

Abstract
Identifying the location of a specific RNA in a cell, tissue, or embryo is essential to understand its function. Here we use echinoderm embryos to demonstrate the power of fluorescence in situ RNA hybridizations to localize sites of specific RNA accumulation in whole mount embryo applications. We add to this technology the use of various probe-labelling technologies to co-label multiple RNAs in one application and we describe protocols for incorporating immunofluorescence approaches to maximize the information obtained in situ. We offer alternatives for these protocols and troubleshooting advice to identify steps in which the procedure may have failed. Overall, echinoderms are wonderfully suited for these technologies, and these protocols are applicable to a wide range of cells, tissues, and embryos.
Keywords: Fluorescence, confocal microscopy, multiplexing, troubleshooting, immunolabeling, experimental controls
1. Introduction
RNA-sequencing has become a routine and powerful technology to identify genes and their activity. An investigator can isolate RNA, prepare a cDNA library, sequence the cognate RNAs within several days, and enjoy costs that are routinely decreasing. The advantage of this technique is sensitivity (theoretically one can detect a single RNA molecule), definitive identification (sequences of several hundred nucleotides allows definitive identification in most cases), quantitation (based on known amounts of spiked RNA in the sample, one can estimate absolute abundance of RNAs) and even differential gene expression (comparing two different samples or treatments of cells enables broad analysis of what RNAs are different between the cells). The software pipelines to quickly and effectively identify gene expression have steadily improved for ease of analysis, so that, relatively novice users are able to extract important results.
The shortcoming of this approach for RNA analysis is that it does not reveal the site within the tissue, embryo, or cell where the RNA accumulates. Does the RNA accumulate uniformly in the tissue? Only in a few cells? Does the RNA stay in the nucleus, accumulate at the leading edge of a motile cell, or in the vegetal pole of the egg? Knowing that information is essential to understanding the functionality of the RNA and any products resulting from it e.g. proteins. Therefore, identifying candidate mRNAs by sequencing, or by orthology to mRNAs with great interest, one may wish to institute an in situ RNA hybridization strategy. This chapter is intended to assist investigators on protocols, troubleshooting, and analysis of such protocols using echinoderm embryos and larvae. Having identified an mRNA of interest for hybridization enables definitive results within 2–2.5 weeks, from PCR of the initial candidate to imaging the hybridized cells. Alternatives to this approach for gene activity include genetically tagging a gene of interest with e.g. green fluorescent protein, and imaging the time and place where that gene is active based on the GFP-reporter expression. These sort of techniques are not yet available for echinoderms, making RNA hybridizations in situ a critical tool for gene expression analysis.
Fifty years ago upon this writing, Pardue and Gall [1] published the first results of in situ hybridization (Figure 1; for wonderful historical perspectives of these developments, see [2]; and a personal account by [3]. They immobilized a nucleus from a Xenopus oocyte on a microscope slide using an agar coating, denatured its DNA with alkaline pH, and hybridized the specimen with an rRNA probe that had been radioactively labeled by culturing Xenopus cells in 3H-Uridine. The 3H-rRNA probe was isolated through sucrose gradient centrifugation, and by use of newly identified hybridization kinetics [4, 5] these investigators found conditions for specificity in the RNA-DNA hybridization on a slide. Through this advancing technology Pardue and Gall were able to identify when and where, within the nucleus, ribosomal gene amplification occurred. This leap in technology opened a rush of advancements that included 1) synthesis of high specific activity radioactive probes, 2) hybridization on tissue and embryo sections, 3) colorimetric substrates that obviated the need for radioactivity and emulsion as the detection system, 4) whole mount preparations that enabled 3-dimensional rendering, and 5) the development of fluorescent probes that enabled confocal technology to assist in identifying sites of mRNA accumulation. Now in 2019 it is relatively standard to label multiple RNAs at once in a whole embryo or tissue, and through use of confocal microscopy, identify where the various RNAs accumulate.
Figure 1.

Three large Xenopus oocyte nuclei and three small follicle cell nuclei hybridized with 3H-radiolabeled rRNA. This is one of the first images on in situ hybridization. Note the small intense black spots (silver grains) indicating sites of probe binding to rDNA that becomes progressive more abundant with growth of the oocyte (seen by increased size of the nuclei [4]. The follicle cell nuclei do not have significant rDNA amplification and are largely signal negative. The image was a re-photograph of the slide originally made in 1969.
The sea urchin embryo has been important in the development of in situ hybridization technologies. Lynne and Bob Angerer relied on this embryo and the rapidly expanding datasets of differentially expressed mRNAs identified to optimize their protocols using radioactive probes and sectioned embryos [6, 7]. Their conditions of fixing and stabilizing tissue sections on slides, synthesizing high specific activity RNA-probes, and stringent criteria for hybridization, were all instrumental in advancing the technology in general. Here we make use also of sea urchin and sea star embryos to document the optimization of whole mount multi-fluorescence in situ RNA hybridization. Sea urchin and sea star embryos are excellent for this approach, being largely transparent and sufficiently robust to withstand the fixing and hybridizing conditions. The embryos also are well characterized with many RNA-sequencing and differential RNA-analysis datasets and models of gene regulatory networks that make them a rich paradigm of differential gene expression.
Here we document the protocols for colorimetric (whole mount in situ hybridization,WMISH) and multi-color fluorescence (fluorescent in situ hybridization, FISH) in situ hybridization of whole mount embryos and incorporate troubleshooting, and co-labeling with antibodies into the description to attempt to make this technology routine for the general user.
2. Materials
Anti-acetylated alpha tubulin (mouse monoclonal SIGMA T67930)
Anti-ANP2 (rabbit polyclonal, [8])
Anti-DIG AP antibody, Roche cat # 11093274910
Anti-DIG-POD antibody, Roche cat #11207733
Anti-TRH (rabbit polyclonal, Arnone Lab, GenScript #lot:656547–1)
Anti-DNP-HRP antibody is included in the Perkin Elmer TSA Plus DNP system, cat # NEL747B
Blocking solution 1 (for WMISH): 0.1M MOPS pH7, 0.5M NaCl, 0.1% Tween-20, 10 mg/ml BSA.
Blocking solution 2 (for WMISH): 0.1M MOPS pH7, 0.5M NaCl, 0.1% Tween-20, 1 mg/ml BSA and 10% sheep serum.
BSA, Sigma A4503, AlbumFin, bovine, (dissolved in DEPC-H2O to a final concentration of 100 mg/ml).
Fixation solution for sea urchins: 4% paraformaldehyde, 0.1M MOPS pH 7, 0.5M NaCl 0.1%, in nuclease free water (See Note 1).
Fixation solution for sea stars: 4% paraformaldehyde, 32.5% seawater, 32.5mM MOPS pH 7, 162.5mM NaCl.
Goat Anti-Mouse Alexa Fluor 488 (Invitrogen)
Goat Anti-Rabbit Alexa Fluor 488 (Invitrogen)
Hybridization buffer: 70% formamide, 100 mM MOPS pH 7, 500 mM NaCl, 0.1%Tween 20, 1 mg/ml BSA.
Label It DNP labeling Kit, Cat. # MIR 3800, Mirus corporation, (www.genetransfer.com).
Maleic acid buffer: 0.1M maleic acid pH 7.4, 0.15M NaCl, 0.1% tween-20.
MOPS buffer: 0.1M MOPS pH7, 0.5M NaCl, 0.1% Tween-20 in nuclease free water.
Paraformaldehyde, Electron Microscopy Sciences, Cat.#15710, Paraformaldehyde 16% solution.
PerkinElmer blocking reagent (for FISH): the kit provides the blocking reagent as a powder that is used to make 0.5% blocking reagent in MOPS buffer.
Perkin Elmer Blocking reagent, amplification reagents, amplification diluent are all contained in the Perkin Elmer TSA Palette system (cat # NEL760)
PBS 10x: Dissolve 9g of NaCl, 1.44g of Na2HPO4, 0.24g of KH2PO4 in 80 ml of RNAse free water. Adjust pH to 7.4. Adjust volume to 100 ml with additional RNAse free water. Sterilize by autoclaving.
Sigma FAST TR/Napthol AS-MX Tablets (Sigma, #F4648)
3. Methods
3.1. Fixation protocol
Fixation is a key step where embryo morphology and mRNA are preserved for future analysis. Once fixed, the embryos can be kept stably for months or years at −20°C, and therefore it is convenient to start large embryo cultures and fix all the stages of interest that can be used for many future experiments. Large embryo cultures (106 embryos grown at 0.1% density until larval stage) are grown in 2L glass or plastic beakers using gentle rotary stirring. Small cultures (hundreds of embryos) can be grown in a 6-well dish (with 5–6 ml of seawater), or in a 50ml conical tubes (thousands) on a gentle rotation device. While of general use for sea urchin and sea star embryos, the protocol herby described were tailored for two sea urchins, Strongylocentrotus purpuratus and Paracentrotus lividus and one sea star, Patiria miniata, species. S. purpuratus and P. miniata are kept at 15–16°C, while P. lividus is cultured at 18–20 °C. To grow embryos up to late larval stages, larvae should be fed with algae such as Rhodomonas lens (3000 cells ml−1), Isocrysis galbana (10000 cells ml−1) or Dunaliela tertiolecta (1000–7000 cells ml−1) as soon as the mouth forms. With more advanced larvae, the density of the culture should decrease to eventually only at 3–4 larvae per ml of seawater (a detailed protocol on alga feeding for sea urchin larvae is reported in [9]).
- Harvest of embryos and larvae
-
1.1Pre-hatching embryos collection. For cleavage stage embryos the fertilization membrane must be removed before fixation since this structure prevents optimized probe penetration. Fertilization membranes can be weakened by fertilizing eggs in 10mM para-aminobenzoioc acid (pABA) in pH 8.0 sea water and then removed mechanically by passing the embryos through a fine Nitex mesh (60μm for S. purpuratus and P. lividus) two or three times.
-
1.2Post-hatching embryos and larvae collection. Blastula to larval stages can be collected by simply pouring them onto a Nitex mesh slightly smaller than the embryonic or larval stage of interest (ranging from 60 to 120 μm mesh). Once collected, reverse wash the embryos into a tube for gentle centrifugation (500 rpms/30 seconds, or optimally, by hand centrifugation for 15 seconds. Embryos can be resuspended in a small amount of sea water and then fixed.
-
1.1
-
Fixation
Embryos and larvae of a variety of stages can be quickly collected from the culture beakers using nylon mesh (e.g. Nitex) with an opening slightly smaller than the embryos.-
2.1Pour the embryos onto the mesh, allowing the seawater to pass, and then promptly backwash the embryos into a 15ml plastic centrifuge tube.
-
2.2Spin the embryos down on a low speed (~500g) clinical centrifuge and decant the seawater.
-
2.3Promptly resuspend the embryos in 1x fixative and invert the tube gently 2–3 times to uniformly mix the embryos in the fixative.
-
2.4Fix the embryos/larvae overnight at 4°C (See Note 2).
-
2.5The day after, wash the embryos with 1x MOPS buffer three times to remove all the fixative (See Note 3).
-
2.6Gradually exchange the embryos into increasing ethanol solutions (30%, 50% and 70% ethanol/water or ethanol/MOPS buffer solutions).
-
2.7After the last 70% ethanol wash add another wash in 70% ethanol to ensure the ethanol concentration is accurate. Embryos in 70% ethanol can be stored at −20°C for months.
-
2.1
3.2. Riboprobe synthesis
Antisense riboprobes (here referred to as probes) are labeled-RNA sequences complementary to the target mRNA. RNA is synthesized by in vitro transcription with DNA-dependent RNA polymerase. Labeled ribonucleotides can be incorporated in the reaction, or the RNA can be labeled after synthesis. The labeled probe binds the endogenous mRNA to form a stable RNA-RNA duplex that is marked by the labeled moiety. An antibody conjugated to an enzyme that is used to specifically bind the label on the RNA duplex and catalyze a color/fluorescent reaction. The steps to design and make probes are as follows:
Probe design. For optimal results, probes should be 500–2000 bp long. Ideally the sequence should include exons only. To design primers for cloning such preferred regions from cDNA, online bioinformatics tools like Primer-3 (http://bioinfo.ut.ee/primer3/) are very helpful.
DNA amplification and cloning. The probe sequence can be amplified from genomic DNA or cDNAs and cloned into a vector that contains Sp6, T7 or T3 RNA polymerase binding sites, like the Topo-TA cloning vector (LifeTechnologies) or pGEM®-T Easy Vector (Promega). After cloning and transformation, the plasmid is extracted from bacteria with conventional miniprep protocols.
Plasmid sequencing. We recommend sequencing the plasmid to verify that the inserted sequence is correct, and to determine its orientation so that one can select the RNA polymerase for antisense probe synthesis. To this end, the plasmid can be sequenced using, for instance, primers that anneal to the T7, Sp6 or T3 sequences on the plasmid.
- DNA linearization. The template for the RNA polymerase is linear DNA. You may choose between two ways to achieve this goal from a plasmid:
-
4.1Digest the plasmid using a restriction enzyme that cuts at the end of the insert. After purification with commercial DNA sephadex columns, a concentration of at least 100 ng/ul is recommended.
-
4.1Alternatively, linear DNA can be made by performing a PCR reaction of the plasmid with the M13 forward and reverse primers. The M13 sites flank the RNA polymerase sites, so that the PCR product will include those sites (check the map of the vector used to clone DNA fragments to determine if these sites are available).
-
4.1
- Labelled-Riboprobe synthesis. The preferred method to label riboprobes is to use digoxigenin (DIG) labeled ribonucleotides in the transcription reaction. DIG-labelled RNAs are stable for years and have very high sensitivity.
-
5.1We recommend to use a DIG-RNA labelling kit (e.g. Roche), and to follow the manufacturer’s instructions for the transcription.
-
5.2Between 500–1000 ng of linear DNA is used as a template for the reaction, and the reaction is carried out at 37°C for 2 hours.
-
5.3RNase free DNase I is added (1U/μl), to remove the DNA template and the mix is incubated for another 20 minutes at 37 °C.
-
5.4To remove unincorporated nucleotides, DIG-riboprobes are purified either using Sephadex columns like the Mini Quick Spin RNA Columns G-50 Sephadex (Roche), or by precipitation (1/10 Vol of ammonium acetate and 2.5 Vol of ethanol added to the probe and left at −20°C overnight).
-
5.5The following day, spin the precipitated probe for 30 min at maximum microcentrifuge speed (14,000 rpm, then wash the pellet with 1ml of 70% ethanol, and finally resuspend the probe in nuclease free water (store probes at −80 °C) (See Note 4).
-
5.1
- Riboprobe quantification and determination of specific activity. Riboprobe concentration is evaluated spectrophotometrically (260 nm absorbance) and by dot-blot immunostaining with anti-DIG antibody alkaline phosphatase (anti-DIG-AP) conjugated (Roche) by comparing the riboprobe of interest to a known standard (DIG-RNA control, Roche).
-
6.1RNA dilutions are prepared using the dilution buffer [nuclease-free H2O: 20X SSC: formaldehyde (5:3:2)].
-
6.2Typically, dilutions of the DIG-labeled probes are blotted to Hybond N membrane (Amersham) along with serial dilutions of the standard RNA and UV-crosslinked to the membrane with ultraviolet light (e.g. Stratalinker) for 30 seconds.
-
6.3The filter is incubated for 30 minutes in blocking solution (5% BSA in 0.1 M maleic acid pH 7.5) and 1 hour at RT in the same solution containing the anti-DIG-AP antibody (0.15 U/ml).
-
6.4To remove unbound antibodies, the blot is washed twice in a solution containing 0.1 M maleic acid pH 7.5 and 0.15 M NaCl.
-
6.5The filter is equilibrated in the detection solution (100mM Tris-HCl pH 9.5; 100 mM NaCl; 50mM MgCl2) and then incubated in the dark in the same solution containing 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) (50mg/ml) and nitroblue tetrazolium (NBT) (50mg/ml).
-
6.6The AP enzyme produces an insoluble blue substrate in the presence of these enzymatic substrates. The colored compound precipitates in minutes and can be seen by the naked eye. The reaction is stopped when optimized by washing the filter with H2O. The concentrations of experimental riboprobes are estimated by comparing spot intensities of the standard control and the experimental dilutions.
-
6.1
- DNP-labelled probe synthesis. To perform double FISH (probes against two different RNA targets), a second tag can be dinitrophenol (DNP) [10]. DNP-labelled RNA probes are obtained in two steps:
-
7.1Synthesis of unlabeled riboprobe. Unlabeled RNA is synthetized with a RNA synthesis kit (Ambion), following the manufacturer’s instructions. Purified linear DNA of between 500–1000 ng is used as a template for the reaction.
-
7.1DNP labeling. We use the DNP Labelling kit (Mirus) to attach the DNP group to the unlabeled RNA.The labeling reagent is composed of the DNP label, a linker which facilitates electrostatic interactions with nucleic acids, and the reactive alkylating group that covalently attaches the reagent to non-base-pairing sites on the guanine ring (https://www.mirusbio.com/products/labeling/label-it-nucleic-acid-labeling-reagents#figure1048). Purified unlabeled riboprobe of between 2.5 – 5μg is used as a template for the labelling reaction. The RNA probe labelling reactions are conducted at 37 °C for 2 hours following manufacturer’s instructions. Purification of DNP-riboprobes can be accomplished with Mini Spin RNA Columns G-50 Sephadex provided by the same kit. Probes are stored at −80 °C.
-
7.1
3.3. Sea urchin WMISH protocols
Here we describe two protocols for in situ hybridization: the colorimetric whole mount in situ hybridization (WMISH) and the fluorescent in situ hybridization (FISH). The hybridization steps are the same for WMISH and FISH, the differences are in the detection methods. For both techniques, the main steps include hybridization (the exogenous labeled-RNA binds the complementary endogenous mRNA), high stringency washes to remove nonspecific interactions, antibody binding to the labeled-RNA:mRNA duplex, color detection. All steps can be performed in microcentrifuge tubes or in 96-well plates, depending on the volume of embryos needed. Note that many different stages of embryos can be combined for this analysis and are thus treated identically for better interpretation of comparative labeling. The WMISH protocol described here is modified from the original Arenas-Mena and Minokawa method [11, 12]. Detection of probes with the pink precipitate was popularized by Tara Fresques [13]. Examples of embryos stained with these methods are in Figure 2 A and B.
Figure 2. Comparisons of WMISH and FISH in sea urchin and sea star species performed as described in the text.
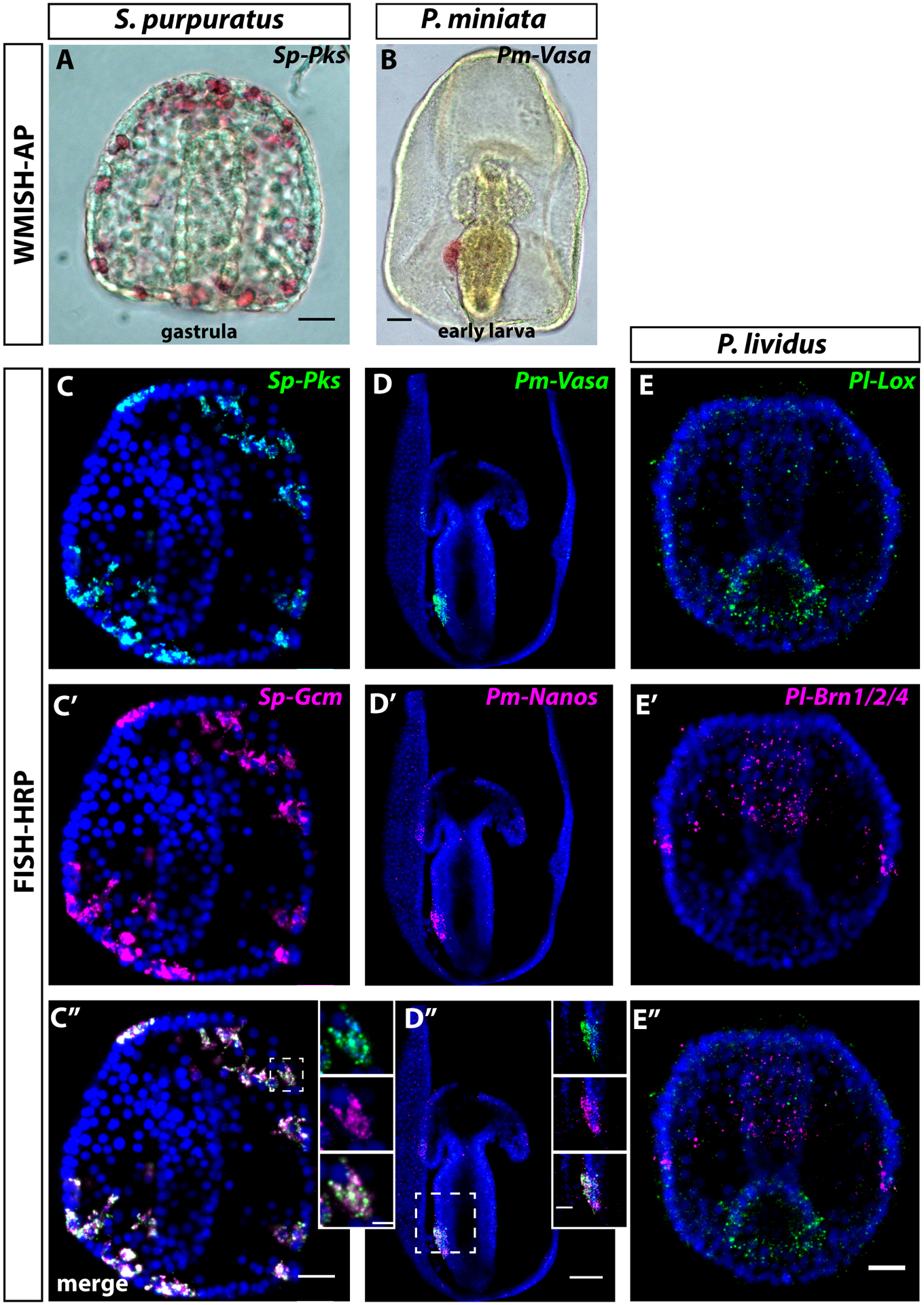
A) and B) show WMISH for the sea urchin S. purpuratus and the sea star P. miniata using DIG-labelled Pks probe (A) that marks pigmented cells in sea urchin gastrulae, and a DIG-labelled Vasa probe (B) that marks the posterior enterocoel in sea star larvae. C-C”) show double FISH of a S. purpuratus gastrula performed using and a DNP-labelled Pks (C) and a DIG-labelled Gcm probe (C’), and a merge of the two channels (C”). D-D”) Show a double FISH of P. miniata larva performed using a DNP-labelled Vasa probe (D) and a DIG-labelled Nanos probe (D”), and a merge of the two channels (D”). In C-D” the two genes detected are expressed in the same cell type. Dashed boxes indicate the region magnified in the white box. Note that RNA localization for the two transcripts can be seen. E-E” show double FISH for the sea urchin P. lividus using a DIG-labelled Lox probe (hindgut) and a DNP-labelled Brn1/2/4 probe (foregut). Scalebars are 20 μm for sea urchins, 50 μm for sea star, and 5μM for the magnifications in the white boxes.
Embryo rehydration and pre-hybridization. The embryos are stored in 70% ethanol to preserve the morphology and endogenous mRNA. In the following step, all the ethanol is removed and the embryos gradually transitioned into the hybridization buffer. All steps of transition use settling of embryos (not centrifuged).
The 70% ethanol is removed from a tube of settled embryos, and the embryos resuspended in 50% ethanol (x1), then 30% ethanol (x1), and then washed x3 with MOPS buffer (15 minutes each wash).
Embryos are then washed x2 in hybridization buffer pre-warmed at 50°C (prepared fresh) and left in a 50°C incubator for 3h or to overnight to pre-hybridize.
Hybridization. This step uses a buffer that favors the stable, sequence-specific hybridization of the probe to the endogenous RNA over time.
Generally, the DIG- and DNP-probes are added at a final concentration of 0.1–0.5 ng/ul. FISH is very sensitive to probe concentration, therefore it is a good practice to use a few probe concentrations when testing a new probe. In a microcentrifuge tube, prepare the optimized probe dilution in hybridization buffer. The final volume of the hybridization solution should be at least ten times the embryo volume. The tube with the probe dilution is then heated at 65–70°C for 10 minutes, spun briefly to get all the probe solution to the bottom of the tube, and left on ice for 5’ min before adding the probe dilution to the pre-hybridized embryos. Gently mix the probe with the embryos by inversion a few times (See Note 5).
Leave the embryos with the probe in the hybridization oven for 1 week at 50°C optimally with gentle rotation. During this time the labeled RNA binds the endogenous RNA of interest.
Post-hybridization washes. This step removes all probes non-specific interactions.To remove the probe not bound, the embryos are washed in the pre-hybridization buffer for at least 3h at 50 °C, replacing the buffer twice with prewarmed (50 °C) buffer.
After the post-hybridization washes, the embryos are washed x3 with MOPS buffer at room temperature. Depending on whether a single or double FISH was going to be performed, or a single one using alkaline phosphatase-conjugated enzyme, different procedures are used (See Note 6).
Blocking and antibody incubation. This step is critical to reduce background due to antibody nonspecific binding. A mix of serum proteins is used to block the nonspecific sites. The binding of the anti-DIG antibodies to the DIG epitope (that now marks the DIG-RNA:endogenous mRNA duplex) can be carried out overnight at 4°C or 1h at room temperature.
Incubate the embryos in blocking solution 1 20 min at room temperature, followed by incubation in blocking solution 2 for 30 min at 37°C. Remove the buffer an incubate with a 1:1500 dilution of anti-DIG alkaline phosphatase (AP) Fab fragments (Roche) at 4°C overnight.
To remove excess of antibody, wash in MOPS buffer x5 (settling embryos only, no centrifugation) at room temperature.
Color detection. This step visualizes the site of the endogenous mRNA, in situ. The enzyme conjugated to the anti-DIG antibody catalyzes a reaction that yields either a colored precipitate (AP enzyme) that can be observed with a conventional stereoscope. Develop the colorimetric signal using SigmaFast-FastRed TR/Naphthol AS-MX tablets (Sigma), a reagent that produces an insoluble intense red end product. This kit includes a buffer that contains Levamisole (0.15 mg/mL) to block endogenous alkaline phosphatase activity. Dissolve one Trizma buffer tablet in 1 mL of deionized water and vortex until dissolved.
Add one FastRed TR/Naphthol AS-MX tablet in the Trizma buffer and vortex until dissolved.
Incubate the embryos in this solution (10x volume) (See Note 7).
Check if the signal has developed after 1h incubation with a stereoscope. The incubation time varies for every probe. If the signal doesn’t develop after a few hours, leave the tube with the embryos at 4°C overnight. If after 3 days there is still no signal, remove the buffer and add fresh Trizma buffer and FastRed tablets.
Once a red signal is visible, stop the reaction by dilution in MOPS buffer. (Further tips on how to perform the WMISH protocol are in Notes 8–13).
3.4. Sea urchin FISH protocol
The FISH is performed using the Tyramide Signal Amplification Kit from PerkinElmer. The Tyramide Signal Amplification (TSA) reagents allow the detection of single types of mRNA molecules. The HRP enzyme conjugated to the anti-DIG antibody transforms the TSA reagent to TSA free radicals that forms covalent bonds with tyrosine residues proximal to the HRP, while unbound TSA radicals form dimers that are washed away. The TSA reagents used in this protocol are cyanine 3 (cy3, excitation 550nm, emission 570nm) or cyanine 5 (cy5, excitation 648nm, emission 667nm). Single and double FISH techinques were first used in echinoderms by Cole et al. [14, 15], and later the protocol was modified to include MOPS buffer and a lower hybridization temperature [16]. The double FISH protocol can also be modified to co-label a gene of interest and microRNAs [17]. Examples of embryos and larvae of three different echinoderm species stained with FISH are in Figure 2C–E”.
Following the MOPS washes in step 8 (previous section 3.3) incubate the embryos in the PerkinElmer blocking reagent that comes with the kit for 30 minutes at room temperature. Remove the entire buffer and incubate in a dilution of 1:1500 anti-DIG horseradish peroxidase (HRP) Fab fragments (Roche) in the PerkinElmer blocking buffer at 4°C overnight.
To remove excess of antibody wash in MOPS buffer x5 at room temperature.
The buffer is then changed to the amplification diluent (a buffer that comes with the PerkinElmer kit) with x2 washes 15 minutes.
The signal is detected by staining for 15–30 minutes in 1:400 cy3 in amplification diluent (See Note 14).
To remove unbound cy3, wash in MOPS buffer x5.
If this is a single FISH, add 1:1000 Hoechst or 1:10000 DAPI in MOPS buffer for 15 minutes to image nuclei.
3.5. Second probe detection for double-FISH
The signal of the DNP probe is detected after the DIG-probe signal has been amplified. Figure 2 C–D” shows examples of double FISH (white boxes show magnification of single cell where one can appreciate the RNA localization; DNP probes are shown in magenta). To detect the DNP-probe, we repeat the same steps as for the DIG-probe following step 5 previous section 3.4, as summarized below:
To inactivate the HRP conjugate to the anti-DIG antibody, wash the samples x2 with 1% H2O2 in MOPS buffer for 30 min.
Wash in MOPS buffer x5 to eliminate all the H2O2.
Block again the embryos with the PerkinElmer blocking reagent for 30 min.
Incubate embryos with 1:1500 anti-DNP-HRP antibody (PerkinElmer) in blocking buffer overnight at 4°C.
Wash embryos 5x in MOPS buffer to remove excess antibody.
Wash in amplification diluent x2.
The signal is detected by staining for 15–30 minutes in 1:400 cy5 (from the TSA kit) in amplification diluent.
Wash x5 in MOPS buffer.
Add 1:1000 Hoechst or 1:10000 DAPI (from a 10mg/ml stock prepared in water) in MOPS buffer for 15 minutes to image nuclei.
3.6. P. miniata WMISH and FISH protocols
The in situ RNA hybridization protocol for the sea star P. miniata follows the same steps of the protocol we present here for sea urchin. An example of a sea star larva stained with FISH and WMISH is in Figure 2 B, D–D”. This protocol is based on the Hinman protocol [18] with several modifications. Here we highlight the few differences with the sea urchin protocol:
The fixation solution we use for sea star embryos and larvae has a high salt buffer (see solutions and reagents section).
Because staining of sea star embryos shows high background, pre-hybridization, hybridization and post-hybridization is performed at 60°C to increase probe specificity. The hybridization buffer is the same that we described for S. purpuratus.
All the washes after the post-hybridization are performed in maleic acid buffer. Maleic acid doesn’t contain phosphates and it improves probe stability.
All the antibodies (anti-DIG HRP, anti-DNP HRP and anti-DIG AP) are used at 1:2000 to maximize signal to noise ratios.
3.7. Positive and negative controls
Controls for both FISH and WMISH are needed to conclude signal specificity. Unfortunately, the presence of fluorescence does not always mean the probe hybridized to the intended mRNA. For instance, FISH on fed embryos may result in a strong signal from the gut due to the autofluorescence of the algae, or poor permeabilization might result in strong ectodermal signal only. Thus, negative controls are always required to determine if the signal is specific to the probe binding the complementary mRNA. The following are different ways to perform negative controls:
Perform the in situ RNA hybridization protocol following the normal protocol except do not add any probe. Any signal from this control would likely be the result of nonspecific antibody interactions. As always with a control, this reaction should be performed in parallel with the experimental in situs with the probe added in order to ensure that both treatments use the same reagents and buffers.
Generate a labeled sense probe. Plasmid sequencing allows for the determination of which RNA polymerase may be used to synthesize the antisense probe. A sense probe may be generated with an alternate RNA polymerase and used as a control. The sense probe will not bind to the mRNA of interest, so it should be removed in washes. A signal from an in situ with a sense probe would demonstrate a nonspecific interaction.
Use a probe against antibiotics, GFP or mCherry mRNAs. These mRNAs are not endogenously present in the cell, and should result in no staining.
Control probes in your experiment may do more than demonstrate a specific interaction (or lack thereof). Positive controls are needed to test the quality of the reagents used and can also be useful as a troubleshooting tool. As an example, an in situ hybridization that produces no signal despite the fact that qPCR or RNA seq data suggest that the gene is expressed at this stage. It is impossible to know at which step an error occurred. However, a positive control may help narrow the options down. By performing an in situ hybridization with a probe whose expression pattern is known, it becomes possible to determine whether the lack of a signal was due to an issue with the probe or the in situ itself and how specific the conditions were for the probe. If the control alone produces a signal, we recommend checking for probe degradation or resynthesizing the probe. If neither the control nor the treatment produce a signal, we recommend making new buffers to enhance proper conditions and no RNAse contamination.
3.8. Combination of immunolabeling and in situ hybridization
In situ hybridization is a powerful tool to study gene expression patterns, detect possible gene interactions and RNA localization throughout embryonic development. It could be possible that apart from the localization data more information is needed about the relative localization of the transcripts in respect to the fully functional proteins. Furthermore, additional staining of the same embryos with different markers might be required either to provide better characterization of the cells of interest or to label different territories of the embryo. FISH and double FISH experiments can be combined with indirect immunofluorescence enabling the user to further characterize those cells, by adding information provided by the fluorescent signal of a specific antibody to a protein of interest.
The FISH protocol described in this chapter is fully compatible with immunofluorescent staining as long as the antigen recognized by the antibody of interest is preserved after the in situ fixation or hybridization process and this can be determined simply by testing the antibody on your fixed embryos following hybridization. Note that the hybridization protocol itself is harsh on protein epitopes so it is important to test the protein following a hybridization protocol. One of the advantages of this technique is that it can be performed prior to, during, or after the in situ hybridized embryos.
In the following protocol, three antibodies were used to stain different cell types of the S. purpuratus and P. lividus larvae (see Figure 3 A–C”). Anti-acetylated alpha tubulin was used to label cilia and stable microtubules (MTs), anti-TRH to label neuron-like cells adjacent to the apical organ (Arnone lab, unpublished) and anti-ANP2 to label post oral, lateral ganglion and apical organ pre-pancreatic neurons [8].
Figure 3. Examples of Combination of immunolabeling and in situ hybridization in two sea urchin species.

FISH and IHC can be used to study RNA and protein localization for the same gene of interest. A-A”) show that the RNA for the neuropeptide ANP2 is everywhere in the cell, while the ANP2 protein is enriched at the cell apical side (see magnification in white boxes in C”). B-B”) show stable microtubule staining paired with a FISH for the stomach gene ManRC1A. C-C”) show immunolabeling of the neuropeptide Trh and orthopedia (Otp). Dashed boxes indicate the region magnified in the white box. Scalebars are 20 μm for sea urchins, 50 μm for sea star, and 5μM for the magnifications in the white boxes.
If performing a double FISH, the primary antibody of interest can be added (in the manufacturer’s suggested dilution) and incubated together with the anti-DIG horseradish peroxidase (HRP) Fab fragments (Roche). Then the desired Alexa Fluor (Thermo Fisher Scientific) secondary antibody can be added in a dilution of 1:1000 while adding the anti-DNP-HRP antibody (PerkinElmer). Apart from those two additional steps the double FISH protocol proceeds as described.
Another strategy is to do Immunohistochemistry following FISH. One may use this strategy if it is important to see the FISH results prior to spending antibody on a sample. To prevent embryo loss or damage keep the embryos in the same tubes in which FISH was performed (See Note 15).
At the end of the FISH protocol, remove MOPS Buffer and resuspend the embryos in 1x Phosphate buffered saline (PBS) or 1x PBST (0.1% Tween).
Wash 4 times with 1x PBS or 1x PBST to make sure that all of the MOPS buffer residues have been removed.
To block non-specific sites, remove 1x PBS or 1x PBST and replace it with blocking solution (4% sheep serum, 1% BSA in 1x PBS).
Incubate for 1 hour at room temperature or overnight at 4°C.
Prepare the primary antibody working solution. Dilute the antibody in blocking solution according to the manufacturer’s suggested or experimentally tested dilution.
Add the primary antibody working solution and incubate for 1.5 hours at 37°C or overnight at 4°C (primary antibody dilution: anti-acetylated alpha tubulin 1:200, anti-AN 1:250, anti-TRH 1:400).
Remove the antibody solution and wash x5 with 1x PBS or 1x PBST to remove the primary antibody excess.
Do not overstain the embryos. After a signal appears, split the sample in two tubes. Stop the color reaction in one tube and image the embryos. To test whether there is more signal that still hasn’t developed, leave the second tube with fresh staining solution for several hours to overnight and check it for stain every few hours.
Prepare the appropriate (according to the organism in which the primary antibody has been raised) secondary Alexa Fluor antibody working solution of preference (in terms of excitation and emission spectra) by diluting it 1:1000 in 1x PBS.
Add the secondary antibody working solution and incubate for 1 hour at room temperature (See Note 16).
Wash x5 with 1x PBS or 1x PBST to remove the secondary antibody excess.
If DAPI or Hoechst have already been added proceed to mounting the embryos. If not, add it in the dilution described in the in situ section.
3.9. Conclusions
A summary of the techniques here described is in Figure 4. Development of technologies for in situ hybridizations have not stalled. New approaches in this goal of identifying RNAs in situ are documented regularly and enhance ease of use, sensitivity, and multiplexing capabilities. A recent advance enables enhanced multiplexing based on in situ hybridization chain reaction (HCR). By use of DNA hybridization initiators that target specific mRNAs, and then detecting the sequence-specific initiators with metastable DNA hairpins that are differentially labeled for each RNA target, enable extremes in multiplexing, ease of use, and cross-species approaches. The protocol is costly to start, but has been tested successfully on a variety of animal embryos, including sea urchins [19].
Figure 4.
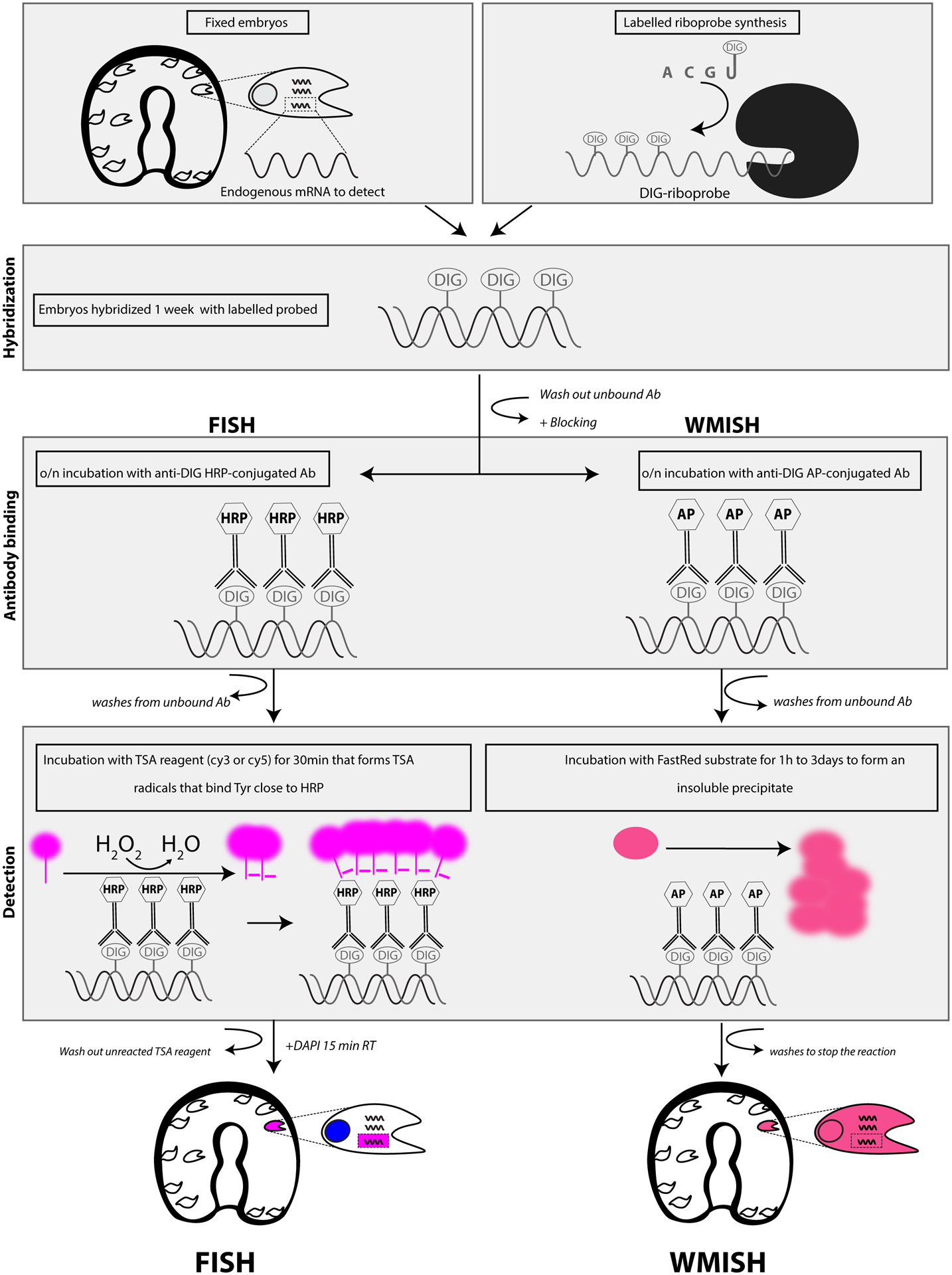
Summary of FISH and WMISH techniques described in this chapter
Single cell sequencing identifies cell types based on mRNA presence. This strategy enables distinguishing different cell types within a tissue or embryo that are otherwise hidden within a bulk sequence dataset, or requiring in situ hybridization. This procedure requires dissociation of tissues and thereby organizational schemes are lost. These results need instead to be back-calculated by some other means such as in situ hybridization of marker gene sets.
RNA sequencing in situ has now shifted from a dream to an emerging technology in which mRNAs are sequenced on the tissue, directly on the slide [20, 21]. Although still in its infancy, this approach enables identification of cell types based on RNA-seq profiles identified in place, overcoming the drawbacks of disrupting cellular distribution of single cell sequencing. It is not clear yet how many sequences can be read in this approach, since the technology is only just developing.
Neither of these emerging technologies, however, are likely to replace in situ hybridization technologies anytime soon. Instead, we anticipate a collaborative technology whose foundation of multi-probe in situ RNA hybridization enables identification of key elements of cellular function within the cell in situ. Needed for this are simply more and specific detectable fluorochromes or chromophores. Recent documentation for use of quantum dots (Q-dots) in the labeling of probes for mRNA hybridization may enable such advances [22]. Such technology will likely transition in the next several years and should yield a dynamic enhancement of what was once – a stretch. From silver grains on Xenopus nuclei to multicolor RNA identification in 50 years. Given the rapid advances in microscopy, nucleic acid labeling, sensitivity of detecting hybrids, and sequencing, even the next five years should be exciting for this field.
4. Notes
To preserve the RNA, all the recipes are prepared with DEPC-treated water or nuclease free water.
The embryos can be preserved in the fixation solution up to several days without affecting the hybridization efficiency.
After the embryos are fixed, washes will refer to allowing the embryos to settle at 1 × g (no centrifuging), decanting the solution, adding the next solution gently, and inverting the tube 2–3 times to uniformly mix the embryos in the next solution.
Always keep the riboprobes on ice to avoid RNA degradation. Add 0.2μl of glycogen (1 micogram/microliter) to see the probe as a white pellet during precipitation.
For double-FISH add both DIG and DNP probes to the hybridization solution.
Embryos are transparent in hybridization buffer and slowly become visible again during the MOPS washes. Check if the morphology of the embryos has been retained under a stereoscope.
Cover the tubes with aluminum foil to keep them in the dark.
After fixation, only settle embryos at 1xg during washes to ensure they are not damaged. Centrifugation of fixed embryos can damage them irreparably.
To avoid embryos drying out, always keep embryos in excess buffer volumes (at least 2x the embryo pellet)
The advantage of using microcentrifuge tubes is that all the washes can be perfromedin big volumes (1–1.5ml) to mix solutions completely.
The volume ratios of embryos to buffer should always be 1:10.
When washing embryos from buffer to buffer, remember to close and invert the tubes a few times to ensure that all embryos are uniformly mixed in the solution.
Embryos in ethanol are dehydrated and when checked under the stereoscope appear shriveled. Once returned to hybridization buffer they will relax and return to normal morphologies.
From this step on, protect the tube with the embryos from light.
At all times the tubes should be covered with aluminum foil to keep the embryos in the dark.
Make sure that the excitation and emission spectra of the secondary antibody are not overlapping with the cy3 and cy5 profiles to avoid falsely overlapping signals.
Table 1.
Troubleshooting
| Problem | Potential Solution(s) |
|---|---|
| High background signal | Use smaller volumes of conjugated antibodies or substrate. Try running multiple in situs with various amounts of antibody or substrate in parallel to see which volumes produce the best results. |
| Embryos stay suspended in buffer | This sometimes occurs in the first wash after posthybridization, especially in P. miniata. Try to remove as much hybridization buffer as possible before adding MOPS or maleic acid buffer (depending on species). Alternatively, pipet the suspended embryos into a larger container such as a 15mL Falcon tube and add additional MOPS or maleic acid buffer to dilute the hybridization buffer even further. |
| Embryos are sticky or lost | Double check your MOPS and/or maleic acid buffer to ensure enough Tween-20 was added. Not enough detergent may cause embryos to stick to pipet tips and thus be lost during washes. |
| No or low signal | In the case of low signal, try using more substrate or antibody. If there is no signal, see the previous section on using a positive control to determine whether the issue is with the probe or some other factor. |
| Damaged embryos or embryos with poor morphology | Damaged embryos may result from being too aggressive when pipetting or handling them. Ensure you are always gentle as it is very easy to damage fixed embryos and larvae. Embryos should never be centrifuged. If embryos still have poor morphologies, try leaving them an additional day or two in fixative solution before dehydrating to 70% EtOH. |
References
- 1.Pardue ML, Gall JG (1969) Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proc Natl Acad Sci USA 64:600–604. doi: 10.1073/pnas.64.2.600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hennig W (1973) Molecular hybridization of DNA and RNA in situ. Int Rev Cytol 36:1–44. [DOI] [PubMed] [Google Scholar]
- 3.Gall JG (2016) The origin of in situ hybridization -A personal history. Methods 98:4–9. doi: 10.1016/j.ymeth.2015.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gall JG (1968) Differential synthesis of the genes for ribosomal RNA during amphibian oögenesis. Proc Natl Acad Sci USA 60:553–560. doi: 10.1073/pnas.60.2.553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Britten RJ, Kohne DE (1968) Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science 161:529–540. [DOI] [PubMed] [Google Scholar]
- 6.Venezky DL, Angerer LM, Angerer RC (1981) Accumulation of histone repeat transcripts in the sea urchin egg pronucleus. Cell 24:385–391. [DOI] [PubMed] [Google Scholar]
- 7.Angerer LM, Angerer RC (1981) Detection of poly A+ RNA in sea urchin eggs and embryos by quantitative in situ hybridization. Nucleic Acids Res 9:2819–2840. doi: 10.1093/nar/9.12.2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perillo M, Paganos P, Mattiello T, et al. (2018) New Neuronal Subtypes With a “Pre-Pancreatic” Signature in the Sea Urchin Stongylocentrotus purpuratus. Front Endocrinol (Lausanne) 9:650. doi: 10.3389/fendo.2018.00650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perillo M, Arnone MI (2014) Characterization of insulin-like peptides (ILPs) in the sea urchin Strongylocentrotus purpuratus: insights on the evolution of the insulin family. Gen Comp Endocrinol 205:68–79. doi: 10.1016/j.ygcen.2014.06.014 [DOI] [PubMed] [Google Scholar]
- 10.Long S, Rebagliati M (2002) Sensitive two-color whole-mount in situ hybridizations using digoxygenin- and dinitrophenol-labeled RNA probes. BioTechniques 32:494–496–498 passim. doi: 10.2144/02323bm04 [DOI] [PubMed] [Google Scholar]
- 11.Arenas-Mena C, Cameron AR, Davidson EH (2000) Spatial expression of Hox cluster genes in the ontogeny of a sea urchin. Development 127:4631–4643. [DOI] [PubMed] [Google Scholar]
- 12.Minokawa T, Rast JP, Arenas-Mena C, et al. (2004) Expression patterns of four different regulatory genes that function during sea urchin development. Gene Expr Patterns 4:449–456. doi: 10.1016/j.modgep.2004.01.009 [DOI] [PubMed] [Google Scholar]
- 13.Fresques TM, Wessel GM (2018) Nodal induces sequential restriction of germ cell factors during primordial germ cell specification. Development dev.155663. doi: 10.1242/dev.155663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole AG, Rizzo F, Martinez P, et al. (2009) Two ParaHox genes, SpLox and SpCdx, interact to partition the posterior endoderm in the formation of a functional gut. Development 136:541–549. doi: 10.1242/dev.029959 [DOI] [PubMed] [Google Scholar]
- 15.Cole AG, Arnone MI (2009) Fluorescent in situ hybridization reveals multiple expression domains for SpBrn1/2/4 and identifies a unique ectodermal cell type that co-expresses the ParaHox gene SpLox. Gene Expr Patterns 9:324–328. doi: 10.1016/j.gep.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 16.Andrikou C, Iovene E, Rizzo F, et al. (2013) Myogenesis in the sea urchin embryo: the molecular fingerprint of the myoblast precursors. Evodevo 4:33. doi: 10.1186/2041-9139-4-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perillo M, Wang YJ, Leach SD, Arnone MI (2016) A pancreatic exocrine-like cell regulatory circuit operating in the upper stomach of the sea urchin Strongylocentrotus purpuratus larva. BMC Evol Biol 16:117. doi: 10.1186/s12862-016-0686-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinman VF, Nguyen AT, Davidson EH (2003) Expression and function of a starfish Otx ortholog, AmOtx: a conserved role for Otx proteins in endoderm development that predates divergence of the eleutherozoa. Mech Dev 120:1165–1176. [DOI] [PubMed] [Google Scholar]
- 19.Choi HMT, Calvert CR, Husain N, et al. (2016) Mapping a multiplexed zoo of mRNA expression. Development 143:3632–3637. doi: 10.1242/dev.140137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitra RD, Shendure J, Olejnik J, et al. (2003) Fluorescent in situ sequencing on polymerase colonies. Anal Biochem 320:55–65. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH, Daugharthy ER, Scheiman J, et al. (2014) Highly multiplexed subcellular RNA sequencing in situ. Science 343:1360–1363. doi: 10.1126/science.1250212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Le P, Lim SJ, et al. (2018) Enhanced mRNA FISH with compact quantum dots. Nat Commun 9:4461. doi: 10.1038/s41467-018-06740-x [DOI] [PMC free article] [PubMed] [Google Scholar]