

Graphical abstract
Keywords: Coronavirus, COVID-19, SARS-CoV-2, SARS-CoV-2 main protease, Hypericin, Cyanidin-3-O-glucoside, Molecular docking, Molecular dynamics simulations
Highlights
-
•
The SARS-CoV-2 Mpro is an important viral target for the ongoing COVID-19 pandemic.
-
•
Initial screen by docking 300 small molecules to the Mpro active site.
-
•
Preferential binding of 3 leads to the Mpro active site revealed by blind docking.
-
•
Stability of lead compounds with the Mpro active site confirmed by MD simulations.
-
•
Hypericin and cyanidin-3-O-glucoside inhibit Mpro in vitro in micromolar range.
Abstract
Caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the COVID-19 pandemic is ongoing, with no proven safe and effective vaccine to date. Further, effective therapeutic agents for COVID-19 are limited, and as a result, the identification of potential small molecule antiviral drugs is of particular importance. A critical antiviral target is the SARS-CoV-2 main protease (Mpro), and our aim was to identify lead compounds with potential inhibitory effects. We performed an initial molecular docking screen of 300 small molecules, which included phenolic compounds and fatty acids from our OliveNet™ library (224), and an additional group of curated pharmacological and dietary compounds. The prototypical α-ketoamide 13b inhibitor was used as a control to guide selection of the top 30 compounds with respect to binding affinity to the Mpro active site. Further studies and analyses including blind docking were performed to identify hypericin, cyanidin-3-O-glucoside and SRT2104 as potential leads. Molecular dynamics simulations demonstrated that hypericin (ΔG = -18.6 and -19.3 kcal/mol), cyanidin-3-O-glucoside (ΔG = -50.8 and -42.1 kcal/mol), and SRT2104 (ΔG = -8.7 and -20.6 kcal/mol), formed stable interactions with the Mpro active site. An enzyme-linked immunosorbent assay indicated that, albeit, not as potent as the covalent positive control (GC376), our leads inhibited the Mpro with activity in the micromolar range, and an order of effectiveness of hypericin and cyanidin-3-O-glucoside > SRT2104 > SRT1720. Overall, our findings, and those highlighted by others indicate that hypericin and cyanidin-3-O-glucoside are suitable candidates for progress to in vitro and in vivo antiviral studies.
1. Introduction
Since being declared a pandemic in early March, COVID-19 has spread rapidly throughout the world and is currently ongoing (World Health Organization, 2020). There are currently no approved human coronavirus vaccines (Amanat and Krammer, 2020). As a result, there is an urgent need to investigate, identify and repurpose small molecules with potential antiviral effects (Rosa and Santos, 2020). To date the RNA-dependent RNA polymerase inhibitor, remdesivir, is the only antiviral drug that has been approved; it has been granted emergency use authorisation for the compassionate management of severe COVID-19 (Mullard, 2020).
In addition to the spike glycoprotein, the coronavirus main protease (Mpro) is an important target for antiviral therapy (Pillaiyar et al., 2016). The replicase gene of SARS-CoV-2 encodes the replicase polyproteins, pp1a and pp1ab (Pillaiyar et al., 2016; Zhu et al., 2020; Xue et al., 2008). Mpro is the enzyme that is predominantly responsible for the proteolytic processing of these polyproteins into functional polypeptides (Pillaiyar et al., 2016; Zhu et al., 2020; Xue et al., 2008). Thus, Mpro plays an important role in viral replication and infection (Zhu et al., 2020). There is a growing body of literature on lead compounds that could be used to target the Mpro enzyme of SARS-CoV-2. This includes peptidomimetics, such as α-ketoamide inhibitors (Zhu et al., 2020; Yan et al., 2020; Zhang et al., 2020a). Aside from synthetic ligands, researchers are also investigating the antiviral properties of natural compounds and dietary polyphenols have been of particular interest (Pillaiyar et al., 2016; Denaro et al., 2020; Zakaryan et al., 2017; Mohammadi Pour et al., 2019a; Yi et al., 2004).
Extra-virgin olive oil (EVOO), which is the primary source of dietary fat within the Mediterranean diet, is rich in phenolic compounds (Lăcătușu et al., 2019). The OliveNet™ library, which was previously created by our laboratory, is a curated database of 676 compounds from Olea europaea and 222 phenolic compounds are divided into 13 subclasses (Bonvino et al., 2018). Crystal structures of the SARS-CoV-2 Mpro enzyme have been made available on the RCSB Protein Data Bank (PDB) and can be used for in silico screening (Zhu et al., 2020; Zhang et al., 2020a). Utilising a combination of targeted molecular docking and blind docking, we aimed to identify ‘hit’ compounds from a selection of 300 ligands that could potentially inhibit Mpro. This included 211 phenolic compounds and 13 fatty acids from our OliveNet™ library, known protease inhibitors, several antibiotics for comparison and the α-ketoamide inhibitor as a control (Bonvino et al., 2018). Molecular dynamics (MD) simulations were subsequently performed to evaluate the top three candidates and ultimately determine the lead compounds.
2. Materials and methods
2.1. Protein structure and ligands
The crystal structures of SARS-CoV-2 Mpro was obtained from the RSCB Protein Data Bank (PDB ID: 6LU7, 6Y2G, 6M03) (Zhang et al., 2020a; Jin et al., 2020; Berman et al., 2000; Zhang et al., 2020b). 300 compounds were selected for screening against Mpro. This comprised of 211 phenolic compounds and 13 fatty acids sourced from the OliveNet™ Library (Bonvino et al., 2018), and an additional 76 ligands based on known protease inhibitors and antibiotics, as well as compounds with antiviral, anti-inflammatory, anti-parasitic, anti-malarial, antioxidant and anti-ageing properties (Yan et al., 2020; Zakaryan et al., 2017; Agbowuro et al., 2018; Agostini et al., 2018; Amici et al., 2006; Chan et al., 2017; Chen et al., 2019a; Davies, 2010; Gbinigie and Frie, 2020; Huemer, 2015; Ichikawa et al., 2013; Khaerunnisa et al., 2020; Kim et al., 2019a, 2018; Krueger et al., 2015; Liang et al., 2020a; Lin et al., 2018, 2017; Lu et al., 2017; Moghadamtousi et al., 2014; Moghaddam et al., 2014; Mohammadi Pour et al., 2019b; Richardson et al., 2020; Rogosnitzky et al., 2020; Vankadari, 2020; Zhou et al., 2020a, b). A full list of the ligands that were screened can be found in the supplementary information (Table S1). Ligand structures were obtained from the National Centre for Biotechnology Information (NCBI) PubChem database (Kim et al., 2019b), or drawn using Chem3D 19.0 (Perkin Elmer, Massachusetts, USA) if they weren’t available.
2.2. Docking to the active site of the SARS-CoV-2 main protease monomer
Structure preparation and molecular docking was performed using the quantum-mechanics-polarised ligand docking (QPLD) protocol of the Schrödinger Suite (version 2018−1 and 2020−2) molecular modelling package (Maestro, 2018; Madhavi Sastry et al., 2013; Dong et al., 2020; Phua et al., 2020; Cho et al., 2005) as previously described (Liang et al., 2020b). A 20 × 20 × 20 Å receptor grid was generated centred around active site residues Thr24, Thr25, His163, Pro168 and Gly143 (Zhu et al., 2020).
Compounds were also docked to the active site of Mpro using AutoDock Vina (Trott and Olson, 2010), following the processing of protein and ligand structures using PyRx (Dallakyan and Olson, 2015) to generate their corresponding pdbqt files. The protein structure was assumed to be rigid, and rotatable torsions of the ligands were activated. A receptor grid with dimensions of 25 × 25 × 25 Å was generated around the same active site residues. Docking was performed with an exhaustiveness of 128.
Docking calculations were performed on a Windows 10 workstation equipped with an Intel Core i7 (2.90 GHz) and 8.00 GB of RAM.
2.3. Blind docking to the SARS-CoV-2 main protease dimer
As the SARS-CoV-2 Mpro is known to function as a homodimer (Jin et al., 2020), this complex was assembled using the Proteins, Interfaces, Structures and Assemblies (PDBePISA) server (Krissinel and Henrick, 2007) for blind docking to identify potential binding sites. Structures were processed in PyRx, and docking performed AutoDock Vina (Trott and Olson, 2010) using a receptor grid encompassing the entire protein surface at an exhaustiveness of 128. For selected top binding compounds, blind docking was also performed at an exhaustiveness of 2000 using cloud computing services provided by Galileo (Hypernet Labs).
2.4. Molecular dynamics simulations
Classical MD simulations were performed using GROMACS 2018.2 software (Berendsen et al., 1995; Abraham et al., 2015) with the CHARMM27 force field (Bjelkmar et al., 2010; Vanommeslaeghe et al., 2010) using docked ligands as starting structures as previously described (Liang et al., 2020b). Ligand topologies were generated using SwissParam (Zoete et al., 2011). For cyanidin-3-O-glucoside, Lennard-Jones parameters for the oxonium ion were assumed to be similar to those for the ether group (Sagnella and Voth, 1996). Simulations were performed with a time-step of 2 fs in triplicate for 100 ns.
Molecular Mechanics-Poisson Boltzmann Surface Area (MM-PBSA) was employed for the quantification of free energy calculations (Baker et al., 2001) using the g_mmpbsa tool (Kumari et al., 2014), as previously described (Liang et al., 2020b). Calculations were performed in triplicate on 1 ns segments, from 99 to 100 ns, of the stabilised trajectories (Hou et al., 2011).
Visual Molecular Dynamics 1.9.3 (Humphrey et al., 1996) was used for visualisation and analysis. Calculations were performed on a Dual Intel Xeon “Cascade Lake” Silver 4215 processor cluster (Topaz) at the Pawsey Supercomputing Centre, and an Intel Xeon E5−2650 v4 processor cluster (Spartan) hosted at the University of Melbourne (Bernard et al., 2017).
2.5. Enzyme-linked immunosorbent assay
To confirm inhibition of the SARS-CoV-2 Mpro in vitro, an enzyme-linked immunosorbent assay (ELISA), was performed. The BPL 3CL protease (SARS-CoV-2) assay kit (BPS Bioscience, San Diego, CA, USA), was used, and the assay performed according to the manufacturer’s instructions. The internal positive control was the broad-spectrum antiviral GC376 and was tested (n = 9 determinations), at a final concentration of 50 μM. The small molecule test inhibitors that we tested were hypericin (89 %, HWI pharma services GmbH, Germany), cyanidin-3-O-glucoside (reference standard, PhytoLab, Germany), SRT2104 and SRT1720 (>99 %, AdooQ® Bioscience, Irvine, CA, USA), resveratrol (>99 %), and l-sulforaphane (>95 %, Sigma-Aldrich, St Luis, MO, USA). The inhibitors were prepared as 20 mM stock solutions and stored at -80 °C until use. For the ELISA assay, doubling dilutions of each of the test inhibitors were performed to achieve final concentrations ranging from 0.25–128 μM; each concentration of the test inhibitors was assayed in triplicate. Following addition of the substrate solution the plate was read using an excitation wavelength of 360 nm and detection of emission at a wavelength of 460 nm. Fluorescence intensities were measured, and the % protease inhibition was calculated as the ratio of fluorescence intensity observed with each test inhibitor and the total activity (n = 9 determinations), taking background (n = 9 determinations) into account. The IC50 values for applicable test inhibitors (hypericin, cyanidin-3-O-glucoside, and SRT2104), were also calculated.
3. Results and discussion
3.1. Identification of key compounds with relatively high affinity to the active site of the SARS-CoV-2 main protease
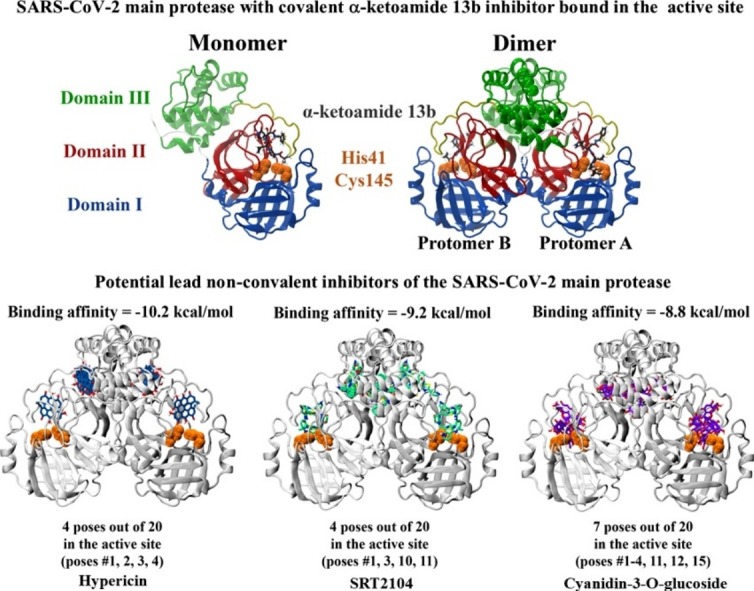
The α-ketoamide ligand (13b) was previously identified by Zhang et al. to inhibit Mpro with a half maximal inhibitory concentration (IC50) value of 0.67 ± 0.18 μM (Zhang et al., 2020a). As a result, this compound was used as a control. Each protomer of the Mpro enzyme consists of three domains and the active site is located between domain I and domain II (Zhu et al., 2020). When docked to the substrate-binding site of the crystal structure, the α-ketoamide ligand formed hydrogen bonds with the protein residues and this included Glu166, His164 and Gln189. The inhibitor was predominantly surrounded by hydrophobic and polar residues including Cys145, Asn142, Tyr54, Thr190 and Pro168 (Fig. 1 ). The binding affinities of this compound were -65.7 and -7.7 kcal/mol in Schrödinger and AutoDock Vina, respectively. Our lab has also recently verified the interaction of the α−ketoamide inhibitor with the active site of Mpro from SARS-CoV-2 (Liang et al., 2020b).
Fig. 1.

Structure of the SARS-CoV-2 Mpro dimer in complex with the α-ketoamide 13b ligand within the substrate-binding site. Mpro consists of three domains, with the catalytic core located between domains I and II. Catalytic dyad residues His41 and Cys145 are highlighted in orange, and the α-ketoamide 13b ligand is shown in grey. The α-ketoamide 13b was docked to the catalytic core using the QPLD protocol of Glide, and interactions with residues are depicted. Hydrogen bonds are shown as dashed yellow lines. Hydrophobic residues are green, polar uncharged residues are cyan, and negatively charged residues are shown in red.
The control compound was positioned in a similar site as the co-crystallised ligand (N3) (Zhu et al., 2020). Previously, it has been found that the negatively charged residue Glu166 plays an important role in forming the S1 pocket of the binding site (Zhu et al., 2020). The hydrophobic residue Cys145 is also involved in the mechanisms of action of N3 and the α-ketoamide inhibitor (Zhu et al., 2020; Zhang et al., 2020a). The Cys145 and His41 residues in the SARS-CoV-2 main protease form a catalytic dyad (Zhu et al., 2020). The Mpro enzyme is a cysteine protease and the inhibitors specifically interact with Cys145 covalently (Zhu et al., 2020; Zhang et al., 2020a). Covalent docking tools have been made available however, the success of this screening approach depends on a number of factors (Carlesso et al., 2019; Scarpino et al., 2018). This includes the contribution of non-covalent interactions and the mechanism of covalent bonding (Carlesso et al., 2019; Scarpino et al., 2018). In the current study, conventional docking methods were used and there is evidence to suggest that noncovalent docking is successful in elucidating the interactions that are occurring within protein-ligand complexes at the molecular level (Saikia and Bordoloi, 2019; Meng et al., 2011; Wang et al., 2016).
Drug repositioning has become one of the most important strategies for combating COVID-19 and virtual screening approaches continue to play a major role in this (Ahn et al., 2020). With this in mind, 300 compounds were docked to the catalytic core of the SARS-CoV-2 Mpro enzyme. Binding to the active site of the protomer using the QPLD protocol in Schrödinger yielded Glide energies ranging from -16.3 to -82.0 kcal/mol. All 300 compounds were predicted to bind using AutoDock Vina and the binding affinities ranged from -3.7 to -10.7 kcal/mol. When examining the effect of molecular weight of the small molecules and when comparing the binding affinities from both programs, the correlation coefficients were found to be approximately 0.7 for both parameters (Figure S1 and S2). Previous studies have evaluated a number of molecular docking programs that are available for use and have found that Glide is more accurate in predicting the crystallographic pose of ligands (Perola et al., 2004). For comparative purposes, we used a combination of binding energies derived from Schrödinger QPLD and AutoDock Vina and considered the orientation and position within the SARS-CoV-2 Mpro active site. For example, hypericin with a Glide energy of -51.7 kcal/mol using Schrödinger QPLD and docking score of -10.2 kcal/mol with AutoDock Vina, was selected on the basis of its consistent interactions with key residues in the active site.
In regards to the 211 phenolic compounds from the OliveNet™ database, it was evident that the flavonoid, glucoside and secoiridoid subclasses were binding strongly to the active site (Table S1). Conversely, the simple phenols, hydroxyphenylacetic acids, hydroxybenzoic acids and methoxyphenols had weaker binding affinities (Table S1). The biological activities of flavonoids have been extensively investigated over the years and there are studies that have examined the inhibitory activity of certain flavonoid compounds against the Mpro enzyme of SARS-CoV. This includes luteolin, tetra-O-galloyl-β-d-glucose and baicalin to name a few (Yi et al., 2004; Chen et al., 2004; Peiris et al., 2004). Since being declared a pandemic, several papers that have assessed the ability of natural compounds to target SARS-CoV-2 proteins have been made available (Ibrahim et al., 2020; Das et al., 2020). In a recent study conducted by Ul Qamar et al. in silico techniques were used to detect lead molecules from a medical plant library and they highlighted how their study may contribute to the development of natural antiviral agents in the future (Ul Qamar et al., 2020). Although there are hurdles that are yet to be overcome, Thomford et al. emphasise that advancements in predictive computational methods have made it possible for the properties of natural products and their derivatives to be explored and for novel therapeutic moieties to be discovered (Thomford et al., 2018).
Saquinavir and ritonavir were the protease inhibitors that were binding more strongly than the α-ketoamide ligand, while nelfinavir had a similar Glide energy as the control compound. This is in accordance with a paper published by Pant et al., as their molecular docking analysis revealed that these three antivirals scored well and interacted with important residues (Pant et al., 2020). In addition to the phenolic compounds from OliveNet™ and the protease inhibitors, some of the other top binding ligands included (-)-epicatechin gallate, remdesivir, d,l-sulforaphane glutathione, SRT2104, SRT1720, hypericin, curcumin, demethoxycurcumin, baricitinib and baicalin.
Based on this initial screen, 30 compounds were selected for further examination. To select the 30 compounds from the initial screen of the 300 compounds, a number of criteria were applied. Firstly, we selected compounds on the basis of binding affinity; compounds that were significantly below the positive controls, in this case, α-ketoamide 13b, were the first group to be eliminated. We then took into account compound availability. Given that we are trying to identify potential lead compounds for the current ongoing pandemic, it is important that the compounds are at least commercially available, thus allowing potential further investigation in vitro and in vivo. Many of our compounds would require synthesis and a complete evaluation of bioactivity before they can used in further experiments. Therefore, the more obscure less-well characterized compounds were the next group to be eliminated. It should be noted that the screening data obtained for these less well-studied compounds would still be useful, for potential structure activity-related studies, and for potential synthesis and evaluation at a more appropriate time. Compounds with relatively high binding affinity for the active site, that have been studied in animal models, and preferably in humans were the first to be selected for further evaluation. A description of these compounds and their binding affinities can be found in Table 1 . The structures of the ligands not found in the OliveNet™ database are provided in the supplementary information (Table S2).
Table 1.
Binding affinities (kcal/mol) of the compounds that were selected for further investigation.
| Compound | Classification | Glide energies (kcal/mol) |
|---|---|---|
| Ritonavir | Protease inhibitor | −71.5 |
| Saquinavir | Protease inhibitor | −77.7 |
| Lopinavir | Protease inhibitor | −56.2 |
| Indinavir | Protease inhibitor | −52.0 |
| Nelfinavir | Protease inhibitor | −65.2 |
| Darunavir | Protease inhibitor | −58.2 |
| Simeprevir | Protease inhibitor | −59.4 |
| Remdesivir | Nucleotide analog | −58.0 |
| Amikacin | Antibiotic | −64.5 |
| Ceftazidime | Antibiotic | −60.5 |
| Baricitinib | Janus kinase inhibitor | −48.8 |
| Suramin | Antiparasitic | −66.3 |
| Hypericin | Anthraquinone derivative | −51.7 |
| Ebselen | Organoselenium drug | −32.2 |
| Ivermectin | Anti-parasitic agent | −40.7 |
| SRT2104 | Sirtuin activator | −60.5 |
| SRT1720 | Sirtuin activator | −60.5 |
| D,L-Sulforaphane glutathione | Isothiocyanate analog | −61.7 |
| (-)-Epicatechin gallate | Natural flavonoid compound | −64.1 |
| Quercitrin | OliveNetTM | −60.7 |
| β-Hydroxy verbascoside | OliveNetTM | −71.1 |
| β-Hydroxy acteoside | OliveNetTM | −67.7 |
| Isoacteoside | OliveNetTM | −75.1 |
| Verbascoside | OliveNetTM | −76.3 |
| Oxidized verbascoside | OliveNetTM | −71.2 |
| Quercetin 3-O-rutinoside | OliveNetTM | −77.0 |
| Hesperidin | OliveNetTM | −64.0 |
| Rutin | OliveNetTM | −69.3 |
| Luteolin-7,4-O-diglucoside | OliveNetTM | −66.3 |
| Cyanidin-3-O-glucoside | OliveNetTM | −62.7 |
In order to narrow down the list and identify lead compounds, blind docking was conducted on the crystal structure of the Mpro dimer using the α-ketoamide 13b inhibitor and selection of 30 compounds at an exhaustiveness of 128. Overall, there were 19 ligands that had poses within the substrate-binding sites of the enzyme protomers (protomer A and protomer B). They were β-hydroxy verbascoside, ceftazidime, cyanidin-3-O-glucoside, d,l-sulforaphane glutathione, ebselen, (-)-epicatechin gallate, hypericin, indinavir, isoacteoside, nelfinavir, oxidized verbascoside, quercetin-3-O-rutinoside, quercitrin, remdesivir, saquinavir, simeprevir, SRT1720, SRT2104 and verbascoside.
Based on the results, three compounds were chosen for further study and they were hypericin, SRT2104 and cyanidin-3-O-glucoside (Fig. 2 ). Blind docking was performed for these three compounds at an exhaustiveness of 2000. Hypericin and SRT2104 had 4 poses each within the active sites of the dimer whereas cyanidin-3-O-glucose had 7 poses (Fig. 2 & Tables S3-S6). When docked to the active site of the Mpro monomer, these compounds were binding with relatively high affinity to the active site, with Glide energies of -51.7, -60.5 and -62.7 kcal/mol, respectively (Table 1). SRT2104 and cyanidin-3-O-glucoside formed a π−π interaction with His41 of Mpro, while hypericin formed a hydrogen bond with His164. Residues Asn142, Leu141, Glu166 and Thr190 were also involved in inter-atomic contacts with cyanidin-3-O-glucoside. In addition to His41, SRT2104 formed hydrogen bonds with Thr26 and Gly143 (Fig. 3 ). Although all three compounds were positioned in a similar manner within the catalytic core, the conformations of hypericin and cyanidin-3-O-glucoside were closer to that of the α-ketoamide inhibitor.
Fig. 2.

Blind docking and docking of hypericin, SRT2104, and cyanidin-3-O-glucoside to the active site of the SARS-CoV-2 Mpro dimer. Blind docking was performed using AutoDock Vina to produce 20 poses. Catalytic dyad residues His41 and Cys145 are highlighted in orange. Docking to the active site was performed using the QPLD protocol of Glide to each protomer of the Mpro dimer.
Fig. 3.

Residue interactions between the Mpro monomer and hypericin, SRT2104, and cyanidin-3-O-glucoside at 4.0 Å. Hydrogen bonds and π−π interactions are depicted by the yellow and cyan lines, respectively. The green residues are hydrophobic, the cyan residues are polar, and the red residues are negatively charged.
We extended our docking studies with these compounds to include additional dimeric structures of the SARS-CoV-2 Mpro, including 6Y2G (Zhang et al., 2020c) and 6M03 (Zhang et al., 2020b). Our results indicate docking to these protein structures yielded similar results, with ligands consistently binding in proximity to the key active site residues (Figure S3). It is noted that out of the top three ligands, cyanidin-3-O-glucoside consistently binds with the strongest affinity across all protease structures.
3.2. Molecular dynamic simulations highlight the stability of cyanidin-3-O-glucoside, SRT2104, and hypericin compounds with the main protease complex
To assess the stability of the ligands in complex with the main protease dimer, classical MD simulations were performed. Each system comprised of two ligands bound to the active sites on each protomer, as shown in Movies S1−4. Root mean square deviation (RMSD) analysis in Fig. 4 shows that the systems reached equilibrium after 50 ns, with subsequent analysis performed after this timepoint. Analysis of RMSD during the stabilised trajectory indicated that SARS-CoV-2 Mpro bound to ligands yielded slightly more stable protein complexes. With an average RMSD of 0.30 nm in the apo protein, the top ligands bound to the protease complex yielded slightly lower values compared to apo: 0.27 nm for hypericin, 0.28 nm for SRT2104, and 0.25 nm for cyanidin-3-O-glucoside. Root mean square fluctuation (RMSF) was also analysed, indicating that the main fluctuations were occurring at Leu50 in domain I and at Gln189 located in the connecting loop, which are located in proximity to the active site (Fig. 4). A more prominent fluctuation also occurred at residue Tyr154 in domain II. The largest fluctuation occurred at the C-terminal Gln306 across both protomers. When the RMSF values for the apo protease were subtracted from the ligand-bound values in Fig. 4, peaks in residue fluctuation diminished, with the exception of the C-terminal Gln306. This indicates that the binding of hypericin, SRT2104, and cyanidin-3-O-glucoside to the active site of the protease does not influence residue fluctuation in the overall protein.
Fig. 4.

Stability of SARS-CoV-2 Mpro complex in the presence of hypericin, SRT2104, and cyanidin-3-O-glucoside. The protein-ligand complexes comprised of a single compound bound to the active site of each protomer of Mpro, with cyanidin-3-O-glucoside depicted in purple (A) as an example. Average root mean square deviation (RMSD) for protein fit to backbone (B) for 100 ns, and average root mean square fluctuation of whole protein (C) following stabilisation. (D) shows the RMSF values the apo form subtracted from ligand bound forms of the protein. For the graphs (B – D), Mpro in its apo form is shown in blue. Mpro bound to hypericin, SRT2104, and cyanidin-3-O-glucoside is shown in red, green, and purple respectively.
Utilising trajectory segments spanning the final nanosecond of each system in triplicate, MM-PBSA was performed to determine binding free energy, as well as residue energy contributions to ligand binding (Fig. 5 ). Out of the three ligands, cyanidin-3-O-glucoside demonstrated the strongest ΔG to the active site of both protomers of Mpro, with values of -50.8 kcal/mol in protomer A and -42.1 kcal/mol in protomer B. ΔG values of approximately -20 kcal/mol were observed for hypericin bound to both protomers of Mpro and for SRT2104 bound to the active site of protomer B. It is noted that SRT2104 bound to the active site of protomer A demonstrates a noticeably weaker ΔG of -8.7 kcal/mol. This is due to unbinding events occurring in two out of three replicate simulations, with one of these shown in Movie S3. Consequently, while residue energy contributions to SRT2104 binding in the active site of protomer B are confined to active site residues, energy contributions in protomer A binding are also apparent in Val72, Arg76, and Asp92 in domain I, where SRT2104 attaches towards the end of the trajectory (Movie S3).
Fig. 5.

Average energy contributions from MM-PBSA analysis were decomposed into a per-residue basis for binding of SARS-CoV-2 Mpro with A) hypericin, B) SRT2104, and C) cyanidin-3-O-glucoside to the active site in protomer A (blue), and active site of protomer B (purple). Energy contributions were calculated in triplicate on 1000 ps segments of stabilised trajectories.
For SRT2104 bound to protomer B and hypericin bound to the active site of both protomers of Mpro, the main peaks in residue energy contributions were confined to active site residues surrounding the bound ligand in its corresponding protomer. With favourable energy contributions defined as peaks below the y-axis, active site residues Met49 and Met165 were relatively consistent in producing strong favourable energy contributions ranging from -0.5 to -1.3 kcal/mol. Glu166 produced unfavourable contributions, with values of +0.8 kcal/mol for hypericin bound to protomer A, +1.0 kcal/mol to protomer B, and +0.5 kcal/mol for SRT2104 bound to the active site of protomer B.
In contrast to hypericin and SRT2104, residue energy contributions for cyanidin-3-O-glucoside demonstrates large fluctuations across the entire protein, regardless of the protomer to which the ligand is bound. Energy peaks were nevertheless present at active site residues Ser1, Asp187, and Arg188. Peaks for Glu166 across both protomers were conversely favourable contributions. While more rigorous free energy methods may be required for further investigation, the dominating favourable energy contributions yield a strong ΔG of cyanidin-3-O-glucoside to the active site of Mpro, which is observed to remain stable throughout the trajectory (Movie S4).
3.3. Hypericin and cyanidin-3-O-glucoside as lead compounds for further evaluation
Cyanidin-3-O-glucoside belongs to the flavonoid subclass and is classified as an anthocyanin (Wongwichai et al., 2019). In general, anthocyanins are water-soluble pigments and they are present in a variety of plants and fruits (Wongwichai et al., 2019; Díaz-Mula et al., 2019). Their antioxidant, anti-inflammatory and neuroprotective properties have gained the attention of researchers and different methodologies are being trialled to assess how the bioavailability of these compounds could be increased (Mohammadi Pour et al., 2019a; Amin et al., 2017; Pojer et al., 2013). Pour et al. have also published a comprehensive review on the antiviral properties of anthocyanins and emphasised the need for novel drugs to be rapidly developed (Mohammadi Pour et al., 2019a). We also identified the sirtuin 1 (SIRT1) activator SRT2104 as a potential hit ligand (Hoffmann et al., 2013).
Furthermore, hypericin is a compound that is naturally found in the perennial plant Hypericum perforatum (St. John’s wort) and its mechanisms of action require further elucidation (Barnes et al., 2001; Schmidt and Butterweck, 2015; Brockmann et al., 1939; Jendželovská et al., 2016). Hypericin is considered to be a potent photosensitising agent and its potential use in cancer therapy has been investigated (Jendželovská et al., 2016). The antidepressant effects of St. John’s wort have also been reported in the literature (Schmidt and Butterweck, 2015). Likewise, the protective properties of hypericin against enveloped and non-enveloped viruses have been explored and previous studies have focused on hepatitis C, infectious bronchitis virus and human immunodeficiency virus 1 (HIV-1) (Hudson et al., 1993; Shih et al., 2018; Chen et al., 2019b; Jacobson et al., 2001; Chen et al., 2019c; Tang et al., 1990). It is important to note that drug-drug interactions have been described for hypericin and medications such as HIV protease inhibitors (Patel et al., 2004; Murtaza et al., 2017). Therefore, it is imperative that the pharmacokinetics of these compounds are taken into consideration. Hypericin and cyanidin-3-O-glucoside were consequently identified as the lead dietary compounds in this study and this is also consistent with a recently published paper by Islam et al. (Islam et al., 2020).
To confirm potential inhibition of the SARS-CoV-2 in vitro, we performed an ELISA using a commercially available 3CL protease (SARS-CoV-2) assay kit. The IC50 for the GC376 positive antiviral control used in this ELISA has been calculated to be 0.46 μM (as provided the kit supplier; BPS Bioscience). Our findings indicate that hypericin results in a concentration-dependent inhibition of Mpro activity, with an IC50 value calculated to be 63.6 ± 5.7 μM (Fig. 6 and Table 2 ). Similarly, cyanidin-3-O-glucoside and SRT2104 resulted in concentration-dependent inhibition of the Mpro with IC50 values calculated to be 65.1 ± 14.6 μM and 85.0 ± 16.8 μM, respectively (Fig. 6 and Table 2). Notably, IC50 values for SRT1720, resveratrol and l-sulforaphane could not be determined (up to 128 μM, Table 2), consistent with the lower binding energies observed for these compounds in the in silico work (Table S1). Similarly, the percentage protease inhibition at 50 μM GC376 was determined to be 97.9 ± 1.8 % in our experiments (n = 9 determinations, Fig. 6). As shown in Table 2, the percentage protease inhibition at 50 μM for our test inhibitors was lower with 42.8 ± 8.2 % and 22.6 ± 4.3 % inhibition of Mpro activity observed for hypericin and cyanidin-3-O-glucoside, respectively. Therefore, our findings clearly highlight a distinct order of potency of GC376 (covalent inhibitor) >>> hypericin and cyanidin-3-O-glucoside > SRT2104 > SRT1720 > >> resveratrol and l-sulforaphane.
Fig. 6.

Concentration-dependent inhibition of the SARS-CoV-2 Mpro by hypericin and cyanidin-3-O-glucoside. To confirm in vitro inhibition a 3CL protease ELISA assay was performed and fluorescence intensities at an emission of wavelength of 460 nm for concentrations of hypericin and cyanidin-3-O-glucoside up to 128 μM (doubling dilutions), were measured. The average background (n = 9 determinations), total enzymatic activity (n = 9 determinations), and inhibition by the covalent internal positive control, GC376 at 50 μM (n = 9 determinations), are highlighted (horizontal dashed lines). Hypericin (closed circles) and cyanidin-3-O-glucoside (open circles) were assayed in triplicate and the average ± SEM values are depicted. Concentration-dependent inhibition of the SARS-CoV-2 Mpro by hypericin and cyanidin-3-O-glucoside. To confirm in vitro inhibition a 3CL protease ELISA assay was performed and fluorescence intensities at an emission of wavelength of 460 nm for concentrations of hypericin and cyanidin-3-O-glucoside up to 128 μM (doubling dilutions), were measured. The average background (n = 9 determinations), total enzymatic activity (n = 9 determinations), and inhibition by the covalent internal positive control, GC376 at 50 μM (n = 9 determinations), are highlighted (horizontal dashed lines). Hypericin and cyanidin-3-O-glucoside were assayed in triplicate and the average ± SEM values are depicted.
Table 2.
Inhibition of Mpro activity by small molecules. Percentage inhibition at a ligand concentration of 50 μM and IC50 values calculated using an ELISA.
| Compound | IC50 (μM) Average ± SEM | % Protease inhibition at 50 μM ligand concentration |
|---|---|---|
| Hypericin | 63.6 ± 5.7 | 42.8 ± 8.2 |
| Cyanidin-3−0-glucoside | 65.1 ± 14.6 | 22.6 ± 4.3 |
| SRT2104 | 85.0 ± 16.8 | 19.4 ± 6.4 |
| SRT1720 | NA* | 9.3 ± 2.2 |
| Resveratrol | NA* | 1.8 ± 0.9 |
| L-sulforaphane | NA* | 8.4 ± 2.7 |
Compound concentrations of up to 128μM were used in our experiments; the IC50 could not be reached for the ligands noted.
4. Conclusion
Overall, our in silico findings and in vitro ELISA assay results indicate that, although not as potent as the covalent GC376 broad-spectrum antiviral, hypericin and cyandin-3-O-glucoside, may be considered as potential lead compounds as inhibitors of the SARS-CoV-2 Mpro. Given the considerable previous work with hypericin and cyandin-3-O-glucoside in models of disease, including previously reported antiviral effects, further investigation of these compounds in the context of COVID-19 is warranted.
Author contributions
TCK, KN, and AH conceptualized the aims and methodology, contributed to writing the original draft, and were involved in supervision. EP and JL performed formal data analysis, was involved in data curation, and contributed to writing the original draft. CK and KV performed formal data analysis and was involved in data curation. KKD performed formal data analysis and validation. All authors contributed to editing and reviewing the manuscript.
Declaration of Competing Interest
The authors report no declarations of interest.
Acknowledgements
We would like to acknowledge intellectual and financial support by McCord Research (Iowa, USA). JL and KKD are supported by an Australian Government Research Training Program Scholarship. We are indebted to Alfonso Perez Escudero and the team at Crowdfight COVID-19 for enabling access to supercomputing facilities, and to Matthew Gasperetti and the team at Hypernet Labs; Galileo, for enabling cloud computing for this project. We thank the National Computing Infrastructure (NCI), and the Pawsey Supercomputing Centre in Australia (funded by the Australian Government). Further, we thank the Spartan High Performance Computing service (University of Melbourne), and the Partnership for Advanced Computing in Europe (PRACE) for awarding the access to Piz Daint, hosted at the Swiss National Supercomputing Centre (CSCS), Switzerland.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.compbiolchem.2020.107408.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. [Google Scholar]
- Agbowuro A.A., Huston W.M., Gamble A.B., Tyndall J.D.A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018;38(4):1295–1331. doi: 10.1002/med.21475. [DOI] [PubMed] [Google Scholar]
- Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9(2):e00221–18. doi: 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn D.-G., Shin H.-J., Kim M.-H., Lee S., Kim M.-H., Myoung J. Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel Coronavirus Disease 2019 (COVID-19) J. Microbiol. Biotechnol. 2020;30(3):313–324. doi: 10.4014/jmb.2003.03011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat F., Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020 doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amici C., Di Caro A., Ciucci A., Chiappa L., Castilletti C., Martella V. Indomethacin has a potent antiviral activity against SARS coronavirus. Antivir. Ther. (Lond.) 2006;11(8):1021–1030. [PubMed] [Google Scholar]
- Amin F.U., Shah S.A., Badshah H., Khan M., Kim M.O. Anthocyanins encapsulated by PLGA@PEG nanoparticles potentially improved its free radical scavenging capabilities via p38/JNK pathway against Aβ(1-42)-induced oxidative stress. J. Nanobiotechnology. 2017;15(1):12-. doi: 10.1186/s12951-016-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes J., Anderson L.A., Phillipson J.D. St John’s wort (Hypericum perforatum L.): a review of its chemistry, pharmacology and clinical properties. J. Pharm. Pharmacol. 2001;53(5):583–600. doi: 10.1211/0022357011775910. [DOI] [PubMed] [Google Scholar]
- Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91(1):43–56. [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard M., Lev L., Greg S., Daniel T. 2017. Spartan HPC-Cloud Hybrid: Delivering Performance and Flexibility. [Google Scholar]
- Bjelkmar P., Larsson P., Cuendet M.A., Hess B., Lindahl E. Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010;6(2):459–466. doi: 10.1021/ct900549r. [DOI] [PubMed] [Google Scholar]
- Bonvino N.P., Liang J., McCord E.D., Zafiris E., Benetti N., Ray N.B. OliveNet™: a comprehensive library of compounds from Olea europaea. Database (Oxford). 2018;2018:bay016. doi: 10.1093/database/bay016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann H., Haschad M.N., Maier K., Pohl F. Über das Hypericin, den photodynamisch wirksamen Farbstoff aus Hypericum perforatum. Naturwissenschaften. 1939;27(32):550-. [Google Scholar]
- Carlesso A., Chintha C., Gorman A.M., Samali A., Eriksson L.A. Merits and pitfalls of conventional and covalent docking in identifying new hydroxyl aryl aldehyde like compounds as human IRE1 inhibitors. Sci. Rep. 2019;9(1):3407. doi: 10.1038/s41598-019-39939-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.N., Trinité B., Levy D.N. Potent inhibition of HIV-1 replication in resting CD4 T cells by resveratrol and pterostilbene. Antimicrob. Agents Chemother. 2017;61(9):e00408–17. doi: 10.1128/AAC.00408-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Chan K.H., Jiang Y., Kao R.Y.T., Lu H.T., Fan K.W. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J. Clin. Virol. 2004;31(1):69–75. doi: 10.1016/j.jcv.2004.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Muhammad I., Zhang Y., Ren Y., Zhang R., Huang X. Antiviral activity against infectious bronchitis virus and bioactive components of Hypericum perforatum L. Front. Pharmacol. 2019;10(1272) doi: 10.3389/fphar.2019.01272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Feng R., Muhammad I., Abbas G., Zhang Y., Ren Y. Protective effects of hypericin against infectious bronchitis virus induced apoptosis and reactive oxygen species in chicken embryo kidney cells. Poult. Sci. 2019;98(12):6367–6377. doi: 10.3382/ps/pez465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Muhammad I., Zhang Y., Ren Y., Zhang R., Huang X. Antiviral activity against infectious bronchitis virus and bioactive components of Hypericum perforatum L. Front. Pharmacol. 2019;10(1272) doi: 10.3389/fphar.2019.01272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho A.E., Guallar V., Berne B.J., Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005;26(9):915–931. doi: 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S., Olson A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Das P., Majumder R., Mandal M., Basak P. In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (M(pro)) from flavonoid based phytochemical constituents of Calendula officinalis. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1796799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B.E. Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J. Antimicrob. Chemother. 2010;65(Suppl 2):ii5–ii10. doi: 10.1093/jac/dkq015. (Suppl 2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denaro M., Smeriglio A., Barreca D., De Francesco C., Occhiuto C., Milano G. Antiviral activity of plants and their isolated bioactive compounds: an update. Phytother. Res. 2020;34(4):742–768. doi: 10.1002/ptr.6575. [DOI] [PubMed] [Google Scholar]
- Díaz-Mula H.M., Tomás-Barberán F.A., García-Villalba R. Pomegranate fruit and juice (cv. Mollar), rich in ellagitannins and anthocyanins, also provide a significant content of a wide range of proanthocyanidins. J. Agric. Food Chem. 2019;67(33):9160–9167. doi: 10.1021/acs.jafc.8b07155. [DOI] [PubMed] [Google Scholar]
- Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20(5):533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbinigie K., Frie K. Should chloroquine and hydroxychloroquine be used to treat COVID-19? A rapid review. Bjgp Open. 2020;4(2) doi: 10.3399/bjgpopen20X101069. bjgpopen20X101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E., Wald J., Lavu S., Roberts J., Beaumont C., Haddad J. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br. J. Clin. Pharmacol. 2013;75(1):186–196. doi: 10.1111/j.1365-2125.2012.04340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T., Wang J., Li Y., Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011;51(1):69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson J.B., Harris L., Towers G.H.N. The importance of light in the anti-HIV effect of hypericin. Antiviral Res. 1993;20(2):173–178. doi: 10.1016/0166-3542(93)90006-5. [DOI] [PubMed] [Google Scholar]
- Huemer H.P. Possible immunosuppressive effects of drug exposure and environmental and nutritional effects on infection and vaccination. Mediators Inflamm. 2015;2015:349176. doi: 10.1155/2015/349176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. 27-8. [DOI] [PubMed] [Google Scholar]
- Ibrahim M.A.A., Abdeljawaad K.A.A., Abdelrahman A.H.M., Hegazy M.F. Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: in-silico drug discovery. J. Biomol. Struct. Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1790037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa T., Hayashi R., Suzuki K., Imanishi S., Kambara K., Okazawa S. Sirtuin 1 activator SRT1720 suppresses inflammation in an ovalbumin-induced mouse model of asthma. Respirology. 2013;18(2):332–339. doi: 10.1111/j.1440-1843.2012.02284.x. [DOI] [PubMed] [Google Scholar]
- Islam R., Parves M.R., Paul A.S., Uddin N., Rahman M.S., Mamun A.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1761883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson J.M., Feinman L., Liebes L., Ostrow N., Koslowski V., Tobia A. Pharmacokinetics, safety, and antiviral effects of hypericin, a derivative of St. John’s wort plant, in patients with chronic hepatitis C virus infection. Antimicrob. Agents Chemother. 2001;45(2):517–524. doi: 10.1128/AAC.45.2.517-524.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendželovská Z., Jendželovský R., Kuchárová B., Fedoročko P. Hypericin in the light and in the dark: two sides of the same coin. Front. Plant Sci. 2016;7(560) doi: 10.3389/fpls.2016.00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Khaerunnisa S., Kurniawan H., Awaluddin R., Suhartati S., Soetjipto S. 2020. Potential Inhibitor of COVID-19 Main Protease (Mpro) From Several Medicinal Plant Compounds by Molecular Docking Study. [Google Scholar]
- Kim J.Y., Kim Y.I., Park S.J., Kim I.K., Choi Y.K., Kim S.-H. Safe, high-throughput screening of natural compounds of MERS-CoV entry inhibitors using a pseudovirus expressing MERS-CoV spike protein. Int. J. Antimicrob. Agents. 2018;52(5):730–732. doi: 10.1016/j.ijantimicag.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.E., Min J.S., Jang M.S., Lee J.Y., Shin Y.S., Song J.H. Natural bis-benzylisoquinoline alkaloids-tetrandrine, fangchinoline, and cepharanthine, inhibit human coronavirus OC43 infection of MRC-5 human lung cells. Biomolecules. 2019;9(11):696. doi: 10.3390/biom9110696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Chen J., Cheng T., Gindulyte A., He J., He S. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. Protein interfaces, surfaces and assemblies service PISA at European Bioinformatics Institute. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Krueger J.G., Suárez-Fariñas M., Cueto I., Khacherian A., Matheson R., Parish L.C. A randomized, placebo-controlled study of SRT2104, a SIRT1 activator, in patients with moderate to severe psoriasis. PLoS One. 2015;10(11) doi: 10.1371/journal.pone.0142081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Lynn A. g_mmpbsa—a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Lăcătușu C.-M., Grigorescu E.-D., Floria M., Onofriescu A., Mihai B.-M. The Mediterranean diet: from an environment-driven food culture to an emerging medical prescription. Int. J. Environ. Res. Public Health. 2019;16(6):942. doi: 10.3390/ijerph16060942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Mantelos A., Toh Z., Tortorella S., Ververis K., Vongsvivut J. Investigation of potential anti-pneumococcal effects of l-sulforaphane and metabolites: insights from synchrotron-FTIR microspectroscopy and molecular docking studies. J. Mol. Graph. Model. 2020;97 doi: 10.1016/j.jmgm.2020.107568. [DOI] [PubMed] [Google Scholar]
- Liang J., Pitsillou E., Karagiannis C., Darmawan K.K., Ng K., Hung A. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020 doi: 10.1016/j.compbiolchem.2020.107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.-C., Ho C.-T., Chuo W.-H., Li S., Wang T.T., Lin S.-C. Effective inhibition of MERS-CoV infection by resveratrol. BMC Infect. Dis. 2017;17(1):144. doi: 10.1186/s12879-017-2253-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.-H., Moses D.C., Hsieh C.-H., Cheng S.-C., Chen Y.-H., Sun C.-Y. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antiviral Res. 2018;150:155–163. doi: 10.1016/j.antiviral.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.-W., Hsieh P.-S., Lin C.-C., Hu M.-K., Huang S.-M., Wang Y.-M. Synergistic effects of combination treatment using EGCG and suramin against the chikungunya virus. Biochem. Biophys. Res. Commun. 2017;491(3):595–602. doi: 10.1016/j.bbrc.2017.07.157. [DOI] [PubMed] [Google Scholar]
- Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Maestro . LLC; New York, NY: Schrödinger: 2018. Schrödinger Release 2018-1. ed. [Google Scholar]
- Meng X.-Y., Zhang H.-X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Drug Des. 2011;7(2):146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadamtousi S.Z., Kadir H.A., Hassandarvish P., Tajik H., Abubakar S., Zandi K. A review on antibacterial, antiviral, and antifungal activity of curcumin. Biomed Res. Int. 2014;2014 doi: 10.1155/2014/186864. 186864- [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam E., Teoh B.-T., Sam S.-S., Lani R., Hassandarvish P., Chik Z. Baicalin, a metabolite of baicalein with antiviral activity against dengue virus. Sci. Rep. 2014;4(1):5452. doi: 10.1038/srep05452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi Pour P., Fakhri S., Asgary S., Farzaei M.H., Echeverría J. The signaling Pathways, and therapeutic targets of antiviral agents: focusing on the antiviral approaches and clinical perspectives of anthocyanins in the management of viral diseases. Front. Pharmacol. 2019;10:1207-. doi: 10.3389/fphar.2019.01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi Pour P., Fakhri S., Asgary S., Farzaei M.H., Echeverría J. The signaling pathways, and therapeutic targets of antiviral agents: focusing on the antiviral approaches and clinical perspectives of anthocyanins in the management of viral diseases. Front. Pharmacol. 2019;(1207):10. doi: 10.3389/fphar.2019.01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. Hints of hope with remdesivir. Nat. Rev. Drug Discov. 2020;19(6):373. doi: 10.1038/d41573-020-00088-y. [DOI] [PubMed] [Google Scholar]
- Murtaza G., Ullah N., Mukhtar F., Nawazish S., Muneer S., Mariam Phytotherapeutics: the emerging role of intestinal and hepatocellular transporters in dug interactions with botanical supplements. Molecules. 2017;22(10):1699. doi: 10.3390/molecules22101699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S., Singh M., Ravichandiran V., Murty U.S.N., Srivastava H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J., Buddha B., Dey S., Pal D., Mitra A. In vitro interaction of the HIV protease inhibitor ritonavir with herbal constituents: changes in P-gp and CYP3A4 activity. Am. J. Ther. 2004;11:262–277. doi: 10.1097/01.mjt.0000101827.94820.22. [DOI] [PubMed] [Google Scholar]
- Peiris J.S.M., Guan Y., Yuen K.Y. Severe acute respiratory syndrome. Nat. Med. 2004;10(12):S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E., Walters W.P., Charifson P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56(2):235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- Phua J., Weng L., Ling L., Egi M., Lim C.M., Divatia J.V. Intensive care management of coronavirus disease 2019 (COVID-19): challenges and recommendations. Lancet Respir. Med. 2020;8(5):506–517. doi: 10.1016/S2213-2600(20)30161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59(14):6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pojer E., Mattivi F., Johnson D., Stockley C.S. The case for anthocyanin consumption to promote human health: a review. Compr. Rev. Food Sci. Food Saf. 2013;12(5):483–508. doi: 10.1111/1541-4337.12024. [DOI] [PubMed] [Google Scholar]
- Richardson P., Griffin I., Tucker C., Smith D., Oechsle O., Phelan A. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395(10223) doi: 10.1016/S0140-6736(20)30304-4. e30-e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogosnitzky M., Okediji P., Koman I. Cepharanthine: a review of the antiviral potential of a Japanese-approved alopecia drug in COVID-19. Pharmacol. Rep. 2020 doi: 10.1007/s43440-020-00132-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa S.G.V., Santos W.C. Clinical trials on drug repositioning for COVID-19 treatment. Rev. Panam. Salud Publica. 2020;44 doi: 10.26633/RPSP.2020.40. e40-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagnella D.E., Voth G.A. Structure and dynamics of hydronium in the ion channel gramicidin A. Biophys. J. 1996;70(5):2043–2051. doi: 10.1016/S0006-3495(96)79773-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia S., Bordoloi M. Molecular docking: challenges, advances and its use in drug discovery perspective. Curr. Drug Targets. 2019;20(5):501–521. doi: 10.2174/1389450119666181022153016. [DOI] [PubMed] [Google Scholar]
- Scarpino A., Ferenczy G.G., Keserű G.M. Comparative evaluation of covalent docking tools. J. Chem. Inf. Model. 2018;58(7):1441–1458. doi: 10.1021/acs.jcim.8b00228. [DOI] [PubMed] [Google Scholar]
- Schmidt M., Butterweck V. The mechanisms of action of St. John’s wort: an update. Wiener Med. Wochenschr. 2015;165(11):229–235. doi: 10.1007/s10354-015-0372-7. [DOI] [PubMed] [Google Scholar]
- Shih C.-M., Wu C.-H., Wu W.-J., Hsiao Y.-M., Ko J.-L. Hypericin inhibits hepatitis C virus replication via deacetylation and down-regulation of heme oxygenase-1. Phytomedicine. 2018;46:193–198. doi: 10.1016/j.phymed.2017.08.009. [DOI] [PubMed] [Google Scholar]
- Tang J., Colacino J.M., Larsen S.H., Spitzer W. Virucidal activity of hypericin against enveloped and non-enveloped DNA and RNA viruses. Antiviral Res. 1990;13(6):313–325. doi: 10.1016/0166-3542(90)90015-y. [DOI] [PubMed] [Google Scholar]
- Thomford N.E., Senthebane D.A., Rowe A., Munro D., Seele P., Maroyi A. Natural products for drug discovery in the 21st century: innovations for novel drug discovery. Int. J. Mol. Sci. 2018;19(6):1578. doi: 10.3390/ijms19061578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Qamar M.T., Alqahtani S.M., Alamri M.A., Chen L.-L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020 doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankadari N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents. 2020;56(2) doi: 10.1016/j.ijantimicag.2020.105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J. CHARMM General Force Field (CGenFF): a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Sun H., Yao X., Li D., Xu L., Li Y. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. J. Chem. Soc. Faraday Trans. 2016;18(18):12964–12975. doi: 10.1039/c6cp01555g. [DOI] [PubMed] [Google Scholar]
- Wongwichai T., Teeyakasem P., Pruksakorn D., Kongtawelert P., Pothacharoen P. Anthocyanins and metabolites from purple rice inhibit IL-1β-induced matrix metalloproteinases expression in human articular chondrocytes through the NF-κB and ERK/MAPK pathway. Biomed. Pharmacother. 2019;112 doi: 10.1016/j.biopha.2019.108610. [DOI] [PubMed] [Google Scholar]
- World Health Organization . World Health Organization; Geneva: 2020. Coronavirus Disease 2019 (COVID-19): Situation Report - 51.https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200311-sitrep-51-covid-19.pdf Available from: [Google Scholar]
- Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82(5):2515. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L., Li Z., Yuan K., Qu X., Chen J., Wang G. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J. Virol. 2004;78(20):11334–11339. doi: 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakaryan H., Arabyan E., Oo A., Zandi K. Flavonoids: promising natural compounds against viral infections. Arch. Virol. 2017;162(9):2539–2551. doi: 10.1007/s00705-017-3417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020 doi: 10.1126/science.abb3405. eabb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Zhao Y., Jin Z., Liu X., Yang H., Rao Z. 2020. The Crystal Structure of COVID-19 Main Protease in Apo Form. 10.2210/pdb6M03/pdb. [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Fang Y., Xu T., Ni W.-J., Shen A.-Z., Meng X.-M. Potential therapeutic targets and promising drugs for combating SARS-CoV-2. Br. J. Pharmacol. 2020;177(14):3147–3161. doi: 10.1111/bph.15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6(1):14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.