a ΔH of binding > 4 kcal/mol. However, when selected agents were subsequently tested in full isothermal titration calorimetry (ITC) measurements and in the DELFIA displacement assays, we found a poor correlation between the ΔH values and dissociation constant (Kd), indicating that each agent displayed varying entropic contributions to binding to the BIR3 domain of XIAP. Hence, in an attempt to predict whether given combinations of P2 and P3/P4 elements could result in more potent and/or more selective compounds, we computed the enthalpy and entropy contribution of binding to the BIR3 of XIAP of each element with respect to a reference molecule, namely NMe-Ala-Val-Pro-Phe-NH2. In essence, given the thermodynamics of binding of NMe-Ala-Val-Pro-Phe-NH2 (Figure 1b), differential δΔH and δ(−TΔS) values were calculated from experimental values and assigned to each P2 element in the NMe-Ala-P2-Pro-Phe-NH2 compounds (Table 1) and to each P3/P4 elements in the NMe-Ala-Val-P3/P4 agents (Table 2). We found it useful to report the data using a thermodynamic Craig plot of δΔH versus δ(−TΔS) for each agent (Figure 2a). In this representation, compounds that are close to the diagonal would possess similar ΔG values (hence, similar dissociation constants) for XIAP BIR3 as the reference molecule NMe-Ala-Val-Pro-Phe-NH2, while ligands that fall on the dashed lines parallel to this diagonal would have dissociation constants that are approximately either 4 times greater (less potent, grey line) or about 6 times smaller (more potent, light blue line) than the reference compound (Figure 2a). In addition, and based on our previous hypothesis, compounds that possess a greater ΔH of binding could also result more selective for XIAP BIR3, compared to cIAP1 or cIAP2. Hence, to probe the utility of this thermodynamic Craig plot, we selected proper combinations of elements that we predicted being able to confer either the greatest potency or the greatest selectivity towards the BIR3 domain of XIAP (Figure 2b). Hence, merging compound 2 (δΔH = −0.5 kcal/mol, δ(−TΔS) = 0 kcal/mol) with compound 19 (δΔH = 0.3 kcal/mol, δ(−TΔS) = 0.2 kcal/mol), compounds with the best compromise between largest enthalpy without losing too much in potency, resulted in compound 22 that displayed a ΔH value of −8.4 kcal/mol, and a −TΔS value of 0.1 kcal/mol that are remarkably close to the predicted additive values derived from the two compounds (Table2, Figure 2b–d). Likewise, merging compounds with the largest free energy of binding ΔG, namely compound 14 (δΔH = 1.4 kcal/mol, δ(−TΔS) = −2.3 kcal/mol) and compound 17 (δΔH = 1.7 kcal/mol, δ(−TΔS) = −2.4 kcal/mol), resulted in agent compound 31 with a ΔH value of −5.1 kcal/mol, and a −TΔS value of −4.5 kcal/mol that are again remarkably close to the additive values derived from the two compounds (Table 2, Figure 2b–d). A systematic merging of agents reported in Table 1 and Table 2 revealed a remarkable predictive ability of the thermodynamic Craig plot (Figure 2b,c). From these studies, compound 31 was the most potent, but perhaps not particularly selective by virtue of the smaller ΔH of binding for XIAP, while compound 22 was predicted to be the most selective for XIAP BIR3 by virtue of its largest enthalpy of binding for this target. In agreement with these predictions, compound 31 was very potent in displacing a SMAC peptide in the DELFIA assays against XIAP, cIAP1 and cIAP2 with IC50 values of 37.1 nM, 4.5 nM, 15 nM, respectively (Figure 2e, Table 3). Whereas, as predicted, compound 22, was XIAP selective with IC50 values of approximately 190.7 nM, and >1000 nM against XIAP, and cIAP1 and cIAP2, respectively (Figure 2e, Table 3).
Figure 1. Molecular docking and thermodynamic analysis of N-Me-AVPF-NH2, LCL161, and compound 1 followed by selectivity studies against the BIR3 domains of XIAP, cIAP1, and cIAP2.

a) Docking pose of N-Me-AVPF-NH2 into the binding pocket of the BIR3 domain of XIAP (PDB ID 2OPZ). b) Isothermal Titration Calorimetry (ITC) curve for the binding between the BIR3 domain of XIAP and N-Me-AVPF-NH2. c) DELFIA displacement curves relative to the compound N-Me-AVPF-NH2 tested against the BIR3 domains of XIAP, cIAP1, and cIAP2, respectively (IC50 values 108.2 nM, 48.2 nM, and 209 nM, for XIAP, cIAP1, and cIAP2, respectively). d) Docking pose of the clinical compound LCL161 into the binding pocket of the BIR3 domain of XIAP (PDB ID 2OPZ). e) Isothermal Titration Calorimetry (ITC) curve for the interaction between the BIR3 domain of XIAP and LCL161. f) DELFIA displacement curves relative to the compound LCL161 tested against the BIR3 domains of XIAP, cIAP1, and cIAP2 (IC50 values 52.7 nM, 10.4 nM, and 12.9 nM, for XIAP, cIAP1, and cIAP2, respectively). g) Docking pose of the compound 1 into the binding pocket of the BIR3 domain of XIAP (PDB ID 2OPZ). The XIAP BIR3 residue Lys311, interacting with the phosphonate group, is highlighted. h) Isothermal Titration Calorimetry (ITC) curve for the binding between the BIR3 domain of XIAP and compound 1. i) DELFIA displacement curves relative to the compound 1 tested against the BIR3 domain of XIAP, cIAP1, and cIAP2 (IC50 values 35 nM, 197.6 nM, and 364.3 nM, for XIAP, cIAP1, and cIAP2, respectively).
Table 1.
Binding affinities and thermodynamics parameters for different P2 and P3/P4 substituents of XIAP BIR3 targeting agents.
| N-Me-Ala-P2-Pro-Phe-NH2 | ||||||
|---|---|---|---|---|---|---|
| Compd | P2 | ΔH | −TΔS | Kd (nM) | δ(ΔH) | δ(−TΔS) |
| 1 | 4-(phosphonomethyl)Phe | −12.2 ± 0.5 | 3.0 | 206 | −4.4 | 4 |
| 2 | (4SO3−)-Phe | −8.3 ± 0.3 | −1.0 | 155 | −0.5 | 0 |
| 3 | (4-NO2)-Phe | −8.9 ± 0.3 | −0.1 | 280 | −1.1 | 0.9 |
| 4 | (4-sulfomethyl)-Phe | −8.7 ± 0.4 | −0.2 | 346 | −0.9 | 0.8 |
| 5 | (3Cl-4CF3)Phe | −8.3 ± 0.2 | −0.9 | 190 | −0.5 | 0.1 |
| 6 | (3F-4CF3)Phe | −8.1 ± 0.2 | −1.2 | 155 | −0.3 | −0.2 |
| 7 | (3Cl-5CF3)Phe | −8.0 ± 0.1 | −1.3 | 133 | −0.2 | −0.3 |
| 8 | Cyclohexyl Glycine | −7.8 ± 0.4 | −1.4 | 354 | 0 | −0.4 |
| 9 | Dab | −7.7 ± 0.3 | −1.4 | 230 | 0.1 | −0.4 |
| 10 | (2F-4CF3)Phe | −7.4 ± 1.0 | −1.8 | 194 | 0.4 | −0.8 |
| 11 | Phe(4-CF3) | −7.2 ± 0.3 | −1.9 | 213 | 0.6 | −0.9 |
| 12 | (4-OCF3)Phe | −7.1 ± 0.3 | −1.9 | 227 | 0.7 | −0.9 |
| 13 | (p-guanidino)Phe | −6.9 ± 0.2 | −2.3 | 182 | 0.9 | −1.3 |
| 14 | (2F-4CF3-5Me)Phe | −6.4 ± 0.2 | −3.3 | 81 | 1.4 | −2.3 |
| N-Me-Ala-Val-P3/P4 | ||||||
| Compd | P3/P4 | ΔH | −TΔS | Kd (nM) | δ(ΔH) | δ(−TΔS) |
| 15 | Pro-(2-aminoindan) | −4.5 ± 0.2 | −4.0 | 602 | 3.3 | −3 |
| 16 | Pro-(1-aminoindan) | −7.1 ± 0.4 | −1.9 | 269 | 0.7 | −0.9 |
| 17 | Pro-((R)-4-F-2,3-dihydro-1H-inden-1-amine) | −6.1 ± 0.1 | −3.4 | 122 | 1.7 | −2.4 |
| 18 | 2-{5-[(2S)-2-pyrrolidinyl]-1,2,4-oxadiazol-3-yl}pyridine | −6.2 ± 0.5 | −2.0 | 1000 | 1.6 | −1 |
| 19 | 2-benzyl-6-(pyrrolidine-2-yl)pyrazine | −7.5 ± 0.4 | −0.8 | 719 | 0.3 | 0.2 |
| 20 | 2-(2-pyrrolidinyl)-1H-benzamidazole | −5.1 ± 5.0 | −1.4 | 12000 | 2.7 | −0.4 |
ΔH, −TΔS, and Kd were calculated using ITC measurement against the BIR3 domain of XIAP. ΔH values are reported with a confidence interval level 95%. δ(ΔH) and δ(−TΔS) are the difference in ΔH or −TΔS with respect to the thermodynamics of binding between BIR3 of XIAP and the reference peptide N-Me-AVPF-NH2 (ΔH = −7.8 kcal/mol, and −TΔS = −1 kcal/mol). The differences are calculated as: δ(ΔH) = ΔH − ΔHref and δ(−TΔS) = −TΔS − (−TΔSref)
Table 2.
Comparison of experimental and predicted thermodynamics parameters for XIAP BIR3 agents obtained from combinations of P2 and P3/P4 substituents.
| Compd | Structure | Predicted versus experimental parameters (kcal/mol) | |||||
|---|---|---|---|---|---|---|---|
| ΔHexp | −TΔSexp | ΔGexp | ΔHpred | −TΔSpred | ΔGpred | ||
| 21 | 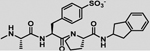 |
−5.8 ± 0.4 | −3 | −8.8 | −5 | −4 | −9 |
| 22 | 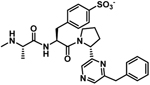 |
−8.4 ± 0.2 | 0.1 | −8.3 | −8 | −0.8 | −8.8 |
| 23 | 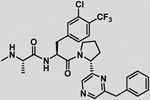 |
−8.0 ± 0.3 | −0.4 | −8.4 | −8 | −0.7 | −8.7 |
| 24 | 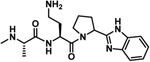 |
−4 ± 2 | −2.5 | −6.5 | −5 | −1.8 | −6.8 |
| 25 | 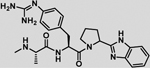 |
−7 ± 2 | 0.3 | −6.7 | −4.2 | −2.7 | −6.9 |
| 26 | 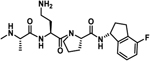 |
−6.2 ± 0.2 | −3.3 | −9.5 | −6 | −3.8 | −9.8 |
| 27 | 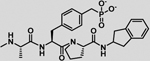 |
−9.3 ± 0.6 | 1.1 | −8.1 | −8.9 | 0 | −8.9 |
| 28 | 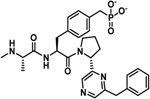 |
−14.0 ± 0.6 | 5.1 | −8.9 | −11.9 | 3.2 | −8.7 |
| 29 | 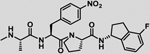 |
−7.2 ± 0.2 | −2.5 | −9.7 | −7.2 | −2.5 | −9.7 |
| 30 |  |
−6.7 ± 0.2 | −3.1 | −9.8 | −6.6 | −3.4 | −10 |
| 31 | 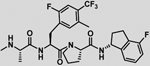 |
−5.1 ± 0.1 | −4.5 | −9.6 | −4.7 | −5.7 | −10.4 |
ΔHexp, −TΔSexp, and ΔGexp were calculated using ITC measurement against the BIR3 domain of XIAP. ΔHexp values are reported with a confidence interval level 95%. ΔHpred, −TΔSpred, and ΔGpred were calculated as: ΔXpred = ΔXref + δ(ΔX)P2 + δ(ΔX)P3/P4, where ΔXref are the thermodynamics parameters of the reference agent N-Me-AVPF-NH2 (ΔH = −7.8 kcal/mol, and −TΔS = −1 kcal/mol), and δ(ΔX) values are calculated as in Table 1.
Figure 2. A Craig plot of thermodynamic parameters guided the design of selective and pan-inhibitors, against the BIR3 domains of XIAP, cIAP1, and cIAP2.
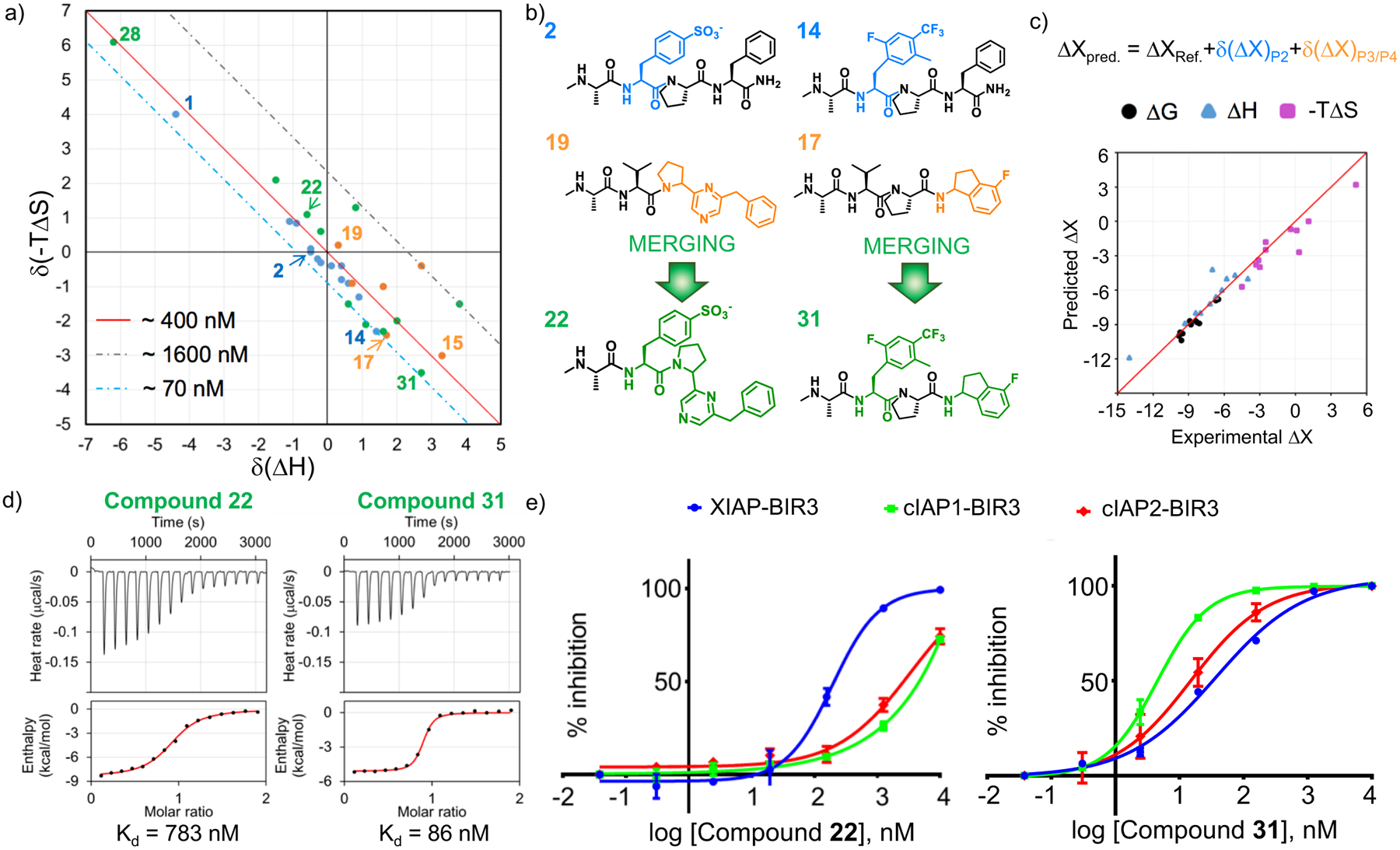
a) Craig plot of δ(−TΔS) as function of δ(ΔH), showing the difference in term of thermodynamics parameters in respect to the reference compound N-Me-AVPF-NH2. Compounds on or near the diagonal (solid red line) are expected to possess a similar affinity of the reference peptide; compounds falling below the diagonal will present an increase in activity, while agents falling above the diagonal will be less potent than N-Me-AVPF-NH2. Compounds that differ from N-Me-AVPF-NH2 in position P2 are depicted in blue, while those that differ at the P3/P4 position are depicted in orange; the compounds resulted from the combination of different P2 and P3/P4 substituents are depicted in green and black. b) Schematic representation of the combination of compounds with P2 and P3/P4 substituents selected based on the thermodynamic Craig plot analysis. On the left, the combination of the P2 element of compound 2 with the P3/P4 element of compound 19 resulted in compound 22 designed to be more selective for XIAP compared to cIAP1/2. On the right, the combination of the P2 element of compound 14 with the P3/P4 element of compound 17 resulted in compound 31 designed to be a pan agent for IAPs. c) Correlation plot between predicted (based on the thermodynamics Craig plot) and experimental thermodynamic values for the compounds synthesized. d) Isothermal Titration Calorimetry (ITC) curve for the binding between the BIR3 domain of XIAP and compound 22 (left panel) or compound 31 (right panel). e) DELFIA displacement curves relative to the compounds compound 22 (left panel) and compound 31 (right panel) tested against the BIR3 domain of XIAP, cIAP1, and cIAP2. The IC50 values for compound 22 are 191 nM, > 1000 nM, and > 1000 nM, against the BIR3 domain of XIAP, cIAP1, and cIAP2, respectively. The IC50 values for compound 31 are 37.1 nM, 4.5 nM, and 15 nM, against the BIR3 domain of XIAP, cIAP1, and cIAP2, respectively.
Table 3.
Relative binding affinities and selectivity for XIAP BIR3 targeting agents designed from combinations of various P2 and P3/P4 substituents.
| Compd | XIAP | cIAP1 | cIAP2 | |||
|---|---|---|---|---|---|---|
| Kd (ITC, nM) | IC50 (DELFIA, nM) | IC50 (DELFIA, nM) | Selectivity cIAP1/XIAP | IC50 (DELFIA, nM) | Selectivity cIAP2/XIAP | |
| 21 | 337 | 229.4 ± 35.9 | > 1000 | > 4.4 | > 1000 | > 4.4 |
| 22 | 783 | 190.7 ± 25.6 | > 1000 | > 5.2 | > 1000 | > 5.2 |
| 23 | 753 | 175.9 ± 0.4 | 352.6 ± 36.4 | 2.0 | > 9000 | > 51.1 |
| 24 | 17000 | > 10000 | > 16000 | > 1.6 | > 10000 | 1.0 |
| 25 | 13000 | 5944 ± 816 | > 5000 | > 0.8 | > 10000 | > 1.7 |
| 26 | 110 | 76.9 ± 3.9 | 17.2 ± 3.8 | 0.2 | 56.6 ± 3.0 | 0.7 |
| 27 | 1100 | 215.3 ± 12.1 | 1203 ± 55 | 5.6 | 1103 ± 126 | 5.1 |
| 28 | 321 | 275.7 ± 12.7 | > 2000 | > 7.2 | > 1500 | > 5.5 |
| 29 | 81 | 86.8 ± 14.9 | 23.9 ± 0.1 | 0.3 | 53.5 ± 4.7 | 0.6 |
| 30 | 70 | 23.2 ± 2.2 | 21.7 ± 0.9 | 0.9 | 30.6 ± 0.4 | 1.3 |
| 31 | 86 | 37.1 ± 1.1 | 4.5 ± 0.7 | 0.1 | 15.0 ± 5.1 | 0.4 |
IC50 values with respective standard errors for the BIR3 domains of XIAP, cIAP1, and cIAP2 were obtained with a DELFIA displacement assay. Selectivity was calculated as a ratio of cIAP1 or cIAP2 IC50 values versus IC50 values for XIAP.
Potent and selective, covalent XIAP antagonists targeting the BIR3 domain residue Lys311
While using the thermodynamic Craig plot was instrumental in deriving novel pan-IAP (compound 31) and moderately selective XIAP BIR3 agents (compound 22), we further investigated the basis for this selectivity using single point mutation analysis as corroborated by our docking studies (Figure 1) and sequence alignment between XIAP, cIAP1 and cIAP2, that had identified residue Lys311 of XIAP as possibly a discriminating feature between these proteins. In cIAP1 and cIAP2 this position is occupied by a glutamic acid,48 while most of other SMAC binding site residues are conserved among the three proteins. The possible involvement of Lys311 in the selectivity of our agents for XIAP is also suggested by the nature of the compounds, with agents presenting a formal negative charge displaying the largest enthalpy of binding and greater selectivity over neutral compounds, indicating a possible salt bridge formation. Hence, given that our most selective ligands are likely juxtaposed across from Lys311, we sought to derive novel P2 derivatives containing an electrophile that could react covalently and specifically with this residue. Among the various possible electrophiles (Table 4), introduction of sulfonyl fluoride placed on the side chain of L-diaminopropionic acid in P2 (L-Dap; Figure 3a) placed the electrophile at proper distance and juxtaposition for reaction with Lys311, without altering the pose of the other substructures. Indeed, we found that this agent efficiently formed a covalent adduct with the BIR3 of XIAP, as clearly appreciable using both SDS gel electrophoresis and mass spectrometry (Figure 3b,c) of the complex between BIR3 of XIAP and the agent NMe-Ala-pSFB-Dap-Pro-Phe-NH2 (compound 32) where pSFB-Dap represent a p-Sulfonyl fluoride benzoic acid coupled via amide bond to the side chain amino group of an L-Dap (L-diaminopropionic acid) in P2. The IC50 value for this compound against XIAP BIR3 was 11.3 nM, in contrary to the IC50 values for cIAP1 and cIAP2 of 181 nM, and 304 nM, respectively (Figure 3d, Table 4). These results confirmed our hypothesis that we can design a compound that can selectively targets the Lys311 is present just in XIAP BIR3. Hence, proper combination of this covalent P2 substituent with the above identified P3/P4 (or in principle any previously identified P3/P4 substituents such as those present in clinical candidates GDC-0152 or LCL161, for example) could lead to potent and selective XIAP antagonists. Several agents were therefore prepared as listed in Table 4. Among these agents, compound 34 (with a 2-(3-fluorobenzyl)-6-(pyrrolidine-2-yl)pyrazine in P3/P4; Figure3e) displayed a remarkable IC50 value for XIAP BIR3 of 16.6 nM, and very modest inhibition of cIAP1 and cIAP2 with IC50 values > 200 nM for both proteins (Figure 3h). SDS gel electrophoresis and mass spectrometry data confirmed the covalent interaction of this agent with XIAP BIR3 (Figure 3f,g) but not with cIAP1 or cIAP2 (Figure 3j). We mutated XIAP BIR3 not only at the Lys311 with a Glu (XIAP BIR3 K311E), but also the nearby Lys322 with an Ala (XIAP BIR3 K322A) to further confirm if the covalent interaction is specific to the Lys311. SDS gel electrophoresis data confirmed that the covalent interaction is present just when the target protein has the Lys311 (Figure 3i). Furthermore, mutating Lys311 with a glutamic acid in BIR3 of XIAP resulted in a drop in affinity also in the DELFIA displacement assays for compound 34 (IC50 values dropped from 16.6 nM with wt-BIR3 to > 1 μM when the agent was tested against the Lys311Glu mutant, while the inhibition is not affected by the mutation of Lys322 with an Ala, IC50 = 19.7 nM; Figure 3k) further clearly substantiating our SDS gel data implicating XIAP BIR3 Lys311 as the target for the covalent compound.
Table 4.
Relative binding affinities and selectivity for XIAP BIR3 targeting covalent agents.
| N-Me-Ala-P2-P3/P4 | ||||||
|---|---|---|---|---|---|---|
| XIAP | cIAP1 | cIAP2 | ||||
| Compd | Structure | IC50 (nM) | IC50 (nM) | Selectivity cIAP1/XIAP | IC50 (nM) | Selectivity cIAP2/XIAP |
| 32 | 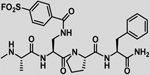 |
11.3 ± 0.8 | 181.0 ± 20.1 | 16.0 | 304.0 ± 60.2 | 27.0 |
| 33 | 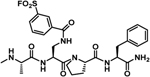 |
47.3 ± 3.3 | 264.1 ± 35.5 | 5.6 | 212.1 ± 8.9 | 4.5 |
| 34 | 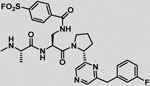 |
16.6 ± 2.1 | >200 | >12.0 | 353.3 ± 118.2 | 17.6 |
| 34* | 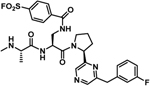 |
189.4 ± 1.3 | >1000 | >5.3 | >1000 | >5.3 |
IC50 values with respective standard errors for the ability of test agents to displace a reference AVPI peptide from the BIR3 domains of XIAP, cIAP1, and cIAP2, were obtained with a DELFIA assay. Selectivity was calculated as the ratio of cIAP1 or cIAP2 IC50 values versus IC50 values for XIAP.
Figure 3. Design and characterization of covalent XIAP BIR inhibitors.
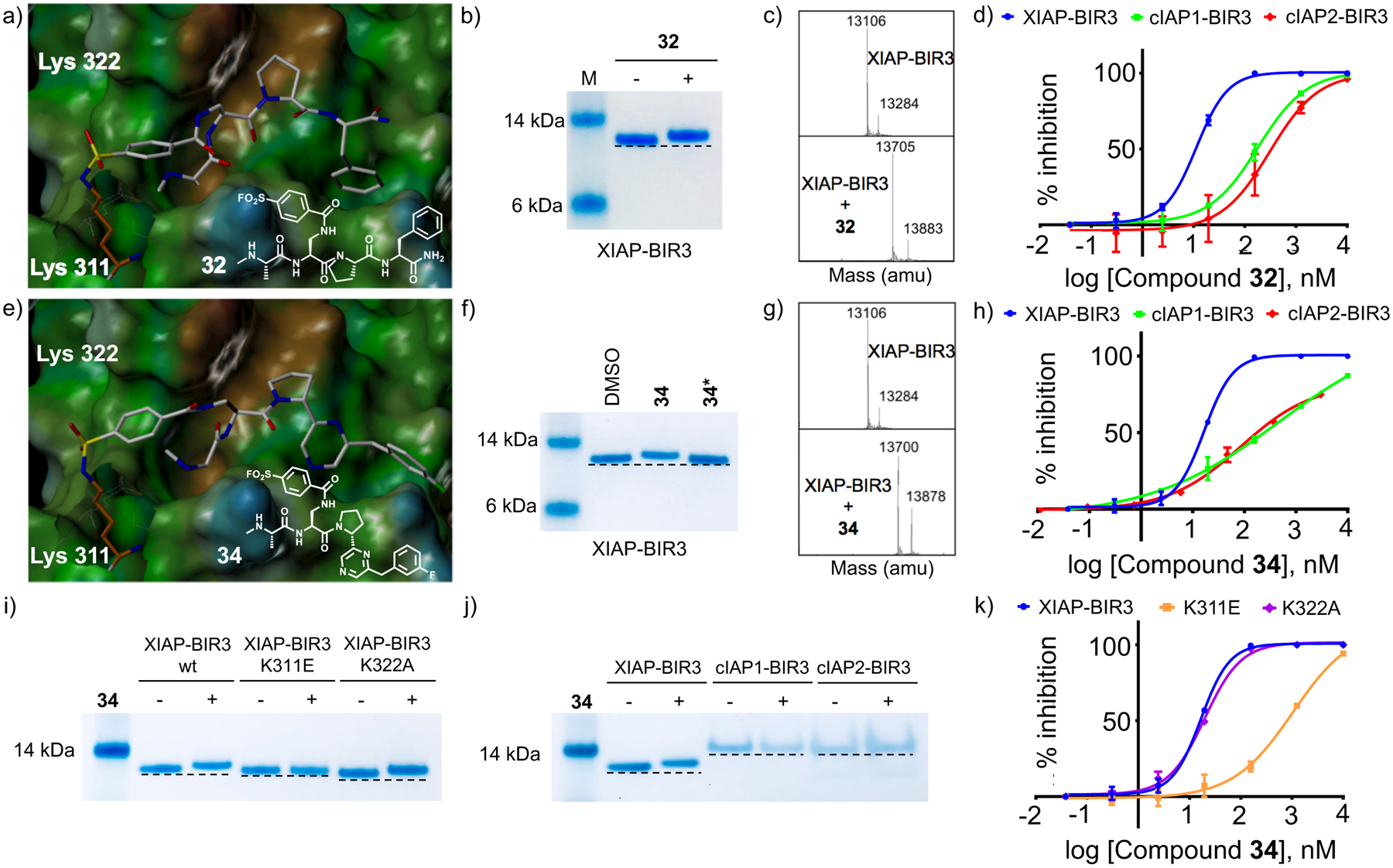
a) Covalent docking pose of compound 32 into the binding pocket of the BIR3 domain of XIAP (PDB ID 2OPZ). The Lysine 311 forming the covalent bond with compound 32 is highlighted. b) SDS-PAGE gel electrophoresis followed by Coomassie staining of the BIR3 domain of XIAP in the absence and presence of compound 32 after 10 minutes incubation at RT and at a protein-ligand ratio 1:2. c) LC-MS spectra of the BIR3 domain of XIAP in the absence (top) and presence (bottom) of compound 32 at a protein-ligand ratio 1:2. d) DELFIA displacement curves relative to the compound 32 tested against the BIR3 domain of XIAP, cIAP1, and cIAP2. The IC50 values for compound 32 are 11.3 nM, 180.4 nM, and 306.7 nM, against the BIR3 domain of XIAP, cIAP1, and cIAP2, respectively. e) Covalent docking pose of compound 34 into the binding pocket of the BIR3 domain of XIAP (PDB ID 2OPZ). The Lysine 311 forming the covalent bond with compound 34 is highlighted. f) SDS-PAGE gel electrophoresis followed by Coomassie staining of the BIR3 domain of XIAP in the absence and presence of compound 34 and the diasteroisomer 34* after 10 minutes incubation at RT and at a protein-ligand ratio 1:2. g) LC-MS spectra of the BIR3 domain of XIAP in absence (top) and in presence (bottom) of compound 34 at a protein-ligand ratio 1:2. h) DELFIA displacement curves relative to the compound 34 tested against the BIR3 domain of XIAP, cIAP1, and cIAP2. The IC50 values for compound 34 are 16.6 nM, >200 nM, and 353.3 nM, against the BIR3 domain of XIAP, cIAP1, and cIAP2, respectively. i) SDS-PAGE gel electrophoresis followed by Coomassie staining of the BIR3 domain of XIAP, XIAP-BIR3 K311E, and XIAP-BIR3 K322A in the absence and presence of compound 34 after 10 minutes incubation at RT and at a protein-ligand ratio 1:2. j) SDS-PAGE gel electrophoresis followed by Coomassie staining of the BIR3 domain of XIAP, cIAP1, and cIAP2 in the absence and presence of compound 34 after 10 minutes incubation at RT and at a protein-ligand ratio 1:2. k) Dose-response curves in DELFIA displacement assays for compound 34 against XIAP-BIR3, XIAP-BIR3 K311E, and XIAP-BIR3 K322A, respectively (IC50 values 16.6 nM, 1039 nM, and 19.7 nM, for XIAP-BIR3, XIAP-BIR3 K311E, and XIAP-BIR3 K322A, respectively).
The novel pan-IAP agent and the XIAP-BIR3 covalent agent are both effective against LCL161 resistant cell lines and sensitize cell lines to chemotherapy.
To further characterize the cellular activity of the identified compounds, we tested them against the LCL161-resistant Acute Lymphoblastic Leukemia (ALL) cell line MOLT-4. Cell viability assay confirmed that LCL161 is not particularly active against this cell line, despite it causes significant degradation of both cIAP1 and cIAP2. On the contrary, both the pan-IAP compound 31 and the XIAP BIR3 covalent compound 34 (Figure 4a) were equally effective with IC50 values in the single digit micromolar range. Interestingly, in this cell line compound 31 was able to induce cIAP1 and cIAP2 degradation, like LCL161 and as expected by its pan-IAP inhibitory activity, while compound 34 (and its inactive diasteroisomer compound 34*) was less effective in inducing degradation of these proteins in agreement with its increased activity against XIAP BIR3 compared to these other two IAPs (Figure 4b). To further corroborate these data, we compared the activity of our agents side by side with LCL161 against a panel of multiple myeloma (MM) cell lines, given the clinical application of LCL161 for this indication in clinical trials. Of the 6 MM cell lines tested, two are known to be LCL161 sensitive (namely, H929 and L363) while 4 others are known to be LCL161 resistant (namely, MM1S, RPMI 8226, LP1, and U266). In agreement, we found that LCL161 was particularly active in the two sensitive cell lines; likewise, both compound 31 and compound 34 were approximately equipotent in these LCL161-sensitive MM cell lines (Figure 4c). However, and in agreement with the data with MOLT-4, compound 31 and 34 (but not its less active enantiomer, 34*) were equally effective against the LCL161-resistant MM cell lines RPMI 8226, LP1 and U266, while only compound 34 was effective against the cell line MM1S (Figure 4c). Finally, to assess whether our agent can restore cancer cell sensitivity in chemoresistant cell lines, we tested LCL161, compound 31, and compound 34 in combination with gemcitabine in various pancreatic cancer cell lines (Figure 4e). In the most gemcitabine-sensitive cell line (MIA PaCa-2 that expresses only XIAP; Figure 4d), the effect of the SMAC mimetics is at best additive, given the efficacy of gemcitabine as a single agent (Figure 4e). However, for less sensitive cell lines such as BxPC3, and to the largest extent the gemcitabine-resistant cell line PANC-1 (both expressing XIAP, cIAP1, and cIAP2; Figure 4d), both LCL161 and to a greater extent compound 31, were able to significantly restore growth inhibition by gemcitabine (Figure 4e). To rule out that the activity of our agents compared to LCL161 could be due to inhibition of the BIR2 domain of XIAP, the BIR2 domain was expressed and a DELFIA assay was further developed, and the compounds tested. The data, reported in supplementary Table S1, indicated that like LCL161, our agents displayed only modest affinity for this domain (in the micromolar range).
Figure 4. Comparative cellular activity of LCL161, compound 31, and compound 34 in ALL, MM, and pancreatic cancer cell lines.

a) Cell viability of ALL cell line MOLT-4 cells was assessed after treating them with the indicated compounds for 48 hrs. Error bars are SD of triplicate readouts. b) IAP inhibitors induce degradation of IAP protein levels. MOLT4 cells were treated for 3 hr with 1 μM and propped for XIAP, cIAP1 or cIAP2. The β-actin blot was detected to ensure equal sample loading. c) Multiple myeloma cell lines were treated for 48 hr with the indicated compounds at 20 μM concentration. Error bars are SD of triplicate readouts. d) Western blot analysis of the basal expression level of XIAP, cIAP1, and cIAP2 in pancreatic cancer cell lines BxPC3, PANC-1 and MIA PaCa-2. The β-actin blot was detected to ensure equal sample loading. e) Compound 34 and compound 31 significantly sensitize pancreatic cancer cell lines to gemcitabine (GEM). Cells were first treated with two doses of GEM or DMSO for 24 hrs. Next day, media was replenished with the co-treatment media containing GEM and 15 μM of the indicated IAP inhibitors for additional 24 hrs. Error bars are SD of quadruplicate readouts. *, P < 0.05; **, P < 0.005, ***, P < 0.0005, and **** P < 10−5.
Discussion and conclusions
In the realm of drug discovery, the design of effective therapeutics often relies on a lengthy and elaborate iterative process known as the hit-to-lead optimization process. In such process, the chemical structure of a hit compound is iteratively modified in an attempt to increase potency and in most cases also selectivity against the given target. In some circumstances, pan-active compounds (i.e. agents that inhibit simultaneously several members of a given class of proteins) are desirable or needed to achieve maximal efficacy. In targeting protein-protein interaction (PPIs), these studies usually rely primarily on assessments of potency of test agents using biochemical assays that can measure the ability of the new molecules to displace a reference compound. Hence, one can usually follow the iterative optimizations of potency by measuring IC50 values of test agents, and the data are interpreted and used to guide next iteration of synthesis and testing. This approach is best suited when supported by structural data of the complex between the test agents and the target(s) that can be used to formulate hypotheses. Recently, the use of biophysical approaches has been introduced at both ends of the hit-to-lead optimization process particularly for the design of PPIs antagonists, first as screening tools to discover initial fragment hits in fragment-based drug discovery, and finally to validate a handful of optimized agents. Traditionally, during the optimization process, biochemical IC50 measurements are usually preferred as these offer a more rapid and cost effective means to rank order agents. However, we and others have recently reported that rapid determination of enthalpy of binding can be achieved for a large number of congeneric agents49 or in combinatorial libraries48 fairly efficiently. Our working hypothesis was that ligands displaying the largest enthalpy of binding would result not only as more potent but also as more selective for a given target.48 We found however that this hypothesis is only partially correct, especially with respect to potency, as unpredictable enthalpy/entropy compensation mechanisms play a major role in determining the binding affinity of a given molecule50. In this study we targeted the BIR3 domain of XIAP given that most known inhibitors discovered to date are usually more potent for two other members of this protein family, namely cIAP1 and cIAP2.51 The binding properties of these tetrapeptide mimetics have been well established, requiring invariably an Ala and a Pro residue (or mimetics) in positions P1 and P3, respectively, while aliphatic and aromatic residues are preferred in P2 and P4, respectively (Figure 1a). Recently, we surprisingly discovered using an enthalpy screening campaign against the BIR3 domain of XIAP that the position P2 can be occupied by a phospho-tyrosine residue, resulting in molecules with a large enthalpy of binding for XIAP BIR3.48 Likewise, replacing the P2 valine residue in AVPF with a non-hydrolysable 4-phosphonomethyl-Phe resulted in an agent (compound 1) with an increased −ΔH of binding (Figure 1g,h), resulting relatively more selective for XIAP BIR3 versus cIAP1/2 in the biochemical displacement assay (IC50 values 35 nM, 198 nM and 364 nM against the BIR3 domains of XIAP, cIAP1, and cIAP2, respectively; Table 1). To assess whether thermodynamic based structure-activity relationship (SAR) studies can be used to optimize these initial agents into more potent and selective, and/or more potent and pan-active compounds, we systematically explored various substitutions in the P2 position with bioisoters of a 4-phosphonomethyl-Phe, and in P3/P4 with bioisosters of pyrrolidine-aromatic moieties (Table 1; supplementary Figure S1). In particular, in an attempt to predict whether given combinations of P2 and P3/P4 elements could result in more potent and/or more selective compounds, we decided to tabulate the enthalpy and entropy contribution of binding to the BIR3 domain of XIAP of each P2 and P3/P4 elements with respect to a reference molecule, namely NMe-Ala-Val-Pro-Phe-NH2. Therefore, differential δΔH and δ(−TΔS) values were tabulated from experimental ITC curves and assigned to each P2 element in the NMe-Ala-P2-Pro-Phe-NH2 compounds and to each P3/P4 elements in the NMe-Ala-Val-P3/P4 agents (Table 1). Reporting these values using a thermodynamic Craig plot of δΔH versus δ(−TΔS) for each agent (Figure 2a) provided a visualization of the entropy-enthalpy compensation phenomenon for each element. Indeed, in this representation, compounds that fall near or on the diagonal would have a similar ΔG of binding (hence, a similar dissociation constant) as the reference molecule, regardless of ΔH and −TΔS values. For example, the 4-Phosphonomethyl-Phe residue in P2 (compound 1, Table 1, Figure 1g) is approximately equipotent with NMe-AVPF-NH2 despite the larger ΔH of binding that was entirely compensated by a concomitant loss in entropy (Table 1, Figure 2a). However, while it is very challenging to alter the entropy/enthalpy compensation of individual substituents,50 we sought to verify if it is possible to predict the thermodynamic profile of combined P2 and P3/P4 elements based on their individual entropy and enthalpy contributions to binding. We found that simple additivity of the thermodynamic parameters resulted in a remarkably close agreement between predicted (ΔHpred and −TΔSpred) and experimental thermodynamic values in agents containing various combinations of P2 and P3/P4 elements (Figure 2c, Table 2). Practically, we could predict ΔHpred, −TΔSpred, and ΔGpred using the simple relation: ΔXpred = ΔXref + δ(ΔX)P2 + δ(ΔX)P3/P4, where ΔXref are the thermodynamics parameters of the reference agent N-Me-AVPF-NH2 (ΔH = −7.8 kcal/mol, and −TΔS = −1 kcal/mol), and δ(ΔX) values are calculated as in Table 1 for each P2 and P3/P4 element. For example, merging the P2 element of compound 2 with the P3/P4 of compound 19 resulted in compound 22 (ΔHpred −8 = kcal/mol, ΔHexp = −8.8; −TΔSpred = −0.8, −TΔSexp = 0.1 kcal/mol), while merging compound 14 P2 with compound 17 P3/P4 resulted in compound 31 (ΔHpred −4.7 = kcal/mol, ΔHexp = −5.1; −TΔSpred = −5.7, −TΔSexp = −4.5 kcal/mol) (Figure 2b). A complete list of combined molecules and their respective predicted and experimental thermodynamic values is reported in Table 2, while a plot illustrating the correlation is reported in Figure 2c. We also found a very good correlation between the thermodynamic Kd values and IC50 values determined using a DELFIA displacement assay (Table 3). Furthermore, to assess the selectivity of these agents, IC50 values were determined also against the BIR3 domains of cIAP1, and cIAP2 (Table 3). From these studies we concluded that nearly additive behavior can be observed in the thermodynamic parameters of various substituents, and that a thermodynamic Craig plot can be useful in selecting suitable combinations of substituents with the predicted desired thermodynamics of binding. For example, selecting for agents that could confer the greatest enthalpy of binding also correlated with largest selectivity as exemplified by compound 28, the agent with the largest ΔH of binding to XIAP BIR3 (ΔH=−14 kcal/mol; Figure 2a, Table 2) that also resulted in the most selective albeit not the most potent agent (Table 3). Likewise, compound 31 was among the most potent agents, while because it displayed a relatively smaller enthalpy of binding (ΔH = −5.1 kcal/mol; Figure 2a, Table 2) it was also anticipated to be less selective, as indeed experimentally observed (Table 3). These studies clearly suggested that thermodynamic measurements aimed at dissecting entropy and enthalpy contributions in various substituents in a hit molecule can be very effective in selecting compounds with the most desired binding profiles during the hit-to-lead optimization process. While the approach was very successful in identifying potent pan-active compounds such as compound 31, attaining even relatively modest selectivity often came at the expense of potency mostly because of the well-known issue of enthalpy/entropy compensation.
Therefore, we next conducted molecular modeling studies to try to rationalize the observed partial selectivity and to design more potent and selective agents. As we recognized in our most recent work, a reasonable responsible residue for the selectivity of the P2 pTyr derivatives was Lys311, which is indeed a Glu residue in both cIAP1 and cIAP2. Recently, a few reports have emerged that successfully demonstrated covalent targeting of Lys residues in active sites of proteins by introduction of appropriately placed electrophiles on an existing ligand.52 These examples include not only targeting active site catalytic and non-catalytic Lys residues53, 54 but, and perhaps most excitingly, also targeting surface exposed Lys residues at protein-protein interfaces, such as in the recent examples of a covalent Mcl-1 inhibitor,55 and a covalent inhibitor of MDM2/P53 interactions.56 Accordingly, and based on our observations that our agents may target Lys311, we introduced a sulfonyl fluoride at various P2 positions (Table 4), and assessed the ability of these resulting agents to form a stable covalent bond with XIAP BIR3 by various means. Using molecular modeling we could anticipate that coupling a p-Sulfonyl fluoride-benzoic acid to the side chain of a P2 diaminopropionic acid (Dap), would juxtapose the electrophile with Lys311, and could form a covalent bond (Figure 3a). Excitingly, agents 32 (P2 p-sulfonyl-benzoic acid Dap; P3/P4, Pro-Phe-NH2) formed a stable covalent bond with XIAP BIR3 as detected by SDS gel electrophoresis and mass spectrometry (Figure 3b,c). In the DELFIA displacement assay panel, compound 32 was significantly more potent against XIAP BIR3 compared to cIAP1 and cIAP2 (Table 4, Figure 3d). The covalent binding was fairly selective as changing the sulfonyl-fluoride from the para to the meta position resulted in an incomplete reaction and diminished activity (Table 4, and supplementary Figure S4). Next we introduced the P3/P4 element of compound 22 into compound 32 to obtain compound 34 (Figure 3e), hence preserving selectivity (Table 4) and reducing the tPSA of the molecule (tPSA values 197 Å and 140 Å for compound 32 and compound 34, respectively). We were able to separate the two diasteroisomers of this agent differing for chirality at the pyrrolidinyl moiety. Testing these two agents, compound 34 and 34*, against XIAP BIR3 using SDS gel clearly revealed that only one agent, compound 34, but not its diasteroisomer compound 34*, formed a covalent adduct with the protein (Figure 3f). Moreover, to further establish Lys311 as the residue targeted by these covalent agents we produced single mutant proteins in which either Lys311 or the nearby Lys322 (Figure 3) are mutated to Glu and Ala, respectively. SDS gel electrophoresis with these proteins in the absence and presence of compound 34 revealed indeed a covalent adduct only with wt BIR3 and for the Lys322Ala mutant, that preserved Lys311, whereas no covalent adduct was observed with the Lys311Glu mutant (Figure 3i). Finally, in similar SDS gel electrophoresis experiments, we also noted that covalent adduct formation occurs only between compound 34 and XIAP BIR3, but not with the BIR3 domains of cIAP1 or cIAP2 (Figure 3j). IC50 values for compound 34 against the panel of BIR3 domains revealed indeed that this molecule is very potent and selective against BIR3 XIAP and compared to cIAP1 and cIAP2 (Figure 3h, Table 4). Accordingly, mutating Lys311 with a glutamic acid in BIR3 of XIAP resulted in a drop in affinity for compound 34, while the activity is unaffected by the mutation of Lys322 (Figure 3k). Hence, the thermodynamic driven approach has identified two classes of possible novel antagonists: compound 31, a novel pan-IAP inhibitor, and compound 34, a covalent XIAP BIR3 inhibitor. Of note and as expected, the activity of compound 31 was not affected by mutating Lys311 or Lys322 (supplementary Figure S2). Perhaps another advantage of the covalent agents is that unlike the charged reversible agents such as compound 1 that possess a relatively large tPSA (197 Å2), replacing the charged moiety with an electrophile reduced the polar surface area, as for example in compound 34 (tPSA = 140 Å2), presumably increasing cell permeability.57 Finally, compound 31 and LCL161 have both a similar tPSA value of 91 Å.
Currently the most advanced agent for these targets is the Novartis clinical candidate LCL161 that in our DELFIA assay presents IC50 values in displacing a reference AVPI peptide of 52.7, 10.4, and 12.9 nM against the BIR3 domains of XIAP, cIAP1, and cIAP2, respectively. The agent is currently in clinical trials for various indications including multiple myeloma and pancreatic cancer. The activity of this agent in multiple myeloma is not fully understood, but it is suspected to be mainly due to its ability to activate an immune response as a consequence to cIAP1/2 inhibition, rather than sensitizing cancer cells to apoptosis via the XIAP inhibition.10, 58 LCL161 is not active against several cell lines and for example it showed limited in vitro and in vivo activity as a single agent against childhood cancer preclinical models.59 Accordingly, the acute lymphoblastic leukemia (ALL; the most common type of childhood cancer) cell line MOLT-4 was reported to be insensitive to the agent.60 Likewise, several MM cell lines were tested in various laboratories, and many of these were resistant to LCL161.61 Because of the different binding and selectivity profiles of our agents, we sought to test them side by side with LCL161 in these cell lines. When testing agents 31 and 34 side by side with and LCL161 against MOLT-4 we noticed that while the LCL161 is very effective in inducing cIAP1 and cIAP2 degradation (Figure 4b), the agent is less potent than both compound 34 and compound 31 in suppressing cell viability (Figure 4a). This may be perhaps attributable to an increased affinity of our agents for the XIAP BIR3, although our data are still speculative in this regards and further investigations will be needed. Likewise, when profiling the agents against a panel of MM cell lines, both LCL161-sensitive (H929 and L363; Figure 4c), and LCL161-resistant (RMPI, LP1, and U266; Figure 4c), we observed again that our compound 31 and compound 34 inhibited cell proliferation in these cell lines equally well (Figure 4c), while only compound 34 was effective against the MMS1 cell line. In all experiments, the less active enantiomer of compound 34 (namely compound 34*) was not effective, possibly ruling out non-specific cell killing effects due to the electrophile.
LCL161 is currently in clinical trials against advanced pancreatic cancers in combination with Abraxane and gemcitabine (https://clinicaltrials.gov/ct2/show/NCT01934634). To assess if our agents could enhance gemcitabine (GEM) activity in both GEM-sensitive BxPC3 and MIA PaCa-2 cell lines (Figure 4e), and the GEM-resistant PANC-1 cell line (Figure 4e), we tested our agents in combination. For the GEM-sensitive MIA-PaCa-2 cell line, we found at best a modest significant additive effect for only compound 31 (Figure 4e). However, when compound 34 or compound 31 were used in combination with cell lines PANC-1 and BxPC3, both expressing all 3 IAPs (Figure 4d), we observed a significant synergism, with compound 31 producing the most remarkable effect against the GEM-resistant PANC-1 cell line (Figure 4e).
Obviously the complex interplay between expression and regulation of the three oncogenes and the different activity of LCL161 compared to our agents can result in one or the other molecule to perform better against certain cell lines or situations (single agent versus combinations). Hence, the full potential of our agents in oncology and for other indications such as pulmonary fibrosis,62 has yet to be fully determined and it will require additional cellular mechanistic studies followed by detailed in vivo pharmacology and efficacy studies, and likely involving further optimizations including evaluating various war-heads for compound 34, exploring further P3/P4 substituents both compound 34 and compound 31, or obtaining homo- or hetero-dimeric versions of these agents.33, 35, 51, 63 Nonetheless, we feel that our work presents several novel insights not only into the inhibition of this important class of targets, but also into the use of thermodynamic parameters to guide the hit-to-lead optimization process, and in targeting Lys residues with covalent agents. Our discoveries and considerations are likely of general applicability to other targets, and in particular those involving protein-protein interactions (PPIs) where ligands of peptide or peptide mimetic nature can be designed in a modular fashion. Given that PPIs represent a largely untapped target space, we believe that our studies provide novel insights into possible effective strategies to guide the identification and the optimization of potent and selective agents against this challenging class of drug targets.
Experimental Section
General Chemistry.
Solvent and reagents were commercially obtained and used without further purification. NMR spectra used to check concentration were recorded on Bruker Avance III 700MHz. High-resolution mass spectral data were acquired on an Agilent LC-TOF instrument. RP-HPLC purifications were performed on a JASCO preparative system equipped with a PDA detector and a fraction collector controlled by a ChromNAV system (JASCO) on a Luna C18 10μ 10 × 250mm (Phenomenex) to > 95% purity. RP-chromatography purification for intermediates was performed using a CombiFlash Rf (Teledyne ISCO). LCL161 was obtained from MedChem Express.
General Peptide Synthesis
Peptides were synthesized by using standard solid-phase synthesis protocols (Innopep, Inc., San Diego) or using standard microwave-assisted Fmoc peptide synthesis protocols with a Liberty Blue Peptide Synthesizer (CEM). For each coupling reaction, 6 eq. of Fmoc-AA, 3 eq. of DIC and 1 eq. of OximaPure in 4.5 ml of DMF were used. The coupling reaction was allowed to proceed for 5 min at 90°C. Fmoc deprotection was performed by treating the resin-bound peptide with 20% piperidine in DMF (2×3ml) for 3 min at 90°C. Peptides were cleaved from Rink amide resin with a cleavage cocktail containing TFA/TIS/water/phenol (94:2:2:2) for 3 h. The cleaving solution was filtered from the resin, evaporated under reduced pressure and the peptides precipitated in Et2O, centrifuged and dried in high vacuum. The crude peptide was purified by preparative RP-HPLC using a Luna C18 column (Phenomenex) and water/acetonitrile gradient (5% to 70%) containing 0.1% TFA. The final compounds were characterized by HRMS. Detailed experimental procedures and analytical data for key compounds are provided as supplementary information.
Protein expression and purification
For the expression of XIAP BIR3, a pET15b vector encoding for the human BIR3 domain of XIAP fragment (residues 253–347) and an N-terminal His tag was transformed into E. coli BL21(DE3) Gold cells. The transformed cells were transferred to LB medium at 37°C with 100 μg/L of ampicillin until reaching an OD600 of 0.6 – 0.7, followed by induction with 1 mM IPTG overnight at 25°C. Bacteria were collected and lysed by sonication at 4°C. The overexpressed protein was purified using Ni2+ affinity chromatography. The buffer of the eluted protein was exchanged with a desalting column into an aqueous buffer composed of 50 mM MES pH = 6.0, 100 mM NaCl, 50 μM Zn(Ac)2, and 1 mM DTT. The BIR3 domain of XIAP where the Lys 311 was mutated to Glu (K311E), was expressed in the same way described previously; while the BIR3 domain of XIAP where the Lys 322 was mutated to Ala (K322A), was expressed as previously described but after Ni2+ affinity chromatography the buffer of the protein was exchange with a desalting column in 25 mM TRIS pH = 7.5, 300 mM NaCl, 50 μM Zn(Ac)2, and 1 mM DTT. The recombinant BIR3 domains of cIAP1 and cIAP2 with N-terminal 6xHis tag were obtained from Reaction Biology Corp. (Malvern, PA).
ITC measurements
Isothermal titration calorimetry measurements were performed using the Affinity ITC Autosampler from TA Instruments (New Castle, DE). The titrations were performed in a reverse fashion by titrating the protein into the ligand solution. All the measurements were performed at 25°C dissolving the agents in buffer 50 mM MES, pH = 6.0, 100 mM NaCl, 50 μM Zn(Ac)2, and 1 mM DTT, and a final DMSO concentration of 1 %. The syringe was filled with a 200 μM solution of XIAP BIR3 domain and 15 injections of 2.5 μL each were performed into the cell containing a 25 μM solution of the compounds. The injections were made at a 200-second interval with a stirring speed of 75 rpm. All the solutions were kept in the autosampler at 4°C in two different 96-well plates for the reaction cell solutions and syringe solutions, respectively. The volume of the reaction cell is 180 μL, but 630 μL were loaded as an excess volume is needed for the cell conditioning and to avoid the introduction of air. The analysis of the thermodynamics signatures and for dissociation constant determination was performed by the NanoAnalyze software (TA Instruments, New Castle, DE), and subsequently exported into Microsoft Excel.
Gel Electrophoresis
10 μM of each protein were incubated for 10 min with 20 μM of each compound in a buffer composed of 25 mM TRIS at pH 8, 150 mM NaCL, 50 μM zinc acetate, and 1 mM DTT. Samples were subjected to gel electrophoresis with SDS-PAGE gel using the NuPAGE 12% bis-tris mini gels (Life Technologies), MES as running buffer, and were stained with SimplyBlue SafeStain (Life Technologies) according to the manufacture’s protocol.
Cell lines and antibodies
Human Acute lymphoblastic leukemia, pancreatic cancer cell lines, and multiple myeloma cell lines were obtained from the American Type Culture Collection (ATCC; www.atcc.org) : MOLT-4 (ATCC® CRL-1582™), BxPC-3 (ATCC® CRL-1687™), PANC-1 (ATCC® CRL-1469™), MIA PaCa-2 (ATCC® CRL-1420™), MM.1S (ATCC® CRL-2974™), RPMI 8226 (ATCC® CCL-155™), U266B1 [U266] (ATCC® TIB-196™), NCI-H929 [H929] (ATCC® CRL-9068™), and from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (DSMZ; www.dsmz.de) : LP-1 (ACC 41), L363 (ACC 49). Cells were cultured according to standard mammalian tissue culture protocols, and sterile technique in RPMI medium 1640 with or DMEM L-glutamine supplemented with 10% fetal bovine serum, 100 units/ml penicillin/100 μg/ml streptomycin. Primary antibody XIAP (Cat. No. 2045), cIAP1 (Cat. No. 7065), and cIAP2 (Cat. No3130) were purchased from Cell Signaling Technology and diluted at 1:1000 concentration. β-actin antibody (Santa Cruz Biotechnologies) was used as a loading control.
MTS assays
MM.1S, U266, L363, H929, LP1, RPMI cells were seeded on 96-well plates in three replicates at 100 μl/well (2.5×105cells/ml) in growing medium and exposed to 20 μM) of different chemical compounds. The effects of the drugs on growth inhibition were measured at 48 h. At the above indicated time points, 20 μl of MTS, (Promega Corporation, Madison, WI CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay), was added to each well, and the plates were incubated for 1–4 h at 37°C in a humidified, 5% CO2 atmosphere. The absorbance was measured in a microtiter plate reader at 492 nm. The ratio of detection reagents to cell culture was selected according to recommendations of a commercially available test kit.
Cell Proliferation assays
On day one, MOLT-4 cells were collected and resuspended in serum-free OPTI-MEM supplemented with 1% Penicillin-Streptomycin, and they were seeded at 20 × 10^3 cells per well in 96-well plates. Compounds or DMSO were added to treated or control wells, respectively and every well had 1% of DMSO. Cells were further incubated for 48 h in a cell culture incubator.
Pancreatic cancer cells co-treatment with gemcitabine (GEM) and IAP inhibitors.
Pancreatic cancer cells were plated at 30 × 10^3 cells per well in 96-well plates. The next day, cells were treated with different concentrations of GEM. After 24-h incubation, media was removed and replenished with the same GEM concentration alone or with 15 μM of IAP inhibitors in serum-free media and cells were further incubated for 24 h.
Cell proliferation assay was determined using ATPlite 1Step Luminescence Assay System (PerkinElmer) according to the manufacturer’s instructions, and luminescence was measured by VICTOR X5 microplate reader (PerkinElmer). Finally, data was plotted, and IC50 values were calculated using Prism GraphPad version 7. IC50 is the concentration of compound that inhibits 50% growth of the treated cells compared to control wells. This experiment was repeated three times, and each concentration was tested in triplicate.
Immunoblot study
Cells were collected and lysed with lysis buffer (20 mM Tris, pH 7.4, 120 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1% IGEPAL, 5 mM EDTA) supplemented with EDTA-free Protease Inhibitor Cocktail and PhosStop (Sigma-Aldrich) for 10 min on cold ice. Lysates were centrifuged and supernatants were collected. Protein content was quantified and samples were prepared using NuPAGE antioxidant and LDS sample buffer (ThermoFisher) and heated for 10 min at 70°C. Each sample containing 16 μg of proteins were loaded into 4–12% NuPAGE Bis-Tris precast gels and transferred to PVDF membranes. The membranes were blocked with 5% milk in TBS and 0.1% Tween (TBST) and incubated with primary antibodies overnight at 4°C. Next day, the membrane was washed with TBST and incubated with goat anti-mouse HRP secondary antibodies. The antigen-antibody complexes were visualized using a Clarity Western ECL kit (BIO-RAD).
Molecular modeling
Compounds N-Me-AVPF-NH2, compound 1, compound 32, LCL161, and compound 34 were docked using Gold [Cambridge Crystallographic Data Center (www.ccdc.cam.ac.uk)] and Protein Data Bank entry 2OPZ. The docking preparation for both protein and ligands were performed using SYBYL-X 2.1.1 (Certara, Princeton, NJ). The surface figures were prepared using MOLCAD as implemented in SYBYL-X 2.1.1.
DELFIA (Dissociation-Enhanced Lanthanide Fluorescent Immunoassay)
A solution containing 100 μL of 100 nM AVPI-Biotin (AVPIAQKSEK-Biotin) was added to each well of the 96-well streptavidin-coated plates (PerkinElmer) and incubated for 1 h, followed by three washing steps to remove the unbound AVPI-Biotin. Subsequently, 89 μL of 1.56 nM (for XIAP BIR3 and cIAP1 BIR3) or 2.08 nM (for cIAP2 BIR3) solutions of Eu-N1-labeled anti-6xHis antibody (PerkinElmer) and a mixture containing 11 μL of the protein and a serial dilution of the test compounds were added to each well. Following 1 h of incubation, the unbound protein-Eu antibody complexes, which were displaced by a test compound, were eliminated through the second washing step and 200 μL of the DELFIA enhancement solution (PerkinElmer) was then added to each well and incubated for 10 min. The fluorescence was measured using the VICTOR X5 microplate reader (PerkinElmer) with excitation and emission wavelengths of 340 and 615 nm, respectively. The final protein concentrations were 30 nM for XIAP BIR3 and cIAP1 BIR3, and 15 nM for cIAP2 BIR3. The final antibody concentrations used for XIAP BIR3 and cIAP1 BIR3 was 22.2 ng/well and 29.7 ng/well for cIAP2 BIR3. DELFIA assay buffer (PerkinElmer) was used to prepare the protein, peptide and antibody solutions and the incubations were done at room temperature. All of the samples were normalized to 1% DMSO and reported as % inhibition. The IC50 values were calculated by GraphPad Prism version 7.
Supplementary Material
Acknowledgements
Financial support was obtained in part by the NIH, with NCI grant CA168517 (to MP) and a City of Hope - UC Riverside Biomedical Research Initiative (CUBRI) grant (to MP and FP). MP holds the Daniel Hays Chair in Cancer Research at the School of Medicine at UCR. PU is a recipient of the 2017-2018 Pease Cancer Fellowship through the Division of Biomedical Sciences, School of Medicine at UCR. Our agents can be distributed in small amounts (1-5mg) for research purposes upon request and signing of a standard material transfer agreement.
Abbreviations used
- IAP
Inhibitor of Apoptosis Proteins
- XIAP
X-linked Inhibitor of Apoptosis Protein
- BIR
baculovirus IAP repeat domains
- cIAP1
cellular Inhibitor of Apoptosis Protein 1
- cIAP2
cIAP1: cellular Inhibitor of Apoptosis Protein 2
- TNF
tumor necrosis factor
- TRAF2
tumor necrosis factor receptor-associated factor 2
- ITC
Isothermal titration calorimetry
- DELFIA
Dissociation-Enhanced Lanthanide Fluorescent Immunoassay
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website.
Experimental synthetic procedures. Figure S1 contains the Chemical structures of 32 compounds synthesized to probe for P3/P4 substituents in NMe-Ala-Val-P3/P4. Figure S2 reports dose-response curves in DELFIA displacement assays for compound 31 against XIAP BIR3, XIAP BIR3 K311E, and XIAP BIR3 K322A, respectively. Figure S3 reports ITC curve for compounds AT-406 and GDC-0152. Figure S4 reports the LC-MS spectra of the BIR3 domain of XIAP in the absence and presence of compound 33. Table S1 reports IC50 values for selected compounds against the BIR2 domain of XIAP.
The authors declare no competing financial interest
References
- 1.Deveraux QL; Reed JC IAP family proteins--suppressors of apoptosis. Genes Dev 1999, 13, 239–252. [DOI] [PubMed] [Google Scholar]
- 2.Salvesen GS; Duckett CS IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol 2002, 3, 401–410. [DOI] [PubMed] [Google Scholar]
- 3.Holcik M; Gibson H; Korneluk RG XIAP: apoptotic brake and promising therapeutic target. Apoptosis 2001, 6, 253–261. [DOI] [PubMed] [Google Scholar]
- 4.Gyrd-Hansen M; Meier P IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer 2010, 10, 561–574. [DOI] [PubMed] [Google Scholar]
- 5.Samuel T; Welsh K; Lober T; Togo SH; Zapata JM; Reed JC Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem 2006, 281, 1080–1090. [DOI] [PubMed] [Google Scholar]
- 6.Varfolomeev E; Blankenship JW; Wayson SM; Fedorova AV; Kayagaki N; Garg P; Zobel K; Dynek JN; Elliott LO; Wallweber HJ; Flygare JA; Fairbrother WJ; Deshayes K; Dixit VM; Vucic D IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [DOI] [PubMed] [Google Scholar]
- 7.Vince JE; Wong WW; Khan N; Feltham R; Chau D; Ahmed AU; Benetatos CA; Chunduru SK; Condon SM; McKinlay M; Brink R; Leverkus M; Tergaonkar V; Schneider P; Callus BA; Koentgen F; Vaux DL; Silke J IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007, 131, 682–693. [DOI] [PubMed] [Google Scholar]
- 8.Lopes RB; Gangeswaran R; McNeish IA; Wang Y; Lemoine NR Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer 2007, 120, 2344–2352. [DOI] [PubMed] [Google Scholar]
- 9.Mizutani Y; Nakanishi H; Li YN; Matsubara H; Yamamoto K; Sato N; Shiraishi T; Nakamura T; Mikami K; Okihara K; Takaha N; Ukimura O; Kawauchi A; Nonomura N; Bonavida B; Miki T Overexpression of XIAP expression in renal cell carcinoma predicts a worse prognosis. Int J Oncol 2007, 30, 919–925. [PubMed] [Google Scholar]
- 10.Nakagawa Y; Abe S; Kurata M; Hasegawa M; Yamamoto K; Inoue M; Takemura T; Suzuki K; Kitagawa M IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes. Am J Hematol 2006, 81, 824–831. [DOI] [PubMed] [Google Scholar]
- 11.Tamm I; Kornblau SM; Segall H; Krajewski S; Welsh K; Kitada S; Scudiero DA; Tudor G; Qui YH; Monks A; Andreeff M; Reed JC Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res 2000, 6, 1796–1803. [PubMed] [Google Scholar]
- 12.Mannhold R; Fulda S; Carosati E IAP antagonists: promising candidates for cancer therapy. Drug Discov Today 2010, 15, 210–219. [DOI] [PubMed] [Google Scholar]
- 13.Fulda S Inhibitor of apoptosis proteins as targets for anticancer therapy. Expert Rev Anticancer Ther 2007, 7, 1255–1264. [DOI] [PubMed] [Google Scholar]
- 14.Fulda S; Vucic D Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 2012, 11, 109–124. [DOI] [PubMed] [Google Scholar]
- 15.LaCasse EC; Mahoney DJ; Cheung HH; Plenchette S; Baird S; Korneluk RG IAP-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [DOI] [PubMed] [Google Scholar]
- 16.Vucic D; Fairbrother WJ The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res 2007, 13, 5995–6000. [DOI] [PubMed] [Google Scholar]
- 17.Huang JW; Zhang Z; Wu B; Cellitti JF; Zhang X; Dahl R; Shiau CW; Welsh K; Emdadi A; Stebbins JL; Reed JC; Pellecchia M Fragment-based design of small molecule X-linked inhibitor of apoptosis protein inhibitors. J Med Chem 2008, 51, 7111–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du C; Fang M; Li Y; Li L; Wang X Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [DOI] [PubMed] [Google Scholar]
- 19.Shiozaki EN; Shi Y Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends Biochem Sci 2004, 29, 486–494. [DOI] [PubMed] [Google Scholar]
- 20.Verhagen AM; Ekert PG; Pakusch M; Silke J; Connolly LM; Reid GE; Moritz RL; Simpson RJ; Vaux DL Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y; Rich RL; Myszka DG; Wu H Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac. J Biol Chem 2003, 278, 49517–49522. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z; Sun C; Olejniczak ET; Meadows RP; Betz SF; Oost T; Herrmann J; Wu JC; Fesik SW Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 2000, 408, 1004–1008. [DOI] [PubMed] [Google Scholar]
- 23.Wu G; Chai J; Suber TL; Wu JW; Du C; Wang X; Shi Y Structural basis of IAP recognition by Smac/DIABLO. Nature 2000, 408, 1008–1012. [DOI] [PubMed] [Google Scholar]
- 24.Cai Q; Sun H; Peng Y; Lu J; Nikolovska-Coleska Z; McEachern D; Liu L; Qiu S; Yang CY; Miller R; Yi H; Zhang T; Sun D; Kang S; Guo M; Leopold L; Yang D; Wang S A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem 2011, 54, 2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen F; Alicke B; Elliott LO; Flygare JA; Goncharov T; Keteltas SF; Franklin MC; Frankovitz S; Stephan JP; Tsui V; Vucic D; Wong H; Fairbrother WJ Orally bioavailable antagonists of inhibitor of apoptosis proteins based on an azabicyclooctane scaffold. J Med Chem 2009, 52, 1723–1730. [DOI] [PubMed] [Google Scholar]
- 26.Flygare JA; Beresini M; Budha N; Chan H; Chan IT; Cheeti S; Cohen F; Deshayes K; Doerner K; Eckhardt SG; Elliott LO; Feng B; Franklin MC; Reisner SF; Gazzard L; Halladay J; Hymowitz SG; La H; LoRusso P; Maurer B; Murray L; Plise E; Quan C; Stephan JP; Young SG; Tom J; Tsui V; Um J; Varfolomeev E; Vucic D; Wagner AJ; Wallweber HJ; Wang L; Ware J; Wen Z; Wong H; Wong JM; Wong M; Wong S; Yu R; Zobel K; Fairbrother WJ Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J Med Chem 2012, 55, 4101–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaither A; Porter D; Yao Y; Borawski J; Yang G; Donovan J; Sage D; Slisz J; Tran M; Straub C; Ramsey T; Iourgenko V; Huang A; Chen Y; Schlegel R; Labow M; Fawell S; Sellers WR; Zawel L A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res 2007, 67, 11493–11498. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto K; Saito B; Miyamoto N; Oguro Y; Tomita D; Shiokawa Z; Asano M; Kakei H; Taya N; Kawasaki M; Sumi H; Yabuki M; Iwai K; Yoshida S; Yoshimatsu M; Aoyama K; Kosugi Y; Kojima T; Morishita N; Dougan DR; Snell GP; Imamura S; Ishikawa T Design and synthesis of potent inhibitor of apoptosis (IAP) proteins antagonists bearing an octahydropyrrolo[1,2-a]pyrazine scaffold as a novel proline mimetic. J Med Chem 2013, 56, 1228–1246. [DOI] [PubMed] [Google Scholar]
- 29.Li L; Thomas RM; Suzuki H; De Brabander JK; Wang X; Harran PG A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 2004, 305, 1471–1474. [DOI] [PubMed] [Google Scholar]
- 30.Ndubaku C; Varfolomeev E; Wang L; Zobel K; Lau K; Elliott LO; Maurer B; Fedorova AV; Dynek JN; Koehler M; Hymowitz SG; Tsui V; Deshayes K; Fairbrother WJ; Flygare JA; Vucic D Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS Chem Biol 2009, 4, 557–566. [DOI] [PubMed] [Google Scholar]
- 31.Oost TK; Sun C; Armstrong RC; Al-Assaad AS; Betz SF; Deckwerth TL; Ding H; Elmore SW; Meadows RP; Olejniczak ET; Oleksijew A; Oltersdorf T; Rosenberg SH; Shoemaker AR; Tomaselli KJ; Zou H; Fesik SW Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 2004, 47, 4417–4426. [DOI] [PubMed] [Google Scholar]
- 32.Peng Y; Sun H; Nikolovska-Coleska Z; Qiu S; Yang CY; Lu J; Cai Q; Yi H; Kang S; Yang D; Wang S Potent, orally bioavailable diazabicyclic small-molecule mimetics of second mitochondria-derived activator of caspases. J Med Chem 2008, 51, 8158–8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheng R; Sun H; Liu L; Lu J; McEachern D; Wang G; Wen J; Min P; Du Z; Lu H; Kang S; Guo M; Yang D; Wang S A potent bivalent Smac mimetic (SM-1200) achieving rapid, complete, and durable tumor regression in mice. J Med Chem 2013, 56, 3969–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun H; Lu J; Liu L; Yi H; Qiu S; Yang CY; Deschamps JR; Wang S Nonpeptidic and potent small-molecule inhibitors of cIAP-1/2 and XIAP proteins. J Med Chem 2010, 53, 6361–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun H; Nikolovska-Coleska Z; Lu J; Meagher JL; Yang CY; Qiu S; Tomita Y; Ueda Y; Jiang S; Krajewski K; Roller PP; Stuckey JA; Wang S Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc 2007, 129, 15279–15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun H; Nikolovska-Coleska Z; Lu J; Qiu S; Yang CY; Gao W; Meagher J; Stuckey J; Wang S Design, synthesis, and evaluation of a potent, cell-permeable, conformationally constrained second mitochondria derived activator of caspase (Smac) mimetic. J Med Chem 2006, 49, 7916–7920. [DOI] [PubMed] [Google Scholar]
- 37.Sun H; Nikolovska-Coleska Z; Yang CY; Qian D; Lu J; Qiu S; Bai L; Peng Y; Cai Q; Wang S Design of small-molecule peptidic and nonpeptidic Smac mimetics. Acc Chem Res 2008, 41, 1264–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun H; Nikolovska-Coleska Z; Yang CY; Xu L; Liu M; Tomita Y; Pan H; Yoshioka Y; Krajewski K; Roller PP; Wang S Structure-based design of potent, conformationally constrained Smac mimetics. J Am Chem Soc 2004, 126, 16686–16687. [DOI] [PubMed] [Google Scholar]
- 39.Sun H; Nikolovska-Coleska Z; Yang CY; Xu L; Tomita Y; Krajewski K; Roller PP; Wang S Structure-based design, synthesis, and evaluation of conformationally constrained mimetics of the second mitochondria-derived activator of caspase that target the X-linked inhibitor of apoptosis protein/caspase-9 interaction site. J Med Chem 2004, 47, 4147–4150. [DOI] [PubMed] [Google Scholar]
- 40.Sun H; Stuckey JA; Nikolovska-Coleska Z; Qin D; Meagher JL; Qiu S; Lu J; Yang CY; Saito NG; Wang S Structure-based design, synthesis, evaluation, and crystallographic studies of conformationally constrained Smac mimetics as inhibitors of the X-linked inhibitor of apoptosis protein (XIAP). J Med Chem 2008, 51, 7169–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun W; Nikolovska-Coleska Z; Qin D; Sun H; Yang CY; Bai L; Qiu S; Wang Y; Ma D; Wang S Design, synthesis, and evaluation of potent, nonpeptidic mimetics of second mitochondria-derived activator of caspases. J Med Chem 2009, 52, 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S Design of small-molecule Smac mimetics as IAP antagonists. Curr Top Microbiol Immunol 2011, 348, 89–113. [DOI] [PubMed] [Google Scholar]
- 43.Zhang B; Nikolovska-Coleska Z; Zhang Y; Bai L; Qiu S; Yang CY; Sun H; Wang S; Wu Y Design, synthesis, and evaluation of tricyclic, conformationally constrained small-molecule mimetics of second mitochondria-derived activator of caspases. J Med Chem 2008, 51, 7352–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zobel K; Wang L; Varfolomeev E; Franklin MC; Elliott LO; Wallweber HJ; Okawa DC; Flygare JA; Vucic D; Fairbrother WJ; Deshayes K Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem Biol 2006, 1, 525–533. [DOI] [PubMed] [Google Scholar]
- 45.Hennessy EJ; Saeh JC; Sha L; MacIntyre T; Wang H; Larsen NA; Aquila BM; Ferguson AD; Laing NM; Omer CA Discovery of aminopiperidine-based Smac mimetics as IAP antagonists. Bioorg Med Chem Lett 2012, 22, 1690–1694. [DOI] [PubMed] [Google Scholar]
- 46.Sun H; Lu J; Liu L; Yang CY; Wang S Potent and selective small-molecule inhibitors of cIAP1/2 proteins reveal that the binding of Smac mimetics to XIAP BIR3 is not required for their effective induction of cell death in tumor cells. ACS Chem Biol 2014, 9, 994–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donnell AF; Michoud C; Rupert KC; Han X; Aguilar D; Frank KB; Fretland AJ; Gao L; Goggin B; Hogg JH; Hong K; Janson CA; Kester RF; Kong N; Le K; Li S; Liang W; Lombardo LJ; Lou Y; Lukacs CM; Mischke S; Moliterni JA; Polonskaia A; Schutt AD; Solis DS; Specian A; Taylor RT; Weisel M; Remiszewski SW Benzazepinones and benzoxazepinones as antagonists of inhibitor of apoptosis proteins (IAPs) selective for the second baculovirus IAP repeat (BIR2) domain. J Med Chem 2013, 56, 7772–7787. [DOI] [PubMed] [Google Scholar]
- 48.Baggio C; Udompholkul P; Barile E; Pellecchia M Enthalpy-based screening of focused combinatorial libraries for the identification of potent and selective ligands. ACS Chem Biol 2017, 12, 2981–2989. [DOI] [PubMed] [Google Scholar]
- 49.Schon A; Freire E Enthalpy screen of drug candidates. Anal Biochem 2016, 513, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fox JM; Zhao M; Fink MJ; Kang K; Whitesides GM The molecular origin of enthalpy/entropy compensation in biomolecular recognition. Annu Rev Biophys 2018, 47, 223–250. [DOI] [PubMed] [Google Scholar]
- 51.Flygare JA; Fairbrother WJ Small-molecule pan-IAP antagonists: a patent review. Expert Opin Ther Pat 2010, 20, 251–267. [DOI] [PubMed] [Google Scholar]
- 52.Pettinger J; Jones K; Cheeseman MD Lysine-targeting covalent inhibitors. Angew Chem Int Ed Engl 2017, 56, 15200–15209. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Q; Ouyang X; Wan X; Gajiwala KS; Kath JC; Jones LH; Burlingame AL; Taunton J Broad-spectrum kinase profiling in live cells with lysine-targeted sulfonyl fluoride probes. J Am Chem Soc 2017, 139, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anscombe E; Meschini E; Mora-Vidal R; Martin MP; Staunton D; Geitmann M; Danielson UH; Stanley WA; Wang LZ; Reuillon T; Golding BT; Cano C; Newell DR; Noble ME; Wedge SR; Endicott JA; Griffin RJ Identification and characterization of an irreversible inhibitor of CDK2. Chem Biol 2015, 22, 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akcay G; Belmonte MA; Aquila B; Chuaqui C; Hird AW; Lamb ML; Rawlins PB; Su N; Tentarelli S; Grimster NP; Su Q Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol 2016, 12, 931–936. [DOI] [PubMed] [Google Scholar]
- 56.Hoppmann C; Wang L Proximity-enabled bioreactivity to generate covalent peptide inhibitors of p53-Mdm4. Chem Commun (Camb) 2016, 52, 5140–5143. [DOI] [PubMed] [Google Scholar]
- 57.Clark DE Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J Pharm Sci 1999, 88, 807–814. [DOI] [PubMed] [Google Scholar]
- 58.Chesi M; Mirza NN; Garbitt VM; Sharik ME; Dueck AC; Asmann YW; Akhmetzyanova I; Kosiorek HE; Calcinotto A; Riggs DL; Keane N; Ahmann GJ; Morrison KM; Fonseca R; Lacy MQ; Dingli D; Kumar SK; Ailawadhi S; Dispenzieri A; Buadi F; Gertz MA; Reeder CB; Lin Y; Chanan-Khan AA; Stewart AK; Fooksman D; Bergsagel PL IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med 2016, 22, 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Houghton PJ; Kang MH; Reynolds CP; Morton CL; Kolb EA; Gorlick R; Keir ST; Carol H; Lock R; Maris JM; Billups CA; Smith MA Initial testing (stage 1) of LCL161, a SMAC mimetic, by the pediatric preclinical testing program. Pediatr Blood Cancer 2012, 58, 636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hass C; Belz K; Schoeneberger H; Fulda S Sensitization of acute lymphoblastic leukemia cells for LCL161-induced cell death by targeting redox homeostasis. Biochem Pharmacol 2016, 105, 14–22. [DOI] [PubMed] [Google Scholar]
- 61.Ramakrishnan V; Painuly U; Kimlinger T; Haug J; Rajkumar SV; Kumar S Inhibitor of apoptosis proteins as therapeutic targets in multiple myeloma. Leukemia 2014, 28, 1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashley SL; Sisson TH; Wheaton AK; Kim KK; Wilke CA; Ajayi IO; Subbotina N; Wang S; Duckett CS; Moore BB; Horowitz JC Targeting inhibitor of apoptosis proteins protects from bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol 2016, 54, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu J; Bai L; Sun H; Nikolovska-Coleska Z; McEachern D; Qiu S; Miller RS; Yi H; Shangary S; Sun Y; Meagher JL; Stuckey JA; Wang S SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res 2008, 68, 9384–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.