

Abstract
Neuronal excitotoxicity is the major cause of alcohol-related brain damage, yet the underlying mechanism remains poorly understood. Using dopaminergic-like PC12 cells, we evaluated the effect of N-methyl-d-aspartate receptors (NMDAR) on acetate-induced changes in PC12 cells: cell death, cytosolic calcium, and expression levels of the pro-inflammatory cytokine tumor necrosis factor alpha (TNFα). Treatment of PC12 cells with increasing concentrations of acetate for 4 h caused a dose-dependent increase in the percentage of cells staining positive for cell death using propidium iodide (PI) exclusion and cytosolic reactive oxygen species (ROS) using cell ROX detection analyzed via flow cytometry. The EC50 value for acetate was calculated and found to be 4.40 mM for PI and 1.81 mM for ROS. Ethanol up to 100 mM had no apparent changes in the percent of cells staining positive for PI or ROS. Acetate (6 mM) treatment caused an increase in cytosolic calcium measured in real-time with Fluo-4AM, which was abolished by coapplication with the NMDAR blocker memantine (10 μM). Furthermore, cells treated with acetate (6 mM) for 4 h had increased expression levels of TNFα relative to control, which was abolished by coapplication of memantine (10 μM). Co-application of acetate (6 mM) and memantine had no apparent reduction in acetate-induced cell death. These findings suggest that acetate is capable of increasing cytosolic calcium concentrations and expression levels of the pro-inflammatory cytokine TNFα through an NMDAR-dependent mechanism. Cell death from acetate was not reduced through NMDAR blockade, suggesting alternative pathways independent of NMDAR activation for excitotoxicity.
Keywords: Excitotoxicity, calcium, flow cytometry, alcohol, NMDAR, ROS
Graphical Abstract
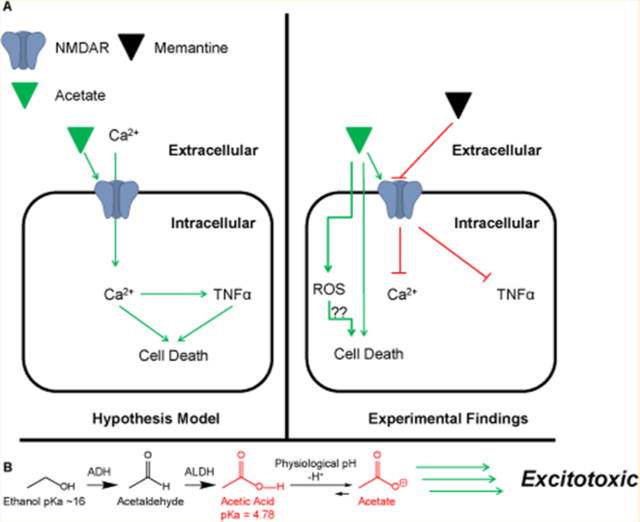
INTRODUCTION
Alcohol-induced neurodegeneration has been well-established in the literature for both chronic and/or binge models in rats1–4 and post-mortem in human brains.5,6 Precisely how alcohol consumption leads to the development of neurodegeneration is still undetermined; however, a majority of research suggests it is at least in part due to excitotoxicity through the N-methyl-d-aspartate receptor (NMDAR).4,7,8 The NMDAR is primarily a calcium-specific, glutamate ligand-gated ion channel, that when activated results in an influx of Ca2+ across the neuronal membrane, depolarizing the neuron. Large influxes of Ca2+ ions are capable of initiating many complex pathways, including apoptosis,9–11 the release of neurotransmitter,12–14 and an influx in large quantities can result in glutamate-induced excitotoxicity.4,10,11
With this in mind, the current consensus among alcohol researchers is that ethanol (EtOH) blocks the NMDAR channel, leading to an increase in NMDAR surface expression15,16 and, upon EtOH clearance, triggers NMDAR inward currents.4,17 Interestingly, a majority of the research has noted that the excitotoxic insult occurs mainly during the withdrawal/clearance phase.18,19 Because these rat models are in vivo models, a confounding factor is EtOH metabolism to acetic acid/acetate, which has yet to be accounted for. A recent study reported blood alcohol concentrations (BAC) and acetate concentrations following ethanol consumption. The major findings were that EtOH BAC and acetate concentrations rise rapidly during alcohol consumption followed by a sharp decline of EtOH BAC, which eventually returns to undetectable levels. Acetate concentrations, however, spike and then remain elevated at a steady state concentration for nearly 12–24 h post EtOH consumption.20,21
Circulating concentrations of acetate in the peripheral system following alcohol consumption have been reported in the range of 1–2 mM21 and substantially higher in the brain.22 Wang and colleagues reported acetate concentrations following EtOH infusion of 4.09 ± 0.24 mM with sustained steady state acetate concentrations of 3.51 ± 0.20 mM in unconditioned alcohol rats.22 This alcohol research data suggests that the main culprit that contributes to excitotoxicity may be the EtOH metabolite acetate.
Indeed, nonalcohol research has established that acetate contributes to an inflammatory response in humans,23,24 and acetic acid is toxic in human cell lines.25 The use of acetate in older hemodialysis solutions was found to increase tumor necrosis factor alpha (TNFα), endothelial nitric oxide synthase (eNOS),23 and interleukin 1-β (IL-1β)24 in humans. It was noted that many of the hemodialysis patients undergoing treatment with acetate-based solutions experienced headache, nausea, vomiting, extreme flushing, and vision disturbances.23,26 These symptoms are very similar to those experienced during an alcohol hangover, and at least in rats, acetate was suggested as being the causative factor.27 Additionally, Kendrick and colleagues reported acetate was the key inflammatory mediator in acute alcohol hepatitis,28 and our lab has reported that acetate microinjection into the central nucleus of the amygdala in anesthetized rats caused a sympathoexcitatory response that was primarily driven through activation of NMDAR.29 We therefore hypothesized that the EtOH metabolite acetate may be the primary contributing agent to excitotoxicity through NMDAR activation.
RESULTS AND DISCUSSION
Acetate Increases Cell Death in NGF-Derived PC12 Cells.
NGF-derived PC12 cells exposed to increasing concentrations of acetate (0, 2, 6, 15, 30 mM) for a duration of 4 h had a significantly (P < 0.05) greater percentage of cells staining positive for propidium iodide (PI) (26.5 ± 0.8, 26.5 ± 1.7, 33.3 ± 0.7, 35.5 ± 1.3, and 36.1 ± 1.9%, respectively) compared to NGF-derived PC12 cells treated without acetate (Figure. 1B). Because PI is a common stain used for DNA and only able to penetrate compromised cell membranes, it is a convenient and well-utilized method for examining cell death through increases in PI-stained cells.30 A dose-response curve was constructed correlating acetate concentrations (0, 2, 6, 15, and 30 mM) with increased percentage of PI staining (Figure 1C). From the dose-response curve, an EC50 value of 4.40 mM acetate was determined. In a separate experiment, we examined differences between control and acetate (4.40 and 6 mM). There was no significant difference between percentage of PI staining between acetate (4.40 and 6 mM); however, acetate (6 mM) significantly increased PI staining compared to control (N = 5, data not shown). Acetate (4.40 mM) increased PI staining (3 out of 5 groups, data not shown) compared to control and acetate (6 mM) (5 out of 5 groups, data not shown). We therefore determined 6 mM acetate to be the Emax value. All subsequent experiments utilized 6 mM acetate for investigation of pro-inflammatory cytokine mRNA expression, real-time calcium imaging, and NMDAR blockade. These cytotoxicity data suggest that the ethanol metabolite acetate is capable of increasing cell death in NGF-derived PC12 cells in a dose-dependent manner at physiologically relevant acetate concentrations post ethanol consumption.22
Figure 1.
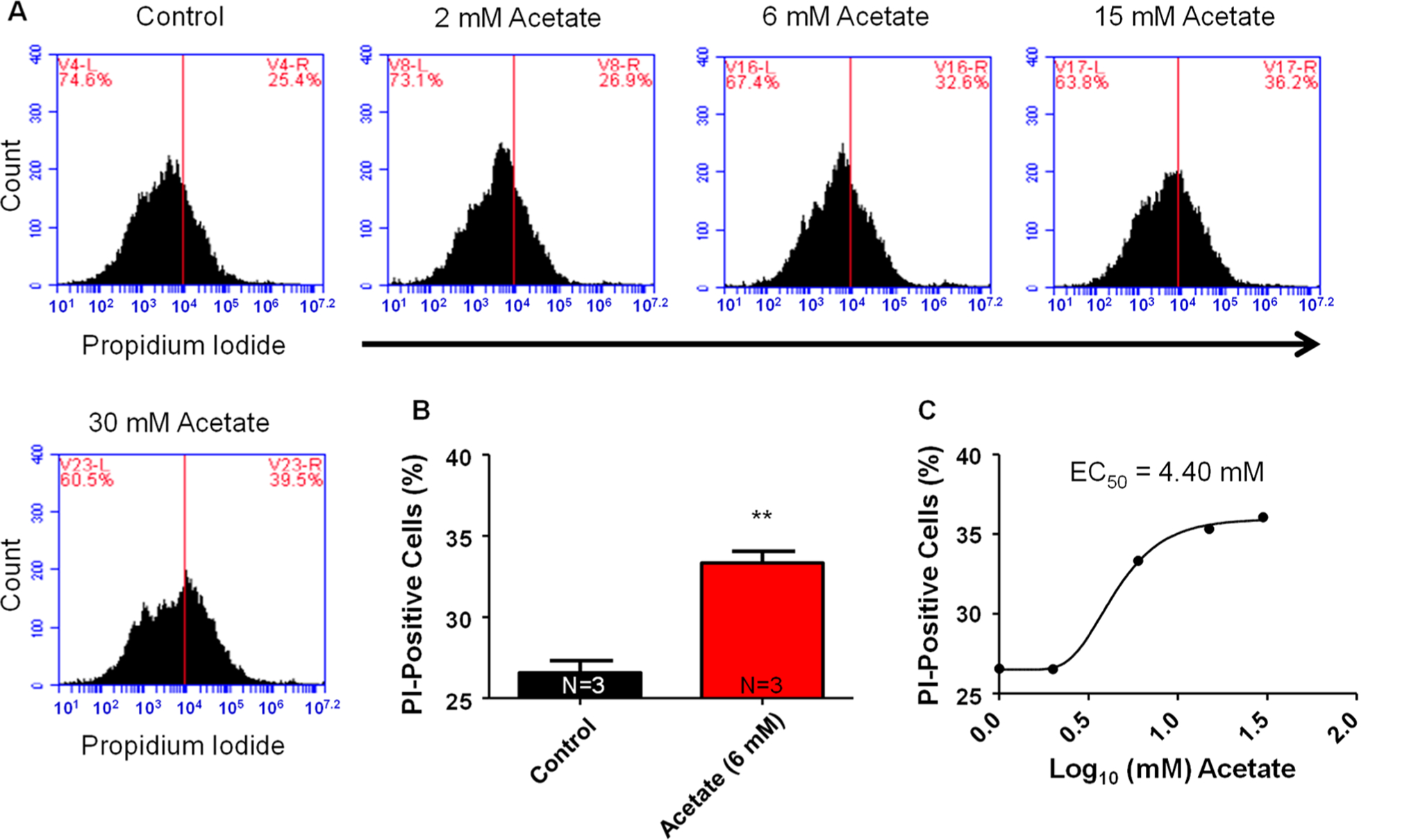
Acetate dose-dependent response to increases in cell death in PC12 cells. (A) Representative flow cytometry charts for varying concentrations of acetate (left to right: 0, 2, 6, 15, 30 mM) and the corresponding percentage of PC12 cells staining positive for PI. Control had a mean PI percentage of 26.6 ± 0.8%. All measurement settings were based off controls (red line on chart). (B) Bar graph summary data for control vs acetate (6 mM) on percentage of PI positive PC12 cells (**P < 0.01). (C) Dose-response curve of acetate in log10 concentration of acetate (mM). On the basis of our dose-dependent response, acetate had an EC50 value of 4.40 mM. (N = number of wells per treatment; each well analyzed for 10,000 events per well.)
Ethanol Has No Effect on Cell Death in NGF-Derived PC12 Cells.
NGF-derived PC12 cells treated with increasing concentrations of ethanol (0, 2, 6, 15, 50, and 100 mM) for 4 h had no increase in PI staining compared to NGF-derived PC12 cells without ethanol (35.8 ± 1.0, 36.2 ± 2, 36.6 ± 0.9, 38.1 ± 1.0, 36.6 ± 0.9, and 37.6 ± 1.4%), respectively (Figure 2B). This data suggests that, at least in dopaminergic-like PC12 cells, 4 h of ethanol exposure has no effect on any apparent increases in PI staining.
Figure 2.
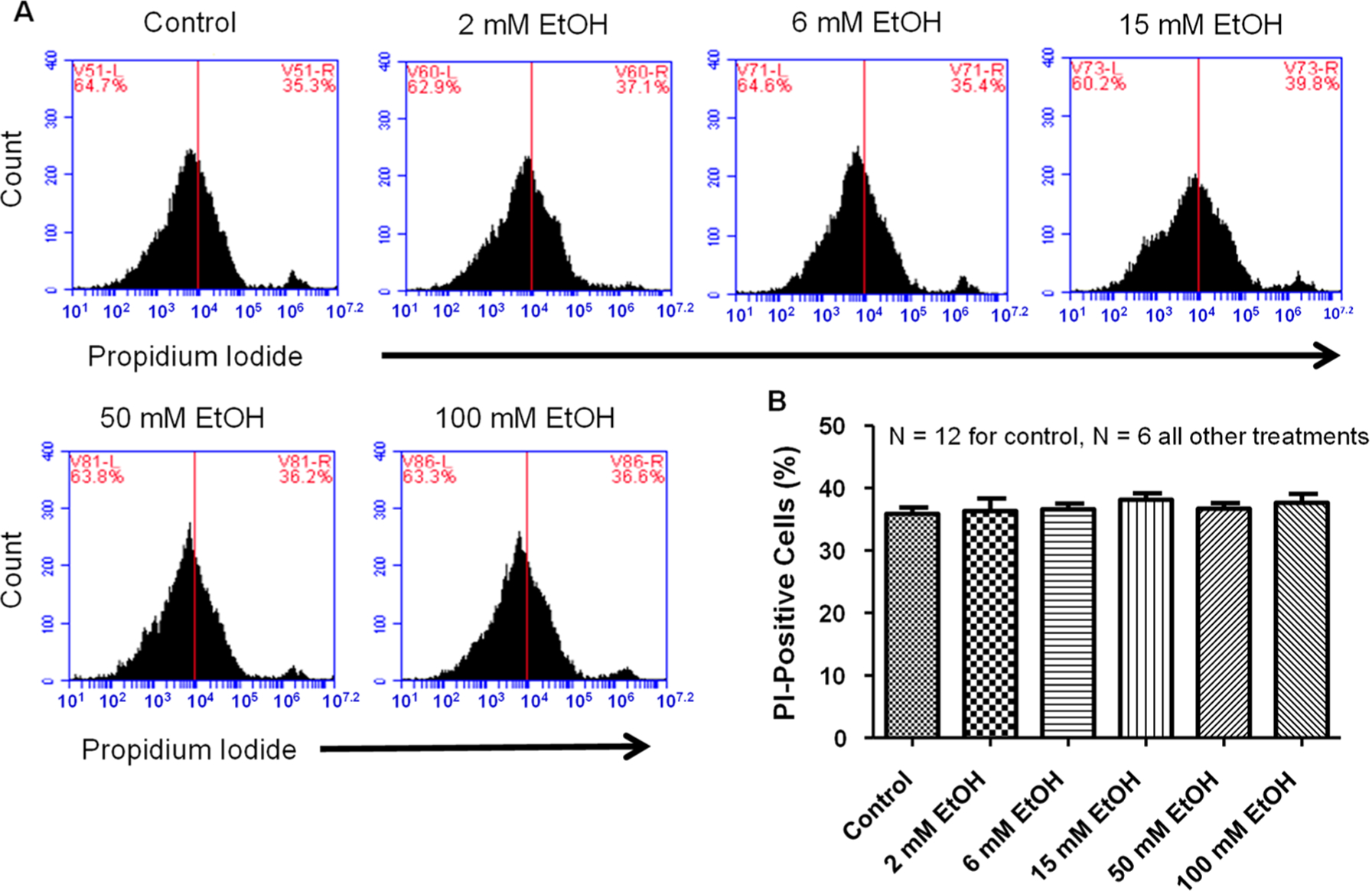
Ethanol has no effect on PC12 cell death. (A) Representative flow cytometry charts for varying concentrations of EtOH (left to right: 0, 2, 6, 15, 50, 100 mM). There was no difference observed between control and any doses of EtOH tested. (B) Summary data for varying concentrations of EtOH. No statistical difference (one-way ANOVA; F5,36 = 0.48) was noted between any EtOH concentrations and control in PC12 cells for PI staining. (N = number of wells per treatment; each well analyzed for 10,000 events per well.)
Acetate Increases Cytosolic Reactive Oxygen Species.
NGF-derived PC12 cells were treated with increasing concentrations of acetate (0, 2, 6, 15, and 30 mM) for 4 h and then stained with Cell ROX Orange and analyzed via flow cytometry. Acetate increased cytosolic reactive oxygen species fluorescence in a dose-dependent manner with an EC50 value of 1.81 mM (Figure 3C). Acetate (6 mM) significantly (**P < 0.01) increased cytosolic reactive oxygen species fluorescence intensity compared to control, 43.1 ± 1.4 vs 33.3 ± 1.5% cells (Figure 3B).
Figure 3.
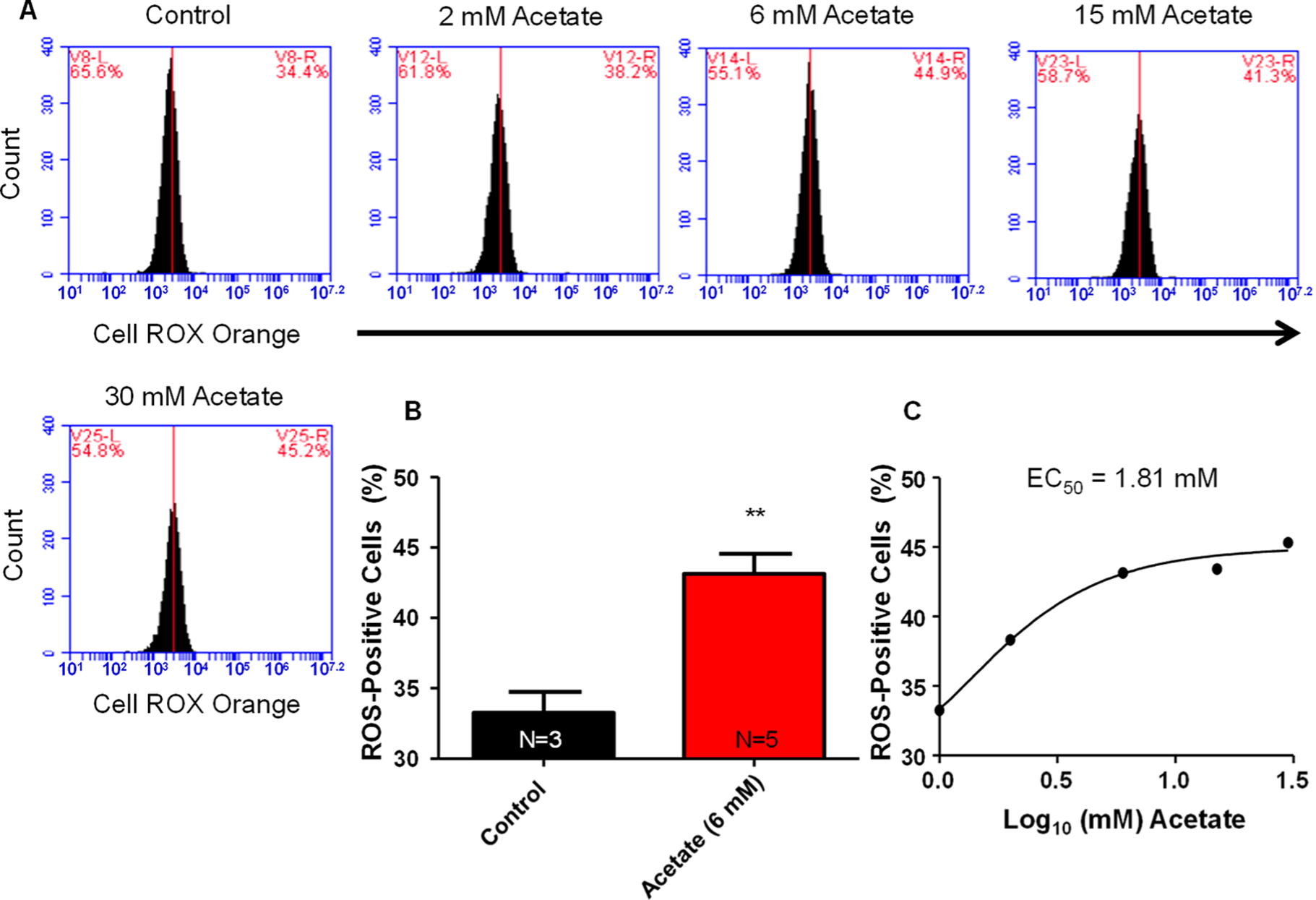
Acetate increases cytosolic reactive oxygen species. (A) Representative flow cytometry charts for cytosolic ROS fluorescence with various concentrations of acetate. (B) Summary data for acetate (6 mM) on increases in cytosolic ROS production. Acetate (6 mM) significantly (**P < 0.01) increased cytosolic ROS percentage, 44.1 ± 1.4%, compared to baseline values of 33.3 ± 1.5%. (C) Dose-response curve for acetate on changes in cytosolic ROS. EC50 value calculated from the dose-response curve indicating a value of 1.81 mM acetate. (N = number of wells per treatment; each well analyzed for 10,000 events per well.)
Ethanol Has No Apparent Increase in Cytosolic Reactive Oxygen Species.
NGF-derived PC12 cells were treated with increasing concentrations of ethanol (0, 2, 6, 15, 50, and 100 mM) for 4 h and then stained with Cell ROX Orange and analyzed via flow cytometry. Ethanol had a trend at lower doses (2, 6, and 15) to lower cytosolic ROS, although this was not statistically significant. There were no apparent increases in cytosolic reactive oxygen species fluorescence compared to control treatment (Figure 4).
Figure 4.
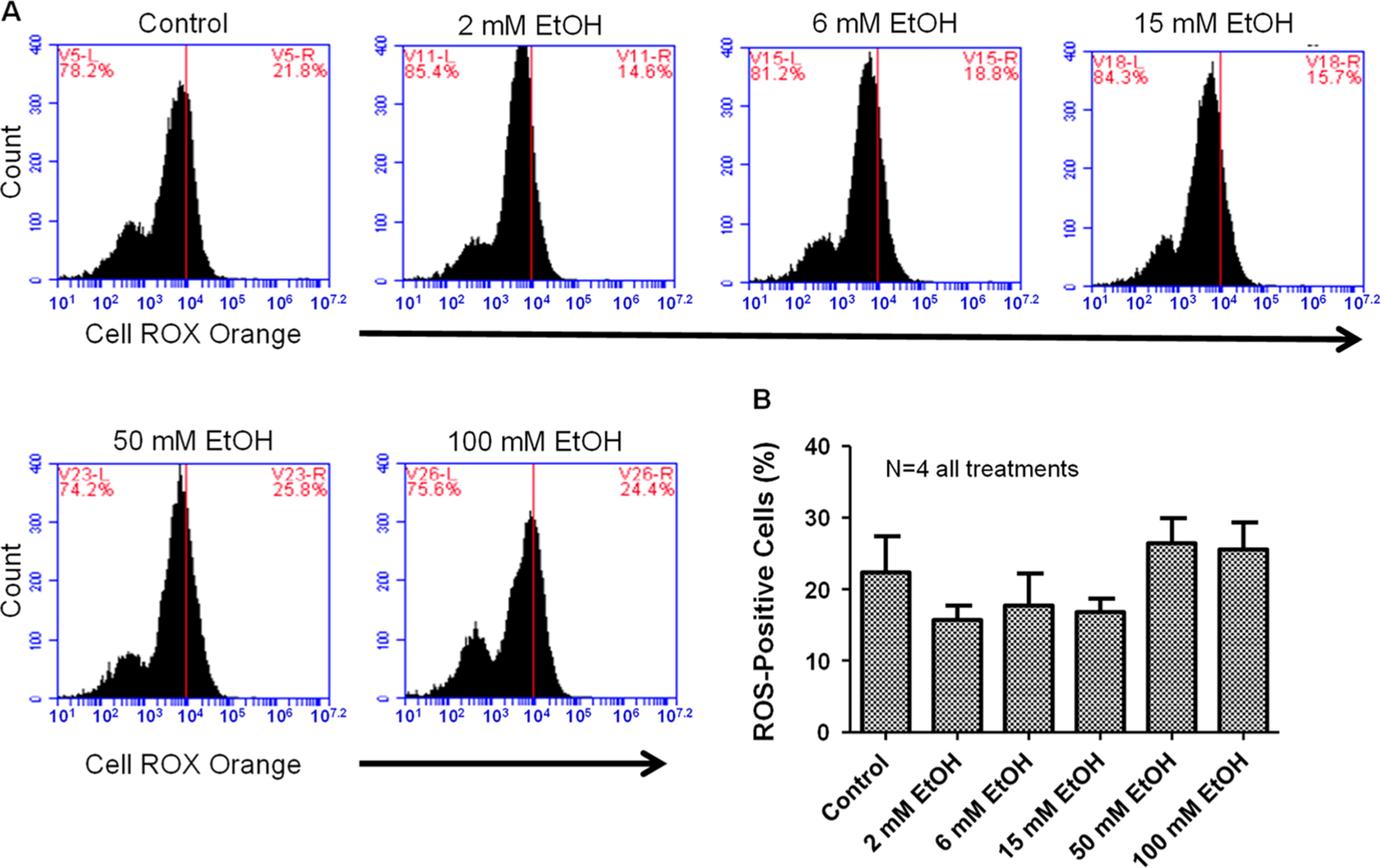
Ethanol has no apparent effect on cytosolic reactive oxygen species. (A) Representative flow cytometry charts for various doses of ethanol on cellular ROS. (B) Summary data for various doses of ethanol on changes in cytosolic ROS. Ethanol had no statistical effect (one-way ANOVA; F5,18 = 1.61) on alterations in cytosolic ROS; however, there was a trend for lower doses (2, 6, and 15 mM) to reduce ROS production. (N = number of wells per treatment; each well analyzed for 10,000 events per well.)
Acetate Increases TNFα mRNA Expression through Activation of NMDAR.
NGF-derived PC12 cells incubated with acetate (6 mM) for 4 h had significantly (p < 0.05) increased mRNA expression levels of the pro-inflammatory cytokine TNFα (Figure 5E) compared to control. Co-application of acetate (6 mM) and memantine (10 μM) completely abolished the acetate-induced increase in TNFα (Figure 5E). Memantine (10 μM) had no effect on TNFα mRNA expression relative to control. This data suggests that acetate increases the mRNA expression levels of the pro-inflammatory cytokine TNFα through an NMDAR-mediated mechanism.
Figure 5.
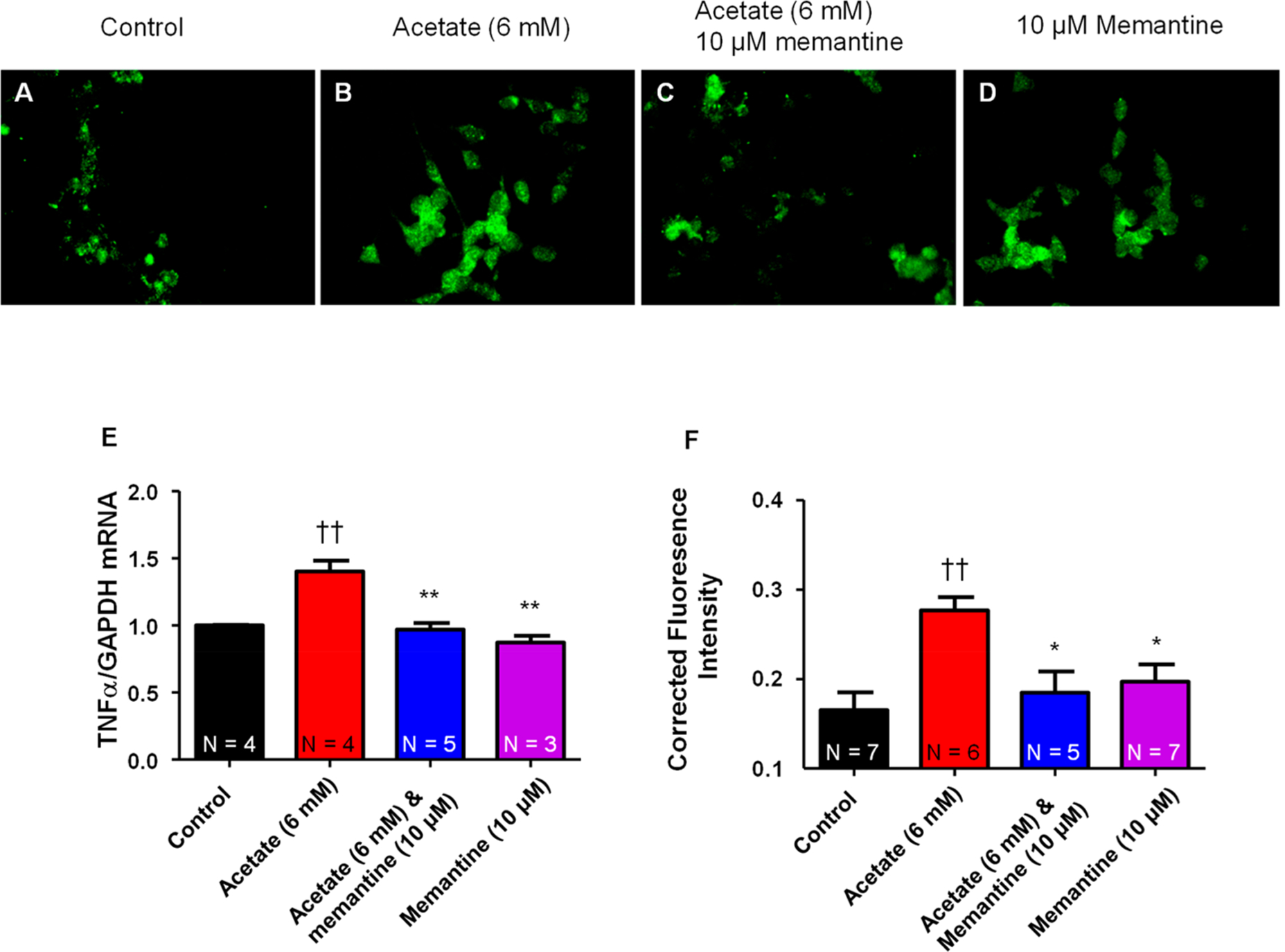
Acetate increases TNFα through activation of NMDAR. Representative immunochemistry staining for TNFα in PC12 cells from (A) control, (B) acetate (6 mM), (C) acetate (6 mM) and memantine (10 μM), and (D) memantine (10 μM). (E) Summary data of varying treatments of acetate (0, 6 mM), acetate (6 mM) and memantine (10 μM), and memantine (10 μM) alone on mRNA expression levels of TNFα. Acetate (6 mM) significantly increased mRNA expression of TNFα compared to control (1.4 ± 0.08 vs 1.0 ± 0.004 fold). Co-application of acetate (6 mM) and memantine (10 μM) abolished (††P < 0.01 vs control, one-way ANOVA; F3,12 = 17.66) the acetate-induced increase in mRNA expression of TNFα. Memantine (10 μM) had no effect on baseline mRNA expression of TNFα compared to control (**P < 0.01 vs acetate (6 mM), one-way ANOVA; F3,12 = 17.66). (F) Summary data for immunochemistry staining fluorescence of TNFα quantified using ImageJ software (††P < 0.01 vs control, *P < 0.05 vs acetate (6 mM), one-way ANOVA; F3,21 = 6.20). (For mRNA summary data (E), N = number of wells per treatment; for fluorescence intensity quantification (F), N = number of cells analyzed.)
In a separate experiment, NGF-derived PC12 cells were incubated with acetate (0 and 6 mM) (Figure 5A and B), acetate (6 mM) and memantine (10 μM) (Figure 5C), or memantine (10 μM) (Figure 5D) for 4 h and then stained for TNFα protein expression using a specific antibody and analyzed for immunochemistry staining. Acetate (6 mM) significantly (P < 0.05) increased TNFα immunochemistry-corrected fluorescence intensity compared to control (0 mM acetate) (Figure 5F). Co-application of acetate and memantine (Figure 5C) significantly (P < 0.05) attenuated TNFα immunochemistry-corrected fluorescence intensity compared to acetate (6 mM) (Figure 5F). Memantine alone had no effect on baseline TNFα immunochemistry relative to control (Figure 5F). This data suggests that acetate increases both mRNA and protein expression of TNFα, which can be abolished by the NMDAR blocker memantine.
Acetate Increases Cytosolic Calcium in PC12 Cells, Which Is Abolished by the NMDAR Antagonist.
NGF-derived PC12 cells incubated with acetate (6 mM) for 4 h had significantly (p < 0.05) higher Fluo-4AM corrected fluorescence intensity (CFIT) compared to that of control (0.3451 ± 0.03869 vs 0.1441 ± 0.01088 CFIT), which is indicative of a greater amount of cytosolic calcium (Figure 6). Co-application of acetate (6 mM) and memantine (10 μM) completely abolished the acetate-induced increase in cytosolic calcium CFIT (0.1681 ± 0.0235 vs 0.3541 ± 0.03869 CFIT) (Figure 6). Memantine (10 μM) alone had no apparent impact on changes in baseline calcium CFIT compared to control (0.09932 ± 0.0240 vs 0.1441 ± 0.01088). This data indicates that acetate is capable of increasing cytosolic calcium through activation of NMDAR.
Figure 6.
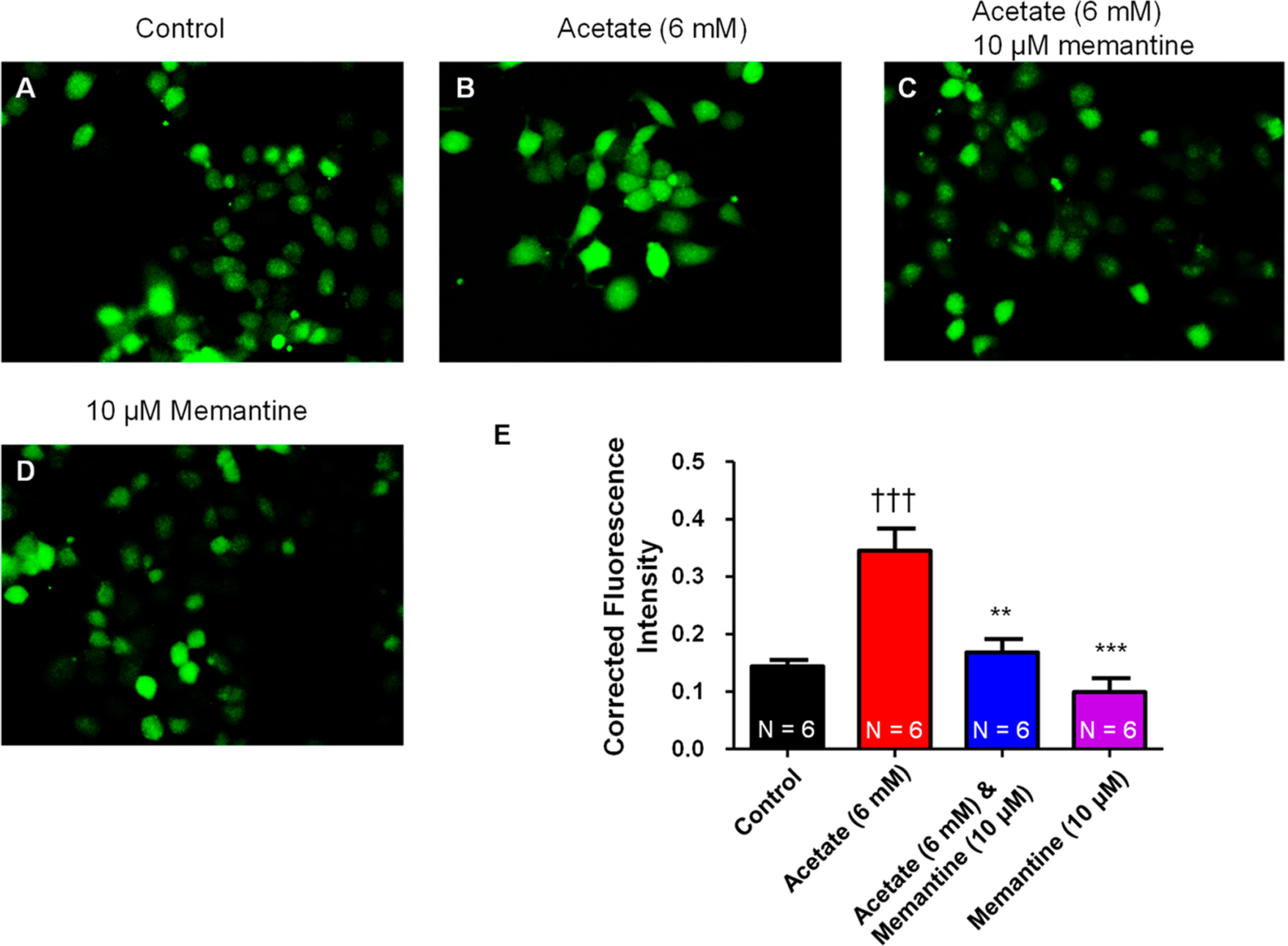
Real-time cytosolic calcium measurements in PC12 cells. Representative cytosolic calcium fluorescence in NGF-derived PC12 cells at (A) control (0 mM acetate), (B) 4 h post acetate (6 mM) treatment, and (C) 4 h post acetate (6 mM) and memantine (10 μM) treatment. (D) Memantine (10 μM) had no effect on baseline cytosolic calcium. (E) Summary data for cytosolic calcium measured with Fluo-4AM and quantified using ImageJ software. Acetate (6 mM) significantly (†††P < 0.05 vs control, one-way ANOVA; F3,20 = 16.91) increased the cytosolic calcium fluorescence after 4 h of treatment, whose response was abolished by coapplication of acetate (6 mM) and memantine (10 μM) (**P < 0.01, ***P < 0.001 vs acetate (6 mM), one-way ANOVA). (N = number of cells analyzed.)
NMDAR Antagonist Memantine Slightly Reduces the Acetate-Induced Increase in Cell Death.
NGF-derived PC12 cells treated with acetate (6 mM) significantly (P < 0.05) increased PI staining compared to control (31.2 ± 1.6 vs 20.5 ± 1.2%). Treatment with both acetate (6 mM) and memantine (10 μM) slightly reduced the acetate-induced increases in PI staining when compared to acetate (6 mM) alone (26.8 ± 0.7 vs 31.2 ± 1.6%) (P < 0.05, unpaired t test) (Figure 7). Memantine (10 μM) also significantly (P < 0.05) increased NGF-derived PC12 PI staining compared to control (31.2 ± 2.3 vs 20.5 ± 1.2%) (Figure 7). This data suggests that acetate increases PI staining, which is reduced in the presence of the NMDAR blocker memantine (10 μM). Furthermore, calcium is extremely important in regulating NGF-derived PC12 cell viability as NMDAR blocker alone also significantly increased PI staining.
Figure 7.
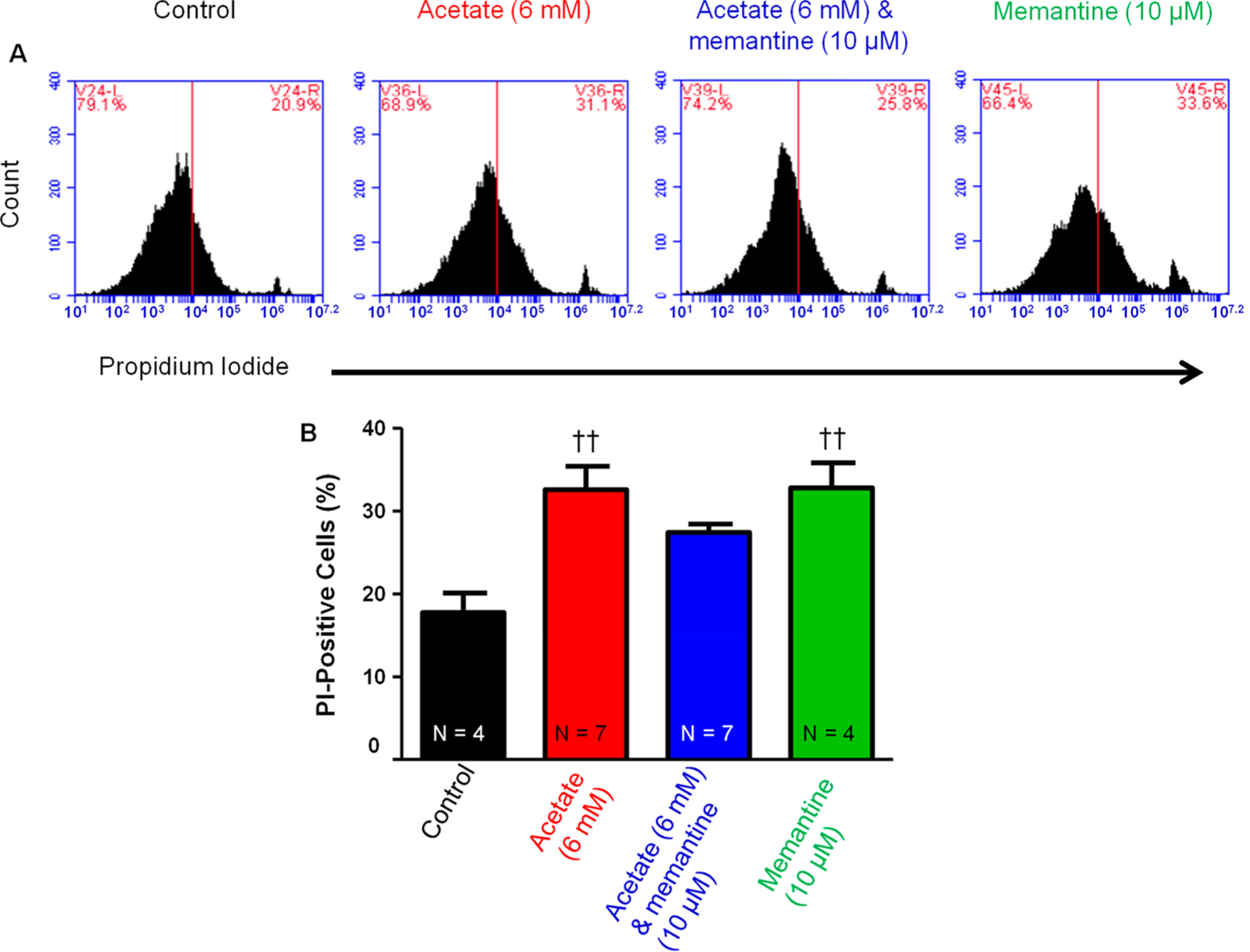
Effect of NMDAR blocker on the acetate-induced increase in cell death in PC12 cells. (A) Representative flow cytometry charts of the effect of (left to right: 0, 6 mM acetate, acetate (6 mM) and memantine (10 μM), and memantine (10 μM). (B) Acetate (6 mM) caused a significant (††P < 0.01 vs control, one-way ANOVA) increase in PI staining compared to that of control (31.2 ± 1.6 vs 20.5 ± 1.2%). Acetate (6 mM) and memantine (10 μM) had a trend for at least partially reducing acetate-induced increases in PI staining (26.8 ± 0.7 vs 31.2 ± 1.6%). Memantine (10 μM) alone had a significant (††P < 0.01 vs control, one-way ANOVA; F3,20 = 6.72) increase in PI staining compared to that of control (31.2 ± 2.3 vs 20.5 ± 1.2%). (N = number of wells per treatment; each well analyzed for 10,000 events per well.)
After seeing that memantine (10 μM) alone caused increases in PI staining, we ran a dose curve to explore what concentrations of memantine were cytotoxic to NGF-derived PC12 cells (Figure 8). What we found was that PC12 cells exposed to concentrations of memantine (0.5–5 μM) for 4 h had no significant increase in PI staining fluorescence. Consistent with our previous experimental findings (Figure 7), memantine (10 μM) for 4 h was cytotoxic to NGF-derived PC12 cells (Figure 8A). We then examined whether nontoxic doses of memantine (0.5–5 μM) with acetate (6 mM) would have an effect on reducing cell death. What we found again was that acetate (6 mM) treatment of NGF-derived PC12 cells for 4 h increased cell death and that coapplication with memantine (0.5–5 μM) had no apparent effect on reducing cell death relative to control (Figure 8B). This data suggests that there are alternative NMDAR independent mechanisms contributing to acetate-induced PC12 cell death.
Figure 8.
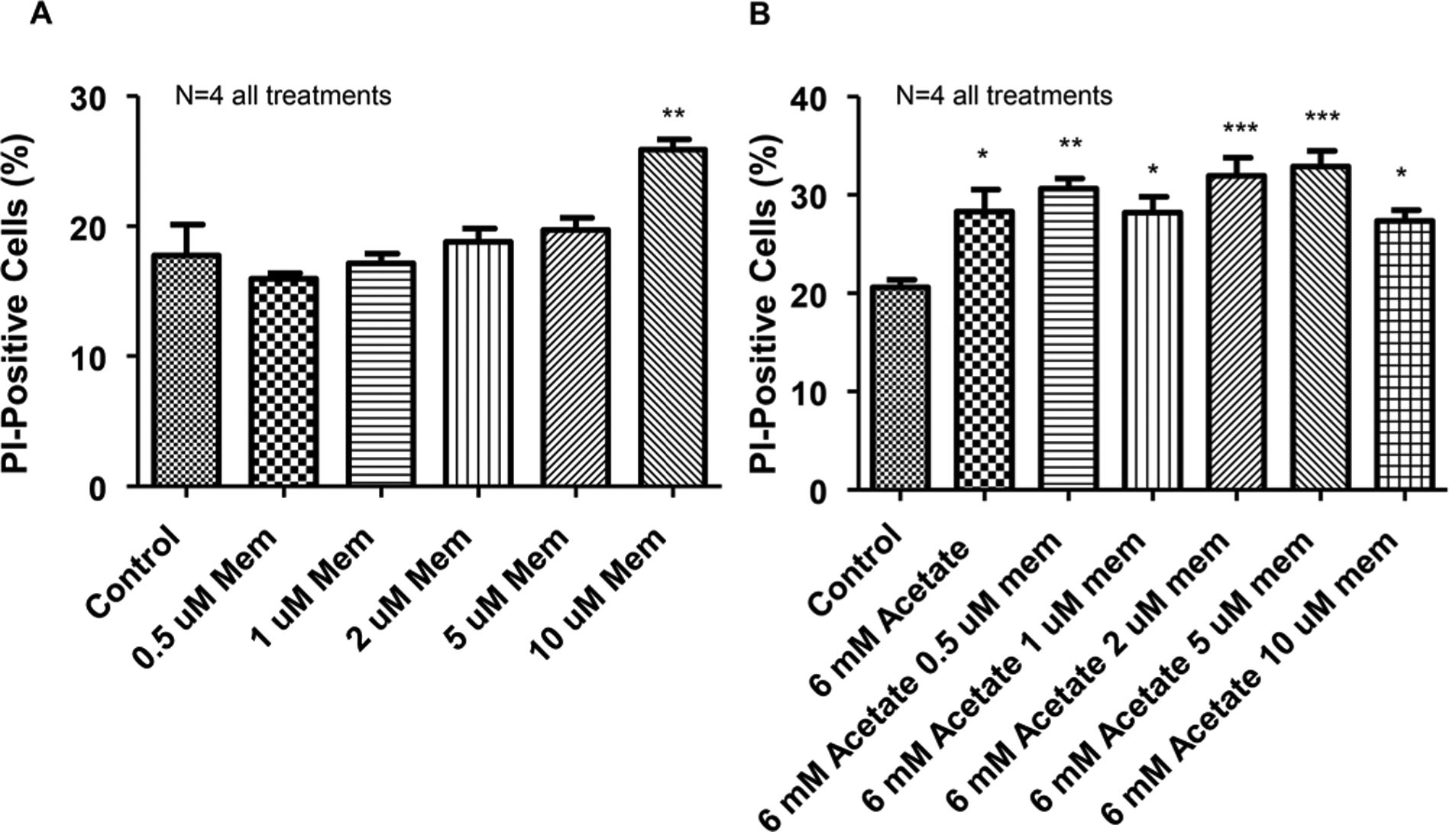
NMDAR blocker dose-response with and without acetate. Summary data for NMDAR antagonist and antagonist with acetate. (A) NMDAR antagonist memantine dose-response curve for cell death. Concentrations of 0.5–5 μM memantine were not cytotoxic (**P < 0.01 vs control; F5,18 = 8.31). (B) Acetate (6 mM) was incubated with varying doses of NMDAR antagonist memantine to determine if lower doses of memantine could decrease acetate-induced cell death. Memantine was unable to abolish the acetate-induced increase in cell death (*P < 0.05, **P < 0.01, ***P < 0.001 vs control; F6,26 = 6.20). (N = number of wells per treatment; each well analyzed for 10,000 events per well.)
DISCUSSION
The present study evaluated the toxic effects of both EtOH and acetate on dopaminergic-like PC12 cells. Although a majority of the literature has investigated the effects of ethanol-induced excitotoxicity, general consensus on a mechanism has yet to be fully understood. Ethanol metabolism to acetate and the effect acetate has on the brain is much less explored. As such, we report for the first time, at least in regard to alcohol use, the cytotoxic and inflammatory effects of acetate on dopaminergic-like PC12 cells.
Using flow cytometry, we confirmed that NGF-derived PC12 cells incubated with acetate for 4 h increased cell death in a dose-dependent manner (Figure 1). The construction of a dose-response curve revealed an EC50 value of 4.40 mM acetate (Figure 1C). Wang and colleagues reported acetate concentrations following EtOH infusion of 4.09 ± 0.24 mM with sustained steady state acetate concentrations of 3.51 ± 0.20 mM in unconditioned alcohol rats.22 Our EC50 concentration for acetate-induced increases in cell death closely corresponds to the apparent peak and steady-state brain concentrations reported by Wang’s group. Because acetate is capable of remaining elevated 12 plus hours post alcohol consumption20,21 while EtOH concentrations return to undetectable levels31,32 suggests that the chemical compound more likely to be contributing to alcohol-induced excitotoxicity is acetate.
To evaluate the effects of EtOH on cytotoxicity, we also treated NGF-derived PC12 cells for 4 h with EtOH concentrations from 0 to 100 mM. The 100 mM concentration physiologically would be considered a near lethal dose in nonheavy drinkers (BAC of ~0.45%, 0.08% is considered legally drunk). What we observed was that, at high EtOH concentrations, there was no apparent change in cell death in PC12 cells (Figure 2). Although this does not mean that EtOH is not cytotoxic, at least in NGF-derived PC12 cells, 4 h exposure was not enough time to produce an effect. Various groups routinely incubate cell cultures with EtOH in the 40–100 mM concentration range for 24 h to observe cytotoxic effects15,33 or physiological effects.34–36
A study published by Jiang and colleagues also reported elevated brain uptake and metabolism of acetate in heavy drinkers.21 It was reasoned that the acetate was an alternative energy source in the brain and may facilitate a reward mechanism in the form of caloric benefit or alteration in adenosinergic signaling.37,38 From a cytotoxic standpoint, perhaps an additional reason for this apparent increased brain uptake and oxidation of acetate in heavy drinkers is to mitigate the excitatory insult from constant brain acetate following alcohol consumption. Moreover, Maxwell’s group was able to show that acetate caused hangovers in rats,27 suggesting that the hangover symptoms are likely a result of excitatory insult to the brain. Our data, at least in PC12 cells, corroborates their data as hangovers are likely due to over excitation of neurons. Behaviorally, acetate is suggested to play a role in reduced locomotion,39 which implies acetate may propagate responses in addition to neurotoxic effects.
Next, we explored the impact both acetate and ethanol had on the production of cytosolic reactive oxygen species following a 4 h exposure. Acetate consistently increased cytosolic reactive oxygen species in a dose-dependent manner (Figure 3). Our dose-response curve for acetate indicated an EC50 value of 1.81 mM acetate (Figure 3C). Oppositely, ethanol had no apparent effect on cytosolic ROS; however, there was a trend for lower concentrations (2, 6, and 15 mM) of ethanol to have a reduction in cytosolic ROS. There is some literature to suggest that low doses of ethanol can inhibit the formation of ROS via inhibition of NLR Family Pyrin Domain Containing 3 (NLRP3) protein. The action was independent of NMDAR or GABA ion channels, which suggests at least a partial alternative pathway for ethanol inhibition of ROS at low doses.40 Another possibility is that low doses of ethanol inhibit NMDAR ion channels, which would inhibit the calcium-calmodulin-NO synthase cascade,41 and that higher doses of ethanol have nonspecific effects. Dizon and colleagues reported that ethanol did increase ROS production in PC12 cells after 24 h treatment at concentrations >50 mM, which would be the equivalent of a sustained BAC of 0.08% for 24 h. Furthermore, they found that 24 h ethanol treatment <25 mM had little or no effect on increased ROS.42 Whether low doses of ethanol inhibit NLRP3, reducing ROS in PC12 cells will need to be examined in the future.
It was not surprising for us to see increases in cytosolic ROS following acetate treatment, as this has been established in the literature from mitochondrial-generated ATP.43,44 Acetate is widely accepted as a precursor for the generation of acetyl-CoA through acetyl-CoA synthetase.45,46 Because a majority of cells express monocarboxylate transporters (transporters of primarily pyruvate, lactate and acetate),47–49 we expected that bath application would result in increased mitochondrial oxidation of acetate and the generation of cytosolic ROS. We were expecting similar responses with ethanol, as it has been documented that ethanol application causes increased ROS generation as well.50–52 This was not the case, at least in PC12 cells.
A majority of ethanol-related ROS are generated in the liver,51,52 at least in part due to liver cells having high amounts of alcohol and acetaldehyde dehydrogenase enzymes.53,54 This generates acetate, which further feeds into the generation of ATP via acetyl-CoA synthetase,45,46 although further oxidation of acetate in the liver does not appear to proceed at an appreciable rate.55,56 This suggests that a majority of the conversion of acetate to CO2 and ATP occurs elsewhere with brain21,57,58 and skeletal muscles59,60 as major sources of metabolism. Thus, we speculate that PC12 cells lack the metabolizing enzymes required for the generation of large quantities of acetate from ethanol.
We next investigated whether acetate was able to increase cytosolic calcium and pro-inflammatory cytokine TNFα through NMDAR activation. Our major findings indicate that, in NGF-derived PC12 cells, acetate was capable of increasing cytosolic calcium after 4 h treatment through activation of NMDAR (Figure 6). Chen and colleagues reported that treatment of developing neuromuscular synapses with acetate increased calcium influx by potentiating NMDAR and increasing spontaneous synaptic currents.61 Similarly, our lab reported that microinjection of acetate and ethanol into the central nucleus of the amygdala increased sympathetic nerve activity through activation of NMDAR.29 Collectively, this suggests that acetate is able to increase cytosolic calcium, thereby increasing neuronal activity. Treatment of NGF-derived PC12 cells with a cocktail of acetate and memantine attenuated the increase in calcium compared to that of acetate alone (Figure 6C). Because there is evidence for a pro-inflammatory cytokine/calcium interaction,62,63 we then explored whether NMDAR activation caused increases in TNFα. What we found, consistent with our calcium imaging, was that acetate (6 mM) significantly increased TNFα mRNA expression levels (Figure 5E) and protein expression (Figure 5F) and was effectively abolished by coapplication of acetate with memantine (Figure 5C). This data suggests that, at least in NGF-derived PC12 cells, acetate-induced increases in cytosolic calcium through NMDAR activation directly increases mRNA and protein expression levels of TNFα. Thus, this supports the notion that acetate is a noxious stimulus in the brain.
The last portion of the study explored what contribution the NMDAR had to the excitotoxic effects from acetate (Figure 7). Co-application of acetate (6 mM) and memantine (10 μM) showed a trend for a slight reduction in cell death; however, this was not significant (one-way ANOVA) (Figure 7B). An interesting observation was that memantine (10 μM) alone also significantly increased cell death (Figure 7B). We therefore conducted a dose-dependent response of memantine alone (Figure 8A). We found that doses of memantine (0.5–5 μM) displayed no apparent cytotoxic effect (Figure 8A). We then used those doses of memantine (0.5, 1, 2, and 5 μM) and coapplied them with acetate (6 mM) (Figure 8B). We found that coapplication of acetate (6 mM) with varying, nontoxic doses of memantine was unable to abolish any acetate-induced increase in cell death (Figure 8B). This suggests that there are alternative mechanisms independent of NMDAR activation that contributes to cell death. On the basis of our preliminary data, we speculate that the generation of cytosolic ROS may be a contributing factor, although future studies will be needed to determine the extent with which ROS contributes to alterations in cell signaling and cell death.
Another possibility for acetate-induced cell death through NMDAR bypass is the monocarboxylate transporter (MCT). MCT, as the name suggests, is a transporter of small organic compounds that contain one carboxylic functional group. It is primarily known for the transport of pyruvate and lactate47 but is also capable of transporting short chain fatty acids such as acetic acid, propionic acid, and butyric acid.47–49,64,65 Once transported across cell membranes, the acidic hydrogen dissociates where it can acidify the intracellular space.14,61 The fluctuations in external and internal pH are capable of modulating many ion channels such as the voltage-gated sodium, calcium, and potassium channels.66,67 Cytosolic acidification is also a common precursor prior to downstream signaling for apoptosis.68,69 It is therefore plausible that, in addition to acetate-induced NMDAR activation, MCT transport of acetic acid is a likely contributor to acetate-induced cell death.
Another interesting finding is that memantine at higher concentrations (10 μM) induces cell death (Figures 7 and 8). Neurite outgrowth is dependent on calcium perturbations in either direction directly affecting neurite outgrowth.70,71 Thus, at higher concentrations of memantine, we may be reducing cytosolic calcium enough to inhibit neurite outgrowth, reducing anchoring and possibly leading to cell death. Although acetate and memantine (10 μM) are both cytotoxic to PC12 cells independently, their combined effects are not additive and in fact show a slight trend to reduce toxicity. This suggests acetate may counteract memantine-induced toxicity, possibly through pH-dependent regulation of voltage-gated ion channels66,67 as mentioned above, although future work will be needed to determine if this is the case.
Taken together, the results of this study demonstrate that acetate at physiologically relevant concentrations produced from alcohol consumption and metabolism are capable of increasing excitotoxic insult in dopaminergic-like PC12 cells. Second, acetate increased cytosolic Ca2+ and upregulated TNFα via an NMDAR-dependent mechanism. NMDAR blockade was unable to abolish the acetate-induced increase in cell death. Furthermore, varying doses of ethanol had no apparent effect at 4 h on PC12 cellular death or cytosolic ROS. Collectively, this suggests that a major contributor in alcohol-induced excitotoxicity is at least in part due to the ethanol metabolite acetate. Future studies will be needed to determine the different cellular responses initiated by (1) acetate-induced increases in cytosolic calcium and (2) acetate-induced increases in cytosolic ROS.
METHODS
Chemicals.
All chemicals and cell culture reagents/supplies were obtained from Thermo Fisher Scientific (USA) except for nerve growth factor (NGF, Sigma-Aldrich).
Cell Culture.
PC12 cells obtained from ATCC (USA) were suspended in plating media (PM) consisting of 50 mL of Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% horse serum (HS), 5% fetal bovine serum (FBS), 1% antibiotics (penicillin and streptomycin), and nerve growth factor (NGF) at 20 ng/mL. Cells were grown to 80% confluency in a 75 cm2 tissue culture flask at which time the cells were detached with 0.25% trypsin (Thermofisher); the trypsin was neutralized with an equal volume of PM, and a portion of the cell suspension was mixed with 10% DMSO (Sigma, USA) and frozen for 2 days in a −80 °C freezer before transfer into liquid N2 for future passages. The cells in PM were transferred to 24-well plates (Costar) and placed in a humidified CO2 incubator (5% CO2) at 37.5 °C and grown to 80% confluency. PC12 cells were passaged for 10 passages before using new cells. Prior to use in experiments, cells were examined for their health and proper neurite outgrowth.
PC12 Cell Treatment.
After checking PC12 cell viability, the PM was aspirated and cells were washed with 12.5 mL of artificial cerebrospinal fluid (ACSF)72 to remove any residual serum (0.5 mL/well). A stock solution of sodium acetate was created (1 M) by dissolving sodium acetate in ultrapure water (Thermofisher) and filter sterilizing through a 0.2 μm sterile syringe filter (Thermofisher). The stock solution of sodium acetate was added into DMEM to the desired acetate concentration, heated to 37.9 °C, and then added to the 24-well plate (0.5 mL/well). The PC12 cells were then placed back into the humidified CO2 incubator (5% CO2) heated at 37.5 °C for 4 h. Similar preparations were used for ethanol treatments and acetate and memantine treatments. After 4 h, the cells were used for either flow cytometry, RT-PCR or real-time imaging experiments.
Cytotoxicity and ROS Assay via Flow Cytometry.
For the effect of acetate on cell death and ROS to be studied, PC12 cells were subjected to a PI exclusion assay for cell death and Cell ROX orange for cytosolic ROS. Both assays were conducted using flow cytometry. After checking PC12 cell viability, the PM was aspirated and cells were washed with 12.5 mL of artificial cerebrospinal fluid (ACSF) to remove any residual serum (0.5 mL/well). After aspirating the ACSF, 12.5 mL of trypsin (at 37.9 °C) was added to the culture dish (0.5 mL/well) and gently agitated to loosen cells from the bottom of the plate. The trypsin was then neutralized by the addition of 12.5 mL of the PC12 cell media (DMEM, horse serum, antibiotics, no NGF), and this mixture was then transferred to individual centrifuge tubes (1.5 mL). Tubes were centrifuged for 2 min at 1000 rpm. The supernatant was aspirated from each tube; cells were resuspended in 12.5 mL (0.5 mL per tube) of live cell imaging solution (LCIS, Thermofisher), and 1 drop of propidium iodide (PI) ReadyProbes (Thermofisher) was added to each individual tube. The tubes were vortexed briefly to break up the cell pellets created by the centrifugation, covered with aluminum foil to protect from light, and allowed to incubate with the PI for 20 min at room temperature before data collection. Flow cytometry was conducted using a BD Accuri C6 (BD Bioscience) flow cytometer. Experiments were conducted in at least triplicate at 10,000 cells per run. Percentages of PI staining were based off controls, and the same parameters for analysis were used for the treatment groups to compare relative to control.
For cytosolic reactive oxygen species, the same protocol above was used except that for PI we used Cell ROX Orange (Thermofisher). The Cell ROX Orange reagent was prepared according to the manufacturer’s instructions in LCIS and added to the cells in each individual 1.5 mL centrifuge tubes (0.5 mL per tube). The cells were incubated at room temperature for 40 min. Following staining, the tubes were centrifuged for 2 min at 1000 rpm, and the staining solution was aspirated. Fresh LCIS was added back into the tubes containing the cells (0.5 mL per tube), vortexed briefly, and then analyzed on the flow cytometer. Single cells were only included in the analysis by gating on forward scatter vs side scatter.
Calcium Imaging.
NGF-derived PC12 cells were incubated with DMEM, acetate (6 mM) in DMEM, or acetate (6 mM) and memantine (10 μM) for 4 h. The treatment solution was aspirated, and the cells then were incubated for 30 min with Fluo-4AM (Thermofisher, 3 μM final concentration) in artificial cerebral spinal fluid containing (in mM) 125 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 10 d-glucose, and 0.4 ascorbic acid (osmolality: 295–302 mosmol L−1; pH 7.3–7.4). Following Fluo-4AM treatment, the solution was aspirated, and 1 mL of LCIS was added to each well and then viewed under an inverted microscope (Leica) equipped with a mercury burner, correct fluorescence filter, and a digital camera (Leica) connected to a computer with image capture software (Leica). Images were captured for each well in a 24-well plate. The backgrounds of images used for fluorescence quantification were normalized across images. Fluorescence intensity was analyzed and quantified using ImageJ software73 on six randomly selected cells for each treatment, and corrected fluorescence intensity was compared between treatment groups. Quadruplicates of each treatment were compared and all showed similar trends.
Proinflammatory Cytokine TNFα mRNA Level Measurement.
NGF-derived PC12 cells were treated with vehicle control, acetate (6 mM), acetate (6 mM) and memantine (10 μM), or memantine (10 μM) for 4 h. Cells were collected, and mRNA was extracted using an RNA extraction kit (Qiagen) according to the manufacturer’s instructions and converted to cDNA using Superscript VILO (Thermofisher). cDNA concentrations were normalized to 250 ng/μL, and real time PCR (Step-one plus real time PCR system, Applied Biosystems) was performed to measure mRNA levels of GAPDH and TNFα using specific primers (Applied Biosystems). Each set of experiments was performed in quadruplicate, and the data were analyzed with Data Assist V. 3.01 (Applied Biosystems) for changes in mRNA expression levels of TNFα using GAPDH as a selected control.
TNFα Immunoreactivity Assessment.
PC12 cells were rinsed with cold PBS and fixed with 4% paraformaldehyde for 10 min and then rinsed with cold PBS and treated with 100% methanol (−20 °C) for 1 min. Following methanol, the cells were rinsed with PBS three times. The cells were then incubated with mouse anti-TNFα (1:200 dilution) (Santa Cruz, Biotechnology, CA, USA) in PBS containing 0.5% Triton X-100 and 5% horse serum overnight at 4 °C. Cells were washed with PBS three times for 5 min each and then incubated with Alexa Fluor 488 donkey antimouse IgG (Thermofisher, MA, USA) for 4 h at room temperature. The cells were observed under an inverted fluorescence microscope (Leica) equipped with a mercury lamp and the correct fluorescent filters and a digital camera (Leica) equipped to a computer with photo software (Leica). Photos were taken and analyzed offline with ImageJ Software (NIH, Bethesda, USA). Image backgrounds were normalized using ImageJ software.
Statistical Analysis.
Data values were reported as means ± SE. Depending on the experiments, group means were compared using either unpaired Student’s t-test or one-way ANOVA. Differences between means were considered significant at p < 0.05. Where differences were found, Bonferroni post hoc tests were used for multiple pairwise comparisons. All statistical analyses were performed with a commercially available statistical package (GraphPad Prism, version 5.0).
Funding
This study was supported by NIHR15HL129213 (Z.S.), NIHR15HL122952 (Q.-H.C.), and AHA16PRE27780121 (A.D.C.).
ABBREVIATIONS
- NMDAR
N-methyl-d-aspartate receptor
- TNFα
tumor necrosis factor alpha
- PI
propidium iodide
- ROS
reactive oxygen species
- EtOH
ethanol
- BAC
blood alcohol concentration
- eNOS
endothelial nitric oxide synthase
- NGF
nerve growth factor
- DMEM
Dulbecco’s modified Eagle media
- FBS
fetal bovine serum
- ACSF
artificial cerebrospinal fluid
- PM
plating media
- LCIS
live cell imaging solution
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Collins MA, and Neafsey EJ (2012) Ethanol and adult CNS neurodamage: oxidative stress, but possibly not excitotoxicity. Front. Biosci., Elite Ed 4, 1358–1367. [DOI] [PubMed] [Google Scholar]
- (2).Corso TD, Mostafa HM, Collins MA, and Neafsey EJ (1998) Brain neuronal degeneration caused by episodic alcohol intoxication in rats: effects of nimodipine, 6,7-dinitro-quinoxaline-2,3-dione, and MK-801. Alcohol.: Clin. Exp. Res 22, 217–224. [PubMed] [Google Scholar]
- (3).Hendricson AW, Maldve RE, Salinas AG, Theile JW, Zhang TA, Diaz LM, and Morrisett RA (2007) Aberrant synaptic activation of N-methyl-D-aspartate receptors underlies ethanol withdrawal hyperexcitability. J. Pharmacol. Exp. Ther 321, 60–72. [DOI] [PubMed] [Google Scholar]
- (4).Lovinger DM (1993) Excitotoxicity and alcohol-related brain damage. Alcohol.: Clin. Exp. Res 17, 19–27. [DOI] [PubMed] [Google Scholar]
- (5).Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, and Persidsky Y (2008) Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radical Biol. Med 45, 1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kril JJ, Halliday GM, Svoboda MD, and Cartwright H (1997) The cerebral cortex is damaged in chronic alcoholics. Neuroscience 79, 983–998. [DOI] [PubMed] [Google Scholar]
- (7).Nelson TE, Ur CL, and Gruol DL (2005) Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Res 1048, 69–79. [DOI] [PubMed] [Google Scholar]
- (8).Reynolds AR, Berry JN, Sharrett-Field L, and Prendergast MA (2015) Ethanol withdrawal is required to produce persisting N-methyl-D-aspartate receptor-dependent hippocampal cytotoxicity during chronic intermittent ethanol exposure. Alcohol (N. Y., NY, U. S.) 49, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, and Hudmon A (2012) Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J. Biol. Chem 287, 8495–8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Young BW, Sengelaub DR, and Steinmetz JE (2010) MK-801 administration during neonatal ethanol withdrawal attenuates interpositus cell loss and juvenile eyeblink conditioning deficits. Alcohol 44, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhou X, Hollern D, Liao J, Andrechek E, and Wang H (2013) NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis 4, e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Davies CH, Clarke VR, Jane DE, and Collingridge GL (1995) Pharmacology of postsynaptic metabotropic glutamate receptors in rat hippocampal CA1 pyramidal neurones. British journal of pharmacology 116, 1859–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Trudeau L-E, Parpura V, and Haydon PG (1999) Activation of Neurotransmitter Release in Hippocampal Nerve Terminals During Recovery From Intracellular Acidification, Vol. 81. [DOI] [PubMed] [Google Scholar]
- (14).Drapeau P, and Nachshen DA (1988) Effects of lowering extracellular and cytosolic pH on calcium fluxes, cytosolic calcium levels, and transmitter release in presynaptic nerve terminals isolated from rat brain. J. Gen. Physiol 91, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chandler LJ, Norwood D, and Sutton G (1999) Chronic ethanol upregulates NMDA and AMPA, but not kainate receptor subunit proteins in rat primary cortical cultures. Alcohol.: Clin. Exp. Res 23, 363–370. [PubMed] [Google Scholar]
- (16).Xiang Y, Kim KY, Gelernter J, Park IH, and Zhang H (2015) Ethanol upregulates NMDA receptor subunit gene expression in human embryonic stem cell-derived cortical neurons. PLoS One 10, e0134907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Churn SB, Limbrick D, Sombati S, and DeLorenzo RJ (1995) Excitotoxic activation of the NMDA receptor results in inhibition of calcium/calmodulin kinase II activity in cultured hippocampal neurons. J. Neurosci 15, 3200–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Obernier JA, Bouldin TW, and Crews FT (2002) Binge Ethanol Exposure in Adult Rats Causes Necrotic Cell Death. Alcohol.: Clin. Exp. Res 26, 547–557. [PubMed] [Google Scholar]
- (19).Qin L, He J, Hanes RN, Pluzarev O, Hong JS, and Crews FT (2008) Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflammation 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sarkola T, Iles MR, Kohlenberg-Mueller K, and Eriksson CJ (2002) Ethanol, acetaldehyde, acetate, and lactate levels after alcohol intake in white men and women: effect of 4-methylpyrazole. Alcohol.: Clin. Exp. Res 26, 239–245. [PubMed] [Google Scholar]
- (21).Jiang L, Gulanski BI, De Feyter HM, Weinzimer SA, Pittman B, Guidone E, Koretski J, Harman S, Petrakis IL, Krystal JH, and Mason GF (2013) Increased brain uptake and oxidation of acetate in heavy drinkers. J. Clin. Invest 123, 1605–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang J, Du H, Jiang L, Ma X, de Graaf RA, Behar KL, and Mason GF (2013) Oxidation of ethanol in the rat brain and effects associated with chronic ethanol exposure. Proc. Natl. Acad. Sci. U. S. A 110, 14444–14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Amore A, Cirina P, Mitola S, Peruzzi L, Bonaudo R, Gianoglio B, and Coppo R (1997) Acetate intolerance is mediated by enhanced synthesis of nitric oxide by endothelial cells. Journal of the American Society of Nephrology: JASN 8, 1431–1436. [DOI] [PubMed] [Google Scholar]
- (24).Bingel M, Koch K, Lonnemann G, Dinarello C, and Shaldon S Enhancement of in-vitro human interleukin-1 production by sodium acetate, The Lancet, 329, 14–16. [DOI] [PubMed] [Google Scholar]
- (25).Lestari F, Hayes AJ, Green AR, and Markovic B (2005) In vitro cytotoxicity of selected chemicals commonly produced during fire combustion using human cell lines. Toxicol. In Vitro 19, 653–663. [DOI] [PubMed] [Google Scholar]
- (26).Graefe U, Milutinovich J, Follette WC, Vizzo JE, Babb AL, and Scribner BH (1978) Less dialysis-induced morbidity and vascular instability with bicarbonate in dialysate. Ann. Intern. Med 88, 332–336. [DOI] [PubMed] [Google Scholar]
- (27).Maxwell CR, Spangenberg RJ, Hoek JB, Silberstein SD, and Oshinsky ML (2010) Acetate Causes Alcohol Hangover Headache in Rats. PLoS One 5, e15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kendrick SF, O’Boyle G, Mann J, Zeybel M, Palmer J, Jones DE, and Day CP (2010) ) Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology (Hoboken, NJ, U. S.) 51, 1988–1997. [DOI] [PubMed] [Google Scholar]
- (29).Chapp AD, Gui L, Huber MJ, Liu J, Larson RA, Zhu J, Carter JR, and Chen QH (2014) Sympathoexcitation and pressor responses induced by ethanol in the central nucleus of amygdala involves activation of NMDA receptors in rats. American journal of physiology. Heart and circulatory physiology 307, H701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dengler WA, Schulte J, Berger DP, Mertelsmann R, and Fiebig HH (1995) Development of a propidium iodide fluorescence assay for proliferation and cytotoxicity assays. Anti-Cancer Drugs 6, 522–532. [DOI] [PubMed] [Google Scholar]
- (31).Jones AW, Lindberg L, and Olsson SG (2004) Magnitude and time-course of arterio-venous differences in blood-alcohol concentration in healthy men. Clin. Pharmacokinet 43, 1157–1166. [DOI] [PubMed] [Google Scholar]
- (32).Kiyoshi A, Weihuan W, Mostofa J, Mitsuru K, Toyoshi I, Toshihiro K, Kyoko K, Keiichi N, Iwao I, and Hiroshi K (2009) Ethanol metabolism in ALDH2 knockout mice - Blood acetate levels. Leg. Med 11 (Supplement 1), S413–S415. [DOI] [PubMed] [Google Scholar]
- (33).Chandler LJ, Newsom H, Sumners C, and Crews F (1993) Chronic ethanol exposure potentiates NMDA excitotoxicity in cerebral cortical neurons. J. Neurochem 60, 1578–1581. [DOI] [PubMed] [Google Scholar]
- (34).Bajo M, Madamba SG, Roberto M, Blednov YA, Sagi VN, Roberts E, Rice KC, Harris RA, and Siggins GR (2014) Innate Immune Factors Modulate Ethanol Interaction with GABAergic Transmission in Mouse Central Amygdala. Brain, Behav., Immun 40, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Roberto M, Bajo M, Crawford E, Madamba SG, and Siggins GR (2006) Chronic Ethanol Exposure and Protracted Abstinence Alter NMDA Receptors in Central Amygdala. Neuropsychopharmacology 31, 988–996. [DOI] [PubMed] [Google Scholar]
- (36).Lovinger D, White G, and Weight F (1990) NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J. Neurosci 10, 1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Carmichael FJ, Orrego H, and Israel Y (1993) Acetate-induced adenosine mediated effects of ethanol. Alcohol and alcoholism (Oxford, Oxfordshire) No. Supplement 2, 411–418. [PubMed] [Google Scholar]
- (38).Phillis JW, O’Regan MH, and Perkins LM (1992) Actions of ethanol and acetate on rat cortical neurons: Ethanol/adenosine interactions. Alcohol 9, 541–546. [DOI] [PubMed] [Google Scholar]
- (39).Pardo M, Betz AJ, San Miguel N, López-Cruz L, Salamone JD, and Correa M (2013) Acetate as an active metabolite of ethanol: studies of locomotion, loss of righting reflex, and anxiety in rodents. Front. Behav. Neurosci 7, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hoyt LR, Ather JL, Randall MJ, DePuccio DP, Landry CC, Wewers MD, Gavrilin MA, and Poynter ME (2016) Ethanol and Other Short-Chain Alcohols Inhibit NLRP3 Inflammasome Activation through Protein Tyrosine Phosphatase Stimulation. J. Immunol 197, 1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Shimohama S, Akaike A, and Kimura J (1996) Nicotine-induced protection against glutamate cytotoxicity. Nicotinic cholinergic receptor-mediated inhibition of nitric oxide formation. Ann. N. Y. Acad. Sci 777, 356–361. [DOI] [PubMed] [Google Scholar]
- (42).Dizon M, Brennan L, and Black S (2006) Ethanol Induces Cytotoxic Oxidative Stress in PC12 Cells: Protection by Reactive Oxygen Species Scavengers. J. Pharmacol. Toxicol 1, 418. [Google Scholar]
- (43).Zorov DB, Juhaszova M, and Sollott SJ (2014) Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev 94, 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Webster LT Jr. (1963) Studies of the acetyl coenzyme A synthetase reaction. I. Isolation and characterization of enzyme-bound acetyl adenylate. J. Biol. Chem 238, 4010–4015. [PubMed] [Google Scholar]
- (45).Webster LT Jr. (1965) Studies of the acetyl coenzyme A synthetase reaction. 3. Evidence of a double requirement for divalent cations. J. Biol. Chem 240, 4164–4169. [PubMed] [Google Scholar]
- (46).Mews P, Donahue G, Drake AM, Luczak V, Abel T, and Berger SL (2017) Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Debernardi R, Pierre K, Lengacher S, Magistretti PJ, and Pellerin L (2003) Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. J. Neurosci. Res 73, 141–155. [DOI] [PubMed] [Google Scholar]
- (48).Rae C, Fekete AD, Kashem MA, Nasrallah FA, and Broer S (2012) Metabolism, compartmentation, transport and production of acetate in the cortical brain tissue slice. Neurochem. Res 37, 2541–2553. [DOI] [PubMed] [Google Scholar]
- (49).Waniewski RA, and Martin DL (1998) Preferential utilization of acetate by astrocytes is attributable to transport. J. Neurosci 18, 5225–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bao H-F, Song JZ, Duke BJ, Ma H-P, Denson DD, and Eaton DC (2012) Ethanol stimulates epithelial sodium channels by elevating reactive oxygen species. American Journal of Physiology - Cell Physiology 303, C1129–C1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kukielka E, Dicker E, and Cederbaum AI (1994) Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch. Biochem. Biophys 309, 377–386. [DOI] [PubMed] [Google Scholar]
- (52).Cahill A, Cunningham CC, Adachi M, Ishii H, Bailey SM, Fromenty B, and Davies A (2002) Effects of Alcohol and Oxidative Stress on Liver Pathology: The Role of the Mitochondrion. Alcohol.: Clin. Exp. Res 26, 907–915. [PMC free article] [PubMed] [Google Scholar]
- (53).Cederbaum AI (2012) ALCOHOL METABOLISM. Clinics in liver disease 16, 667–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Edenberg HJ (2007) The Genetics of Alcohol Metabolism: Role of Alcohol Dehydrogenase and Aldehyde Dehydrogenase Variants. Alcohol Research & Health 30, 5–13. [PMC free article] [PubMed] [Google Scholar]
- (55).Yamashita H, Kaneyuki T, and Tagawa K (2001) Production of acetate in the liver and its utilization in peripheral tissues. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1532, 79–87. [DOI] [PubMed] [Google Scholar]
- (56).Knudsen CT, Quistorff B, and Grunnet N (1995) Ethanol inhibits acetate metabolism in rat hepatocytes. Pharmacol. Toxicol 76, 133–135. [DOI] [PubMed] [Google Scholar]
- (57).Deelchand DK, Shestov AA, Koski DM, Uğurbil K, and Henry P-G (2009) Acetate transport and utilization in the rat brain. J. Neurochem 109, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kiselevski Y, Oganesian N, Zimatkin S, Szutowicz A, Angielski S, Niezabitowski P, Uracz W, and Gryglewski RJ (2003) Acetate metabolism in brain mechanisms of adaptation to ethanol. Medical science monitor: international medical journal of experimental and clinical research 9, BR178–182. [PubMed] [Google Scholar]
- (59).Lundquist F, Sestoft L, Damgaard SE, Clausen JP, and Trap-Jensen J (1973) Utilization of acetate in the human forearm during exercise after ethanol ingestion. J. Clin. Invest 52, 3231–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Bertocci LA, Jones JG, Malloy CR, Victor RG, and Thomas GD (1997) Oxidation of lactate and acetate in rat skeletal muscle: analysis by 13C-nuclear magnetic resonance spectroscopy. J. Appl. Physiol 83, 32–39. [DOI] [PubMed] [Google Scholar]
- (61).Chen YH, Wu ML, and Fu WM (1998) Regulation of presynaptic NMDA responses by external and intracellular pH changes at developing neuromuscular synapses. J. Neurosci 18, 2982–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jara JH, Singh BB, Floden AM, and Combs CK (2007) Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. J. Neurochem 100, 1407–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Hopp SC, D’Angelo HM, Royer SE, Kaercher RM, Crockett AM, Adzovic L, and Wenk GL (2015) Calcium dysregulation via L-type voltage-dependent calcium channels and ryanodine receptors underlies memory deficits and synaptic dysfunction during chronic neuroinflammation. J. Neuroinflammation 12, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Dringen R, Wiesinger H, and Hamprecht B (1993) Uptake of l-lactate by cultured rat brain neurons. Neurosci. Lett 163, 5–7. [DOI] [PubMed] [Google Scholar]
- (65).Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, and Alberini CM (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Tombaugh GC, and Somjen GG (1996) Effects of extracellular pH on voltage-gated Na+, K+ and Ca2+ currents in isolated rat CA1 neurons. J. Physiol 493, 719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Han J-E, Cho J-H, Choi I-S, Kim D-Y, and Jang I-S (2017) Effects of acidic pH on voltage-gated ion channels in rat trigeminal mesencephalic nucleus neurons. Korean J. Physiol. Pharmacol 21, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ding D, Moskowitz SI, Li R, Lee SB, Esteban M, Tomaselli K, Chan J, and Bergold PJ (2000) Acidosis induces necrosis and apoptosis of cultured hippocampal neurons. Exp. Neurol 162, 1–12. [DOI] [PubMed] [Google Scholar]
- (69).Furlong IJ, Ascaso R, Lopez Rivas A, and Collins MK (1997) Intracellular acidification induces apoptosis by stimulating ICE-like protease activity. J. Cell Sci 110 (Pt 5), 653–661. [DOI] [PubMed] [Google Scholar]
- (70).Solem M, McMahon T, and Messing RO (1995) Depolarization-induced neurite outgrowth in PC12 cells requires permissive, low level NGF receptor stimulation and activation of calcium/calmodulin-dependent protein kinase. J. Neurosci 15, 5966–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zhaleh H, Azadbakht M, and Pour AB (2011) Effects of extracellular calcium concentration on neurite outgrowth in PC12 cells by staurosporine. Neurosci. Lett 498, 1–5. [DOI] [PubMed] [Google Scholar]
- (72).Chen Q-H, and Toney GM (2009) Excitability of paraventricular nucleus neurones that project to the rostral ventrolateral medulla is regulated by small-conductance Ca2+-activated K+ channels. J. Physiol 587, 4235–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Schneider CA, Rasband WS, and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]