

SUMMARY
Vesicle budding for Golgi-to-plasma membrane trafficking is a key step in secretion. Proteins that induce curvature of the Golgi membrane are predicted to be required, by analogy to vesicle budding from other membranes. Here, we demonstrate that GOLPH3, upon binding to the phosphoinositide PI4P, induces curvature of synthetic membranes in vitro and the Golgi in cells. Moreover, efficient Golgi-to-plasma membrane trafficking critically depends on the ability of GOLPH3 to curve the Golgi membrane. Interestingly, uncoupling of GOLPH3 from its binding partner MYO18A results in extensive curvature of Golgi membranes, producing dramatic tubulation of the Golgi, but does not support forward trafficking. Thus, forward trafficking from the Golgi to the plasma membrane requires the ability of GOLPH3 both to induce Golgi membrane curvature and to recruit MYO18A. These data provide fundamental insight into the mechanism of Golgi trafficking and into the function of the unique Golgi secretory oncoproteins GOLPH3 and MYO18A.
INTRODUCTION
Vesicle trafficking allows for protein and lipid movement between organelles and to the extracellular space. Key to vesicle trafficking is the process by which vesicles bud from their originating membrane. Vesicle budding is well-studied in endocytosis at the plasma membrane (PM) and trafficking from the endoplasmic reticulum (ER). These examples demonstrate a critical dependence on proteins that sculpt the lipid bilayer (Daumke et al., 2014; Lee et al., 2005; McMahon and Boucrot, 2011; Saheki and De Camilli, 2012). The implication is that vesicle budding, in general, requires proteins with the ability to induce membrane curvature to promote bud formation.
It seems likely that other, less understood, trafficking events also involve proteins that induce membrane curvature. Exit of vesicles from the trans-Golgi is a key step in the secretory pathway that remains poorly understood (Farhan, 2015; Kienzle and von Blume, 2014; Makowski et al., 2017; Mironov et al., 2016). A deficiency in our understanding of Golgi-to-PM trafficking is a lack of identification of membrane-curving proteins that function to promote Golgi vesicle exit.
The GOLPH3 protein promotes efficient trafficking from the Golgi to the PM (Buschman et al., 2015; Makowski et al., 2017). GOLPH3 is an abundant protein (Dippold et al., 2009; Ng et al., 2013) that localizes to the cytosolic face of the trans-Golgi and to vesicles that bud from the trans-Golgi (Bell et al., 2001; Dippold et al., 2009; Snyder et al., 2005; Wu et al., 2000). GOLPH3 localizes to the Golgi via its tight interaction with phosphatidylinositol-4-phosphate (PI4P; Dippold et al., 2009), found in abundance in the cytosolic leaflet of the trans-Golgi (Godi et al., 1999, 2004; Wang et al., 2003). GOLPH3 further interacts with the unconventional myosin, MYO18A (Buschman and Field, 2018b; Dippold et al., 2009; Ng et al., 2013; Taft et al., 2013), linking the Golgi to the F-actin cytoskeleton (Dippold et al., 2009; Lazaro-Dieguez et al., 2006; Ng et al., 2013). PI4P, GOLPH3, MYO18A, and F-actin are all required for exit of vesicles from the trans-Golgi for efficient forward trafficking, as detected by trafficking of PI4P-positive, cargo-positive vesicles (Dippold et al., 2009). Accordingly, unbiased pulse-chase analysis of total secretory trafficking reveals that knockdown of GOLPH3 or MYO18A arrests overall secretion nearly as completely as treatment with Golgi poisons Brefeldin A or Golgicide A (Ng et al., 2013). Likewise, GOLPH3, MYO18A, and F-actin are required for efficient Golgi-to-PM trafficking of the vesicular stomatitis virus G glycoprotein (VSVG; Dippold et al., 2009; Farber-Katz et al., 2014; Hirschberg et al., 1998; Lazaro-Dieguez et al., 2007), and exit of Hepatitis C virus from infected cells (Bishe et al., 2012). Overall, the data indicate that the PI4P/GOLPH3/MYO18A/F-actin complex plays a role in vesicle exit from the Golgi for efficient forward trafficking to the PM. The discovery of the GOLPH3 pathway has led to the identification of regulatory pathways that converge on the GOLPH3 complex to modulate Golgi function (Blagoveshchenskaya et al., 2008; Farber-Katz et al., 2014; Kuna and Field, 2019; Makowski et al., 2017; Ng et al., 2013).
Interference with the GOLPH3 complex results in impaired trafficking, and also changes in the shape of the Golgi (as observed by light and electron microscopy [EM]), suggesting that the complex normally exerts a tensile force on the Golgi (Dippold et al., 2009; Farber-Katz et al., 2014; Ng et al., 2013; Xie et al., 2018). Previously, we proposed a role for the tensile force that is generated by the PI4P/GOLPH3/MYO18A/F-actin complex in the process of vesicle budding (Buschman et al., 2015; Dippold et al., 2009; Ng et al., 2013).
Here, we demonstrate that GOLPH3 alone (independently of MYO18A) drives PI4P-dependent membrane curvature both in vitro and in cells, and that the ability of GOLPH3 to induce membrane curvature is required for efficient Golgi-to-PM trafficking. However, GOLPH3-induced curvature of the Golgi, while necessary, is not sufficient to enable trafficking. Rather, efficient trafficking also depends on GOLPH3 interaction with MYO18A. Interference with the engagement of MYO18A by GOLPH3 causes accumulation of highly curved Golgi membranes, manifested as tubules, and accumulation of cargo in the Golgi. These results provide important insight into the mechanism of trafficking from the Golgi and identify GOLPH3 as a long-sought, major membrane-curving protein required for efficient Golgi forward trafficking.
RESULTS
GOLPH3 Binding Causes Tubulation of PI4P-Containing Liposomes
Upon docking the x-ray crystal structure of GOLPH3 (Wood et al., 2009) to a lipid bilayer containing PI4P, we observed that the β-loop comprising amino acids 190–201 became embedded in the lipid bilayer (Figure 1A; see also Dippold et al., 2009). Consistent with this being a physiologic interaction with the membrane, the β-loop is highly hydrophobic (amino acid sequence Phe-Leu-Leu-Phe at the tip). Notably, the length of this protruding loop, ~12 Å, is sufficient for insertion half-way into the lipid bilayer, affecting the proximal (cytosolic-facing) leaflet, but not the distal (luminal-facing) leaflet. Partial membrane insertion is known to generate membrane curvature by increasing the area of one leaflet relative to the other (Daumke et al., 2014; Hu et al., 2011; McMahon and Boucrot, 2015). Thus, we predicted that GOLPH3 binding to a lipid bilayer might induce curvature.
Figure 1. GOLPH3 Binding to PI4P Causes Liposomal Tubulation.
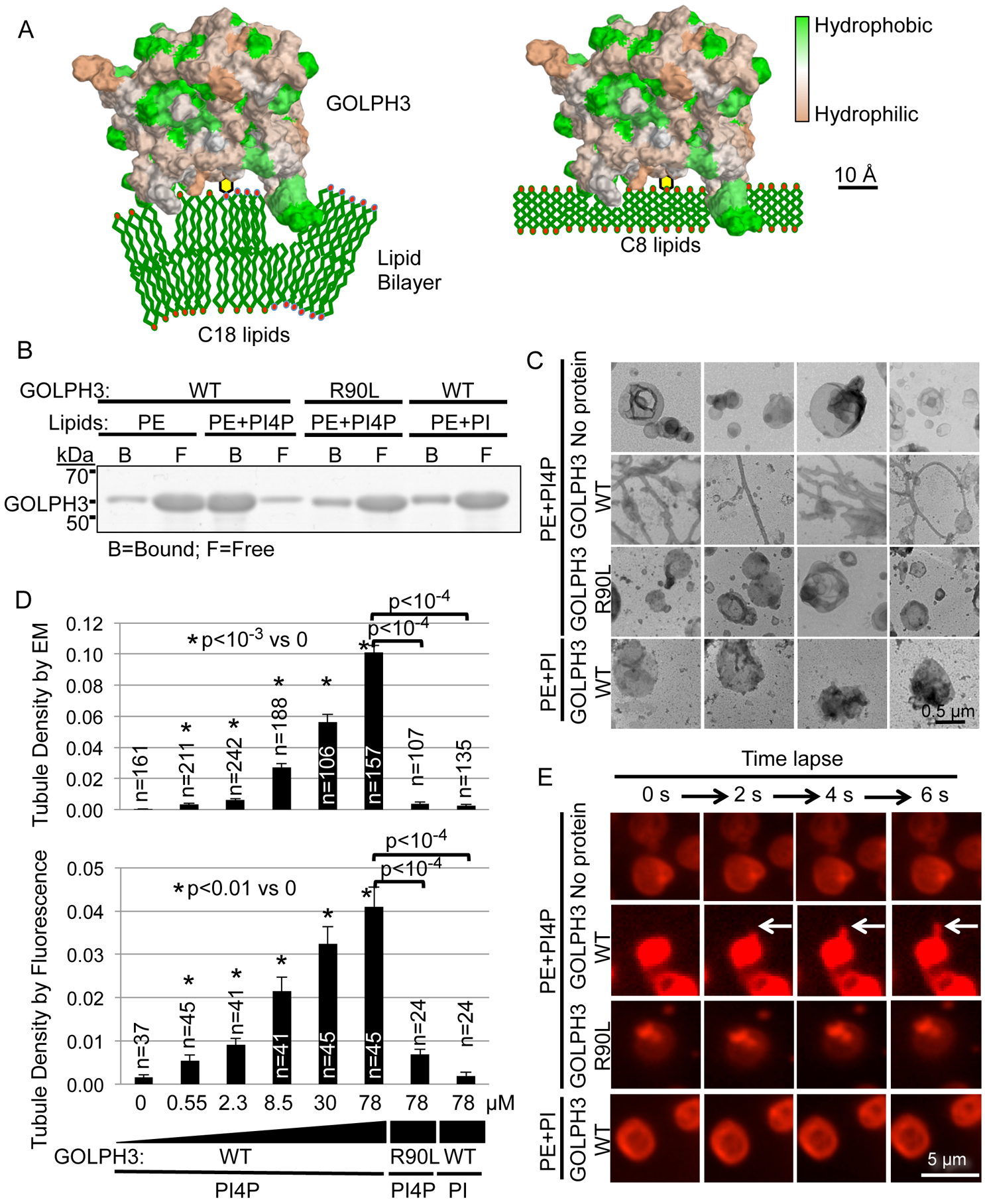
(A) Model of GOLPH3 (hydrophobicity/hydrophilicity surface map from PDB 3KN1) binding to PI4P in natural liposomes (C18 and higher) causing membrane curvature (left), whereas binding to short-chain (C8) liposomes does not (right; see also Figure 3).
(B) Liposomes that contain 1.8 mol% PI4P, PI, or no phosphoinositide were incubated with 14 μM bacterially expressed GST-GOLPH3 (WT or the R90L mutant that does not bind PI4P). Liposome-bound and free protein were separated by ultracentrifugation and detected by SDS-PAGE/Coomassie Brilliant Blue.
(C) Aliquots of the binding reactions from (B) were examined by EM. Four representative images are included for each condition. See also Figures S1A–D for comparison to the EPSIN ENTH domain and examples of vesicles containing PS or PC.
(D) Quantification of (C) and (E), measuring tubulation in response to increasing concentrations of GOLPH3 (WT or the R90L mutant). Graphs indicate mean ± SEM of tubule density observed by EM (upper graph) or by fluorescence imaging (lower graph). The number (n) of randomly selected fields imaged is indicated, pooled from four (EM) or four (fluorescence) independent experiments. p values are indicated, calculated by t test.
(E) Nile Red-labeled liposomes containing 1.8 mol% PI4P, PI, or no phosphoinositide were examined by confocal fluorescence time-lapse imaging (2 seconds per frame) upon addition of the indicated proteins (at 14 μM). See also Movie S1.
We examined binding of bacterially expressed, purified GOLPH3 to liposomal vesicles in vitro. As reported previously (Dippold et al., 2009), GOLPH3 bound specifically to PI4P-containing vesicles, but the R90L mutation within the PI4P-binding pocket impaired binding (Figure 1B). Next, we used EM to observe vesicle morphology. Strikingly, upon binding to GOLPH3, the liposomal vesicles were reshaped into extended tubules (Figure 1C). Quantification indicated highly significant tubulation dependent on PI4P and an intact PI4P-binding pocket in GOLPH3 (Figure 1D). Notably, this tubulation occurred at physiological GOLPH3 concentrations (Dippold et al., 2009; Ng et al., 2013). Comparison to a bona fide membrane-curving protein domain, the EPSIN ENTH domain (Ford et al., 2001), revealed that when the two bound similarly to their cognate lipids (PI(4,5)P2 for ENTH, PI4P for GOLPH3; Figure S1A), GOLPH3 was at least as efficient as EPSIN ENTH at inducing membrane curvature (Figure S1B and S1C). We also examined whether phosphatidylethanolamine (PE) in the vesicles was playing a role, or could be substituted by other lipids, such as phosphatidylcholine (PC) or phosphatidylserine (PS). GOLPH3 produced dramatic tubulation of vesicles containing these lipids, again dependent on its ability to bind to PI4P (Figure S1D). Thus, GOLPH3 binding to PI4P-containing vesicles efficiently produces highly curved membranes.
Observation of lipid vesicles by EM requires staining and dehydration, potentially introducing artefactual structures. Therefore, we examined the effect of GOLPH3 on liposomal vesicles under physiological aqueous conditions. We performed time-lapse fluorescence microscopy of Nile Red-labeled vesicles as we added GOLPH3. Within seconds we observed prominent tubule formation, but not in the absence of PI4P or in response to GOLPH3-R90L (Figure 1E, Movie S1). Quantification of tubule density demonstrated results similar to those observed by EM (Figure 1D). We conclude that GOLPH3 binding alone is sufficient to induce curvature of PI4P-containing lipid membranes.
Overexpression of GOLPH3 Causes Tubulation of the Golgi in Cells
In cells, GOLPH3 localizes to the trans-Golgi (Dippold et al., 2009; Snyder et al., 2005) in a PI4P-dependent manner (Dippold et al., 2009). We wondered whether GOLPH3 binding to the PI4P-rich trans-Golgi membrane generates curvature and therefore induces tubulation of the Golgi. Although ultimately (in data presented below) we will test the role of endogenous GOLPH3, we first examined the effect of overexpression of GOLPH3 tagged with the fluorescent protein mKO2. We performed time-lapse confocal fluorescence microscopy of HeLa cells co-expressing a Golgi marker, the minimal Golgi-localizing region of α-mannosidase II fused to GFP (ManII-GFP; Cole et al., 1996; Velasco et al., 1993), and mKO2-GOLPH3. Compared to cells expressing mKO2 alone, cells expressing mKO2-GOLPH3 exhibited large numbers of tubules dynamically extending from the Golgi (Figure 2A, Movie S2). These tubules were marked by both ManII-GFP and by mKO2-GOLPH3, demonstrating that these tubules arise from the Golgi and that mKO2-GOLPH3 is bound to them. The ability of mKO2-GOLPH3 to induce Golgi tubulation was dependent on its ability to bind PI4P, as the R90L mutant did not drive Golgi tubulation. Quantification indicated that mKO2-GOLPH3 significantly drove tubulation of the Golgi in HeLa cells (Figure 2B). Since overexpressed ManII-GFP is found at both medial- and trans-Golgi, to further confirm the nature of the tubules, we examined the effect of mCherry-GOLPH3 on a marker highly localized to the trans-Golgi, α−2,6-sialyltransferase-GFP (SialT-GFP; Berger et al., 1987; Roth et al., 1985). We observed Golgi tubules decorated with both mCherry-GOLPH3 and SialT-GFP (Figure S1E), demonstrating GOLPH3-driven tubulation of the trans-Golgi. To determine if the ability of mKO2-GOLPH3 to drive tubulation of the Golgi is idiosyncratic to HeLa cells, or is more generally true, we also examined HEK293 cells. In HEK293 cells, mKO2-GOLPH3 also drove Golgi tubulation, dependent on its ability to bind PI4P (Figure 2C).
Figure 2. Expression of mKO2-GOLPH3 Drives Golgi Tubulation.
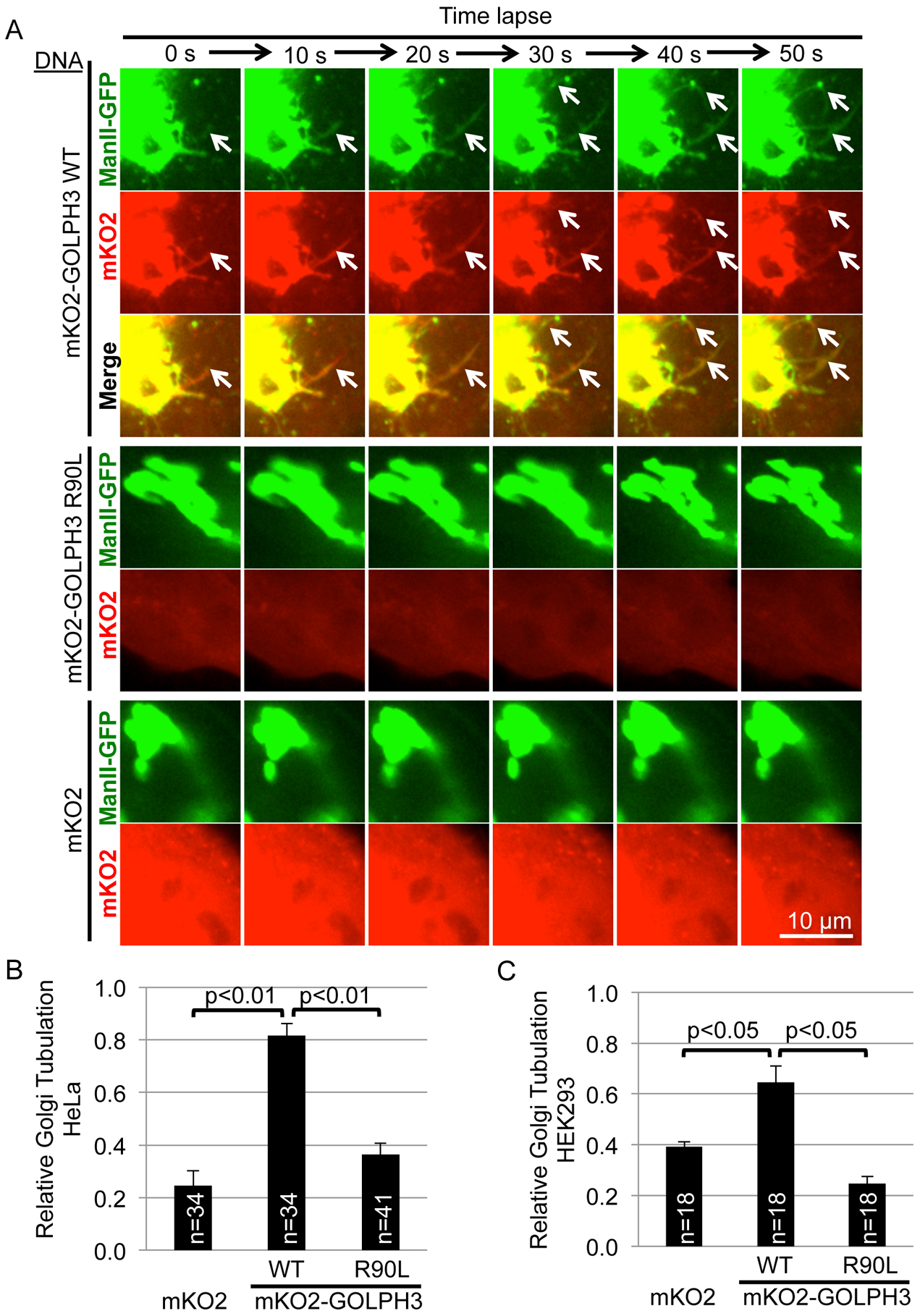
(A) HeLa cells stably overexpressing ManII-GFP to mark the Golgi were transfected to express mKO2-GOLPH3 (WT), mKO2-GOLPH3-R90L, or mKO2 and imaged every 10 seconds. White arrows indicate Golgi tubulation. See also Movie S2 (time-lapse) and Figure S1E (using trans-Golgi marker SialT-GFP).
(B and C) Quantification of (A) and a similar experiment performed in HEK293 cells (C). Graphs in (B) and (C) indicate mean ± SEM of relative Golgi tubulation for n randomly chosen fields pooled from five (B) or three (C) independent experiments. p values are indicated, calculated by t test.
Membrane Insertion of the Hydrophobic β-Loop of GOLPH3 Drives Tubulation
Modeling predicted that insertion of the hydrophobic β-loop of GOLPH3 into the proximal leaflet would drive membrane curvature (Figure 1A, left panel). If the model is correct, then GOLPH3 binding to PI4P-containing liposomes should protect the hydrophobic β-loop from solvent, detectable by decreased hydrogen-deuterium exchange when placed in deuterated buffer. Therefore, we performed deuterium exchange mass spectrometry (DXMS) to compare deuterium exchange rates of GOLPH3 alone versus GOLPH3 bound to PI4P-containing liposomes (Figure 3A). Validating the assay, we observed significant protection from solvent (reduced exchange) of R90, R171, and R174, amino acid residues involved in binding PI4P (Dippold et al., 2009). Furthermore, the hydrophobic β-loop was protected from solvent, indicative of insertion into the lipid bilayer. To further test this model, we eliminated the hydrophobicity of the β-loop by mutating two leucine residues at the tip to glutamic acid, generating the GOLPH3-L195E/L196E mutant. As shown in Figure S1F, this mutation strongly interfered with GOLPH3 binding to PI4P-containing vesicles, consistent with the model that the hydrophobic β-loop embeds into the bilayer upon GOLPH3 binding to PI4P.
Figure 3. GOLPH3-Induced Membrane Tubulation Depends on Insertion of the Hydrophobic β-Loop Partially into the Bilayer.
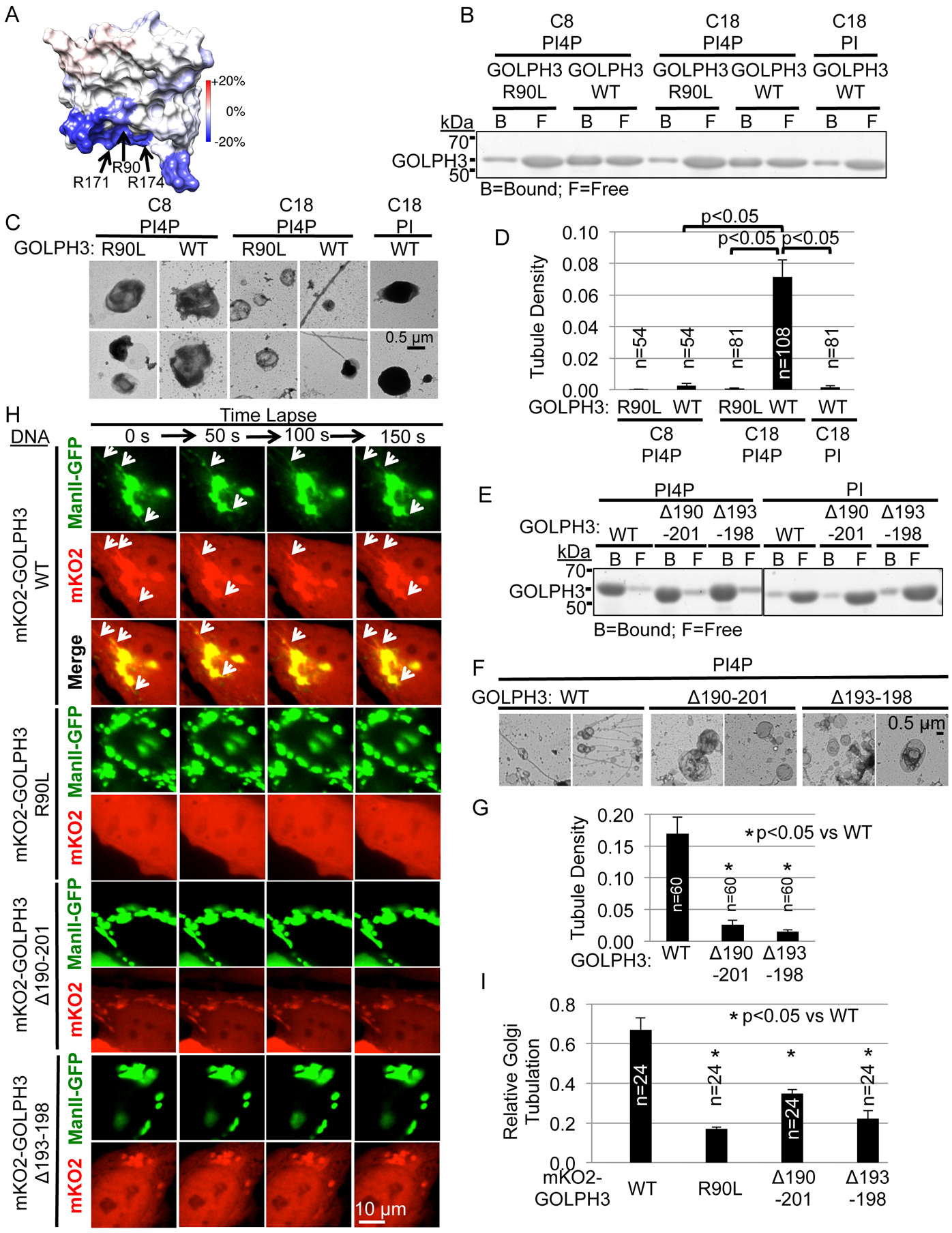
(A) Deuterium exchange mass spectrometry (DXMS) comparing solvent accessibility of GOLPH3 alone versus bound to PI4P-containing liposomes. Changes in deuterium exchange are mapped onto the solvent-accessible surface of GOLPH3 (PDB 3KN1). Residues R90, R171, and R174, previously implicated in ligating PI4P, are protected upon binding (decreased exchange), providing validation. The hydrophobic β-loop is strongly protected.
(B) GOLPH3 binds to both natural (C18 or greater) and short-chain (C8) lipid vesicles containing PI4P. To compensate for intrinsically reduced binding to C8 liposomes, PI4P in C18 vesicles was reduced (414 μM for C8 [2.7 mol%], 23 μM for C18 [0.15 mol%]) to produce similar binding of GOLPH3 to C8 and C18 vesicles to allow fair comparison of tubulation. Binding of GST-GOLPH3 (WT or R90L mutant) at 14 μM to the liposomes was assessed by separation into lipid-bound and free fractions by ultracentrifugation, quantified by SDS-PAGE/Coomassie Brilliant Blue (Figure S1G).
(C) Aliquots of the binding reactions from (B) were examined by EM. Two representative examples are provided for each. Tubulation was observed upon GOLPH3 binding to long-chain, but not short-chain liposomes.
(D) Quantification of liposomal tubulation for (C).
(E) Deletion of the hydrophobic β-loop does not significantly affect binding to PI4P. PI4P- or PI-containing liposomes (at 4.2 mol%) were incubated with 1.4 μM GST-GOLPH3, WT or the Δ190–201 or Δ193–198 mutants that delete the hydrophobic β-loop, separated from free protein by ultracentrifugation, and analyzed by SDS-PAGE/Coomassie Brilliant Blue.
(F) Aliquots of the binding reactions from (E) were examined by EM. Deletion of the hydrophobic β-loop significantly impairs GOLPH3-induced tubulation of lipid vesicles.
(G) Quantification of liposomal tubulation for (F).
(H) Confocal fluorescence time-lapse imaging of HeLa cells expressing ManII-GFP and mKO2-GOLPH3 WT or mutants. White arrows indicate Golgi tubulation. Deletion of the hydrophobic β loop impaired GOLPH3-driven Golgi tubulation. See also Movie S3.
(I) Quantification of (H). All graphs indicate mean ± SEM and the number of fields examined (n), pooled from four (D), three (G), or four (I) independent experiments. p values are indicated, calculated by t test.
The length of the protrusion of the hydrophobic β-loop relative to the thickness of the lipid bilayer establishes the geometry such that in natural membranes with phospholipid chain lengths of C18 (or greater), the hydrophobic β-loop would extend into the proximal leaflet, without entering the distal leaflet. The model predicts that a bilayer of short chain-length lipids would allow protrusion of the loop into the distal leaflet, resulting in less membrane curvature (Figure 1A, right panel). To test this prediction, we compared GOLPH3 tubulation of vesicles composed of lipids with chain lengths characteristic of the trans-Golgi (C18 or longer) (Mitra et al., 2004) versus short chain-length lipids (C8). By including a high concentration of C8 PI4P, C8 lipids produced vesicles that bound to GOLPH3 at least as well as do the C18 vesicles (Figures 3B and S1G). Binding of GOLPH3 to these vesicles showed specificity, as the R90L mutation significantly impaired binding. We then examined vesicle morphology using EM (Figure 3C and 3D). While GOLPH3 induced tubulation of vesicles composed of C18 lipids, it was unable to induce tubulation of vesicles composed of C8 lipids, despite equal binding by GOLPH3. These data support the model that partial membrane insertion of the hydrophobic β-loop of GOLPH3 is responsible for generating the membrane curvature that produces tubules.
The Hydrophobic β-Loop Is Required for GOLPH3-Induced Tubulation of Membranes in Vitro and in Cells
To test directly the role of the hydrophobic β-loop, we generated mutants of GOLPH3 in which the entire loop is deleted (GOLPH3 Δ190–201) or the hydrophobic tip of the loop is deleted (GOLPH3 Δ193–198). Both mutants still bound to PI4P-containing liposomes (Figure 3E), demonstrating that the proteins are properly folded. However, these mutant GOLPH3 proteins failed to induce tubulation of the liposomes (Figure 3F and 3G). Thus, the ability of GOLPH3 to generate membrane curvature of liposomes in vitro is dependent on the hydrophobic β-loop.
We next examined whether deletion of the hydrophobic β-loop of GOLPH3 affected its ability to drive Golgi tubulation. We expressed mKO2-tagged GOLPH3 in HeLa cells, comparing the wild-type protein to the R90L, Δ190–201, and Δ193–198 mutants (Figures 3H and 3I, and Movie S3). As previously shown, wild-type GOLPH3 localized to the Golgi and drove Golgi tubulation. As expected, the R90L mutant, unable to bind to PI4P, did not localize to the Golgi and did not induce Golgi tubulation. Furthermore, the Δ190–201 and Δ193–198 mutants did localize to the Golgi but were unable to drive Golgi tubulation (Figure 3H and 3I). Thus, the ability of GOLPH3 to generate curvature of the Golgi membrane is dependent on GOLPH3’s hydrophobic β-loop.
The GOLPH3 Hydrophobic β-Loop Is Not Required for Normal Golgi Morphology
GOLPH3 and its interaction with MYO18A are required to maintain the normal shape of the Golgi as a ribbon that extends around the nucleus (Dippold et al., 2009; Farber-Katz et al., 2014; Ng et al., 2013; examined in more detail below). To test whether deletion of the hydrophobic β-loop interferes with the ability of GOLPH3/MYO18A to stretch the Golgi around the nucleus, we performed GOLPH3 siRNA knockdown/rescue experiments. Immunofluorescence (IF) to the Golgi marker GM130 and to GOLPH3 allowed assessment of Golgi morphology (Figures 4A and 4B) and GOLPH3 expression levels (Figures 4A and S2A). As expected, siRNA knockdown of GOLPH3 (or MYO18A) caused compaction of the Golgi. Expression of siRNA-resistant wild-type GOLPH3 restored GOLPH3 expression levels and Golgi morphology, while the R90L mutant, unable to localize to the Golgi, did not restore Golgi morphology. Likewise, the L195E/L196E mutant, placing charged residues at the tip of the hydrophobic β-loop, prevented Golgi localization, and was ineffective at restoring Golgi morphology. By contrast, the hydrophobic β-loop deletion mutants, Δ190–201 and Δ193–198, both localized partly to the Golgi and both significantly restored Golgi morphology to its normal extended form. We conclude that while the hydrophobic β-loop of GOLPH3 is required for GOLPH3 to induce membrane curvature, the β-loop and the ability to induce membrane curvature are dispensable for localization to the Golgi and for interaction with MYO18A to enable stretching of the Golgi around the nucleus.
Figure 4. GOLPH3’s Hydrophobic β-Loop is Required for Forward Trafficking from the Golgi to the Plasma Membrane.
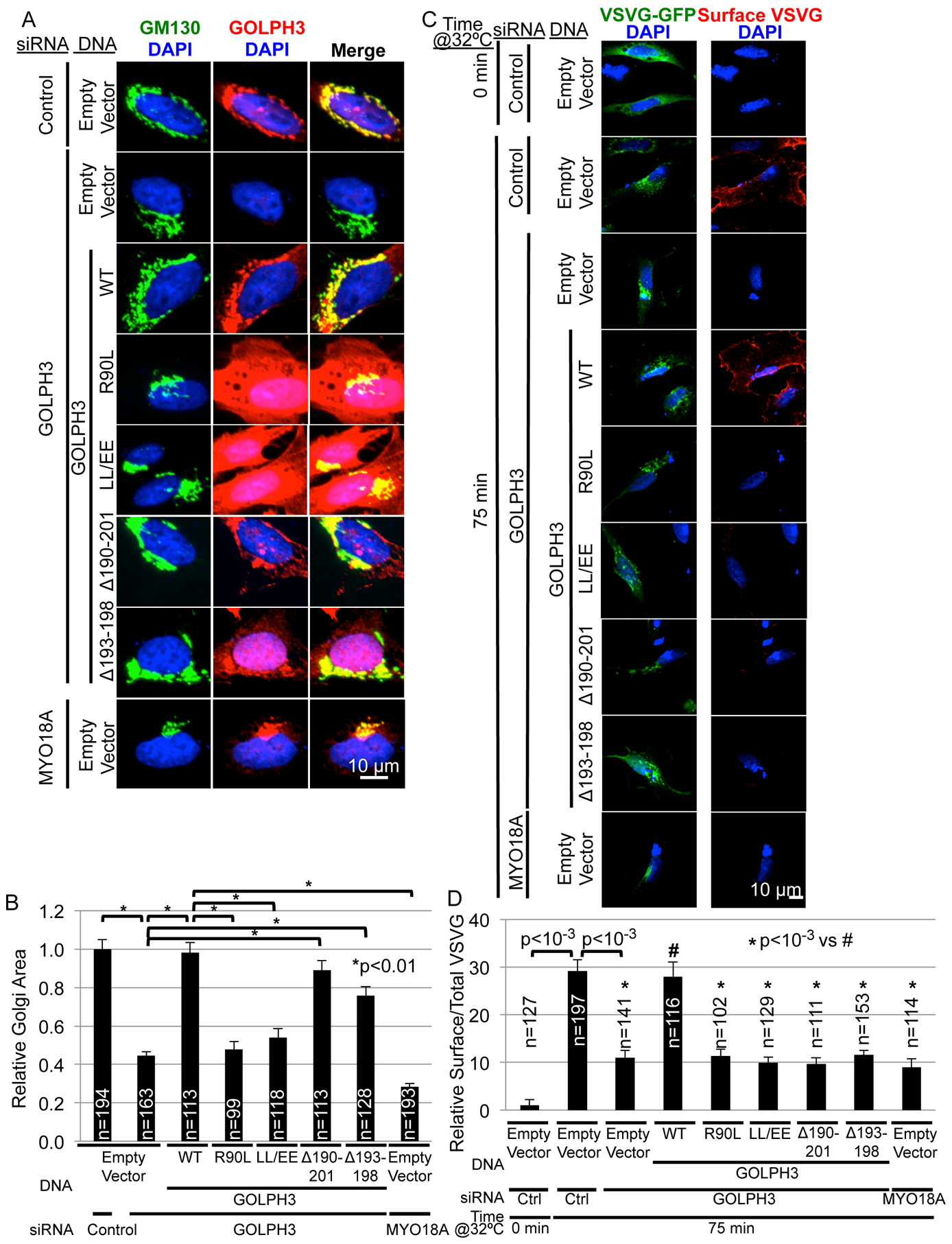
(A) The hydrophobic β-loop is not required for GOLPH3-dependent extended Golgi morphology. GOLPH3 knockdown/rescue experiments were performed in HeLa cells by transfection of GOLPH3-specific siRNA (or control or MYO18A siRNA) followed by transfection of expression vectors for siRNA-resistant GOLPH3 (WT), the indicated mutants, or the empty vector. Cells were examined by IF to the Golgi (GM130) and GOLPH3.
(B) Quantification of Golgi area for (A). Quantification of GOLPH3 expression is provided in Figure S2A.
(C) Trafficking of ts045-VSVG-GFP to the PM was examined in a GOLPH3 knockdown/rescue experiment in HeLa cells (see Figure S2B for data demonstrating that VSVG-GFP is found in mKO2-GOLPH3-driven Golgi tubules and Figures S2C–F for assessment of ER-to-Golgi and Golgi-to-ER trafficking). HeLa cells were transfected with indicated siRNA and expression vector for siRNA-resistant GOLPH3 (WT), the indicated mutants, or empty vector, together with ts045-VSVG-GFP. Trafficking was arrested at the ER at 40°C (0 minutes at 32°C), followed by release at 32°C. IF was used to examine surface VSVG in unpermeabilized cells, followed by permeabilization to stain for GOLPH3 (Figure S2G).
(D) Quantification of relative surface VSVG normalized to total VSVG per cell (C). Graphs in (B) and (D) show mean ± SEM, with the number of cells (n) indicated, pooled from four (B) or six (D) experiments. p values are indicated, calculated by t test. In (A) and (C) maximum projections of z-stacks are shown.
GOLPH3-Induced Membrane Curvature is Required for Efficient Golgi-to-Plasma Membrane Trafficking
To assess whether GOLPH3-induced curvature of the Golgi plays a role in Golgi-to-PM trafficking, we first assessed whether cargo is present in GOLPH3-driven Golgi tubules. We co-expressed in HeLa cells mKO2-GOLPH3 to drive Golgi tubulation and the experimental cargo ts045-VSVG-GFP (Presley et al., 1997). At the permissive temperature VSVG-GFP colocalized with mKO2-GOLPH3 on tubules emanating from the Golgi (Figure S2B). Thus, cargo is found within the GOLPH3-driven Golgi tubules.
We next examined trafficking of ts045-VSVG-GFP. As expected, in control cells maintained at 40°C, we observed ts045-VSVG-GFP at the ER, with minimal levels detectable at the PM (Figures 4C and 4D). Shift to the permissive temperature of 32°C for 75 minutes, to allow ts045-VSVG-GFP to fold properly and traffic, resulted in detection of high levels at the cell surface. Furthermore, siRNA knockdown of GOLPH3 or MYO18A prevented trafficking to the PM, with ts045-VSVG-GFP accumulating at the Golgi, as expected (Dippold et al., 2009).
Next, we examined ER-to-Golgi trafficking of ts045-VSVG-GFP, by performing a shift to 22°C, which enables ER-to-Golgi trafficking, with arrest at the Golgi (Matlin and Simons, 1983). Compared to the siRNA control, knockdown of GOLPH3 or MYO18A resulted in an increase in VSVG at the Golgi (Figure S2C). In control cells at 22°C, some VSVG was still delivered to the PM (Figure S2D) via the Golgi (blocked by Golgicide A), and dependent on GOLPH3 and MYO18A. Thus, knockdown of GOLPH3 or MYO18A impairs Golgi-to-PM trafficking, while still allowing normal ER-to-Golgi trafficking. Next, we assessed Golgi-to-ER recycling using mCherry-KDEL. mCherry-KDEL levels at the Golgi were low in control cells and remained similarly low upon knockdown of GOLPH3 or MYO18A (Figures S2E and S2F), indicating that recycling to the ER remained intact.
Having established that knockdown of GOLPH3 or MYO18A specifically interferes with Golgi-to-PM trafficking, we next performed GOLPH3 rescue experiments. Our data demonstrate that the GOLPH3 hydrophobic β-loop deletion mutants, Δ190–201 and Δ193–198, are still able to bind to PI4P, localize to the Golgi, and drive MYO18A-dependent extension of the Golgi ribbon. However, these mutants are defective in inducing membrane curvature, seen as a lack of tubulation of vesicles in vitro and of the Golgi in cells. Therefore, these mutants allowed us to ask whether the ability of GOLPH3 to drive membrane curvature is required for Golgi-to-PM trafficking. In the GOLPH3 knockdown cells, expression of siRNA-resistant, wild-type GOLPH3 restored normal levels of GOLPH3 (Figure S2G) and restored trafficking to the PM (Figures 4C and 4D). As expected, the R90L and L195E/L196E mutants that do not localize to the Golgi, did not rescue Golgi-to-PM trafficking. Finally, the GOLPH3 Δ190–201 and Δ193–198 mutants were both unable to rescue Golgi-to-PM trafficking. We conclude that efficient Golgi-to-PM trafficking depends on GOLPH3-induced membrane curvature, which depends on GOLPH3’s hydrophobic β-loop.
Tagged GOLPH3 Drives Tubulation More Effectively than Untagged GOLPH3
As shown above, expression of mKO2-GOLPH3 induced striking Golgi tubulation. To examine whether the mKO2 tag contributed to this tubulation, we compared mKO2-GOLPH3 to mCherry-GOLPH3, tags that differ by sequence and structure. Both were similarly effective at inducing Golgi tubulation (Figures S3A and S3B). Furthermore, 3xHA-GOLPH3 or 3xMYC-GOLPH3, with small amino-terminal epitope tags, efficiently tubulated the Golgi (Figure S3C and Movie S4).
We next compared Golgi tubulation by untagged and amino-terminally tagged GOLPH3. Since mCherry and mKO2 behaved interchangeably, we compared mKO2-GOLPH3 to bicistronic GOLPH3-IRES-mCherry (using an internal ribosome entry site (IRES) to co-express untagged GOLPH3 and mCherry). Tagged GOLPH3 induced tubulation of the Golgi significantly more efficiently than did untagged GOLPH3 (Figures 5A and 5B and Movie S5). We conclude that although the nature of the tag is unimportant, placing an amino-terminal tag on GOLPH3 enhances its ability to drive Golgi tubulation.
Figure 5. An N-Terminal Tag Impairs GOLPH3’s Interaction with MYO18A.
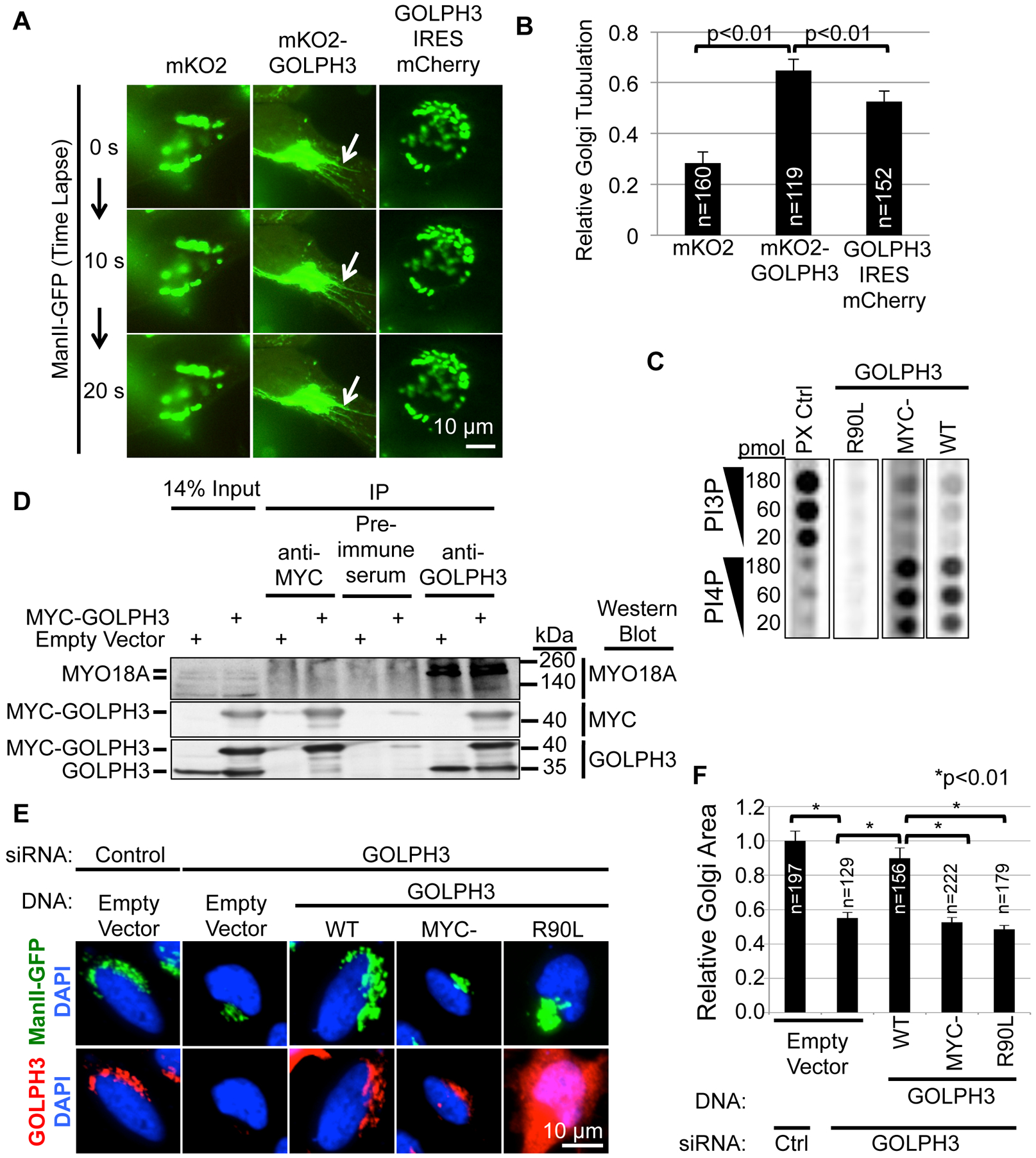
(A) Overexpression of N-terminally tagged GOLPH3 significantly increases Golgi tubulation compared to untagged GOLPH3. Confocal time-lapse images were obtained every 10 seconds of HeLa cells expressing ManII-GFP and mKO2, mKO2-GOLPH3, or GOLPH3-IRES-mCherry. White arrows indicate Golgi tubulation. See also Movie S5.
(B) Quantification of Golgi tubulation for (A). See also Figures S3A–C, and Movie S4.
(C) 3xMYC-tagged GOLPH3 binds PI4P similarly to untagged (WT) GOLPH3. 35S-Met-labeled proteins produced by in vitro transcription and translation (expression validated by SDS-PAGE, see Figure S3D) were used for lipid blotting to detect binding to a dilution series of PI3P and PI4P. The PX control protein (CG4960) bound preferentially to PI3P, as expected (Dippold et al., 2009).
(D) MYO18A co-IP’d with GOLPH3, but not with 3xMYC-GOLPH3. HeLa cells were transfected with either expression vector for 3xMYC-GOLPH3 or empty vector. IPs using pre-immune serum (negative control), anti-GOLPH3, or anti-MYC antibodies were examined by Western blotting against MYC and GOLPH3 (to validate the IP) and MYO18A (to detect co-IP). While IP of GOLPH3 robustly co-IPs MYO18A, IP of 3xMYC-GOLPH3 does not.
(E) 3xMYC-GOLPH3 is unable to restore normal Golgi extended ribbon morphology in cells depleted of endogenous GOLPH3. HeLa cells stably expressing ManII-GFP were transfected with control or GOLPH3-specific siRNA and expression vectors for siRNA-resistant GOLPH3 (WT), 3xMYC-GOLPH3, GOLPH3-R90L, or empty vector. IF was performed to assess GOLPH3 levels (see Figure S3E) and Golgi morphology. Maximum projections of z-stacks are shown.
(F) Quantification of Golgi area for (E). See also Figure S3E for GOLPH3 expression. Graphs in (B) and (F) show mean ± SEM, with the number of cells (n) indicated, pooled from three (B) or four (F) experiments. p values are indicated, calculated by t test.
Tagging GOLPH3 Disrupts its Interaction with MYO18A
To determine why an amino-terminal tag enhances GOLPH3-driven Golgi tubulation, we examined the effect of the tag on GOLPH3’s known interactions with PI4P and MYO18A (Dippold et al., 2009; Farber-Katz et al., 2014; Ng et al., 2013; Taft et al., 2013; Wood et al., 2009). First, GOLPH3 and 3xMYC-GOLPH3 bound similarly to PI4P, without significant binding to PI3P (Figures 5C and S3D). Likewise, tagged GOLPH3 localized to the Golgi (e.g., Figures 2A, 3H, 5E, and 6A), which is known to reflect binding to PI4P (Dippold et al., 2009).
Figure 6. Golgi Tubulation is Driven by Endogenous GOLPH3, but MYO18A Reduces the Appearance of Tubules.
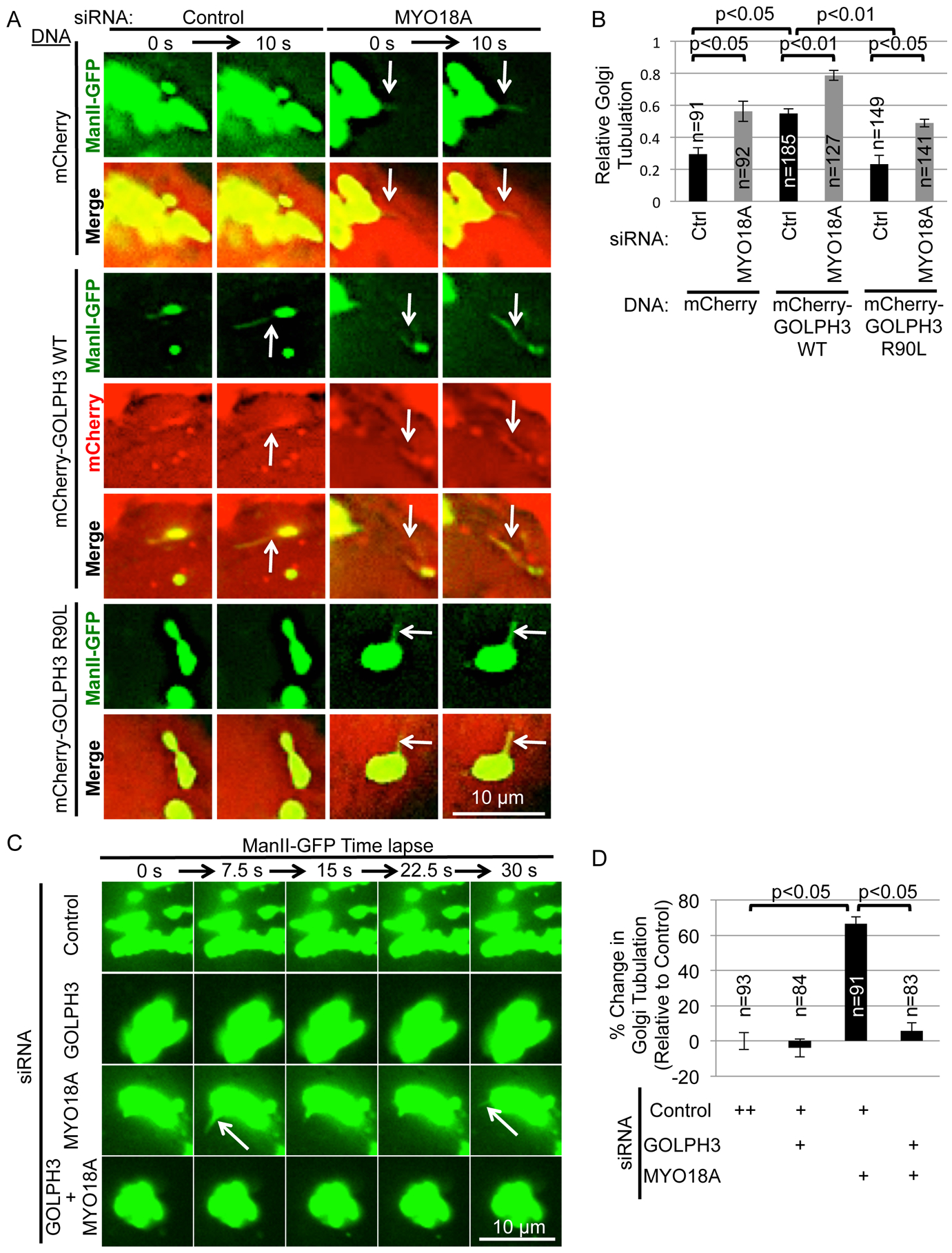
(A) Golgi tubulation is significantly increased in cells depleted for MYO18A. HeLa cells stably expressing ManII-GFP were transfected with either control or MYO18A-specific siRNA oligonucleotides and expression vectors for mCherry, mCherry-GOLPH3 (WT), or mCherry-GOLPH3-R90L. Protein expression and knockdown were validated by Western blotting of parallel coverslips (Figure S4A). Confocal time-lapse images were captured every 10 seconds. See also Movie S6.
(B) Quantification of Golgi tubulation for (A). Depletion of MYO18A results in enhanced Golgi tubulation, even without overexpression of tagged GOLPH3. See Figure S4B and Movie S7 for demonstration that loss of F-actin also drives Golgi tubulation, and Figure S4C–D for demonstration that endogenous GOLPH3 is found on Golgi tubules that occur upon depletion of MYO18A, but that ARF1 is generally not on GOLPH3-driven tubules.
(C) Golgi tubulation seen in MYO18A-depleted cells is dependent on endogenous GOLPH3. HeLa cells stably expressing ManII-GFP were transfected with combinations of control, GOLPH3, and MYO18A-specific siRNA oligonucleotides. Protein knockdown was validated by Western blotting of parallel coverslips (Figure S4E). Confocal time-lapse images were captured every 7.5 seconds. See also Movie S8. Figures S5A and B and Movie S9 demonstrate that rescue with wild-type GOLPH3 restores Golgi tubulation, dependent on GOLPH3’s ability to bind PI4P and to induce membrane curvature.
(D) Quantification of Golgi tubulation for (C). Graphs in (B) and (D) show mean ± SEM, with the number of cells (n) indicated, pooled from six (B) or four (D) independent experiments. p values are indicated, calculated by t test. White arrows indicate Golgi tubulation.
Next, we examined the interaction of GOLPH3 with MYO18A. As expected, IP of endogenous GOLPH3 robustly co-precipitated MYO18A (Figure 5D). When 3xMYC-GOLPH3 was expressed, anti-GOLPH3 also IP’d 3xMYC-GOLPH3, but without a significant change in the co-IP of MYO18A. Specific IP of 3xMYC-GOLPH3 using an anti-MYC antibody did not co-IP MYO18A. These data indicate that 3xMYC-GOLPH3 does not interact with MYO18A.
Tagged GOLPH3 Does Not Rescue Normal Extended Golgi Morphology
Since the GOLPH3/MYO18A complex is required to maintain the normal extended morphology of the Golgi (Dippold et al., 2009; Ng et al., 2013), we predicted that the inability of amino-terminally tagged GOLPH3 to interact with MYO18A would render it ineffective to rescue normal Golgi morphology. We performed GOLPH3 siRNA knockdown/rescue experiments observing the Golgi marker ManII-GFP and GOLPH3 levels by IF (Figures 5E, 5F, and S3E). As expected, siRNA knockdown of GOLPH3 caused a reduction of GOLPH3 levels (Figure S3E) and compaction of the Golgi (Figures 5E and 5F). Furthermore, expression of siRNA-resistant, wild-type GOLPH3 restored GOLPH3 levels and normal Golgi morphology, while the R90L mutant did not. Unlike GOLPH3, 3xMYC-GOLPH3, despite restoration of GOLPH3 protein levels at the Golgi, was unable to rescue Golgi morphology. Thus, while amino-terminally tagged GOLPH3 binds to PI4P, localizes to the Golgi, and induces Golgi tubulation, it does not interact with MYO18A, and therefore is unable to restore the Golgi’s normal extended morphology.
MYO18A is Not Required for, and Instead Reduces the Appearance of GOLPH3-Dependent Golgi Tubules
The observation that amino-terminally tagged GOLPH3 can induce Golgi tubulation, but does not interact with MYO18A, raised the question as to whether MYO18A contributes to GOLPH3-induced tubulation of the Golgi. Indeed, in all published data the phenotype of GOLPH3 depletion is indistinguishable from the phenotype of MYO18A depletion, and further, MYO18A depletion interferes with phenotypes caused by overexpression of GOLPH3 (Bishe et al., 2012; Dippold et al., 2009; Farber-Katz et al., 2014; Ng et al., 2013; Xie et al., 2018; Xing et al., 2016). Therefore, we examined the effect of knockdown of MYO18A on the ability of GOLPH3 to drive Golgi tubulation. HeLa cells expressing the Golgi marker ManII-GFP were transfected with siRNA to MYO18A (or control), and with expression vectors for mCherry, mCherry-GOLPH3 (WT), or mCherry-GOLPH3-R90L. As expected, expression of mCherry-GOLPH3 increased Golgi tubulation compared to mCherry or mCherry-GOLPH3-R90L (Figures 6A and 6B, and Movie S6). Unexpectedly, siRNA knockdown of MYO18A (confirmed by Western blot, Figure S4A) resulted in increased tubulation of the Golgi, even in control cells expressing only mCherry (or mCherry-GOLPH3-R90L). Thus, loss of MYO18A is sufficient to cause increased tubulation of the Golgi.
Since MYO18A’s function is thought to depend on the actin cytoskeleton (Buschman and Field, 2018a), we tested whether depolymerization of F-actin would cause increased tubulation of the Golgi. We examined cells expressing the Golgi marker ManII-GFP and either mKO2 or mKO2-GOLPH3 (Figure S4B and Movie S7). As expected, prior to treatment, cells expressing mKO2-GOLPH3 exhibited increased Golgi tubulation compared to cells expressing mKO2. In both cases, treatment with cytochalasin B resulted in an increase in Golgi tubulation, consistent with MYO18A acting together with F-actin to suppress Golgi tubulation.
We wondered whether Golgi tubulation due to loss of MYO18A is driven by endogenous GOLPH3. First, IF to observe endogenous GOLPH3 demonstrated that, indeed, the tubules that form upon knockdown of MYO18A are decorated with GOLPH3 (Figure S4C). For comparison, we examined whether ARF1 is found on GOLPH3-driven Golgi tubules, observing that, in general, it is not present (Figure S4D).
Next, we examined the effect of knockdown of GOLPH3 on Golgi tubulation observed in response to knockdown of MYO18A. Increased Golgi tubulation caused by knockdown of MYO18A was ablated by knockdown of GOLPH3 (Figures 6C, 6D, S4E, and Movie S8). We conclude that endogenous GOLPH3 is sufficient to drive tubulation of the Golgi, but that endogenous MYO18A and F-actin reduce the appearance of tubules.
To validate the knockdown and further interrogate the role of GOLPH3, we performed GOLPH3 knockdown/rescue experiments in the context of MYO18A knockdown (Figures S5A and S5B and Movie S9). Expression of siRNA-resistant WT GOLPH3 (together with tdTomato to mark transfected cells) restored tubulation caused by MYO18A knockdown. As a control, GOLPH3-R90L was ineffective. Furthermore, the Δ190–201 and Δ193–198 hydrophobic β-loop deletion mutants were unable to support Golgi tubulation. Thus, the tubulation that occurs upon knockdown of MYO18A depends on GOLPH3, the ability of GOLPH3 to bind to PI4P, and the ability of GOLPH3 to induce membrane curvature.
GOLPH3 Interaction with MYO18A is Required for Golgi Forward Trafficking
Our data indicate that endogenous MYO18A reduces the appearance of Golgi tubules driven by endogenous GOLPH3. This could either reflect a role for MYO18A to prevent GOLPH3-driven trafficking, or the tubulation could represent the accumulation of a trafficking intermediate that depends on MYO18A for progression to productive trafficking. Having shown that GOLPH3-induced membrane curvature is required for Golgi forward trafficking, we tested whether the ability of GOLPH3 to induce Golgi tubulation is sufficient for Golgi forward trafficking or if GOLPH3 interaction with MYO18A is also required. 3xMYC-GOLPH3 binds to PI4P, localizes to the Golgi, and induces Golgi tubulation, but does not interact with MYO18A, providing a means to test whether PI4P-binding, Golgi localization, and Golgi tubulation by GOLPH3 are sufficient for forward trafficking. Therefore, we performed GOLPH3 siRNA knockdown/rescue experiments to assess ts045-VSVG-GFP trafficking to the PM (Figures 7A and 7B). In control cells, shift to the permissive temperature allowed trafficking to the PM that was blocked at the Golgi by knockdown of GOLPH3 or MYO18A. The defect in trafficking due to knockdown of GOLPH3 was rescued by expression of untagged, siRNA-resistant GOLPH3 (wild-type), but not GOLPH3-R90L. Finally, 3xMYC-GOLPH3 was unable to restore trafficking to the PM. Quantification revealed a profound defect in the ability of 3xMYC-GOLPH3 to restore trafficking compared to untagged, wild-type GOLPH3 (Figure 7B). We conclude that the ability of GOLPH3 to induce tubulation, although required, is not sufficient to allow Golgi forward trafficking, but that forward trafficking also requires that GOLPH3 interact with MYO18A.
Figure 7. GOLPH3 Interaction with MYO18A is Required for Golgi-to-PM Trafficking.
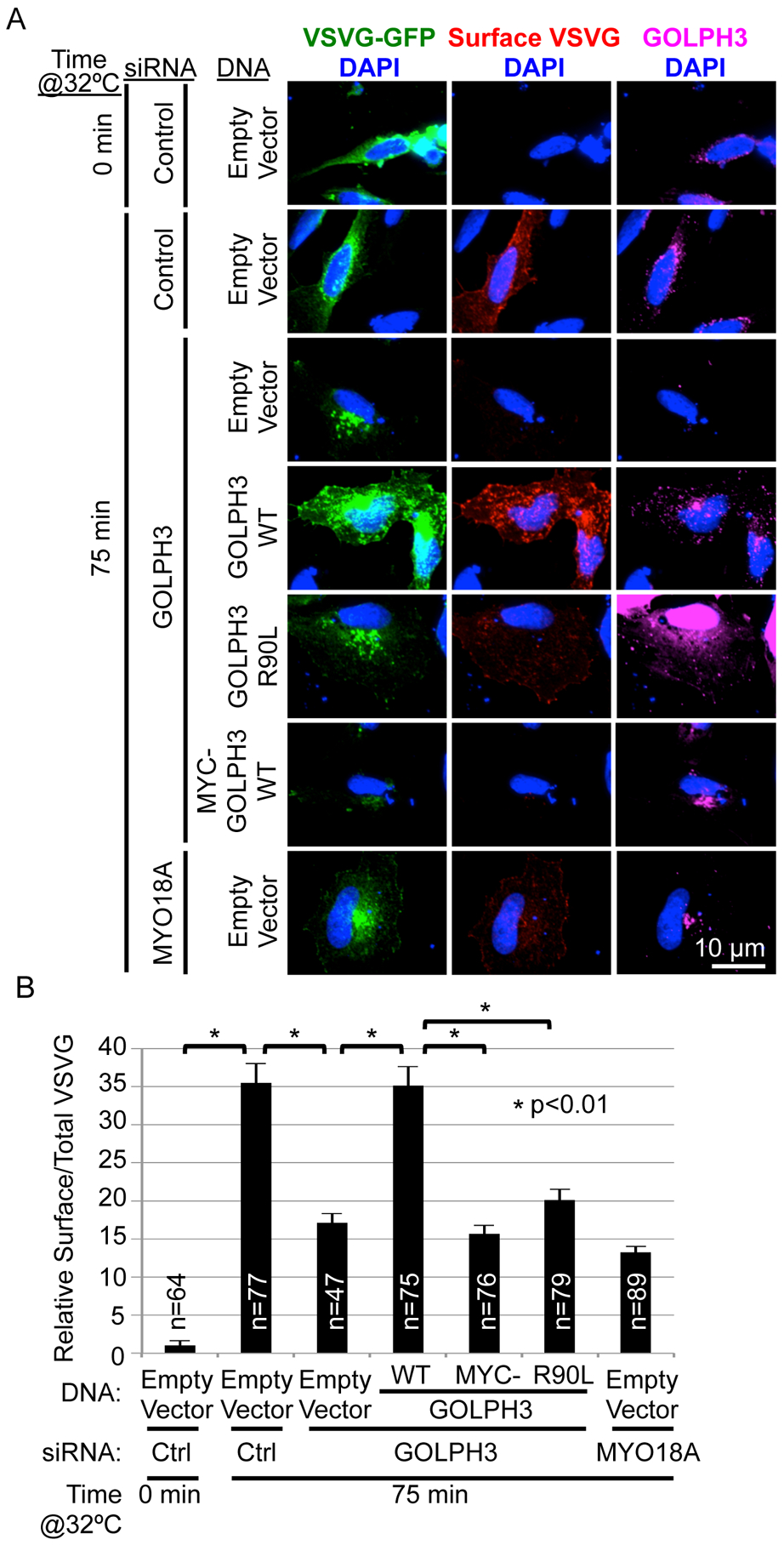
(A) A GOLPH3 knockdown/rescue experiment was performed to examine trafficking of the experimental cargo ts045-VSVG-GFP. HeLa cells were transfected with control, GOLPH3, or MYO18A-specific siRNA oligonucleotides and expression vectors for ts045-VSVG-GFP and for siRNA-resistant GOLPH3 (WT), GOLPH3-R90L, 3xMYC-GOLPH3 (WT), or empty vector. After incubation at 40°C overnight, cells were shifted to 32°C for 75 min to allow trafficking, and then fixed and stained for surface VSVG, followed by permeabilization and staining for GOLPH3 and DAPI. Maximum projections of z-stacks are shown.
(B) Quantification of VSVG-GFP trafficking for (A). Graph shows mean ± SEM, with the number of cells (n) indicated, pooled from three experiments. p values are indicated, calculated by t test.
DISCUSSION
A large and growing body of data establish a role for GOLPH3 in the process of budding of vesicles from the trans-Golgi for trafficking to the PM (Bishe et al., 2012; Dippold et al., 2009; Farber-Katz et al., 2014; Ng et al., 2013). Analogies to other, better understood mechanisms of vesicle budding during endocytosis (Daumke et al., 2014; McMahon and Boucrot, 2011; Saheki and De Camilli, 2012) or trafficking from the ER to the Golgi (Lee et al., 2005) raise the suspicion that Golgi-to-PM trafficking must also involve mechanisms to induce Golgi membrane curvature. Here we find that GOLPH3 serves that role, and that the ability of GOLPH3 to induce membrane curvature is required for normal Golgi-to-PM trafficking.
Nevertheless, the ability of GOLPH3 to induce membrane curvature is insufficient for forward trafficking. Expression of amino-terminally tagged GOLPH3, unable to interact with MYO18A, or knockdown of MYO18A results in both excessive tubulation of the Golgi and ineffective trafficking. Thus, the tubulation observed, rather than a component of productive trafficking, occurs with arrest of trafficking, and thus reflects a frustrated intermediate. We conclude that efficient forward trafficking from the Golgi to the PM depends on both the induction of membrane curvature by GOLPH3, and GOLPH3’s recruitment of MYO18A, which has been observed to apply a tensile force to the Golgi membrane (Dippold et al., 2009; Farber-Katz et al., 2014; Ng et al., 2013).
Analogy to Endocytosis
The role of GOLPH3 in trafficking at the Golgi bears a striking resemblance to the mechanism of trafficking observed in endocytosis. Endocytosis involves proteins that induce membrane curvature to promote vesicle budding (Daumke et al., 2014; McMahon and Boucrot, 2011; Saheki and De Camilli, 2012), proteins such as SNX9 that link the membrane of the budding endocytic vesicle to the generation of actin scaffolds (Yarar et al., 2007), and myosin motors that act on actin to drive the endocytic vesicle away from the PM (Buss et al., 2001; Chandrasekar et al., 2014; Spudich et al., 2007; Yu et al., 2009). Thus, endocytosis requires proteins to induce membrane curvature and to link to a myosin motor to enable effective vesicle formation. We observe marked similarity at the Golgi, where GOLPH3 serves both to drive membrane curvature and to link the membrane to MYO18A, both of which are required for productive vesicle trafficking.
Conservation Through Evolution
In budding yeast, it has been proposed that secretory trafficking from the Golgi to the PM involves an unknown adaptor protein that binds to PI4P and also binds to the type 5 myosin, MYO2 (Santiago-Tirado et al., 2011). The analogy to the role of GOLPH3 in mammalian cells is striking. Indeed, the yeast ortholog of GOLPH3, VPS74, binds PI4P and localizes to the late Golgi (Dippold et al., 2009). The structure of the PI4P binding domain of VPS74 is remarkably similar to that of human GOLPH3, including conservation of the hydrophobic β-loop, suggesting that the yeast protein will also induce curvature of the Golgi membrane upon binding to PI4P (Dippold et al., 2009; Ng et al., 2013; Schmitz et al., 2008; Wood et al., 2009). Furthermore, comparison of GOLPH3 paralogs across species suggests that myosin binding is likely to be conserved in lower organisms (Ng et al., 2013). Thus, we would propose that VPS74 is likely to be the “unknown” adaptor protein that is postulated in yeast to bridge PI4P and MYO2.
We cannot discount the possibility (some might argue likelihood) that there exist multiple mechanisms for Golgi-to-PM trafficking. Indeed, redundancy is a common theme in biology. Nevertheless, the defect in overall secretory trafficking observed upon loss of GOLPH3 or MYO18A (Bishe et al., 2012; Dippold et al., 2009; Ng et al., 2013; and this study) argues that the PI4P/GOLPH3/MYO18A/F-actin complex plays an important role to allow efficient Golgi-to-PM trafficking in mammalian cells.
Role of the GOLPH3 Complex in Cancer
Both GOLPH3 and MYO18A are drivers of cancer in humans (Buschman et al., 2015; Makowski et al., 2017; Sanchez-Garcia et al., 2014; Scott et al., 2009). Mechanisms identified for GOLPH3/MYO18A-driven oncogenic transformation are dependent on their role in Golgi-to-PM trafficking (Farber-Katz et al., 2014; Halberg et al., 2016; Xing et al., 2016). Interference with the GOLPH3 complex may offer an attractive target for novel cancer therapeutic agents with mechanisms of action orthogonal to all current treatment options. Clearly, better understanding of the mechanism by which the GOLPH3 complex promotes Golgi secretory function, in part illuminated here, will be necessary to develop and understand rationally targeted therapies against the GOLPH3 complex.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests may be directed to and will be fulfilled by the corresponding author, Seth J. Field (sjfield@ucsd.edu).
EXPERIMENTAL MODELS
HeLaS3 and HEK293 (AD293 variant) cell lines were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum and Penicillin/Streptomycin at 37°C in a humidified, 10% carbon dioxide atmosphere. The HeLaS3 cell line that stably expresses ManII-GFP was grown under similar conditions, using media supplemented with 800 μg/mL G418.
METHOD DETAILS
Cloning/Plasmids
Mammalian expression vectors pcDNA3.1(+)-GOLPH3, pcDNA3.1(+)-GOLPH3-R90L, and mCherry-GOLPH3, and the bacterial expression vector pGEX4T3-GOLPH3 have been described previously (Dippold et al., 2009). pcDNA3.1–3xHA-GOLPH3 and pcDNA3.1–3xMYC-GOLPH3 mammalian expression vectors were produced by cloning GOLPH3 into the BamHI and EcoRI restriction sites of pcDNA3.1(+)-3xHA and pcDNA3.1(+)-3xMYC empty vectors (gifts from Dr. Michael G. Rosenfeld). To make mCherry-GOLPH3, mCherry (Shaner et al., 2005) was PCR amplified from pcDNK mCherry (a gift from Dr. Roger Tsien) with primers: forward (adding NheI site) 5’ GTCCGCTAGCGCCACCATGGTGAGCAAGGGCGAG 3’ and reverse (adding BspEI site) 5’ GTGTTCCGGACTTGTACAGCTGGTCCATGCC 3’. EGFP was cut from pEGFP-C1-GOLPH3 with NheI and BspEI for insertion of the mCherry PCR product. Sequences of primers used to clone human GOLPH3 into pGEX 4T3 (GST-GOLPH3) were: forward (adding EcoRI site) 5’ GGTCAGGAATTCCATGGAGGACGACGCGCAGAGC 3’ and reverse (adding XhoI site) 5’ CGTGCTCGAGTTACTTGGTGAACGCCGCCAC 3’. To make mKO2-GOLPH3, mKO2 was PCR amplified from alpha-synuclein-mKO2-miniSOG-HA construct (a gift from Dr. Roger Tsien) and cloned into GOLPH3 in pcDNA3.1(+) with primers: forward (adding BamHI site) 5’ CGCGGATCCATGGTGAGCGTGATCAAGCC 3’ and reverse (adding EcoRI site) 5’ CCGGAATTCCTGCCGCCACCACTGCCGCCACCATC 3’. ts045-VSVG-GFP was a gift from Dr. Jennifer Lippincott-Schwartz (Presley et al., 1997). mCherry-KDEL was a gift from Dr. Gia Voeltz (Zurek et al., 2011). pGEX4T1-EPSIN ENTH amino acids 1–146 was a gift from Dr. Peter McPherson (Hussain et al., 2003).
Site-directed mutagenesis for generation GOLPH3 Δ190–201, GOLPH3 Δ193–198, and GOLPH3 L195E L196E constructs was performed using QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Sequences of oligos for making GOLPH3 Δ190–201 were: forward 5’ GAAAAGGGTGTATTGACAACACATCCCCTCACCAATAACAAC 3’ and reverse 5’ GTTGTTATTGGTGAGGGGATGTGTTGTCAATACACCCTTTTC 3’. Sequences of oligos for making GOLPH3 Δ193–198 were: forward 5’ TATTGACAACAGAGAAACAGATGACAACACATCCCCTCAC 3’ and reverse 5’ GTGAGGGGATGTGTTGTCATCTGTTTCTCTGTTGTCAATA 3’. Sequences of oligos for making GOLPH3 L195E L196E were: forward 5’ CAGAGAAACAGAACTTCGAAGAGTTTGACATGACAACACATCC 3’ and reverse 5’ GGATGTGTTGTCATGTCAAACTCTTCGAAGTTCTGTTTCTCTG 3’.
siRNA oligonucleotides and knockdown
siRNA oligonucleotides containing Stealth modifications were purchased from Invitrogen. Negative control siRNA oligonucleotides with 48% GC content were designed by the manufacturer to have no cognate sequences in mammalian transcriptomes (Invitrogen Catalog #’s 12935300).
The sequences of specific oligonucleotides were as follows:
| Name | Sequence |
|---|---|
| GOLPH3 | AAAUGAUGUGUAACCCUCGCGGUCCGGACCGCGAGGGUUACACAUCAUUU |
| MYO18A | UGGAGGUUAUGGAAAUGGAGGUGAUAUCACCUCCAUUUCCAUAACCUCCA |
To perform siRNA knockdown, Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) was used according to the manufacturer’s instructions. We note that robust phenotypes occur only with >75% knockdown of GOLPH3. Since GOLPH3 has a half-life of ~24 hours, only after 48 hours do we observe significant phenotypes. All experiments described here occur 55–65 hours after knockdown, measuring the effect of acute loss of GOLPH3.
Immunofluorescence
Cells were fixed in 3.7% paraformaldehyde in phosphate buffered saline (PBS), pH 7.5 at room temperature for 20 minutes. Cells were then washed with PBS, permeabilized with 0.1% TX-100 in PBS for 10 min, washed with PBS, and blocked with 8% (w/v) BSA and 0.02% (v/v) Tween-20 in PBS. Next, cells were immunostained with primary antibody in blocking solution overnight at 4° C, washed with PBS, immunostained with secondary antibody in blocking solution for 1 h at room temperature, washed with PBS, and stained with DAPI (Invitrogen, Carlsbad, CA), and where appropriate, HCS CellMask (Invitrogen, Carlsbad, CA) to allow cell segmentation for automated image analysis.
Fluorescence Microscopy
Fluorescence microscopy was performed with an Olympus IX81-ZDC spinning-disk confocal microscope using a 40x/NA 1.30 or 60x/NA 1.40 objective lens. Images were captured using a Photometrics CoolSnap HQ2 cooled CCD camera. The microscope was controlled by, and images captured using Slidebook software (Intelligent Imaging Innovations). Automated image segmentation and analysis were performed using CellProfiler software (Carpenter et al., 2006). Additional image analysis was performed using ImageJ software (Schneider et al., 2012).
Golgi Tubulation Imaging and Quantification
Imaging of Golgi tubulation was complicated by two issues. First, we observed that most of the Golgi tubules were lost upon fixation. Second, because the tubules are much less intense than the Golgi itself, illumination and exposure parameters required to observe the tubules resulted in gross over-exposure of the Golgi itself. However, by imaging live cells stably expressing a GFP-tagged Golgi marker (ManII-GFP, or transient expression of the trans-Golgi marker SialT-GFP), with appropriate illumination and exposure parameters, the tubules were readily observed in response to overexpression of GOLPH3 or knockdown of MYO18A. For live imaging, cells were grown on MatTek window dishes. For imaging, the cells were maintained at 37 °C in humidified 10% CO2 in a Warner DH40i stage-top incubator with a TC124 objective lens warmer. The Olympus ZDC system was used to maintain the focal plane over time. Image capture frequency is indicated in each figure legend. The fraction of cells producing Golgi tubules during the period of imaging was quantified.
Measurement of Golgi Area
Immunofluorescence images of cells stained with a Golgi marker were measured by automated segmentation to identify individual cells, the Golgi within each cell, and calculation of the area of the Golgi per cell, as determined by the area of an ellipse with the same second moment as the Golgi (essentially equivalent to the area of the smallest encircling ellipse). Automated analysis was performed using CellProfiler software (Carpenter et al., 2006). Data produced was analyzed using Excel spreadsheet software (Microsoft).
Western Blotting
Cells were lysed in boiling SDS sample buffer (4% [w/v] SDS, 10% [v/v] glycerol, 125 mM Tris-Cl pH 6.8, and 1 mM DTT), run on SDS-PAGE, and then wet transferred onto polyvinylidene fluoride (PVDF) membrane at 100 mV for 3 hours. The membranes were blocked with 5% (w/v) non-fat milk in 150 mM NaCl/14 mM Tris-Cl pH 7.5/0.02% (v/v) Tween-20 (TBST) for 30 min. The membrane was then incubated with primary antibody in 5% (w/v) milk in TBST at 4°C overnight, washed with TBST, and incubated with the horse radish peroxidase-conjugated secondary antibody at room temperature for 1 h, washed, incubated with enhanced chemiluminescence substrates, and exposed to film.
Immunoprecipitation
Immunoprecipitation experiments were performed as described previously (Dippold et al., 2009). Cells were lysed in buffer containing 100 mM NaCl/10 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)/ 5 mM dithiothreitol (DTT)/ 100 μM leupeptin/ 1 mM 4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF)/ 1 μg/mL pepstatinA/ 40 μM bestatin/ protease inhibitor tablets (complete, Ultra, EDTA-free, Roche, Basel, Switzerland)/ 1 mM sodium orthovanadate/ 20 mM sodium fluoride/ 5 mM β-glycerophosphate/ 2.5 mM sodium pyrophosphate. Lysates were precleared and incubated with anti-GOLPH3, pre-immune serum or anti-MYC antibodies, bound to Protein A Sepharose (GE Healthcare, Little Chalfont, UK), washed extensively, and eluted by boiling in SDS sample buffer, followed by Western blotting, as described above.
ts045-VSVG-GFP Trafficking Experiments
Cells were transfected with either GOLPH3, MYO18A or control siRNA oligonucleotides, then transfected using TransIT-LT1 transfection reagent (Mirrus, Bio LLC, Madison, WI) with expression vector for ts045-VSVG-GFP, and with either GOLPH3 (WT), GOLPH3-R90L, 3xMYC-GOLPH3, GOLPH3 Δ190–201, GOLPH3 Δ193–198, GOLPH3 L195E/L196E, or empty vector. Cells were incubated at the non-permissive temperature of 40°C for 16 hours prior to shifting to the permissive temperature of 32°C for 75 min to allow trafficking of VSVG to the cell surface, or, alternatively, to 22°C for 40 min to allow trafficking to the Golgi. Cells were fixed in 3.7% paraformaldehyde and non-permeabilized cells were immunostained with the exofacial monoclonal antibody 8G5F11 followed by AlexaFluor 594-conjugated anti-mouse secondary antibody. For assessment of trafficking to the Golgi, cells were permeabilized and IF performed to detect GM130, using AlexaFluor 594-conjugated anti-mouse secondary antibody. Cells were then permeabilized with 0.1% TX-100 in PBS for 10 min and immunostained with anti-GOLPH3 antibody followed with AlexaFluor 647-conjugated anti-rabbit secondary antibody. Images were analyzed using CellProfiler to segment images to identify individual cells and to measure exofacial antibody staining (or Golgi-localized GFP) relative to total GFP signal per cell, limiting the analysis to GFP-positive cells. Integrated intensity of GOLPH3 IF per cell was also measured to allow comparison of GOLPH3 expression levels.
Lipid Blots
Lipid blot experiments were performed as described previously (Dippold et al., 2009). Lipids (Cell Signals, Columbus, OH, Echelon Biosciences, Salt Lake City, UT and Avanti Polar Lipids, Alabaster, AL) were dissolved in DMSO/20% CHCl3/50 mM HCl and validated by TLC, and the indicated amounts were spotted on PVDF. Blots were blocked overnight in 3% fatty acid-free BSA in TBST (150 mM NaCl, 50 mM tris [pH 7.5], and 0.03% Tween-20) and were probed in refreshed block with 35S-labeled protein from a 20 μl in vitro transcription/translation (IVT) mix (Promega TNT Gold) with 33 μCi Trans-label (PerkinElmer, Boston, MA). 1 μl of the IVT product was reserved for SDS-PAGE. The remaining 19 μl was applied to the blot for 2 hours, followed by washing five times with TBST, and exposure to phosphorimager (GE Healthcare, Little Chalfont, UK).
Lipid Vesicle Pull-Down
Lipid vesicle pull-down experiments were described previously (Dippold et al., 2009). Lipid mixtures of PE, or PE plus PI or PI4P (at the concentrations indicated in each figure legend) were suspended in 30 μl of buffer (100 mM NaCl, 20 mM HEPES [pH 7.2], 2 mM EGTA, and 100 ng/mL BSA). For Hydrogen-Deuterium Exchange Mass Spectrometry, BSA was omitted from the lipid vesicle pull-down buffer. Liposomal vesicles were prepared by sonicating for 1 h followed by 10 cycles of freeze/thaw alternating between freezing in liquid nitrogen and thawing at 60°C. Purified, bacterially expressed proteins (at the concentrations indicated in each figure legend) were pre-cleared and incubated with lipid vesicles for 20 min at room temperature, pelleted at 100,000×g for 25 min at 4°C, and the pellet and supernatant fractions were boiled in SDS sample buffer, run on SDS-PAGE, and visualized by staining with Coomassie Brilliant Blue.
Hydrogen-Deuterium Exchange Mass Spectrometry
Prior to performing DXMS, optimal GOLPH3 digestion conditions were established, as previously described (Zhang et al., 2018). For DXMS, GOLPH3 (36 μM) was incubated alone or with PI4P-containing liposomes (384 μM PI4P), as per the Lipid Vesicle Pull-Down experiments. After 30 minutes, deuterated buffer was added, and quenched at pH 2.5 at 10, 100, 1000, 10,000, and 100,000 sec. Quenched samples were immediately frozen, prior to LC/MS analysis, along with un-deuterated and equilibrium-deuterated control samples (Tsalkova et al., 2012). Samples were proteolyzed with immobilized pepsin, separated using a Michrom C18 reverse phase analytical column, and analyzed using an OrbiTrap Elite Mass Spectrometer (ThermoFisher Scientific, San Jose, CA) with settings optimized to minimize back-exchange. Proteome Discoverer software identified peptides, centroids of the mass envelopes of deuterated peptides were calculated with HDXaminer (Sierra Analytics Inc., Modesto, CA), and converted to corresponding deuterium incorporation with corrections for back-exchange (Zhang and Smith, 1993). Mean changes in deuterium exchange over all timepoints were calculated and mapped onto PDB structure 3KN1.
Electron Microscopy
Liposomal vesicles, alone or after incubation for 20 minutes with GOLPH3 protein (WT or mutant) were bound to EM grids, stained with 2% uranyl acetate, viewed using a JEOL 1200EX II transmission electron microscope, and photographed using a Gatan Orius 600 digital camera.
Quantification of Liposomal Tubulation
For EM experiments, randomly chosen fields were imaged and each image divided into a 10 × 10 grid of 100 squares. Each grid square was scored for the presence or absence of tubules, providing a quantized estimate of overall tubule density (tubule-positive squares / total squares). The results from all of the randomly chosen fields from multiple independent experiments were pooled, as indicated in the relevant figure legends.
For fluorescence imaging experiments (Figure 1D), four randomly selected fields per MatTek dish were imaged. Each image was divided into a 9 × 11 grid of 99 squares, with each grid square scored for the presence or absence of liposomal tubules, providing a quantized estimate of overall tubule density (tubule-positive squares / total squares). The results from all of the randomly chosen fields from multiple independent experiments were pooled, as indicated in the relevant figure legend.
Molecular Modeling
Molegro Molecular Viewer and Virtual Docker software (Qiagen) was used to visualize the atomic structure of human GOLPH3 (PDB file 3KN1), to produce the van der Waals surface shaded for hydrophobicity/hydrophilicity, and to dock to PI4P. Chimera software (Pettersen et al., 2004) was used to visualize DXMS data by application to a surface model of GOLPH3 (PDB file 3KN1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM. Sample numbers and experimental repetitions are indicated in figures and figure legends. Where indicated, a two-tailed Student’s unpaired (or paired, when noted) t test was calculated using Microsoft Excel. Differences were considered statistically significant at p<0.05. Where appropriate, multiple comparisons were corrected for family-wise Type 1 error using the Holm-Bonferroni correction (Holm, 1979).
Supplementary Material
Movie S1. GOLPH3 Binding to PI4P Drives Tubulation of Liposomes. Related to Figure 1. Nile Red stained liposomes that contain 1.8 mol% PI4P, PI, or no phosphoinositide were incubated with 14 μM purified, bacterially expressed GOLPH3 (WT or the R90L mutant that does not bind PI4P) and examined by fluorescence time-lapse imaging (frames every 2 seconds). The indicated protein (or buffer-only) was added at time zero. Two examples are provided for each condition. Still frames and quantification of liposome tubulation are provided in Figures 1E and 1D.
Movie S2. Expression of mKO2-GOLPH3 causes Golgi tubulation. Related to Figure 2. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for mKO2-GOLPH3 (WT, top row), mKO2-GOLPH3-R90L (middle row), or mKO2 (bottom row) and subsequently imaged every 10 seconds. mKO2 is shown in red and ManII-GFP in green. Still frames and quantification of Golgi tubulation are provided in Figures 2A and 2B.
Movie S3. The Hydrophobic β-Loop is Required for GOLPH3-Driven Golgi Tubulation. Related to Figure 3. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for either WT or mutant mKO2-GOLPH3 and confocal fluorescence time-lapse imaging was performed on live cells, capturing images every 7.5 seconds. mKO2 is shown in red and ManII-GFP in green. Still frames and quantification of Golgi tubulation are provided in Figures 3H and 3I.
Movie S4. 3xMYC-GOLPH3 and 3xHA-GOLPH3 Induce Golgi Tubulation. Related to Figure 5. HeLa cells stably expressing ManII-GFP and were transfected with expression vectors for 3xMYC-GOLPH3 (left) or 3xHA-GOLPH3 (right). Subsequently confocal fluorescence time-lapse imaging was performed on live cells, capturing images every 29 seconds (3xMYC-GOLPH3) or 38 seconds (3xHA-GOLPH3). ManII-GFP is shown in green. Still frames are shown in Figure S3C, with further data in Figure 5.
Movie S5. Expression of Tagged GOLPH3 Significantly Increases Golgi Tubulation Compared to Untagged GOLPH3. Related to Figure 5. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for mKO2 (left), mKO2-GOLPH3 (center), or GOLPH3-IRES-mCherry (right). Subsequently confocal fluorescence time-lapse imaging was performed on live cells, capturing images every 7 seconds. ManII-GFP is shown in green. Still frames and quantification of Golgi tubulation are provided in Figures 5A and 5B.
Movie S6. MYO18A Depletion Induces Golgi Tubulation. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with either control (left) or MYO18A-specific (right) siRNA oligonucleotides, and also transfected with expression vectors for mCherry (top row), mCherry-GOLPH3 (WT, middle row), or mCherry-GOLPH3-R90L (bottom row). Confocal fluorescence time-lapse imaging was performed on live cells, capturing frames every 10 seconds. mCherry is shown in red and ManII-GFP in green. Still frames and quantification of Golgi tubulation are provided in Figures 6A and 6B. Western blots are shown in Figure S4A.
Movie S7. Inhibition of Actin Polymerization Increases Golgi Tubulation. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for mKO2 (control) or mKO2-GOLPH3. Live-cell confocal images were captured every 34 seconds to observe Golgi tubulation before and after addition of 30 μM cytochalasin B (indicated at time t=0 s). ManII-GFP is shown in green and mKO2 in red. Quantification of Golgi tubulation is shown in Figure S4B. Golgi tubulation significantly increases after treatment with cytochalasin B.
Movie S8. Golgi Tubulation Due to Loss of MYO18A is Dependent on GOLPH3. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with combinations of control, GOLPH3, or MYO18A-specific siRNA oligonucleotides, as indicated. Confocal fluorescence time-lapse imaging was performed on live cells, capturing frames every 7.5 seconds. ManII-GFP is shown in green. Still frames and quantification of Golgi tubulation are provided in Figures 6C and 6D. Western blots validating the knockdowns are shown in Figure S4E.
Movie S9. Golgi Tubulation Driven by Depletion of MYO18A Requires GOLPH3, Its Ability to Bind PI4P and to Induce Membrane Curvature. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with either control, GOLPH3, and/or MYO18A-specific siRNA oligonucleotides, as indicated. Cells also were transfected with expression vectors for tdTomato (to mark transfected cells) together with expression vectors for siRNA-resistant wild-type GOLPH3, GOLPH3-R90L, GOLPH3 Δ190–201, GOLPH3 Δ193–198, or empty vector. Confocal fluorescence time-lapse imaging was performed on live cells that were positive for tdTomato (not shown), capturing frames every 50 seconds. ManII-GFP is shown in green. Golgi tubulation driven by depletion of MYO18A depends on GOLPH3 and the ability of GOLPH3 to bind PI4P and to induce membrane curvature. Quantification of Golgi tubulation is provided in Figure S5A. Western blots to confirm knockdowns and protein expression are provided in Figure S5B.
Figure S1. GOLPH3 Binding to PI4P Causes Efficient Liposomal Tubulation. Related to Figures 1, 2, and 3. (A) EPSIN-ENTH and GOLPH3 (each at 5μM) bind similarly to liposomes containing their cognate phosphoinositides, PI(4,5)P2 and PI4P (each at 4.2 mol%), respectively. After incubation with the indicated proteins, liposomes were sedimented by ultracentrifugation, and bound and free protein assessed by Coomassie Brilliant Blue stained SDS-PAGE of the pellet and supernatant fractions.
(B) Aliquots of the binding reactions from (A) were processed and imaged by EM. Four example images are shown for each.
(C) Quantification of (B). Graph shows mean ± SEM, with the number of randomly selected fields (n) indicated, pooled from three independent experiments. p values are indicated, calculated by t test.
(D) Liposomes were generated using natural (brain-derived) PI4P (1.8 mol%) and either PC or PS (at 15 mM), and incubated with purified, bacterially expressed GOLPH3 (WT) or GOLPH3-R90L (at 14 μM). They were then visualized by EM. Five example images are provided for each. Similar results are obtained with liposomes containing PE (Figure 1C).
(E) SialT-GFP marks trans-Golgi-derived tubules driven by mCherry-GOLPH3. Shown are successive frames from time-lapse images of HeLa cells co-expressing SialT-GFP and mCherry-GOLPH3. A dynamic tubule is marked by an arrowhead. Similar results are obtained with ManII-GFP (Figure 2).
(F) Hydrophilic residues placed at the tip of the hydrophobic β-loop (GOLPH3 L195E/L196E) interfere with GOLPH3 binding to PI4P-containing liposomes. WT or mutant GST-GOLPH3 protein (at 1.4 μM) was allowed to bind to PI4P- or PI- (control) containing liposomes (at 4.2 mol%), separated into lipid-bound and free fractions by ultracentrifugation, and assessed by Coomassie Brilliant Blue staining of SDS-PAGE.
(G) GOLPH3 binds to natural (C18 or greater) and short-chain (C8) lipid vesicles containing PI4P. Vesicles were produced with short-chain (C8) or natural long-chain (C18 or greater) lipids including PI4P or PI (control) and incubated with equal amounts of purified, bacterially expressed GOLPH3 (WT) or GOLPH3-R90L. To compensate for intrinsically reduced binding of GOLPH3 to C8 liposomes, the amount of PI4P (or PI) in the C18 liposomes was reduced (414 μM for C8 [2.7 mol%], 23 μM for C18 [0.15 mol%]) to produce similar binding of GOLPH3 to both the C8 and C18 vesicles to allow fair comparison of tubulation. Vesicle-bound and free protein were separated by ultracentrifugation and assessed by SDS-PAGE (Figure 3B). The graph indicates quantification of the amount of GOLPH3 bound to the liposomes, which is similar for C8 and C18 PI4P liposomes. Binding is specific, as observed by loss of binding with the R90L mutation or substitution of PI for PI4P. Graph shows mean ± SEM, with the number of independent experiments (n) indicated. p values are indicated, calculated by t test.
Figure S2. GOLPH3’s Hydrophobic β-Loop is Required for Forward Trafficking from the Golgi to the Plasma Membrane. Related to Figure 4. (A) Quantification of relative GOLPH3 expression levels for Figures 4A and 4B. GOLPH3 knockdown/rescue experiments were performed in HeLa cells by transfection of GOLPH3-specific siRNA (or control or MYO18A-specific siRNA) followed by transfection of expression vectors for siRNA-resistant GOLPH3 (WT), the indicated mutants, or the empty vector. Cells were fixed and IF performed to examine the Golgi (GM130) and GOLPH3 (see Figure 4A) to allow measurement of Golgi area (Figure 4B) and, in this graph, GOLPH3 expression levels relative to the control.
(B) mKO2-GOLPH3-driven Golgi tubules contain the cargo VSVG-GFP. HeLa cells were co-transfected with expression vector for mKO2-GOLPH3 (to drive Golgi tubulation) and ts045-VSVG-GFP (a fluorescently labeled cargo). After overnight incubation at 40°C, cells were shifted to 32°C for 75 min to allow trafficking. Successive frames (interval of 10 s) from live-cell imaging are shown, demonstrating dynamic Golgi tubules from two independent experiments. White arrows indicate co-localization of VSVG-GFP with mKO2-GOLPH3 on Golgi tubules.
(C) Knockdown of GOLPH3 or MYO18A results in increased accumulation of ts045-VSVG-GFP at the Golgi at 22°C. Cells expressing ts045-VSVG-GFP transfected with siRNA to GOLPH3, MYO18A, or control were kept at 40°C for 16 hours, and then shifted to 22°C for 40 min, as indicated. Cells were fixed and the amount of GFP co-localizing with IF to GM130 (to mark the Golgi) was normalized to total GFP per cell. Shift to 22°C resulted in significant trafficking from ER to Golgi, with increased accumulation at the Golgi in GOLPH3 and MYO18A knockdown cells.
(D) In parallel to (C), cells at 22°C for 40 min were left unpermeabilized, and IF performed with antibody to the extracellular domain of VSVG to assess delivery to the PM. Delivery to the PM detected in control siRNA-transfected cells was strongly impaired by treatment with 5 μM Golgicide A (GCA). Knockdown of GOLPH3 or MYO18A also reduced delivery to the PM, accounting for the increased accumulation at the Golgi observed in (C).
(E) Golgi-to-ER recycling was assessed by measuring accumulation of mCherry-KDEL at the Golgi, co-localizing with IF to GM130. HeLa cells expressing mCherry-KDEL were transfected with siRNA to GOLPH3, MYO18A, or control, as indicated.
(F) Representative Western blot for (C), (D), and (E) demonstrating >70% knockdown of GOLPH3 or MYO18A. GAPDH serves as a loading control. Cell lysate from transfection with control siRNA was loaded at decreasing amounts to allow semi-quantitative comparisons.
(G) Quantification of relative GOLPH3 expression levels for Figures 4C and 4D. Trafficking of ts045-VSVG-EGFP was examined in a GOLPH3 knockdown/rescue experiment in HeLa cells. siRNA to GOLPH3 (or MYO18A or control) was transfected, followed by transfection of expression vector for siRNA-resistant GOLPH3 (WT), the indicated mutants, or empty vector. Overnight incubation at 40°C arrested trafficking at the ER, followed by incubation at 32°C for 75 minutes to allow trafficking. Cells were fixed and IF performed to examine exofacial (surface) VSVG in unpermeabilized cells (Figure 4C), followed by permeabilization to allow staining for GOLPH3 to assess relative expression levels. Graphs show mean and SEM with the number of cells (n) indicated, pooled from four (A), three (C, D), two (E), or six (G) independent experiments. p values are indicated, calculated by t test.
Figure S3. Expression of Tagged GOLPH3 Significantly Increases Golgi Tubulation, Regardless of the Nature of the Tag. Related to Figure 5. (A) Cells expressing either mCherry-GOLPH3 or mKO2-GOLPH3 display significantly increased Golgi tubulation compared to cells expressing mCherry or mKO2. HeLa cells stably expressing ManII-GFP as a Golgi marker were transfected with expression vectors for mCherry-GOLPH3, mKO2-GOLPH3, mCherry, or mKO2, and confocal time-lapse images were captured every 10 seconds. Also see Figures 5A and 5B for comparisons of GOLPH3 with and without an amino-terminal tag.
(B) Quantification of Golgi tubulation for (A).
(C) Expression of either 3xMYC-GOLPH3 or 3xHA-GOLPH3 causes Golgi tubulation. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for 3xMYC-GOLPH3 or 3xHA-GOLPH3 and confocal images were captured every 29 or 38 seconds. After live imaging, cells were fixed and examined by immunofluorescence with anti-MYC or anti-HA antibodies to verify expression of 3xMYC-GOLPH3 or 3xHA-GOLPH3. Also see Figures 5A and 5B, and Movie S4.
(D) Expression levels of proteins used for lipid blotting in Figure 5C. GOLPH3 (WT), GOLPH3-R90L, 3xMYC-GOLPH3, and PX protein (Drosophila CG4960; Dippold et al., 2009) were synthesized by in vitro transcription and translation, and labeled by inclusion of 35S-methionine in the translation mix. Protein levels were compared using SDS-PAGE, followed by fixation and detection by phosphorimager. Also see Figure 5C for the lipid blot.
(E) Quantification of relative GOLPH3 expression levels for Figures 5E and 5F. Images from Figure 5E underwent automated segmentation and analysis to measure integrated GOLPH3 IF per cell. Graphed is mean GOLPH3 expression relative to the control. Graphs in (B) and (E) show mean ± SEM, with the number of cells (n) indicated, pooled from four (B) or four (E) independent experiments. p values are indicated, calculated by t test. White arrows indicate Golgi tubulation.
Figure S4. Golgi Tubulation Occurs in Response to Depletion of MYO18A or Depolymerization of F-actin. Related to Figure 6. (A) Protein expression levels in cells depleted for MYO18A. HeLa cells stably expressing ManII-GFP were transfected with either control or MYO18A-specific siRNA oligonucleotides. Cells were then transfected with expression vectors for mKO2-GOLPH3 (WT), mKO2-GOLPH3-R90L, or mKO2. Parallel coverslips were used for IF (Figures 6A and 6B) or Western blotting (shown here), using antibodies to GOLPH3, MYO18A, and GAPDH (as a loading control). The control siRNA-transfected samples are loaded at different relative amounts to allow semi-quantitative assessment of the knockdown of MYO18A, observed to be ~75–90% for each.
(B) HeLa cells stably expressing ManII-GFP to mark the Golgi were transfected with expression vectors for either mKO2-GOLPH3 or mKO2 and confocal images were captured every 34 seconds. Live-cell imaging was performed to observe Golgi tubulation before and after addition of cytochalasin B (Movie S7). Graphs show quantification of relative Golgi tubulation of the same fields prior to, or 34 seconds after treatment with 30 μM cytochalasin B. Graphed are mean ± SEM, with the number of cells (n) indicated, pooled from three independent experiments. p values are indicated, calculated by paired (before vs. after) or unpaired (mKO2 vs. mKO2-GOLPH3) t test.
(C) Endogenous GOLPH3 is observed to localize to Golgi tubules induced by depletion of MYO18A. HeLa cells stably expressing ManII-GFP were transfected with siRNA to deplete MYO18A (as in Figure 6C and S4D). Cells on gridded coverslips were imaged live to unambiguously identify active tubulation, fixed for IF, and the grid coordinates used to return to the same cell to observe the subcellular localization of endogenous GOLPH3. As shown, endogenous GOLPH3 is observed to localize to the Golgi tubules that form in response to loss of MYO18A.
(D) Most mCherry-GOLPH3-driven Golgi tubules do not contain detectable ARF1. HeLa cells stably expressing ManII-GFP were transfected to express mCherry-GOLPH3, fixed, and IF performed to detect endogenous ARF1. Tubules emanating from the Golgi are observed that are positive for both ManII-GFP and mCherry-GOLPH3, as expected. Endogenous ARF1 is observed at the Golgi, as expected. However, ARF1 is not observed on most of the mCherry-GOLPH3-driven tubules. Arrows in (C) and (D) indicate Golgi tubules.
(E) Protein expression levels in cells depleted for GOLPH3, MYO18A, or both. HeLa cells stably expressing ManII-GFP were transfected with combinations of control, GOLPH3, and MYO18A-specific siRNA oligonucleotides, as indicated. Parallel coverslips were used for IF (Figures 6C and 6D) or Western blotting (shown here), using antibodies to GOLPH3, MYO18A, and GAPDH (as a loading control). The control siRNA-transfected samples were loaded at different relative amounts to allow semi-quantitative assessment of the knockdown of GOLPH3 and MYO18A, observed to be 75–90% for each.
Figure S5. Golgi Tubulation Driven by Depletion of MYO18A Requires Endogenous GOLPH3, Its Ability to Bind PI4P and to Induce Membrane Curvature. Related to Figure 6. (A) Measurement of Golgi tubulation in HeLa cells stably expressing the Golgi marker ManII-GFP and transfected with siRNA to knockdown GOLPH3 and MYO18A was performed as indicated in Figure 6C and 6D. Transfection of siRNA-resistant GOLPH3 expression vectors was performed, as indicated. All constructs were co-transfected with an expression vector for tdTomato to mark transfected cells. Live confocal fluorescent imaging of tdTomato-positive cells was performed to assess Golgi tubulation, as observed by the dynamic behavior of the ManII-GFP Golgi marker (Movie S9). WT GOLPH3 rescued Golgi tubulation caused by depletion of MYO18A. However, GOLPH3 mutants defective in binding PI4P (R90L) or in inducing membrane curvature (Δ190–201 and Δ193–198) were unable to rescue Golgi tubulation. Graph shows mean and SEM, with the number of cells (n) indicated, pooled from five independent experiments. p values are indicated, calculated by t test.
(B) Western blotting of lysates from parallel coverslips from (A) to assess MYO18A and GOLPH3 expression. GAPDH is assessed as a loading control. The control lysate is loaded at different relative amounts to allow semi-quantitative assessment of knockdown of MYO18A (75–90%) and GOLPH3 (>90%).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GOLPH3 polyclonal antibody | Dippold et al., 2009 | N/A |
| Anti-GOLPH3 polyclonal antibody | Sigma | Cat# SAB4200341; RRID: AB_10897241 |
| Anti-MYO18A polyclonal antibody | Dr. Zissis Chroneos; Yang et al., 2005 | N/A |
| Anti-VSVG exofacial monoclonal antibody 8G5F11 | KeraFAST | Cat# EB0010 |
| Anti-GAPDH monoclonal antibody D16H11 | Cell Signaling Technology | Cat# 5174S; RRID:AB_10622025 |
| Anti-ARF1 | Sigma | Cat # SAB1412690 |
| Anti-MYC monoclonal antibody 9B11 | Cell Signaling Technology | Cat# 2276; RRID:AB_331783 |
| Horseradish peroxidase-conjugated mouse anti-rabbit secondary antibody | Jackson ImmunoResearch | Cat# 211-035-109; RRID:AB_2339150 |
| AlexaFluor 594-conjugated secondary antibodies | Invitrogen | Cat# A-11005; RRID:AB_141372 |
| AlexaFluor 647-conjugated secondary antibodies | Invitrogen | Cat# A-21244; RRID:AB_141663 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phosphatidylinositol diC16 (PI diC16) | Echelon Biosciences | Cat# P-0016 |
| Phosphatidylinositol diC8 (PI diC8) | Echelon Biosciences | Cat# P-0008 |
| Phosphatidylinositol 4-phosphate diC16 (PI(4)P diC16) | Echelon Biosciences | Cat# P-4016 |
| Phosphatidylinositol 4-phosphate diC8 (PI(4)P diC8) | Echelon Biosciences | Cat# P-4008 |
| L-α-D-myo-Phosphatidylinositol 3-monophosphate, 3-O-phospho linked, D(+)-sn-1,2-di-O-hexadecanoylglyceryl | CellSignals | Cat#910 |
| L-α-phosphatidylinositol-4-phosphate (from porcine brain) | Avanti Polar Lipids, Inc | Cat# 840045P |
| L-a-phosphatidylethanolamine (from bovine liver) | Avanti Polar Lipids, Inc | Cat# 840026P |
| 1,2-dioctanoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | Cat# 850699P |
| Roche inhibitor tablet (complete, ultra, EDTA-free) | Roche | Cat# 05892953001 |
| HCS CellMask Deep Red Stain | Invitrogen | Cat# H32721 |
| EasyTag Express 35S Protein Labeling Mix | PerkinElmer | Cat# NEG772007MCSBF4 |
| Lipofectamine RNAiMax | Thermo Fisher | Cat# 13778–150 |
| Lipofectamine LTX with Plus Reagent | Thermo Fisher | Cat# 15338100 |
| TransIT-LT1 Transfection Reagent | Mirrus | Cat# MIR 2300 |
| Cytochalasin B | VWR | Cat# 89164–968 |
| Experimental Models: Cell Lines | ||
| Human: HeLa S3 | ATCC | RRID: CVCL_0058 |
| Human: HeLa S3 subclone stably expressing Golgi marker ManII-GFP | This study | N/A |
| Human: HEK293 (AD-293 variant) | Stratagene | RRID: CVCL_9804 |
| Oligonucleotides | ||
| siRNA sequence for MYO18A: Forward 5’-UGGAGGUUAUGGAAAUGGAGGUGAU-3’ Reverse 5’-AUCACCUCCAUUUCCAUAACCUCCA-3’ |
Invitrogen, Dippold et al., 2009 | N/A |
| siRNA sequence for GOLPH3: Forward 5’-AAAUGAUGUGUAACCCUCGCGGUCC-3’ Reverse 5’-GGACCGCGAGGGUUACACAUCAUUU-3’ |
Invitrogen, Dippold et al., 2009 | N/A |
| Negative control siRNA, 48% GC content, designed to have no cognate site in mammalian transcriptomes | Invitrogen | Cat# 12935300 |
| Primer sequences to clone mKO2 in-frame
upstream of GOLPH3 (WT or R90L) in pcDNA3.1 (+): Forward: CGCGGATTCCATGGTGAGCGTGATCAAGCC Reverse: CCGGAATTCCTGCCGCCACCACTGCCGCCACCATC |
Integrated DNA Technologies, this study | N/A |
| Primer sequences to generate GOLPH3
Δ190–201 by site-directed mutagenesis: Forward: GAAAAGGGTGTATTGACAACACATCCCCTCACCAATAACAAC Reverse: GTTGTTATTGGTGAGGGGATGTGTTGTCAATACACCCIIIIC |
Integrated DNA Technologies, this study | N/A |
| Primer sequences to generate GOLPH3
Δ193–198 by site-directed mutagenesis: Forward: TATTGACAACAGAGAAACAGATGACAACACATCCCCTCAC Reverse: GTGAGGGGATGTGTTGTCATCTGTTTCTCTGTTGTCAATA |
Integrated DNA Technologies, this study | N/A |
| Primer sequences to generate
GOLPH3-L195E/L196E by site-directed mutagenesis: Forward: CAGAGAAACAGAACTTCGAAGAGTTTGACATGACAACACATCC Reverse: GGATGTGTTGTCATGTCAAACTCTTCGAAGTTCTGTTTCTCTG |
Integrated DNA Technologies, this study | N/A |
| Recombinant DNA | ||
| Plasmid: ManII-GFP, Golgi localization domain of α-mannosidase II (amino acids 1–90)-GFP | Dr. Vivek Malhotra | N/A |
| Plasmid: mKO2 | Dr. Roger Tsien; Sakaue-Sawano et al., 2008 | N/A |
| Plasmid: tdTomato | Dr. Roger Tsien; Shaner et al., 2005 | N/A |
| Plasmid: ts045-VSVG-GFP | Dr. Jennifer Lippincott-Schwartz; Presley et al., 1997 | N/A |
| Plasmid: mCherry-KDEL | Dr. Gia Voeltz; Zurek et al., 2011 | N/A |
| Plasmid: pcDNA3.1(+)-GOLPH3 (WT and R90L, siRNA-resistant, mammalian expression vectors) | Dippold et al., 2009 | N/A |
| pGEX4T3-GOLPH3 (E. coli expression vector) | Dippold et al., 2009 | N/A |
| pGEX4T1-EPSIN ENTH amino acids 1–146 | Hussain et al., 2003 | N/A |
| Software and Algorithms | ||
| Cell Profiler | Carpenter et al., 2006 | RRID:SCR_007358 |
| Slidebook | Intelligent Imaging Innovations | RRID:SCR_014300 |
| Chimera | Pettersen et al., 2004 | RRID:SCR_004097 |
| Molegro Molecular Viewer and Virtual Docker | Qiagen | RRID:SCR_000190 |
| Excel | Microsoft | RRID:SCR_016137 |
| ImageJ | Schneider et al., 2012 | RRID:SCR_003070 |
ACKNOWLEDGMENTS
We thank Susan Ferro-Novick and members of the Field lab for constructive criticism, and Karen Messer, Geoffrey Cumming, and David Vaux for advice on statistical analyses. This work was supported by NIH T32 CA009523 (J.R.), American Cancer Society Postdoctoral Fellowships PF-t3-367-01-CDD (M.D.B.) and 115095-PF-08–228-01-CSM (M.M.N.), and support to S.J.F. by NIH grants (R01 GM120055 and R01 CA201303), a Scholar-Innovator Award from the Harrington Discovery Institute, an Era of Hope Scholar Award from the Department of Defense Breast Cancer Research Program (W81XWH-10-1-0822), and a Burroughs Wellcome Fund Career Award in the Biomedical Sciences. S.L. was supported by NIH grants 1U19AI117905, R01GM020501, R01NS070899, and R01GM121964.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bell AW, Ward MA, Blackstock MP, Freeman HN, Choudhary JS, Lewis AP, Chotai D, Fazel A, Gushue JN, Paiement J, et al. (2001). Proteomics characterization of abundant Golgi membrane proteins. J Biol Chem 276, 5152–5165. [DOI] [PubMed] [Google Scholar]
- Berger E, Thurnher M, and Muller U (1987). Galactosyltransferase and sialyltransferase are located in different subcellular compartments in HeLa cells. Exp. Cell Res 173, 267–273. [DOI] [PubMed] [Google Scholar]
- Bishe B, Syed GH, Field SJ, and Siddiqui A (2012). Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J Biol Chem 287, 27637–27647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagoveshchenskaya A, Cheong FY, Rohde HM, Glover G, Knödler A, Nicolson T, Boehmelt G, and Mayinger P (2008). Integration of Golgi trafficking and growth factor signaling by the lipid phosphatase SAC1. J. Cell Biol 180, 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschman M, and Field S (2018a). MYO18A: An unusual myosin. Adv Biol Regul 67, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschman MD, and Field SJ (2018b). MYO18A: An unusual myosin. Adv. Biol. Regul 67, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschman MD, Xing M, and Field SJ (2015). The GOLPH3 pathway regulates Golgi shape and function and is activated by DNA damage. Front Neurosci 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F, Arden SD, Lindsay M, Luzio JP, and Kendrick-Jones J (2001). Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J 20, 3676–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler:image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar I, Goeckeler ZM, Turney SG, Wang P, Wysolmerski RB, Adelstein RS, and Bridgman PC (2014). Nonmuscle Myosin II is a critical regulator of clathrin-mediated endocytosis. Traffic 15, 418–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Smith CL, Sciaky N, Terasaki M, Edidin M, and Lippincott-Schwartz J (1996). Diffusional mobility of Golgi proteins in membranes of living cells. Science 273, 797–801. [DOI] [PubMed] [Google Scholar]
- Daumke O, Roux A, and Haucke V (2014). BAR domain scaffolds in dynamin-mediated membrane fission. Cell 156, 882–892. [DOI] [PubMed] [Google Scholar]
- Dippold HC, Ng MM, Farber-Katz SE, Lee S-K, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith K, Madhavarapu S, et al. (2009). GOLPH3 Bridges Phosphatidylinositol-4-Phosphate and Actomyosin to Stretch and Shape the Golgi to Promote Budding. Cell 139, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing MC, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM, et al. (2014). DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 156, 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan H (2015). Systems biology of the secretory pathway: what we learned so far? Biol Cell 107, 205–217. [DOI] [PubMed] [Google Scholar]
- Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, and McMahon HT (2001). Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291, 1051–1055. [DOI] [PubMed] [Google Scholar]
- Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Lurisci C, Luini A, Corda D, and De Matteis MA (1999). ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol 1, 280–287. [DOI] [PubMed] [Google Scholar]
- Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, and De Matteis MA (2004). FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol 6, 393–404. [DOI] [PubMed] [Google Scholar]
- Halberg N, Sengelaub CA, Navrazhina K, Molina H, Uryu K, and Tavazoie SF (2016). PITPNC1 Recruits RAB1B to the Golgi Network to Drive Malignant Secretion. Cancer Cell 29, 339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, and Lippincott-Schwartz J (1998). Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J Cell Biol 143, 1485–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S (1979). A simple sequentially rejective multiple test procedure. Scand. J. Stat 6, 65–70. [Google Scholar]
- Hu J, Prinz WA, and Rapoport TA (2011). Weaving the web of ER tubules. Cell 147, 1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain NK, Yamabhai M, Bhakar AL, Metzler M, Ferguson SSG, Hayden MR, McPherson PS, and Kay BK (2003). A Role for Epsin N-terminal Homology/AP180 N-terminal Homology (ENTH/ANTH) Domains in Tubulin Binding. J Biol Chem 278, 28823–28830. [DOI] [PubMed] [Google Scholar]
- Kienzle C, and von Blume J (2014). Secretory cargo sorting at the trans-Golgi network. Trends Biol 24, 584–593. [DOI] [PubMed] [Google Scholar]
- Kuna RS, and Field SJ (2019). GOLPH3: a Golgi phosphatidylinositol(4)phosphate effector that directs vesicle trafficking and drives cancer. J. Lipid Res 60, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro-Dieguez F, Jimenez N, Barth H, Koster AJ, Renau-Piqueras J, Llopis JL, Burger KN, and Egea G (2006). Actin filaments are involved in the maintenance of Golgi cisternae morphology and intra-Golgi pH. Cell Motil Cytoskelet. 63, 778–791. [DOI] [PubMed] [Google Scholar]
- Lazaro-Dieguez F, Colonna C, Cortegano M, Calvo M, Martinez SE, and Egea G (2007). Variable actin dynamics requirement for the exit of different cargo from the trans-Golgi network. FEBS Lett 581, 3875–3881. [DOI] [PubMed] [Google Scholar]
- Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, and Schekman R (2005). Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 122, 605–617. [DOI] [PubMed] [Google Scholar]
- Makowski SL, Tran TT, and Field SJ (2017). Emerging themes of regulation at the Golgi. Curr Opin Cell Biol 45, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin K, and Simons K (1983). Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell 34, 233–243. [DOI] [PubMed] [Google Scholar]
- McMahon HT, and Boucrot E (2011). Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12, 517–533. [DOI] [PubMed] [Google Scholar]
- McMahon HT, and Boucrot E (2015). Membrane curvature at a glance. J Cell Sci 128, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Sesorova IS, Seliverstova EV, and Beznoussenko GV (2016). Different Golgi ultrastructure across species and tissues: Implications under functional and pathological conditions, and an attempt at classification. Tissue Cell pii: S0040–8166, 30256–30257. [DOI] [PubMed] [Google Scholar]
- Mitra K, Ubarretxena-Belandia I, Taguchi T, Warren G, and Engelman DM (2004). Modulation of the bilayer thickness of exocytic pathway membrane proteins rather than cholesterol. Proc Natl Acad Sci U A 101, 4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng MM, Dippold HC, Buschman MD, Noakes CJ, and Field SJ (2013). GOLPH3L antagonizes GOLPH3 to determine Golgi morphology. Mol Biol Cell 24, 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, and Lippincott-Schwartz J (1997). ER to Golgi transport visualized in living cells. Nature 389, 81–85. [DOI] [PubMed] [Google Scholar]
- Roth J, Taatjes D, Lucocq J, Weinstein J, and Paulson J (1985). Demonstration of an extensive trans-tubular network continuous with the Golgi apparatus that may function in glycosylation. Cell 43, 287–295. [DOI] [PubMed] [Google Scholar]
- Saheki Y, and De Camilli P (2012). Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol 4, a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia F, Villagrasa P, Matsui J, Kotliar D, Castro V, Akavia U-D, Chen B-J, Saucedo-Cuevas L, Barrueco R, Llobet-Navas D, et al. (2014). Integration of genomic data enables selective discovery of breast cancer drivers. Cell 159, 1461–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Tirado FH, Legesse-Miller A, Schott D, and Bretscher A (2011). PI4P and Rab inputs collaborate in myosin-V-dependent transport of secretory compartment in yeast. Dev Cell 20, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KR, Liu J, Li S, Setty TG, Wood CS, Burd CG, and Ferguson KM (2008). Golgi localization of glycosyltransferase requires a Vps74p oligomer. Dev Cell 14, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K, Kabbarah O, Liang M-C, Ivanova E, Anagnostou V, Wu J, Dhakal S, Wu M, Chen S, Feinberg T, et al. (2009). GOLPH3 modulates mTOR signaling and rapamycin sensitivity in cancer. Nature 459, 1085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, and Tsien RY (2005). A guide to choosing fluorescent proteins. Nat Methods 2, 905–909. [DOI] [PubMed] [Google Scholar]
- Snyder CM, Mardones GA, Ladinsky MS, and Howell KE (2005). GMx33 associates with the trans-Golgi matrix in a dynamic manner and sorts within tubules exiting the Golgi. Mol Biol Cell 17, 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich G, Chibalina MV, Au J, Arden SD, Buss F, and Kendrick-Jones J (2007). Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to Disabled-2 and PtdIns(4,5)P2. Nat Cell Biol 9, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft MH, Behrmann E, Munske-Weidemann LC, Thiel C, Raunser S, and Manstein DJ (2013). Functional characterization of human myosin-18A and its interaction with F-actin and GOLPH3. J Biol Chem 288, 30029–30041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei F, Li S, Chepurny O, Leech C, Liu T, Holz G, Woods V Jr, and Cheng X (2012). Isoform-specific antagonists of exchange proteins directly activted by cAMP. Proc Natl Acad Sci U A 109, 18613–18618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco A, Hendricks L, Moremen KW, Tulsiani DR, Touster O, and Farquhar MG (1993). Cell type-dependent variations in the subcellular distribution of alpha-mannosidase I and II. J Cell Biol 122, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, and Yin HL (2003). Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114, 299–310. [DOI] [PubMed] [Google Scholar]
- Wood CS, Porter KR, Bessman NJ, Setty TG, Ferguson KM, and Burd CG (2009). PtdIns4P recognition by Vps74/GOLPH3 links PtdIns 4-kinase signaling to retrograde Golgi trafficking. J Cell Biol 187, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Taylor RS, Lane DR, Ladinsky MS, Weisz JA, and Howell KE (2000). GMx33: a novel family of trans-Golgi proeins identified by proteomics. Traffic 1, 963–975. [PubMed] [Google Scholar]
- Xie Z, Hur SK, Zhao L, Abrams CS, and Bankaitis VA (2018). A Golgi Lipid Signaling Pathway Controls Apical Golgi Distribution and Cell Polarity during Neurogenesis. Dev. Cell 44, 725–740.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Peterman MC, Davis RL, Oegema K, Shiau AK, and Field SJ (2016). GOLPH3 drives cell migration by promoting Golgi reorientation and directional trafficking to the leading edge. Mol Biol Cell 27, 3828–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C-H, Szeliga J, Jordan J, Faske S, Sever-Chroneos Z, Dorsett B, Christian RE, Settlage RE, Shabanowitz J, Hunt DF, et al. (2005). Identification of the surfactant protein A receptor 210 as the unconventional myosin 18A. J. Biol. Chem 280, 34447–34457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D, Waterman-Storer CM, and Schmid SL (2007). SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Dev Cell 13, 43–56. [DOI] [PubMed] [Google Scholar]
- Yu C, Feng W, Wei Z, Miyanoiri Y, Wen W, Zhao Y, and Zhang M (2009). Myosin VI undergoes cargo-mediated dimerization. Cell 138, 537–548. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Smith D (1993). Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liang W, Bailey L, Tan Y, Wei H, Wang A, Farcasanu M, Woods V, McCord L, Lee D, et al. (2018). Ensemble cryoEM elucidates the mechanism of insulin capture and degradation by human insulin degrading enzyme. ELife 7, e33572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurek N, Sparks G, and Voeltz G (2011). Reticulon short hairpin transmembrane domains are used to shape ER tubules. Traffic 12, 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. GOLPH3 Binding to PI4P Drives Tubulation of Liposomes. Related to Figure 1. Nile Red stained liposomes that contain 1.8 mol% PI4P, PI, or no phosphoinositide were incubated with 14 μM purified, bacterially expressed GOLPH3 (WT or the R90L mutant that does not bind PI4P) and examined by fluorescence time-lapse imaging (frames every 2 seconds). The indicated protein (or buffer-only) was added at time zero. Two examples are provided for each condition. Still frames and quantification of liposome tubulation are provided in Figures 1E and 1D.
Movie S2. Expression of mKO2-GOLPH3 causes Golgi tubulation. Related to Figure 2. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for mKO2-GOLPH3 (WT, top row), mKO2-GOLPH3-R90L (middle row), or mKO2 (bottom row) and subsequently imaged every 10 seconds. mKO2 is shown in red and ManII-GFP in green. Still frames and quantification of Golgi tubulation are provided in Figures 2A and 2B.
Movie S3. The Hydrophobic β-Loop is Required for GOLPH3-Driven Golgi Tubulation. Related to Figure 3. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for either WT or mutant mKO2-GOLPH3 and confocal fluorescence time-lapse imaging was performed on live cells, capturing images every 7.5 seconds. mKO2 is shown in red and ManII-GFP in green. Still frames and quantification of Golgi tubulation are provided in Figures 3H and 3I.
Movie S4. 3xMYC-GOLPH3 and 3xHA-GOLPH3 Induce Golgi Tubulation. Related to Figure 5. HeLa cells stably expressing ManII-GFP and were transfected with expression vectors for 3xMYC-GOLPH3 (left) or 3xHA-GOLPH3 (right). Subsequently confocal fluorescence time-lapse imaging was performed on live cells, capturing images every 29 seconds (3xMYC-GOLPH3) or 38 seconds (3xHA-GOLPH3). ManII-GFP is shown in green. Still frames are shown in Figure S3C, with further data in Figure 5.
Movie S5. Expression of Tagged GOLPH3 Significantly Increases Golgi Tubulation Compared to Untagged GOLPH3. Related to Figure 5. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for mKO2 (left), mKO2-GOLPH3 (center), or GOLPH3-IRES-mCherry (right). Subsequently confocal fluorescence time-lapse imaging was performed on live cells, capturing images every 7 seconds. ManII-GFP is shown in green. Still frames and quantification of Golgi tubulation are provided in Figures 5A and 5B.
Movie S6. MYO18A Depletion Induces Golgi Tubulation. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with either control (left) or MYO18A-specific (right) siRNA oligonucleotides, and also transfected with expression vectors for mCherry (top row), mCherry-GOLPH3 (WT, middle row), or mCherry-GOLPH3-R90L (bottom row). Confocal fluorescence time-lapse imaging was performed on live cells, capturing frames every 10 seconds. mCherry is shown in red and ManII-GFP in green. Still frames and quantification of Golgi tubulation are provided in Figures 6A and 6B. Western blots are shown in Figure S4A.
Movie S7. Inhibition of Actin Polymerization Increases Golgi Tubulation. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for mKO2 (control) or mKO2-GOLPH3. Live-cell confocal images were captured every 34 seconds to observe Golgi tubulation before and after addition of 30 μM cytochalasin B (indicated at time t=0 s). ManII-GFP is shown in green and mKO2 in red. Quantification of Golgi tubulation is shown in Figure S4B. Golgi tubulation significantly increases after treatment with cytochalasin B.
Movie S8. Golgi Tubulation Due to Loss of MYO18A is Dependent on GOLPH3. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with combinations of control, GOLPH3, or MYO18A-specific siRNA oligonucleotides, as indicated. Confocal fluorescence time-lapse imaging was performed on live cells, capturing frames every 7.5 seconds. ManII-GFP is shown in green. Still frames and quantification of Golgi tubulation are provided in Figures 6C and 6D. Western blots validating the knockdowns are shown in Figure S4E.
Movie S9. Golgi Tubulation Driven by Depletion of MYO18A Requires GOLPH3, Its Ability to Bind PI4P and to Induce Membrane Curvature. Related to Figure 6. HeLa cells stably expressing ManII-GFP were transfected with either control, GOLPH3, and/or MYO18A-specific siRNA oligonucleotides, as indicated. Cells also were transfected with expression vectors for tdTomato (to mark transfected cells) together with expression vectors for siRNA-resistant wild-type GOLPH3, GOLPH3-R90L, GOLPH3 Δ190–201, GOLPH3 Δ193–198, or empty vector. Confocal fluorescence time-lapse imaging was performed on live cells that were positive for tdTomato (not shown), capturing frames every 50 seconds. ManII-GFP is shown in green. Golgi tubulation driven by depletion of MYO18A depends on GOLPH3 and the ability of GOLPH3 to bind PI4P and to induce membrane curvature. Quantification of Golgi tubulation is provided in Figure S5A. Western blots to confirm knockdowns and protein expression are provided in Figure S5B.
Figure S1. GOLPH3 Binding to PI4P Causes Efficient Liposomal Tubulation. Related to Figures 1, 2, and 3. (A) EPSIN-ENTH and GOLPH3 (each at 5μM) bind similarly to liposomes containing their cognate phosphoinositides, PI(4,5)P2 and PI4P (each at 4.2 mol%), respectively. After incubation with the indicated proteins, liposomes were sedimented by ultracentrifugation, and bound and free protein assessed by Coomassie Brilliant Blue stained SDS-PAGE of the pellet and supernatant fractions.
(B) Aliquots of the binding reactions from (A) were processed and imaged by EM. Four example images are shown for each.
(C) Quantification of (B). Graph shows mean ± SEM, with the number of randomly selected fields (n) indicated, pooled from three independent experiments. p values are indicated, calculated by t test.
(D) Liposomes were generated using natural (brain-derived) PI4P (1.8 mol%) and either PC or PS (at 15 mM), and incubated with purified, bacterially expressed GOLPH3 (WT) or GOLPH3-R90L (at 14 μM). They were then visualized by EM. Five example images are provided for each. Similar results are obtained with liposomes containing PE (Figure 1C).
(E) SialT-GFP marks trans-Golgi-derived tubules driven by mCherry-GOLPH3. Shown are successive frames from time-lapse images of HeLa cells co-expressing SialT-GFP and mCherry-GOLPH3. A dynamic tubule is marked by an arrowhead. Similar results are obtained with ManII-GFP (Figure 2).
(F) Hydrophilic residues placed at the tip of the hydrophobic β-loop (GOLPH3 L195E/L196E) interfere with GOLPH3 binding to PI4P-containing liposomes. WT or mutant GST-GOLPH3 protein (at 1.4 μM) was allowed to bind to PI4P- or PI- (control) containing liposomes (at 4.2 mol%), separated into lipid-bound and free fractions by ultracentrifugation, and assessed by Coomassie Brilliant Blue staining of SDS-PAGE.
(G) GOLPH3 binds to natural (C18 or greater) and short-chain (C8) lipid vesicles containing PI4P. Vesicles were produced with short-chain (C8) or natural long-chain (C18 or greater) lipids including PI4P or PI (control) and incubated with equal amounts of purified, bacterially expressed GOLPH3 (WT) or GOLPH3-R90L. To compensate for intrinsically reduced binding of GOLPH3 to C8 liposomes, the amount of PI4P (or PI) in the C18 liposomes was reduced (414 μM for C8 [2.7 mol%], 23 μM for C18 [0.15 mol%]) to produce similar binding of GOLPH3 to both the C8 and C18 vesicles to allow fair comparison of tubulation. Vesicle-bound and free protein were separated by ultracentrifugation and assessed by SDS-PAGE (Figure 3B). The graph indicates quantification of the amount of GOLPH3 bound to the liposomes, which is similar for C8 and C18 PI4P liposomes. Binding is specific, as observed by loss of binding with the R90L mutation or substitution of PI for PI4P. Graph shows mean ± SEM, with the number of independent experiments (n) indicated. p values are indicated, calculated by t test.
Figure S2. GOLPH3’s Hydrophobic β-Loop is Required for Forward Trafficking from the Golgi to the Plasma Membrane. Related to Figure 4. (A) Quantification of relative GOLPH3 expression levels for Figures 4A and 4B. GOLPH3 knockdown/rescue experiments were performed in HeLa cells by transfection of GOLPH3-specific siRNA (or control or MYO18A-specific siRNA) followed by transfection of expression vectors for siRNA-resistant GOLPH3 (WT), the indicated mutants, or the empty vector. Cells were fixed and IF performed to examine the Golgi (GM130) and GOLPH3 (see Figure 4A) to allow measurement of Golgi area (Figure 4B) and, in this graph, GOLPH3 expression levels relative to the control.
(B) mKO2-GOLPH3-driven Golgi tubules contain the cargo VSVG-GFP. HeLa cells were co-transfected with expression vector for mKO2-GOLPH3 (to drive Golgi tubulation) and ts045-VSVG-GFP (a fluorescently labeled cargo). After overnight incubation at 40°C, cells were shifted to 32°C for 75 min to allow trafficking. Successive frames (interval of 10 s) from live-cell imaging are shown, demonstrating dynamic Golgi tubules from two independent experiments. White arrows indicate co-localization of VSVG-GFP with mKO2-GOLPH3 on Golgi tubules.
(C) Knockdown of GOLPH3 or MYO18A results in increased accumulation of ts045-VSVG-GFP at the Golgi at 22°C. Cells expressing ts045-VSVG-GFP transfected with siRNA to GOLPH3, MYO18A, or control were kept at 40°C for 16 hours, and then shifted to 22°C for 40 min, as indicated. Cells were fixed and the amount of GFP co-localizing with IF to GM130 (to mark the Golgi) was normalized to total GFP per cell. Shift to 22°C resulted in significant trafficking from ER to Golgi, with increased accumulation at the Golgi in GOLPH3 and MYO18A knockdown cells.
(D) In parallel to (C), cells at 22°C for 40 min were left unpermeabilized, and IF performed with antibody to the extracellular domain of VSVG to assess delivery to the PM. Delivery to the PM detected in control siRNA-transfected cells was strongly impaired by treatment with 5 μM Golgicide A (GCA). Knockdown of GOLPH3 or MYO18A also reduced delivery to the PM, accounting for the increased accumulation at the Golgi observed in (C).
(E) Golgi-to-ER recycling was assessed by measuring accumulation of mCherry-KDEL at the Golgi, co-localizing with IF to GM130. HeLa cells expressing mCherry-KDEL were transfected with siRNA to GOLPH3, MYO18A, or control, as indicated.
(F) Representative Western blot for (C), (D), and (E) demonstrating >70% knockdown of GOLPH3 or MYO18A. GAPDH serves as a loading control. Cell lysate from transfection with control siRNA was loaded at decreasing amounts to allow semi-quantitative comparisons.
(G) Quantification of relative GOLPH3 expression levels for Figures 4C and 4D. Trafficking of ts045-VSVG-EGFP was examined in a GOLPH3 knockdown/rescue experiment in HeLa cells. siRNA to GOLPH3 (or MYO18A or control) was transfected, followed by transfection of expression vector for siRNA-resistant GOLPH3 (WT), the indicated mutants, or empty vector. Overnight incubation at 40°C arrested trafficking at the ER, followed by incubation at 32°C for 75 minutes to allow trafficking. Cells were fixed and IF performed to examine exofacial (surface) VSVG in unpermeabilized cells (Figure 4C), followed by permeabilization to allow staining for GOLPH3 to assess relative expression levels. Graphs show mean and SEM with the number of cells (n) indicated, pooled from four (A), three (C, D), two (E), or six (G) independent experiments. p values are indicated, calculated by t test.
Figure S3. Expression of Tagged GOLPH3 Significantly Increases Golgi Tubulation, Regardless of the Nature of the Tag. Related to Figure 5. (A) Cells expressing either mCherry-GOLPH3 or mKO2-GOLPH3 display significantly increased Golgi tubulation compared to cells expressing mCherry or mKO2. HeLa cells stably expressing ManII-GFP as a Golgi marker were transfected with expression vectors for mCherry-GOLPH3, mKO2-GOLPH3, mCherry, or mKO2, and confocal time-lapse images were captured every 10 seconds. Also see Figures 5A and 5B for comparisons of GOLPH3 with and without an amino-terminal tag.
(B) Quantification of Golgi tubulation for (A).
(C) Expression of either 3xMYC-GOLPH3 or 3xHA-GOLPH3 causes Golgi tubulation. HeLa cells stably expressing ManII-GFP were transfected with expression vectors for 3xMYC-GOLPH3 or 3xHA-GOLPH3 and confocal images were captured every 29 or 38 seconds. After live imaging, cells were fixed and examined by immunofluorescence with anti-MYC or anti-HA antibodies to verify expression of 3xMYC-GOLPH3 or 3xHA-GOLPH3. Also see Figures 5A and 5B, and Movie S4.
(D) Expression levels of proteins used for lipid blotting in Figure 5C. GOLPH3 (WT), GOLPH3-R90L, 3xMYC-GOLPH3, and PX protein (Drosophila CG4960; Dippold et al., 2009) were synthesized by in vitro transcription and translation, and labeled by inclusion of 35S-methionine in the translation mix. Protein levels were compared using SDS-PAGE, followed by fixation and detection by phosphorimager. Also see Figure 5C for the lipid blot.
(E) Quantification of relative GOLPH3 expression levels for Figures 5E and 5F. Images from Figure 5E underwent automated segmentation and analysis to measure integrated GOLPH3 IF per cell. Graphed is mean GOLPH3 expression relative to the control. Graphs in (B) and (E) show mean ± SEM, with the number of cells (n) indicated, pooled from four (B) or four (E) independent experiments. p values are indicated, calculated by t test. White arrows indicate Golgi tubulation.
Figure S4. Golgi Tubulation Occurs in Response to Depletion of MYO18A or Depolymerization of F-actin. Related to Figure 6. (A) Protein expression levels in cells depleted for MYO18A. HeLa cells stably expressing ManII-GFP were transfected with either control or MYO18A-specific siRNA oligonucleotides. Cells were then transfected with expression vectors for mKO2-GOLPH3 (WT), mKO2-GOLPH3-R90L, or mKO2. Parallel coverslips were used for IF (Figures 6A and 6B) or Western blotting (shown here), using antibodies to GOLPH3, MYO18A, and GAPDH (as a loading control). The control siRNA-transfected samples are loaded at different relative amounts to allow semi-quantitative assessment of the knockdown of MYO18A, observed to be ~75–90% for each.
(B) HeLa cells stably expressing ManII-GFP to mark the Golgi were transfected with expression vectors for either mKO2-GOLPH3 or mKO2 and confocal images were captured every 34 seconds. Live-cell imaging was performed to observe Golgi tubulation before and after addition of cytochalasin B (Movie S7). Graphs show quantification of relative Golgi tubulation of the same fields prior to, or 34 seconds after treatment with 30 μM cytochalasin B. Graphed are mean ± SEM, with the number of cells (n) indicated, pooled from three independent experiments. p values are indicated, calculated by paired (before vs. after) or unpaired (mKO2 vs. mKO2-GOLPH3) t test.
(C) Endogenous GOLPH3 is observed to localize to Golgi tubules induced by depletion of MYO18A. HeLa cells stably expressing ManII-GFP were transfected with siRNA to deplete MYO18A (as in Figure 6C and S4D). Cells on gridded coverslips were imaged live to unambiguously identify active tubulation, fixed for IF, and the grid coordinates used to return to the same cell to observe the subcellular localization of endogenous GOLPH3. As shown, endogenous GOLPH3 is observed to localize to the Golgi tubules that form in response to loss of MYO18A.
(D) Most mCherry-GOLPH3-driven Golgi tubules do not contain detectable ARF1. HeLa cells stably expressing ManII-GFP were transfected to express mCherry-GOLPH3, fixed, and IF performed to detect endogenous ARF1. Tubules emanating from the Golgi are observed that are positive for both ManII-GFP and mCherry-GOLPH3, as expected. Endogenous ARF1 is observed at the Golgi, as expected. However, ARF1 is not observed on most of the mCherry-GOLPH3-driven tubules. Arrows in (C) and (D) indicate Golgi tubules.
(E) Protein expression levels in cells depleted for GOLPH3, MYO18A, or both. HeLa cells stably expressing ManII-GFP were transfected with combinations of control, GOLPH3, and MYO18A-specific siRNA oligonucleotides, as indicated. Parallel coverslips were used for IF (Figures 6C and 6D) or Western blotting (shown here), using antibodies to GOLPH3, MYO18A, and GAPDH (as a loading control). The control siRNA-transfected samples were loaded at different relative amounts to allow semi-quantitative assessment of the knockdown of GOLPH3 and MYO18A, observed to be 75–90% for each.
Figure S5. Golgi Tubulation Driven by Depletion of MYO18A Requires Endogenous GOLPH3, Its Ability to Bind PI4P and to Induce Membrane Curvature. Related to Figure 6. (A) Measurement of Golgi tubulation in HeLa cells stably expressing the Golgi marker ManII-GFP and transfected with siRNA to knockdown GOLPH3 and MYO18A was performed as indicated in Figure 6C and 6D. Transfection of siRNA-resistant GOLPH3 expression vectors was performed, as indicated. All constructs were co-transfected with an expression vector for tdTomato to mark transfected cells. Live confocal fluorescent imaging of tdTomato-positive cells was performed to assess Golgi tubulation, as observed by the dynamic behavior of the ManII-GFP Golgi marker (Movie S9). WT GOLPH3 rescued Golgi tubulation caused by depletion of MYO18A. However, GOLPH3 mutants defective in binding PI4P (R90L) or in inducing membrane curvature (Δ190–201 and Δ193–198) were unable to rescue Golgi tubulation. Graph shows mean and SEM, with the number of cells (n) indicated, pooled from five independent experiments. p values are indicated, calculated by t test.
(B) Western blotting of lysates from parallel coverslips from (A) to assess MYO18A and GOLPH3 expression. GAPDH is assessed as a loading control. The control lysate is loaded at different relative amounts to allow semi-quantitative assessment of knockdown of MYO18A (75–90%) and GOLPH3 (>90%).