

Abstract
Conjugation of peptides with oligonucleotides offers opportunities for combining the strengths of both biopolymer classes. Herein, we show that the combination of a peptide-based module with an antisense oligonucleotide module provides for enhancements of potency and a widened scope of cell delivery options. The peptide unit comprises a Smac mimetic compound (SMCs) which antagonizes the action of inhibitor of apoptosis proteins (IAPs) frequently overexpressed in cancer cells. To counteract SMC resistance, the antisense module downregulates the cellular FLICE-like protein (c-FLIP), a master regulator of the extrinsic apoptosis pathway. We compared c-FLIP antisense units based on oligophosphorothioate (PSO) and peptide nucleic acid (PNA) architectures. Owing to the ease of synthesis, PNA conjugates combined with a cell penetrating peptide (CPP) offer a seemingly ideal solution for cell delivery of dual activity agents. However, our investigations revealed that such congeners have to be handled with care to avoid off-target effects. By contrast, PSO conjugates provided a more robust and specific activity for inducing death of SMC-resistant A549 cells due to a simultaneous activation of caspases and c-FLIP knockdown. We show that lipofection is a convenient approach for delivery of peptide–PSO conjugates into cells. The results highlight that the combination of the peptide and the DNA world confers properties inaccessible by the unconjugated components.
Introduction
Peptides and oligo(ribo)nucleotides are increasingly used as pharmaceutically active compounds. In contrast to traditional small molecules, where high bioactivities typically are the result of extensive high-throughput screenings often followed by laborious manual chemical modification, potent peptides/oligonucleotides are much easier to obtain. They represent the native binding partners of many biological entities and, as such, utilize nature’s pre-existing, highly specific recognition patterns.1−4 In the design of chemical biology probes or pharmaceutically active compounds, peptides and oligonucleotides have typically been treated as separate biopolymer worlds. Yet, in recent years, attention has shifted toward conjugates as the combination of nucleic acid and peptide functions unlocks unique functionalities and may help overcome limitations like cell delivery, which remains a major challenge associated with the development of biopolymer-based therapeutics.5−8
We believe that peptide–oligonucleotide conjugation also provides an opportunity to enhance potency. By combining two biological activities, the peptide unit can be directed against one particular intracellular target, while the oligonucleotide unit targets another distinct cellular process: the resulting dual-mode action synergistically enhancing the efficacy of both components. At the same time, such hybrid molecules may benefit from a facilitated cell delivery. The typically used delivery agents are restricted to a specific biopolymer class and agents that grant cellular entry to oligonucleotides may not work for peptides and vice versa. This limits the potential range of delivery options for native biomolecules. Instead, hybrids comprising both biopolymer classes, peptides and oligonucleotides, should benefit from both of their constituents’ unique delivery methods. Frequently, this has been demonstrated with cell-penetrating peptides mediating cell uptake of antisense oligonucleotides.8−10 The reverse approach, using oligonucleotide conjugation to facilitate cellular delivery of membrane impermeant peptides or proteins, remains less explored11,12 but should present an equally viable approach.
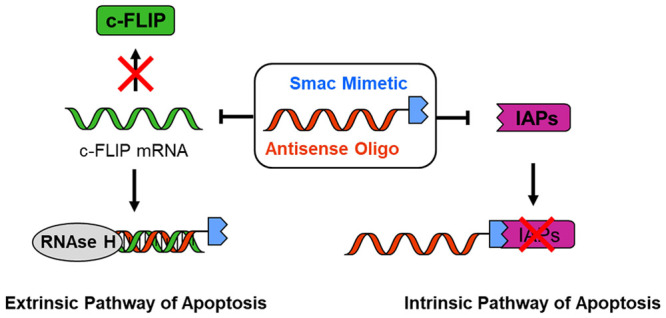
In a previous proof-of-concept study, we have covalently linked a pro-apoptotic, peptidic Smac mimetic compound (SMC) with an anti-c-FLIP phosphorothioate antisense oligonucleotide (ASO) and reported enhanced efficacy of the resulting dual-activity conjugate (SMC-ASO; Figure 1) on cancer cells upon delivery by lipofection.13 The SMC module (blue in SMC-ASO) mimics the N-terminal AVPI tetrapeptide of the Smac protein and antagonizes the antiapoptotic effect of Inhibitor of Apoptosis Proteins (IAPs), frequently overexpressed in cancer.14−17 For numerous cancer cell lines, however, inhibition of IAPs alone has proven insufficient to overcome resistance. SMC unresponsive cells often possess elevated levels of cellular FLICE-like inhibitory protein (c-FLIP), acting as a failsafe. This enzymatically inactive caspase homologue controls the activation state of Caspase-8 and protects the cell against apoptotic stimuli from chemotherapeutics and death receptor agonists.18 The anti-c-FLIP ASO was included (red in SMC-ASO) to silence this anti-apoptotic master switch of the extrinsic pathway of apoptosis and sensitize originally resistant cells to SMC treatment.
Figure 1.

Overview of SMC-ASO conjugate mode of action. The bivalent SMC part inhibits IAPs like XIAP (intrinsic pathway of apoptosis), while the linked c-FLIP antisense oligonucleotide blocks c-FLIP mRNA from translation (extrinsic pathway of apoptosis). Both components work in tandem to synergistically break apotosis resistance.
Previously, we utilized the antisense oligonucleotide platform to attach up to four monovalent SMC units. The SMCs were equipped with strained cycloalyknes and conjugated with antisense oligonucleotides displaying one to four azido groups. The aim was to enable enhanced binding of IAPs such as XIAP, cIAP1, and cIAP2 by simultaneously targeting two of the three baculoviral IAP repeat (BIR).19−21 However, despite improvements over IAP affinity, multivalent SMC presentation did not lead to improved cellular activity of the conjugates. Herein, we describe our efforts for facilitating the synthesis and improving the potency of the SMC-ASO conjugates. One of the aims was to provide unambiguous evidence for the role of the peptide’s mode of action and therefore compared anti-c-FLIP antisense molecules containing high affinity (bivalent) SMC modules with conjugates of low affinity (monovalent) modules.
The chemical nature of the antisense module should play an important role. For an efficient antisense effect, biologically stable molecules with high affinity for complementary RNA are imperative. Nuclease-resistant oligophosphorothioates (PSOs) are an obvious choice given their widespread use as antisense compounds. However, within the frame of dual action agents involving peptide modules, the use of peptide nucleic acids (PNAs) seems even more attractive. Peptide nucleic acids are stable against nucleases and proteases and combine very high binding affinity for complementary RNA with excellent sequence specificity.22,23 PNAs have been successfully applied as steric block-type antisense compounds, but unlike oligophosphorothioates, they are prepared by the same chemistry as peptides.24−28 As a result, peptide–PNA conjugates can be prepared in a linear synthesis without recourse to post-synthetic conjugation chemistry.
In this study, we describe dual activity agents in which bivalent and monovalent peptide modules are combined with antisense modules based on oligophosphorothioates or PNA. We show that care must be taken when SMC-PNA conjugates are endowed with cell-permeabilizing units introduced by the use of guanidino–PNA (GPNA) or cell penetrating peptides. Experiments with the peptide–PSO conjugates point to a robust and specific activity due to a simultaneous activation of caspases and c-FLIP knockdown, showcasing that the application of peptide–ASO conjugates is not limited to the modulation of uptake and/or distribution properties but can also provide for enhanced efficacy.
Results and Discussion
Initial experiments were focused on identifying SMC systems that provide a high enhancement of IAP affinity upon bivalent presentation. The aim was to use the monovalent and bivalency-enhanced SMCs as chemically related low- and high-affinity IAP binders, intending to provide proof for the mode of action by showing a correlation between IAP affinity and cellular activity. We selected BV-6 as a moderately active IAP antagonist.21 BV-6 has frequently been used to sensitize cancer cells to chemotherapeutic agents.29−32 To unambiguously demonstrate that conjugation with antisense modules can provide additional cell delivery options to peptides and peptidomimetics, we intentionally impaired the cell delivery properties of the linked SMC units by introducing a peptide tether (Figure 2). A central (azido)lysine residue was included to allow introduction of oligophosphorothiates by strain-promoted azide–alkyne cycloaddition (see R4 in Figure 2). Monovalent compounds contained the same number of spacer atoms. Replacement of the N-terminal N-methyl alanine, which is crucial for binding to the BIR pocket,33 by N-acetylated serine provided an inactive control compound.
Figure 2.

Structures of Bi- and monovalent Smac mimetic–ASO conjugates. An MV1/BV6 derived, bivalent inhibitor was dimerized and/or tethered to an anti c-FLIP PSO or PNA trough a short tetrapeptide linker. For cellular delivery, PNAs were c-terminally modified with a covalently linked TP10, PDEP-P14, Penetratin, Pip2b, Arg8, CLIP6, or TAT peptide. PSO sequences represent full phosphorothioates. For GPNA4 and scrGPNA4 sequences, underscored bases were replaced with positively charged, γ-guanidinium PNA monomers. Chg = cyclohexylglycine, Dip = diphenylalanine, aea = 5-amino-3-oxapentanoic acid, X = 6-aminohexanoic acid.
The affinity of peptide-dimerized inhibitors (SMC)2-Ln (n = 1, 2, 3) for a XIAP protein construct (XIAP L-Bir2-Bir3) that comprises the BIR2 and BIR3 domains, and the preceding linker region was measured in a competitive fluorescence polarization binding assay using a fluorescein-labeled reference binder (Smac-1F peptide).34 The monovalent inhibitor (H-MeAla-Chg-Pro-Dip-Gly-NH2, SMC) binds the protein with an IC50 value of 4.6 μM, comparable to the literature value for the unmodified AVPI peptide of 10.4 μM.34 Dimerization with linkers L1 to L3 increased the affinity by 1 order of magnitude. The length of the linker appeared to have a minor influence. However, a comparative study with three different Smac mimetics revealed a trend (Supporting Information Figure S1). The highest affinity gain was obtained with the shortest linker L1, resulting in an IC50 = 261 nM for the BV-6 analogous (SMC)2-L1, which was employed in the further experiments.
To access the dual-activity conjugates, we linked the bivalent inhibitors to PSO and PNA oligomers. We used the well-studied antisense sequence ISIS 23296 and scrambled control ISIS 132383.35 PSO conjugates were linked by strain-promoted azide–alkyne cycloaddition reactions between commercially available DBCO-modified oligonucleotides and azido-peptides. Illustrating the advantages of using PNA as an antisense module, the synthesis of SMC-PNA conjugates was performed in a single run on resin by involving Fmoc-protected amino acid building blocks and PNA monomers. ISIS 23296 is an optimized oligophosphorothioate gapmer sequence, which induces RNAs H. Antisense PNAs hinder mRNA translation by steric blockage.24 Literature evidence suggests that inhibition of protein synthesis by steric block antisense compounds such as PNA, morpholino nucleic acids, and 2′-OMe-RNA occurs more effectively near the site of translation initiation rather than in the coding region targeted by ISIS 23296, indicating that interfering with ribosome assembly is easier than perturbing elongation.36,37 For anti c-FLIP morpholinos, positions in the 5′-UTR and near the AUG start codon have been described as efficient target sites.38PNA1–4 conjugates are complementary to different targets around the start codon (−30 to 15, −13 to 3, −5 to 11, and −1 to 15, relative to the start codon). For comparison, we included PNA5, which is designed to bind within the ISIS 23296 target segment in the c-FLIP coding region.
To assess the efficacy of the anti c-FLIP PNA and dual-activity conjugates, we selected human lung adenocarcinoma cell line A549. These cells are highly resistant to Smac mimetics, even when combined with death receptor agonists like TRAIL, unless c-FLIP is silenced.18 The most commonly employed strategy to deliver PNA antisense compounds into cells relies on cell-penetrating peptides (CPPs).39 In recent years, many examples of such PNA-CPP conjugates have been reported to successfully deliver their payload into living eukaryotic cells. For our study, we initially chose octaarginine, as it represents one of the more active CPPs to provide PNA delivery,40−42 while allegedly being less toxic to A549 cells (IC50 > 1 mM) compared with other CPPS like Transportan (2.5 μM) or Penetratin (60.8 μM).43
The A549 cells were incubated with 2.5 μM PNA1–5-Arg8 conjugates and their ability to reduce c-FLIP expression was assessed by Western blotting (Figure 3A). PNA5 complementary to c-FLIP mRNA segment targeted by ISI 23296 (position 530 to 540) was ineffective at knocking down c-FLIP. For oligomers targeting regions at the start codon, efficacy was highly position dependent. PNA4 (−1 to 15) performed best, showing a clear decrease in c-FLIP band intensity. PNA1 (−30 to −15) and PNA3 (−5 to 11) had low to intermediate effect, while c-FLIP knockdown by PNA2 (−13 to 3) failed. We inferred the following: Arg8 delivers anti c-FLIP PNA into A549 cells in a sufficiently high amount for antisense effects to occur, given that a suitable target segment is selected.
Figure 3.

Knockdown of c-FLIP by antisense PNA-Arg8 conjugates (PNA1–5-Arg8) targeted against different mRNA positions. (A) Placement of anti-c-FLIP-PNAs relative to the AUG start codon. (B) Western blot of lysate from A549 cells treated with 2.5 μM PNA conjugates or OptiMEM (C) for 6 h followed by incubation in medium for 18 h. Protein samples were resolved on SDS-PAGE and Western immunoblotted with antibodies against short (C-FLIPS) and long (c-FLIPL) isoforms of c-FLIP and a β-actin loading control.
With the most efficient oligomer PNA4-Arg8, we determined dose–response curves by means of an AlamarBlue viability assay (Figure S7B). The IC50 = 5.6 μM suggested that the 2.5 μM concentration, which enabled knockdown of c-FLIP, was not sufficient to significantly affect cell viability. To facilitate endosomal escape, chloroquine was added, which, according to control experiments (Figure S7A), does not affect the viability of A549 cells up to 200 μM concentration. Addition of 100 μM chloroquine to PNA4 resulted in a small increase of potency (IC50 = 4.1 μM, Figure S7B). Conjugates containing a scrambled PNA4 sequence or PNA5, which proved inefficient in reducing levels of c-FLIP (see Figure 3), showed reduced toxicity (IC50 (scrPNA4-Arg8) = 9.7 μM, Figure S7C; IC50 (PNA5-Arg8) = 17.7 μM, Figure S7D). However, the gain in potency provided by PNA4 was small, and we reasoned that A549 cells cope well with antisense-mediated c-FLIP downregulation and conjugation with a Smac mimetic is required to allow for enhancements of potency.
In the next set of experiments, PNA4 was used for the construction of SMC-PNA conjugates. In addition to octa-arginine, we evaluated alternative CPPs (TP10,44 PDEP-P14,40 Penetratin,45 Pip2b,26 CLIP6,43 and TAT46) reported to deliver PNA into cells. Furthermore, we prepared a conjugate (GPNA4; Figure 2) using γ-modified, guanidinium-based PNA (GPNA47) that has been reported as a less amphiphilic and potentially less toxic alternative to oligoarginine-conjugated PNA. All conjugates were assembled entirely on solid support without the need for a separate ligation step. The panel of test compounds also included conjugates comprising the inactivated Smac mimetics AcSMC and a scrambled PNA oligomer. For delivery, A549 cells were treated with different concentrations of SMC-PNA-CPP conjugates in the presence of 100 mM chloroquine. The most active compounds (SMC)2-PNA4-PDEP-P14 (entry 3) and (SMC)2-PNA4-Pip2b (entry 7) affected A549 survival with an IC50 = 1.74 μM (Table 1). However, with only slightly increased IC50 values, the inactive/scrambled control compounds (AcSMC)2-scrPNA4- P14 (entry 4) and (AcSMC)2-scrPNA4-Pip2b (entry 8) were similarly active. The trend was confirmed with the full panel of CPPs tested. Most exhibited a certain toxicity shift between double active and double inactive conjugate, but in no case did the ΔIC50 value exceed 2.6 μM. CLIP6 and TAT conjugates displayed the widest therapeutic windows (ΔIC50 = 2.32 and 2.63 μM, entries 12–11 and entries 14–13), with the reason being a comparatively low toxicity of the inactive conjugate rather than an enhanced efficacy. Of note, despite a rather short 6 h incubation of cells, toxicity was observed for all conjugates (including the double negative control compounds AcSMC-scrPNA-CPP). In control experiments, we evaluated the viability when chloroquine was excluded. Though marginally reduced, the cytotoxicity of both SMC-PNA-CPP and AcSMC-PNA-CPP conjugates remained similar (Figure S5).
Table 1. IC50 Values of SMC-PNA4-CPP Conjugates Targeting the c-FLIP Start Codon on A549 Cellsa.
| entry | conjugate | SMC | AS PNA | IC50 [μM] | ΔIC50 [μM] |
|---|---|---|---|---|---|
| 1 | (SMC)2-PNA4-TP10 | + | + | 1.89 | –0.05 |
| 2 | (AcSMC)2-scrPNA4-TP10 | - | - | 1.84 | |
| 3 | (SMC)2-PNA4-P14 | + | + | 1.74 | 0.51 |
| 4 | (AcSMC)2-scrPNA4-P14 | - | - | 2.25 | |
| 5 | (SMC)2-PNA4-Pen | + | + | 3.18 | 1.89 |
| 6 | (AcSMC)2-scrPNA4-Pen | - | - | 5.07 | |
| 7 | (SMC)2-PNA4-Pip2b | + | + | 1.74 | 0.68 |
| 8 | (AcSMC)2-scrPNA4-Pip2b | - | - | 2.42 | |
| 9 | (SMC)2-PNA4-Arg8 | + | + | 2.01 | 1.30 |
| 10 | (AcSMC)2-scrPNA4-Arg8 | - | - | 3.31 | |
| 11 | (SMC)2-PNA4-CLIP6 | + | + | 2.66 | 2.32 |
| 12 | (AcSMC)2-scrPNA4-CLIP6 | - | - | 4.98 | |
| 13 | (SMC)2-PNA4-TAT | + | + | 2.27 | 2.63 |
| 14 | (AcSMC)2-scrPNA4-TAT | - | - | 4.90 | |
| 15 | (SMC)2-GPNA4 | + | + | 3.12 | 0.38 |
| 16 | (AcSMC)2-scrGPNA4 | - | - | 3.50 |
Conditions: 24 h after seeding (5 × 103 cells/well), A549 cells were incubated with SMC-PNA-CPP conjugate dilutions in OptiMEM containing 100 μM chloroquine. After 6 h, conjugate solutions were replaced with normal growth medium. After 48 h viability was determined by AlamarBlue assay.
It is surprising that the CPPs reported as highly active (TP10, PDEP14) were the most ineffective at producing a marked difference between active and inactive conjugates. Any potentially superior activity is apparently accompanied by an equally elevated toxicity when coupled to the SMC cargo. Literature precedent suggests that PNA-CPP conjugates are not generally toxic in a low μM concentration range.26,48 Therefore, we speculate that the elevated cytotoxic properties of SMC-PNA-CPP conjugates are caused by the added hydrophobicity of the SMC and the PNA units in combination with the highly charged nature of the CPP function. Similar observations were made by Koppelhus et al. when they modified antisense PNA-CPP conjugates with fatty acids.49 In an attempt to reduce the size of amphiphilic structures, the PNA-CPP unit was replaced by GPNA,47,50 which elegantly combines nucleic acid and CPP functions within one element. Unfortunately, active (entry 15) and inactive (entry 16) conjugates still showed nearly identical dose–response curves.
The factors influencing the toxicity of CPP conjugates remain poorly understood. Studies that systematically investigated the matter found that not only CPP identity and orientation but also cargo type, length, and linkage can have an impact.42,51,52 Of note, a high cytotoxicity has previously been reported for CPPs conjugated with proapoptotic (KLAKLAK)n peptides.53 Optimizing the toxicity of a particular cargo–CPP conjugate thus presents a challenge that might not be easily overcome when the cargo is hydrophobic or amphiphilic by nature.
Oligonucleotides are not as hydrophobic as PNA. Furthermore, unlike PNA, polyanionic DNA-type molecules can be delivered into cells by means of lipofection. In light of the previous experiments with the SMC-PNA-CPP conjugates, we assumed that oligonucleotide conjugation will provide the hydrophobic peptide module with less toxic cell delivery options. For a viability assay, A549 cells were transfected with 100 nM mono- and bivalent SMC-PSO conjugates using Lipofectamine LTX. These concentrations are more than 1 order of magnitude lower than the concentrations required for SMC-PNA-CPP conjugates. Cells treated with the double-active compounds demonstrated a significant loss of viability by up to 50–60% for bivalent ((SMC)2-PSO1; entry 17) and 40% for monovalent (SMC-PSO1; entry 18) SMC conjugates (Figure 4A). Control conjugates in which only the SMC or PSO part was active (SMC-scrPSO1 or AcSMC-PSO1; entries 25 or 26) or ISIS 23296 alone reduced viability by ca. 30%, performing only slightly better than control compounds comprised inactive AcSMC and scrambled ISIS 132383. This confirms a synergistic effect, increasing the general activity of the compound when both components are present and active. Cells that were treated with a double negative control (AcSMC-scrPSO1; entries 23 and 24) or an unconjugated, scrambled PSO (entry 26) also exhibited some decrease in viability which may be explained by the well-studied propensity of phosphorothioate oligonucleotides to nonspecifically bind to cellular proteins and transcription factors.54−58
Figure 4.
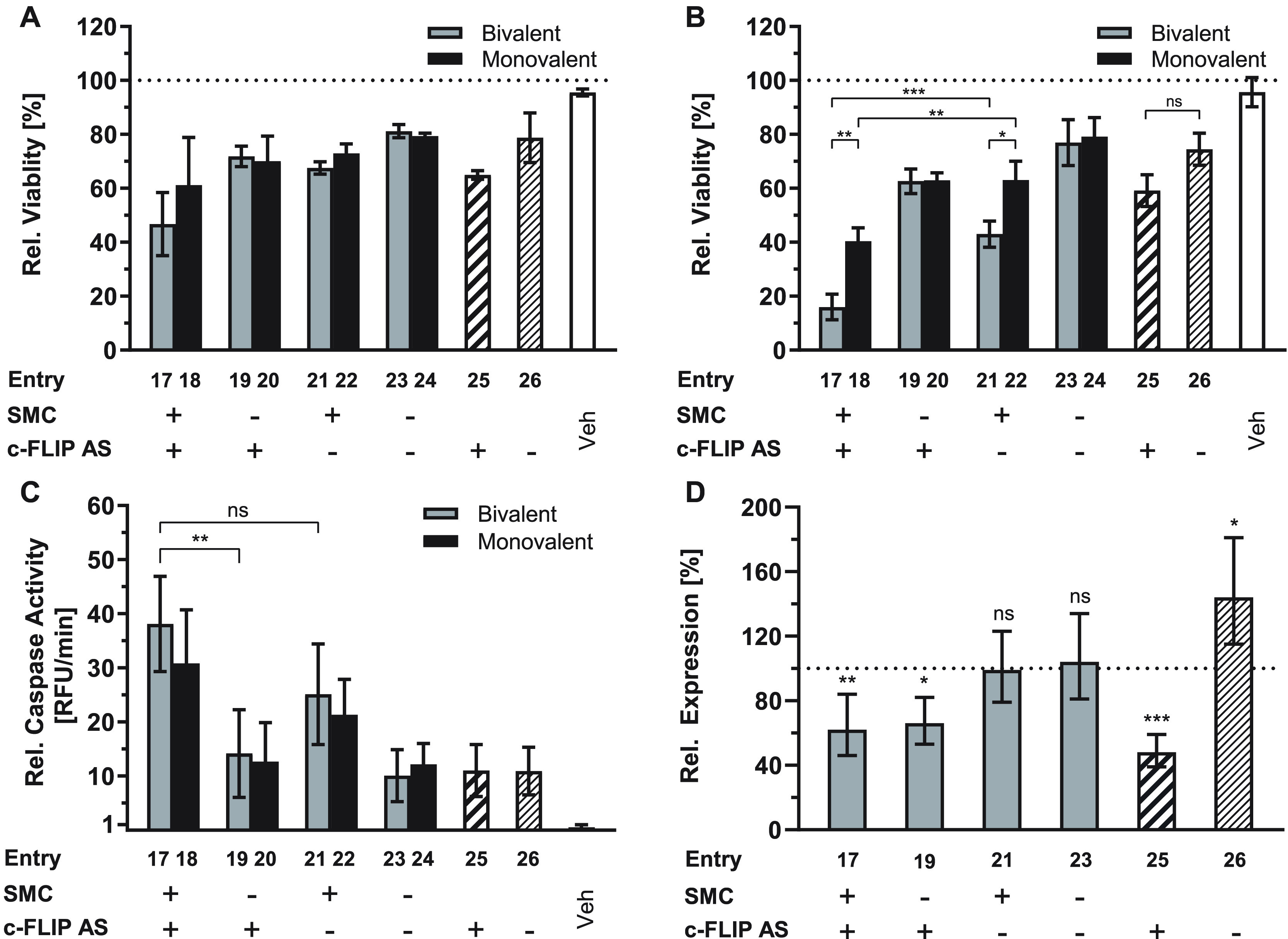
Dual-activity SMC-anti-c-FLIP-PSO conjugates reduce the viability of A549 cells by lowering c-FLIP expression and increasing caspase 3/7 activation. Cell viability after transfection with 100 nM SMC-PSO1 conjugates and incubation for 48 h in the absence (A) or presence (B) of 20 ng/mL SuperKillerTRAIL relative to a medium control. (C) Increase of caspase 3/7 activity based on DEVD-AFC peptide cleavage after transfection with 200 nM compound and incubation for 24 h with 2 ng/mL SuperKillerTRAIL relative to a medium-treated control. Error bars = SD. (D) RT-qPCR analysis of c-FLIP mRNA expression using GAPDH as a reference gene on total RNA isolated from cells transfected with 300 nM bivalent conjugate and incubated for 24 h (n = 3). Error bars = 95CI. Gray bars = bivalent conjugates, black = monovalent conjugates, striped = unconjugated PSO sequences ISIS 23296 and ISIS 132383. (+) indicates the presence of an active SMC peptide or antisense PSO sequence, (−) an inactivated peptide or scrambled PSO sequence. Vehicle (Veh) samples were treated with only transfection agent. * = p ≤ 0.05, ** = p ≤ 0.01 *** = p ≤ 0.001, ns = not significant (ANOVA with posthoc Tukey’s test).
Next, we incubated the cells in the presence of SuperKillerTRAIL, a death receptor agonist expressed by monocytes and B lymphocytes to induce apoptosis of tumor cells, which, however, rapidly develop resistance. Remarkably, the combined action of TRAIL and the SMC-PSO conjugates augmented the synergistic activity of the dual activity agent (Figure 4B). The viability values for cells that received inactive control compounds remained virtually constant but those treated with conjugates bearing an active bivalent inhibitor responded up to three times more strongly. The relative increase in potency was significantly higher when a c-Flip antisense sequence was present in the conjugate (Figure S2E), providing further evidence for the synergistic relationship. The monovalent conjugates followed the same pattern, albeit to a lesser extent (ca. 1.5-fold increase).
Importantly, valence did not matter for conjugates containing the inactivated AcSMC (compare entries 23 and 24). This and the differential response to mono- and bivalent conjugates comprising active SMCs (entry 17 vs entry 18, entry 21 vs entry 23) points to a specific effect of the peptide module that is required to gain activity beyond the potency of the antisense module.
To confirm apoptosis as the underlying cellular mechanism, we measured Caspase 3 and 7 activity. (Figure 4C). Cells treated with 200 nM conjugate in the presence of 2 ng/mL SuperKillerTRAIL were harvested after 24 h and the lysate incubated with the fluorogenic substrate DEVD-AFC. The observed pattern mirrors the trends observed in the viability assays. SMC-bearing conjugates elicited a strong caspase activation, with an increase over background levels of almost 40-fold in the case of (SMC)2-PSO1 (entry 17). The treatment with bivalent conjugates resulted in higher caspase activities than obtained with monovalent counterparts (entry 17 vs entry 18, entry 21 vs entry 23). In conjugates with an inactive peptide module, no significant differences between monovalent and bivalent conjugates could be observed (entry 19 vs entry 20). These results agree with the pattern expected for a Smac mimetic compound.
To verify that the antisense part is mechanistically active, we measured c-FLIP mRNA levels in conjugate-treated cells relative to those of GAPDH (Figure 4D), HPRT, and Tubulin (Figure S3) reference genes by RT-qPCR. Although mRNA levels were subject to strong variation, the experiment demonstrated that the unconjugated antisense PSO (entry 25) reduces c-FLIP levels in A549 cells by up to 50%. This value is in good accordance with the original publication of the ISIS 23296 sequence, which reported a knockdown by 70%, albeit with the use of potentially more efficient 2′-MOE-RNA/DNA gapmer PSOs.35 The SMC-PSO conjugates ((SMC)2-PSO1 and (AcSMC)2-PSO1; entries 17 and 19) demonstrated a slightly reduced efficacy, lowering c-FLIP levels by around 40%. It is possible that the presence of a peptide on the antisense oligonucleotide perturbs its ability to downregulate the target sequence. Cells that were treated with the unconjugated, scrambled PSO showed significant upregulation of c-FLIP levels. Conceivably, this could be the result of a nonspecific response to the phosphorothioate backbone. It has been reported that SP1, a transcription factor involved in the regulation of c-FLIP expression,59−61 is one of the proteins affected by nonspecific interaction with phosphorothioates.58 This would suggest that the correct antisense sequence too causes upregulation to a certain degree, but here, the knockdown may overcompensate this effect. Of note, upregulation of c-FLICE did not occur when the cells were treated with peptide-modified scrambled PSO conjugates (SMC)2-scrPSO1 (entry 21) and (AcSMC)2-scrPSO1 (entry 23). We speculate that the off-target effects might be alleviated by the presence of the linked peptide.
Conclusions
Hybrid conjugates comprising antisense oligonucleotide and peptide modules offer enhanced potency by targeting two cellular processes while potentially benefiting from an improved repertoire of cell delivery methods. This prospect seems particularly attractive for peptides that have moderate activity on intracellular targets. The experiments performed in this exemplary study focused on inducing cancer cell death by treatment with a peptide module reminiscent of the proapoptotic Smac-mimetic (SMC) BV-6, which antagonizes Inhibitor of Apoptosis Proteins. Unlike the parent compound BV-6, our peptide-linked bivalent analogue (SMC)2-L1—despite binding IAPs—does not induce apoptosis (Figure S6), which is most likely due to poor cell uptake. We therefore consider (SMC)2-L1 as a suitable model for moderately active, cell-impermeable peptides. Viability tests with A549 lung cancer cells showed that introduction of the cell penetrating peptide (CPP) octaarginine restored the pro-apoptotic effect (IC50 = 4.4 μM). To improve the potency, we included antisense modules that downregulate the c-FLICE protein: a negative master regulator of the extrinsic apoptosis pathway that confers resistance to SMC treatment. Given the ease of synthesis, the use of peptide nucleic acid (PNA) antisense modules appeared attractive. A screening of five different PNA-Arg8 conjugates directed against different regions of the c-FLICE mRNA afforded PNA4 which, according to Western Blot, enabled nearly complete downregulation of the c-FLICE protein at 2.5 μM concentration. However, the combination of the (SMC)2 module, the PNA module PNA4 and CPPs such as Arg8, TP10, PDEP-P14, penetratin, Pip2b, CLIP6, and TAT exposed major toxicity problems. Despite apparent potency differences between active and inactive conjugates, indicating a successful delivery, the concentration windows with low nonspecific toxicity remained narrow. Given the widespread use of CPPs as a vehicle to deliver PNA into cells, we want to call attention to the pitfalls of combinations which involve the introduction of additional hydrophobic or amphiphilic units.
In contrast, conjugation of the BV-6 analogue (SMC)2-L with an oligophosphorothioate-based antisense module resembling ISIS 23296 provided for substantial enhancements of efficacy. By using Lipofectamine LTX as cell delivery agent, 100 nM of the dual activity (SMC)2-PSO conjugate was sufficient to inhibit growth of A549 cells by more than 50%, which excelled over the activity of the nonconjugated (SMC)2-L1 and PSO units. In the presence of a death receptor agonist, the synergistic effect became even more pronounced. In contrast to our previously reported SMC-PSO conjugates, the bivalent conjugate (SMC)2-PSO provided higher efficacy than monovalent SMC-PSO. The positive correlation between IAP affinity in vitro and cellular activity provides evidence for a specific effect of the peptide module. The observed synergy is a result of the activation of caspases and a concurrent reduction of c-FLIP mRNA levels. Although the oligonucleotide part does not seem to have a direct influence on caspase activity, it sensitizes the cell to the pro-apoptotic effect of SMCs. In conclusion, the results of this study demonstrate that a combination of the peptide and DNA worlds provides opportunities inaccessible by unconjugated components.
Experimental Procedures
Azido-SMC and PNA/GPNA-Conjugate Synthesis
SMC-PNA-CPP and SMC-GPNA conjugates were synthesized by Fmoc Chemistry on TentaGel XV Rink Amide resin (100–200 μm; 0.23 mmol/g) using an automated peptide synthesizer. Modified PNA monomers (GPNA) were prepared analogous to a previously reported procedure starting from Fmoc-Arg(Pbf)-OH.62 Prior to conjugate synthesis, resins were swelled in DMF for 20 min. The Fmoc-protecting group was removed by treatment with 20% (v/v) piperidine in DMF for (2×, 3 and 7 min), and then the resin was washed with DMF (7×). Activated Fmoc-protected PNA/GPNA Monomers (4 equiv) dissolved in NMP (0.2 M) were coupled twice in the presence of HCTU (3.8 equiv) and NMM (8 equiv) for 20 min each at room temperature after 2 min of preactivation. Amino acids (6 equiv) dissolved in NMP (0.6 M) were coupled in the presence of HCTU (5.7 equiv), Oxyma Pure (6 equiv), and NMM (12 equiv). The resin was washed with DMF (3×), and unreacted amino groups were capped by treatment with acetic anhydride/2,6-lutidine/DMF (5:6:89 v/v/v) for 2 min. Afterward, the resin was washed with DMF (5×) and the next coupling cycle started. At the end of the synthesis, the resin was washed with CH2Cl2 (3×), dried under vacuum, and the conjugate cleaved off by adding a TFA/TIS/EDT/water (92.5:2.5:2.5:2.5, v/v/v/v) mixture for 3 h at room temperature. Filtration and precipitation in ice-cold Et2O afforded the crude PNA–conjugate. The solid precipitate was dried under argon, dissolved in water/CH3CN, and purified by semipreparative RP-HPLC (Agilent). Azide-modified Smac peptides were synthesized manually on ChemMatrix resin (35–100 mesh, 0.5–0.7 mmol/g) according to the same protocol. After synthesis, the Mtt protecting group on the C-terminal Dap was removed by 5–7 washes with 2% (v/v) TFA in CH2Cl2 until the solution remained colorless. Peptides were cleaved off the resin by treatment with TFA/TIS/water (95:2.5:2.5, v/v/v) for 2 h, precipitated in ice-cold Et2O and purified by semipreparative RP-HPLC.
SMC-PSO Ligation
For strain-promoted alkyne–azide cycloadditions, DBCO-modified PSOs were dissolved in phosphate buffer (10 mM sodium phosphate, 137 mM NaCl, pH 7.5) to a concentration of 1 mM. Azide-modified SMC (5 equiv) was dissolved in buffer/CH3CN 1:1 (v/v) and added to the reaction mixture. The clear solution was heated to 80 °C for 5 min and then incubated for 24 h at room temperature. The mixture was diluted with water and purified by preparative HPLC. After freeze-drying, product-containing fractions were dissolved in 300 μL water, and sodium acetate (3 M, pH 5.4) was added to a final buffer concentration of 0.3 M. Three volumes of isopropanol were added, and the resulting precipitate centrifuged (10 min, 16 900 rcf, 4 °C). This procedure was repeated once, and the pellet dried at rt for 10 min. The dry precipitate was dissolved in nuclease-free water.
Protein Expression and Purification
XIAP L-Bir2-Bir3 protein was produced in Escherichia coli BL21(DE3) grown at 37 °C in 2xYT medium supplemented with Kanamycin (50 μg/mL). Upon reaching an OD600 of 0.6, protein expression was induced by addition of 0.4 mM IPTG and 100 μM ZnAc2 at 20 °C for 20 h. Cells were lysed by French pressing in buffer (50 mM Tris, 200 mM NaCl, 50 μM ZnAc2, 0.1% β-mercaptoethanol, pH = 7.5) with protease inhibitors. Lysates were centrifuged (45 000 rcf, 30 min, 4 °C) and the supernatant purified on an Äkta pure FPLC system using a 5 mL HisTrapHP Ni column followed by gel filtration on a Superdex 75 column in 20 mM Tris (pH 7.5), 200 mM NaCl, 50 μM ZnAc, and 1 mM DTT. After purification, glycerol and DTT were added to a final concentration of 10% (v/v) and 10 mM.
Fluorescence Polarization Assay
Relative binding affinity was measured as described by Nikolovska et al.34 Fluorescein-labeled probe Smac-1F was synthesized according to the procedure described in the literature. To each well of a black, nonbinding 96-well plate, 1 nM of Smac-1F and 15 nM L-Bir2-Bir3 construct in assay buffer (100 mM sodium phosphate, 110 mM NaCl, 5 mM DTT, 100 μg/mL bovine γ-globulin, 0.02% sodium azide) was added and mixed with different concentrations of Smac mimetics. The plate was incubated for 2 h in the dark at room temperature and the anisotropy values determined using a plate reader (Ex./Em. λ = 485/535 nm).
Cell Culture and Viability Assays
A459 were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FCS, 4 mM Glutamine, and 1% Penicillin/Streptomycin mix at 37 °C in a humidified 5% CO2 atm and subcultivated twice a week. For viability assays, cells were seeded on a 96-well plate in 100 μL medium per well at a density of 5 × 103 cells/well and propagated 24 h to a confluency of ca. 70%. SMC-Phosphorothioate conjugates in OptiMEM were mixed with PLUS Reagent (1 μL/μg PSO), incubated for 5 min and then precomplexed with Lipofectamine LTX (2 μL/μg PSO) in OptiMEM for 20 min at room temperature before being added to the cells in medium without antibiotics to a final concentration of 100 nM. SMC-PNA-CPP conjugates were diluted in OptiMEM containing 100 μM chloroquine and added to the cells. After 6 h incubation at 37 °C and 5% CO2, either 1 volume DMEM + 20% FCS and 2% Penicillin/Streptomycin mix was added to the wells, or the transfection solutions were removed and replaced with normal growth medium. After 24 h, optionally 20 ng/mL SuperKillerTRAIL was added to the wells. Cells were incubated for another 24 h, washed with PBS and 100 μL alamarBlue diluted 1:10 (v/v) in PBS was added to each well. After 2 h incubation at 37 °C fluorescence intensity was measured (Ex./Em. λ = 531/590 nm) on a plate reader. In control experiments, we evaluated the viability when chloroquine was excluded. Though marginally reduced, the cytotoxicity of both SMC-PNA-CPP and AcSMC-PNA-CPP conjugates remained similar (Figure S5).
Caspase 3/7 Assay
Twenty-four hours before treatment, A549 cells were seeded on a 6-well plate and treated with transfection solutions at a final concentration of 200 nM as described above. After 24 h, cells were scraped off, centrifuged (1000 rfc, 5 min), washed with PBS, pelleted (8500 rcf, 3 min), and treated with 40 μL lysis buffer (20 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, pH 7.5) for 20 min on ice with intermittent sonication. Cellular debris was centrifuged off (16 900 rcf, 10 min, 4 °C) and the protein concentration in the supernatant determined by BCA assay according to the manufacturer’s instructions. 35 μg protein was pipetted in each well of a black 96-well assay plate and filled up to 50 μL with lysis buffer. 50 μL 2× caspase assay buffer (20 mM HEPES, 20 mM DTT, 20% glycerol, pH = 7.5) with 50 μM DEVD-AFC were added and the plate incubated for 2 h at 37 °C in the dark. Absorbance (λ = 575 nm) of the released dye was measured on a plate reader.
RT-qPCR
Twenty-four hours before treatment, A549 cells were seeded on a 6-well plate in 2 mL medium per well at a density of 2 × 105 cells/well. For treatment, the medium was replaced with 1800 μL culture medium without antibiotics. SMC-phosphorothioate conjugates were precomplexed with Lipofectamine LTX with PLUS Reagent as described above. Complexes were added to the cells at a final concentration of 300 nM. After 6 h, the medium was replaced with normal growth medium and the cells incubated for another 18 h. Cells were washed with PBS (1×) and total RNA was extracted followed by DNase I treatment according to the corresponding manufacturer’s instructions. The isolated RNA was precipitated from 0.1 volume sodium acetate buffer (3 M, pH 5.4) and 1 volume isopropanol, washed with 70% ethanol, and redissolved in nuclease-free water. The extracted RNA was quantified using its absorption at 260 nm and stored at −80 °C. For the qPCR measurement 1 μg total RNA was reverse transcribed and stored at −20 °C. 2.5 ng cDNA were mixed with 10 μL 2× master mix and 250 nM of forward and reverse primers (c-FLIP [NM_003879.7] forward: 5′-TGTGCCGGGATGTTGCTATA-3′, reverse: 5′-CAGCTTACCTCTTTCCCGTAAAAT-3′; GAPDH [AF261085.1] forward: 5′-GAAGGTGAAGGTCGGAGTC-3′, reverse: 5′-GAAGATGGTGATGGGATTTC-3′; HPRT [NM_000194.3] forward: 5′-CCTGGCGTCGTGATTAGTGAT-3′, reverse: 5′-AGACGTTCAGTCCTGTCCATAA-3′; TUBA1B [NM_006082.3] forward: 5′-CCTGGCGTCGTGATTAGTGAT-3′, reverse: 5′-AGACGTTCAGTCCTGTCCATAA-3′), and diluted with water to a final volume of 20 μL in a 96-well qPCR plate and amplified using the following program: (95 °C, 45 s) × 1 cycle, (95 °C, 10 s ;60 °C, 30 s) × 40 cycles. Subsequently, a melting curve analysis was performed: (85 °C, 15 s) × 1 cycle, (60 °C, 30 s) × 1 cycle, (60–95 °C, + 0.5 °C/cycle, 15 s) × 71 cycles. mRNA expression levels relative to vehicle treated control were calculated by the ΔΔCq method.
Statistical analysis (ANOVA with post hoc Tukey’s test) was performed on viability/ΔΔCq values using GraphPad Prism 8 software.
Acknowledgments
We thank Prof. Shaomeng Wang for providing the XIAP Bir2-Bir3 plasmid. This work was funded by the European Research Council’s Horizon 2020 programme (ERC Advanced Grant, TRIGGDRUG).
Glossary
Abbreviations
- AcS/AcSer
N-acetyl-l-serine
- aeea
8-amino-3,6-dioxaoctanoic acid
- ASO
antisense oligonucleotide
- BIR
baculoviral IAP repeat
- c-FLIP
cellular FLICE-like protein
- Chg
l-2-cyclohexylglycine
- CPP
cell-penetrating peptide
- Dap
l-2,3-diaminopropionic acid
- Dip
3,3-diphenyl-l-alanine
- FCS
fetal calf serum
- GPNA
guanidinium PNA
- IAP
inhibitor of apoptosis protein
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- MeA/MeAla
N-methyl-l-alanine
- NMM
N-methylmorpholine
- PNA
peptide nucleic acid
- PSO
phosphorothioate oligonucleotide
- Smac
second mitochondria-derived activator of caspases
- SMC
Smac mimetic compound
- SPPS
solid-phase peptide synthesis
- TFA
trifluoroacetic acid
- TIS
triisopropylsilane
- TRAIL
TNF-related apoptosis-inducing ligand
- X/Ahx
6-aminohexamoic acid
- XIAP
X-linked inhibitor of apoptosis protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.0c00284.
Synthesis and characterization of building blocks, HPLC and mass spectrometry data of PNA and DNA conjugates, XIAP binding affinities, viability assays with SMC-PSO conjugates, RT-qPCR experiments, dose–response measurements with PNA conjugates (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bennett C. F. (2019) Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 70 (1), 307–321. 10.1146/annurev-med-041217-010829. [DOI] [PubMed] [Google Scholar]
- Fosgerau K.; Hoffmann T. (2015) Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today 20 (1), 122–128. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Lau J. L.; Dunn M. K. (2018) Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 26 (10), 2700–2707. 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- Smith C. I. E.; Zain R. (2019) Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 59 (1), 605–630. 10.1146/annurev-pharmtox-010818-021050. [DOI] [PubMed] [Google Scholar]
- Valeur E.; Guéret S. M.; Adihou H.; Gopalakrishnan R.; Lemurell M.; Waldmann H.; Grossmann T. N.; Plowright A. T. (2017) New Modalities for Challenging Targets in Drug Discovery. Angew. Chem., Int. Ed. 56 (35), 10294–10323. 10.1002/anie.201611914. [DOI] [PubMed] [Google Scholar]
- Taskova M.; Mantsiou A.; Astakhova K. (2017) Synthetic Nucleic Acid Analogues in Gene Therapy: An Update for Peptide–Oligonucleotide Conjugates. ChemBioChem 18 (17), 1671–1682. 10.1002/cbic.201700229. [DOI] [PubMed] [Google Scholar]
- Benizri S.; Gissot A.; Martin A.; Vialet B.; Grinstaff M. W.; Barthélémy P. (2019) Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjugate Chem. 30 (2), 366–383. 10.1021/acs.bioconjchem.8b00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehto T.; Ezzat K.; Wood M. J. A.; El Andaloussi S. (2016) Peptides for Nucleic Acid Delivery. Adv. Drug Delivery Rev. 106, 172–182. 10.1016/j.addr.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Järver P.; Coursindel T.; Andaloussi S. E.; Godfrey C.; Wood M. J.; Gait M. J. (2012) Peptide-Mediated Cell and In Vivo Delivery of Antisense Oligonucleotides and SiRNA. Mol. Ther.--Nucleic Acids 1, e27 10.1038/mtna.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano R.; Alam M. R.; Dixit V.; Kang H. (2008) Mechanisms and Strategies for Effective Delivery of Antisense and SiRNA Oligonucleotides. Nucleic Acids Res. 36 (12), 4158–4171. 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaderer D.; Cashman S. M.; Kumar-Singh R. (2016) G-Quartet Oligonucleotide Mediated Delivery of Proteins into Photoreceptors and Retinal Pigment Epithelium via Intravitreal Injection. Exp. Eye Res. 145, 380–392. 10.1016/j.exer.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harguindey A.; Culver H. R.; Sinha J.; Bowman C. N.; Cha J. N. (2020) Efficient Cellular Uptake of Click Nucleic Acid Modified Proteins. Chem. Commun. 56 (35), 4820–4823. 10.1039/C9CC09401F. [DOI] [PubMed] [Google Scholar]
- Abendroth F.; Seitz O. (2014) Double-Clicking Peptides onto Phosphorothioate Oligonucleotides: Combining Two Proapoptotic Agents in One Molecule. Angew. Chem., Int. Ed. 53 (39), 10504–10509. 10.1002/anie.201406674. [DOI] [PubMed] [Google Scholar]
- Du C.; Fang M.; Li Y.; Li L.; Wang X. (2000) Smac, a Mitochondrial Protein That Promotes Cytochrome c-Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 102 (1), 33–42. 10.1016/S0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Verhagen A. M.; Ekert P. G.; Pakusch M.; Silke J.; Connolly L. M.; Reid G. E.; Moritz R. L.; Simpson R. J.; Vaux D. L. (2000) Identification of DIABLO, a Mammalian Protein That Promotes Apoptosis by Binding to and Antagonizing IAP Proteins. Cell 102 (1), 43–53. 10.1016/S0092-8674(00)00009-X. [DOI] [PubMed] [Google Scholar]
- Srinivasula S. M.; Hegde R.; Saleh A.; Datta P.; Shiozaki E.; Chai J.; Lee R. A.; Robbins P. D.; Fernandes-Alnemri T.; Shi Y.; et al. (2001) A Conserved XIAP-Interaction Motif in Caspase-9 and Smac/DIABLO Regulates Caspase Activity and Apoptosis. Nature 410 (6824), 112–116. 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- Bai L.; Smith D. C.; Wang S. (2014) Small-Molecule SMAC Mimetics as New Cancer Therapeutics. Pharmacol. Ther. 144 (1), 82–95. 10.1016/j.pharmthera.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung H. H.; Mahoney D. J.; Lacasse E. C.; Korneluk R. G. (2009) Down-Regulation of c-FLIP Enhances Death of Cancer Cells by Smac Mimetic Compound. Cancer Res. 69 (19), 7729–7738. 10.1158/0008-5472.CAN-09-1794. [DOI] [PubMed] [Google Scholar]
- Sun H.; Nikolovska-Coleska Z.; Yang C.-Y.; Qian D.; Lu J.; Qiu S.; Bai L.; Peng Y.; Cai Q.; Wang S. (2008) Design of Small-Molecule Peptidic and Nonpeptidic Smac Mimetics. Acc. Chem. Res. 41 (10), 1264–1277. 10.1021/ar8000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott F. L.; Denault J.-B.; Riedl S. J.; Shin H.; Renatus M.; Salvesen G. S. (2005) XIAP Inhibits Caspase-3 and −7 Using Two Binding Sites: Evolutionarily Conserved Mechanism of IAPs. EMBO J. 24 (3), 645–655. 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E.; Blankenship J. W.; Wayson S. M.; Fedorova A. V.; Kayagaki N.; Garg P.; Zobel K.; Dynek J. N.; Elliott L. O.; Wallweber H. J. A.; et al. (2007) IAP Antagonists Induce Autoubiquitination of C-IAPs, NF-KB Activation, and TNFα-Dependent Apoptosis. Cell 131 (4), 669–681. 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- Demidov V. V.; Potaman V. N.; Frank-Kamenetskil M. D.; Egholm M.; Buchard O.; Sönnichsen S. H.; Nlelsen P. E. (1994) Stability of Peptide Nucleic Acids in Human Serum and Cellular Extracts. Biochem. Pharmacol. 48 (6), 1310–1313. 10.1016/0006-2952(94)90171-6. [DOI] [PubMed] [Google Scholar]
- Pellestor F.; Paulasova P. (2004) The Peptide Nucleic Acids (PNAs), Powerful Tools for Molecular Genetics and Cytogenetics. Eur. J. Hum. Genet. 12 (9), 694–700. 10.1038/sj.ejhg.5201226. [DOI] [PubMed] [Google Scholar]
- Knudsen H.; Nielsen P. E. (1996) Antisense Properties of Duplex- and Triplex-Forming PNAs. Nucleic Acids Res. 24 (3), 494–500. 10.1093/nar/24.3.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abes S.; Turner J. J.; Ivanova G. D.; Owen D.; Williams D.; Arzumanov A.; Clair P.; Gait M. J.; Lebleu B. (2007) Efficient Splicing Correction by PNA Conjugation to an R6-Penetratin Delivery Peptide. Nucleic Acids Res. 35 (13), 4495–4502. 10.1093/nar/gkm418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova G. D.; Arzumanov A.; Abes R.; Yin H.; Wood M. J. A.; Lebleu B.; Gait M. J. (2008) Improved Cell-Penetrating Peptide–PNA Conjugates for Splicing Redirection in HeLa Cells and Exon Skipping in Mdx Mouse Muscle. Nucleic Acids Res. 36 (20), 6418–6428. 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Lu Q.; Wood M. (2008) Effective Exon Skipping and Restoration of Dystrophin Expression by Peptide Nucleic Acid Antisense Oligonucleotides in Mdx Mice. Mol. Ther. 16 (1), 38–45. 10.1038/sj.mt.6300329. [DOI] [PubMed] [Google Scholar]
- Soudah T.; Mogilevsky M.; Karni R.; Yavin E. (2017) CLIP6-PNA-Peptide Conjugates: Non-Endosomal Delivery of Splice Switching Oligonucleotides. Bioconjugate Chem. 28 (12), 3036–3042. 10.1021/acs.bioconjchem.7b00638. [DOI] [PubMed] [Google Scholar]
- El-Mesery M.; Shaker M. E.; Elgaml A. (2016) The SMAC Mimetic BV6 Induces Cell Death and Sensitizes Different Cell Lines to TNF-α and TRAIL-Induced Apoptosis. Exp. Biol. Med. (London, U. K.) 241 (18), 2015–2022. 10.1177/1535370216661779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak D. H.; Manton C.; Andreeff M.; Carter B. Z. (2010) SMAC-Mimetic BV-6 Sensitizes Therapeutic Agents-Induced Apoptosis In AML Cells. Blood 116 (21), 2177–2177. 10.1182/blood.V116.21.2177.2177. [DOI] [Google Scholar]
- Panayotopoulou E. G.; Müller A.-K.; Börries M.; Busch H.; Hu G.; Lev S. (2017) Targeting of Apoptotic Pathways by SMAC or BH3Mimetics Distinctly Sensitizes Paclitaxel-Resistant Triple Negative Breast Cancer Cells. Oncotarget 8 (28), 45088–45104. 10.18632/oncotarget.15125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettinger E.; Glatthaar A.; Abhari B. A.; Oelsner S.; Pfirrmann V.; Huenecke S.; Kuçi S.; Kreyenberg H.; Willasch A. M.; Klingebiel T.; et al. (2014) SMAC Mimetic BV6 Enables Sensitization of Resistant Tumor Cells but Also Affects Cytokine-Induced Killer (CIK) Cells: A Potential Challenge for Combination Therapy. Front Pediatr 2, 75. 10.3389/fped.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Sun C.; Olejniczak E. T.; Meadows R. P.; Betz S. F.; Oost T.; Herrmann J.; Wu J. C.; Fesik S. W. (2000) Structural Basis for Binding of Smac/DIABLO to the XIAP BIR3 Domain. Nature 408 (6815), 1004–1008. 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- Nikolovska-Coleska Z.; Meagher J. L.; Jiang S.; Kawamoto S. A.; Gao W.; Yi H.; Qin D.; Roller P. P.; Stuckey J. A.; Wang S. (2008) Design and Characterization of Bivalent Smac-Based Peptides as Antagonists of XIAP and Development and Validation of a Fluorescence Polarization Assay for XIAP Containing Both BIR2 and BIR3 Domains. Anal. Biochem. 374 (1), 87–98. 10.1016/j.ab.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Ackermann E., Bennett C., Zhang H., Watt A., Ricketts W., and Dean N.. Antisense Modulation of FLIP-c Expression. US 20040254137A1, December 16, 2004.
- Nielsen P. E. (2006) The Many Faces of PNA. Peptide Nucleic Acids, Morpholinos and Related Antisense Biomolecules (Janson C. G., and During M. J., Eds.) pp 3–17, Springer US, Boston. [Google Scholar]
- Nielsen P. E. (1999) Antisense Technology Part A: General Methods, Methods of Delivery, and RNA Studies. In Antisense Properties of Peptide Nucleic Acid, Methods in Enzymology (Abelson J. N., Simon M. I., and Phillips M. I., Eds.) pp 156–164, Vol 313, Academic Press, San Diego, CA. [DOI] [PubMed] [Google Scholar]
- Mourich D. V.; Jendrzejewski J. L.; Marshall N. B.; Hinrichs D. J.; Iversen P. L.; Brand R. M. (2009) Antisense Targeting of CFLIP Sensitizes Activated T Cells to Undergo Apoptosis and Desensitizes Responses to Contact Dermatitis. J. Invest. Dermatol. 129 (8), 1945–1953. 10.1038/jid.2009.16. [DOI] [PubMed] [Google Scholar]
- Shiraishi T., and Nielsen P. E. (2014) Cellular Delivery of Peptide Nucleic Acids (PNAs). In Peptide Nucleic Acids: Methods and Protocols, 2nd ed.(Nielsen P. E., and Appella D. H., Eds.) Methods in Molecular Biology, pp 193–205, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Kauffman W. B.; Guha S.; Wimley W. C. (2018) Synthetic Molecular Evolution of Hybrid Cell Penetrating Peptides. Nat. Commun. 9 (1), 1–10. 10.1038/s41467-018-04874-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J.Detailed Comparison of CPP’s Uptake Properties for Pro-Apoptotic Cargo Delivery. [Google Scholar]
- Bendifallah N.; Rasmussen F. W.; Zachar V.; Ebbesen P.; Nielsen P. E.; Koppelhus U. (2006) Evaluation of Cell-Penetrating Peptides (CPPs) as Vehicles for Intracellular Delivery of Antisense Peptide Nucleic Acid (PNA). Bioconjugate Chem. 17 (3), 750–758. 10.1021/bc050283q. [DOI] [PubMed] [Google Scholar]
- Medina S. H.; Miller S. E.; Keim A. I.; Gorka A. P.; Schnermann M. J.; Schneider J. P. (2016) An Intrinsically Disordered Peptide Facilitates Non-Endosomal Cell Entry. Angew. Chem., Int. Ed. 55 (10), 3369–3372. 10.1002/anie.201510518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soomets U.; Lindgren M.; Gallet X.; Hällbrink M.; Elmquist A.; Balaspiri L.; Zorko M.; Pooga M.; Brasseur R.; Langel Ü. (2000) Deletion Analogues of Transportan. Biochim. Biophys. Acta, Biomembr. 1467 (1), 165–176. 10.1016/S0005-2736(00)00216-9. [DOI] [PubMed] [Google Scholar]
- Derossi D.; Calvet S.; Trembleau A.; Brunissen A.; Chassaing G.; Prochiantz A. (1996) Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-Independent. J. Biol. Chem. 271 (30), 18188–18193. 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- Frankel A. D.; Pabo C. O. (1988) Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus. Cell 55 (6), 1189–1193. 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- Dragulescu-Andrasi A.; Rapireddy S.; He G.; Bhattacharya B.; Hyldig-Nielsen J. J.; Zon G.; Ly D. H. (2006) Cell-Permeable Peptide Nucleic Acid Designed to Bind to the 5‘-Untranslated Region of E-Cadherin Transcript Induces Potent and Sequence-Specific Antisense Effects. J. Am. Chem. Soc. 128 (50), 16104–16112. 10.1021/ja063383v. [DOI] [PubMed] [Google Scholar]
- Turner J. J.; Ivanova G. D.; Verbeure B.; Williams D.; Arzumanov A. A.; Abes S.; Lebleu B.; Gait M. J. (2005) Cell-Penetrating Peptide Conjugates of Peptide Nucleic Acids (PNA) as Inhibitors of HIV-1 Tat-Dependent Trans-Activation in Cells. Nucleic Acids Res. 33 (21), 6837–6849. 10.1093/nar/gki991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppelhus U.; Shiraishi T.; Zachar V.; Pankratova S.; Nielsen P. E. (2008) Improved Cellular Activity of Antisense Peptide Nucleic Acids by Conjugation to a Cationic Peptide-Lipid (CatLip) Domain. Bioconjugate Chem. 19 (8), 1526–1534. 10.1021/bc800068h. [DOI] [PubMed] [Google Scholar]
- Sahu B.; Chenna V.; Lathrop K. L.; Thomas S. M.; Zon G.; Livak K. J.; Ly D. H. (2009) Synthesis of Conformationally Preorganized and Cell-Permeable Guanidine-Based γ-Peptide Nucleic Acids (γGPNAs). J. Org. Chem. 74 (4), 1509–1516. 10.1021/jo802211n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Andaloussi S.; Järver P.; Johansson H. J.; Langel Ü. (2007) Cargo-Dependent Cytotoxicity and Delivery Efficacy of Cell-Penetrating Peptides: A Comparative Study. Biochem. J. 407 (Pt 2), 285–292. 10.1042/BJ20070507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bárány-Wallje E.; Gaur J.; Lundberg P.; Langel Ü.; Gräslund A. (2007) Differential Membrane Perturbation Caused by the Cell Penetrating Peptide Tp10 Depending on Attached Cargo. FEBS Lett. 581 (13), 2389–2393. 10.1016/j.febslet.2007.04.046. [DOI] [PubMed] [Google Scholar]
- Vázquez O.; Seitz O. (2014) Cytotoxic Peptide–PNA Conjugates Obtained by RNA-Programmed Peptidyl Transfer with Turnover. Chem. Sci. 5 (7), 2850–2854. 10.1039/C4SC00299G. [DOI] [Google Scholar]
- Migawa M. T.; Shen W.; Wan W. B.; Vasquez G.; Oestergaard M. E.; Low A.; De Hoyos C. L.; Gupta R.; Murray S.; Tanowitz M.; et al. (2019) Site-Specific Replacement of Phosphorothioate with Alkyl Phosphonate Linkages Enhances the Therapeutic Profile of Gapmer ASOs by Modulating Interactions with Cellular Proteins. Nucleic Acids Res. 47 (11), 5465–5479. 10.1093/nar/gkz247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.; Fu W.; Zhang Z.; He Y.; Yu H.; Wang Y.; Wang X.; Zhao Y.-L.; Deng Z.; Wu G.; et al. (2018) Structural Basis for the Recognition of Sulfur in Phosphorothioated DNA. Nat. Commun. 9 (1), 1–12. 10.1038/s41467-018-07093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gura T. (1995) Antisense Has Growing Pains. Science 270 (5236), 575–577. 10.1126/science.270.5236.575. [DOI] [PubMed] [Google Scholar]
- Krieg A. M.; Stein C. A. (1995) Phosphorothioate Oligodeoxynucleotides: Antisense or Anti-Protein?. Antisense Res. Dev. 5 (4), 241. 10.1089/ard.1995.5.241. [DOI] [PubMed] [Google Scholar]
- Perez J. R.; Li Y.; Stein C. A.; Majumder S.; Van Oorschot A.; Narayanan R. (1994) Sequence-Independent Induction of Sp1 Transcription Factor Activity by Phosphorothioate Oligodeoxynucleotides. Proc. Natl. Acad. Sci. U. S. A. 91 (13), 5957–5961. 10.1073/pnas.91.13.5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun H.; Xie J.; Olumi A. F.; Ghosh R.; Kumar A. P. (2015) Activation of AKR1C1/ERβ Induces Apoptosis by Downregulation of c-FLIP in Prostate Cancer Cells: A Prospective Therapeutic Opportunity. Oncotarget 6 (13), 11600–11613. 10.18632/oncotarget.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuz S.; Siegmund D.; Scheurich P.; Wajant H. (2001) NF-KB Inducers Upregulate CFLIP, a Cycloheximide-Sensitive Inhibitor of Death Receptor Signaling. Mol. Cell. Biol. 21 (12), 3964–3973. 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safa A. R. (2012) C-FLIP, a Master Anti-Apoptotic Regulator. Exp. Oncol. 34 (3), 176–184. [PMC free article] [PubMed] [Google Scholar]
- Scheibe C.; Bujotzek A.; Dernedde J.; Weber M.; Seitz O. (2011) DNA-Programmed Spatial Screening of Carbohydrate–Lectin Interactions. Chem. Sci. 2 (4), 770–775. 10.1039/c0sc00565g. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.