

Abstract
Inflammation is a double-edged sword for sterile tissue injury such as in myocardial infarction (MI). After ischemic injury, inflammatory immune responses activate repair processes, clear tissue-debris, form a stable scar and initiate angiogenesis in the myocardium for efficient wound-healing. However, incomplete immune resolution or sustained low-grade inflammation lead to ischemic cardiomyopathy (IC) characterized by maladaptive tissue remodeling and left-ventricular dilatation. It is clear that a delicate balance of cytokines, chemokines, prostaglandins, resolvins, and the innate and adaptive immune systems is critical for adequate healing as both insufficient- or overt-activation of inflammatory responses can either enhance rupture incidence or exacerbate cardiac dysfunction in the long-term. Among all the players, immune cells are the most critical as they are not only a source for all of the inflammatory protein mediators, but are also a target. However, phenotypic complexities associated with different immune subtypes, their interdependence, phasic-activations and varied functionalities often make it difficult to segregate the effects of one immune cell from another. In this review, we briefly summarize the role of several innate and adaptive immune cells to acquaint readers with complex immune-networks that dictate the extent of wound-healing post-MI and maladaptive remodeling during IC.
Keywords: Immune Cells, Inflammation, Myocardial Infarction, Ischemic Cardiomyopathy, Heart Failure
Introduction
Inflammatory responses to myocardial infarction (MI) are the protective mechanisms to initiate and regulate repair processes to maintain homeostasis and regain tissue-function. They are indispensable for the overall restorative process to myocardial injury and determine the extent of infarct-healing and the severity of ischemic cardiomyopathy (IC). Several preclinical studies have established that the inflammatory responses immediately after MI are obligatory to clear the dead and apoptotic cells from the injury site, to initiate scar formation and to promote angiogenesis (Figure 1). Moderation of inflammatory responses at this stage interfere with physiological healing mechanisms and often result in significant loss of cardiac function due to inadequate scar formation. Nonetheless, incomplete immune resolution during this acute inflammatory phase can also precipitate into a 2nd wave of slow immune activation, ranging from months to years, that promotes maladaptive LV remodeling [1]. During this sustained low-grade inflammation heart undergoes distinct physiological, structural and functional changes which eventually lead to hemodynamic insufficiency and progressive IC [2]. Inflammatory responses during this chronic phase are phenotypically different than the acute phase and immune-modulation at this stage often dampens maladaptive remodeling and blunts progressive cardiac dysfunction, if not reversal [3]. Since inflammation and immune activation play a complex role during ischemic injury and maladaptive LV remodeling, in this review we briefly summarize various components of the innate and the adaptive immune system as they pertain to MI and IC.
Figure 1:
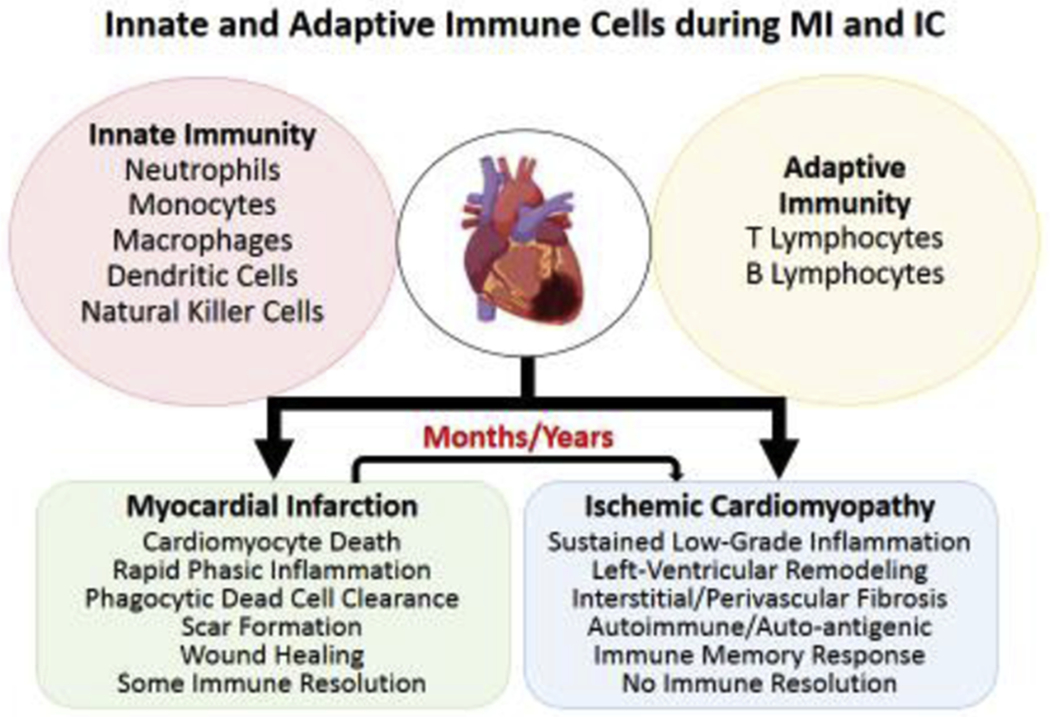
Schematic showing several key players of the innate and adaptive immune system known to play an important role in wound-healing post-myocardial infarction (MI), and mediating left-ventricular remodeling during ischemic cardiomyopathy (IC) in preclinical studies. Both innate and adaptive immune cells function in an interdependent complex network where one cell-type regulates and influences the behavior of others to achieve an overall goal of wound-healing, scar formation and ultimately immune resolution post-MI. However, the same network of immune cells responds differently to sustained low-grade inflammation and undergo a phenotypic change to mediate maladaptive tissue-remodeling during IC. If one player is more pathological/critical than the others is not known. Some of the components were designed using ‘Biorender’.
Innate Immune Reponses
Innate immune cells are the first responders to tissue-injury and rapidly mount an escalated response to clear tissue-debris and invoke repair mechanisms, making them vital for the host-defense (Figure 2). In response to pro-inflammatory cytokines such as IL-1β [4], innate immune responses are activated within minutes to hours of injury and are associated with the activation of resident macrophages and infiltration of more diverse circulating immune components including monocytes/macrophages, neutrophils and dendritic cells [5, 6]. Some of the most important players of the innate immune system and their role in MI and IC are described below.
Figure 2:
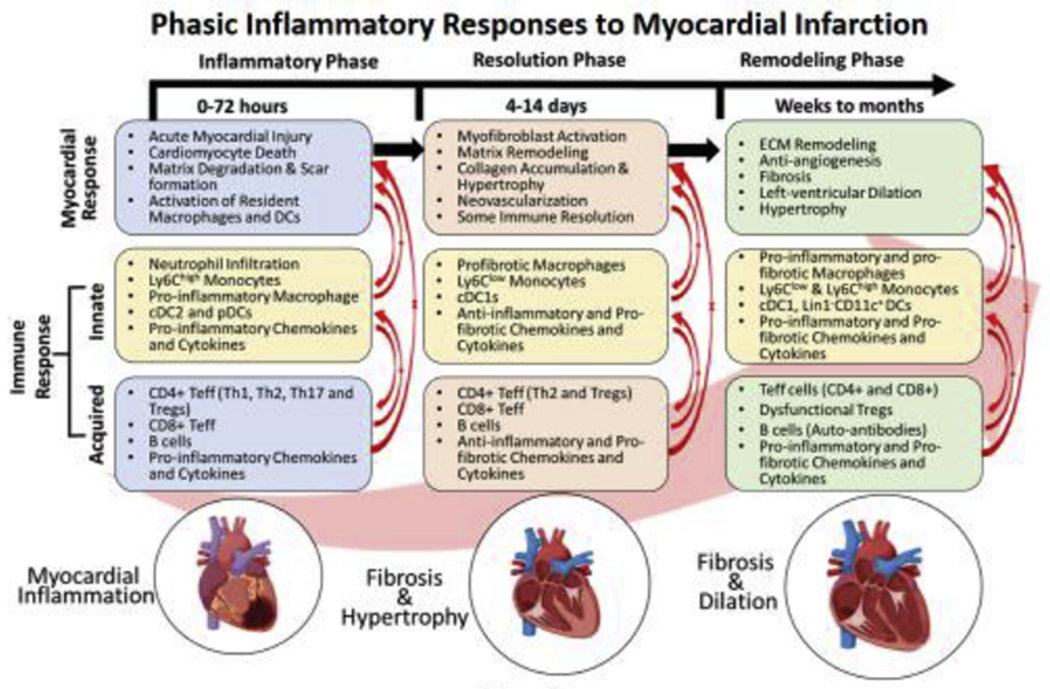
Different phases of immune activation post-MI are temporally regulated to induce intense pro-inflammation from 0-72 h post-injury to clear the damaged cells followed by scar-formation and somewhat immune-resolution from 4-14 days. While the first phase is predominated by the phagocytic and pro-inflammatory cells, 2nd phase is dictated by anti-inflammatory and pro-fibrotic cells. Phase-1 specific chemokines are CXCR2/KC, CXCR4/SDF1α, CCR1/MIP1α, CCR2/MCP1, CXCL1, CXCL12, CXCL13, and CXCL9 whereas cytokines are TNFα, IFNγ, IL-6, and IL-1β. In contrast, phase-II specific chemokines are CCR2/MCP1, CXCR2/CXCR4/MIF, CCR1/MIP1α, CCR5/RANTES, CX3CR1/Fractalkine, CXCL2, CXCL5, CXCL8, and CXCL12, and cytokines are IL-10, TGFβ, CTGF, IL-4, IL-13, IL-6 and eotaxin. Ischemic cardiomyopathy (IC) is associated with low-grade inflammation that over a period of months to years (post-MI) lead to extensive extracellular matrix remodeling, inhibit angiogenesis, and promote LV-remodeling. Both pro- and anti-inflammatory immune responses have been found to be active during this phase suggesting disparate local networks of functionally-similar immune cells. This phase is also associated with several chemokines (CX3CR1/Fractalkine, CXCR4/SDF1α, CCR2/MCP1, CXCL1, CXCL2, CXCL5, and CXCL8) and cytokines (TNFα, IFNγ, TGFβ, IL-10, IL-12, IL-6, and eotaxin). Some of the components were designed using ‘Biorender’.
Neutrophils
Circulating neutrophils constitute the first-line of defense and predominantly dictate initial 24-h post-MI [7] as they follow chemokine gradients to transmigrate across the endothelial walls and infiltrate into the ischemic myocardium [8]. This rapid response to tissue-injury also makes them excellent prognostic markers for acute cardiac injury [9]. Increased G-CSF levels in the circulation (and their concomitant decrease in the bone marrow; BM) facilitate neutrophil emigration into the circulation as well as their differentiation from BM progenitors to ensure constant supply during this period [7]. Once at the site of myocardial injury, neutrophils amplify the resident cardiac immune response and release proteolytic enzymes to either kill evading pathogens or clear dead cells [10, 11]. Activated neutrophils also produce significantly higher levels of reactive oxygen species and IL-1β that are potent chemo-attractants for the recruitment of other immune cells, and hence also regulate subsequent immune responses [7]. During acute inflammation, they undergo significant temporally-determined phenotypic alterations [10] and dynamically modulate their ability to alter macrophage activity and dampen inflammation [12] favoring reparative processes [11]. Thus, early-phase neutrophil response during cardiac injury is critical for efficient wound-healing; failure of which promotes uncontrolled inflammatory response, maladaptive fibrotic myopathy and progressive heart failure [12]. Much is known about acute-phase pro-inflammatory role of neutrophils in myocardial injury, but their direct involvement in ischemic cardiomyopathy remains poorly understood and require detailed temporal immunomodulation studies.
Monocytes
Monocytes are the circulating phagocytic cells that upon injury extravasate into the tissues and differentiate to replenish pro- and anti-inflammatory macrophages and dendritic cells (DCs) [13]. Based on their Ly6C expression, murine monocytes can be characterized to either have high- (Ly6Chigh) or low LY6C (Ly6Clow) expression [13]. Analogues to these, human monocytes can either be classical (CD14+CD16−) or non-classical (CD14−CD16+) monocytes, respectively [13]. Despite having a phagocytic activity similar to the neutrophils, monocytes are much less cytotoxic for tissues and possess specific mechanisms for antigen/pathogen recognition due to which acute injuries, including MI, strongly rely upon their rapid infiltration, and subsequent differentiation into other professional antigen-presenting cells. Studies have shown that monocytes are sequentially recruited into the injured myocardium with predominantly Ly6Chigh monocyte infiltration from 1-3d post-MI (pro-inflammatory phase) followed by Ly6Clow monocytes from 4-7d (reparative/resolution phase) [14]. Ly6Chigh monocytes are recruited using CCR2/CCL2 signaling, express pro-inflammatory cytokines such as TNFα and IL-1β, clear tissue-debris and promote tissue-granulation [14]. Ly6Clow monocytes, on the other hand, infiltrate using CX3CR1-fractalkine mediated pathways, express pro-angiogenic factors such as VEGF [14] and promote TGF-mediated fibrotic scar formation [15]. Reparative phase (4-7 days post-MI) is, thus, marked with the sublimation of pro-inflammation and augmentation of TGFβ and VEGF mediated tissue-regenerative and angiogenic responses [14]. However, a 2nd wave of monocyte influx characterized by heightened Ly6Chigh monocytes, primarily derived from the spleen, has also been reported during IC [16]. Adoptive transfer studies using splenocytes harvested from HF mice further suggest a pathological role of these monocytes in mediating LV remodeling, interstitial fibrosis and progressive cardiac dysfunction [16]. This pathological transition from being protective during MI and pathological during IC could largely be triggered by the sustained inflammatory processes and is associated with continuous cardiomyocyte apoptosis [17], hypertrophy and interstitial fibrosis, thereby emphasizing the necessity of a balanced inflammatory response for reparative cardiac remodeling.
Macrophages
Macrophages are the professional antigen-presenting cells and are either derived from embryonic or hematopoietic lineages. Embryonically-derived macrophages are the tissue-resident macrophages (RMs) and hematopoietic lineages differentiate into ‘infiltrating’ monocyte-derived macrophages (MDMs) [18]. Proliferation of local embryonic progenitor cells maintain RMs during steady-state, and infiltration of Ly6Chigh monocytes during acute inflammation contribute to inflammatory MDMs [19]. While MDMs induce pro-inflammatory milieu, RMs regulate atrio-ventricular conduction [20], and promote myocardial repair [21], neo-vascularization and tissue-regeneration [22] post-MI emphasizing their protective role in tissue-healing. RMs and MDMs, thus, belong to disparate lineages, undergo distinctive activation processes and regulate different signaling cascades to control inflammation [18].
Like monocytes, macrophages also demonstrate a functionally distinct biphasic response during cardiac injury. RMs, owing to their close proximity to the cardiomyocytes, are especially suited to promptly sense myocyte-injury, proliferate to clear tissue debris and activate systemic immune responses [23]. They also release significant amounts of IL-1β to recruit initial wave of neutrophils [23]. Studies have shown that RM activity and proliferation predominate the early phase of injury-response (0-2d post-MI) [23]. However, as monocytes take over the injury site (3-5d post-MI), RMs are replaced (and inhibited) by MDMs marking an ontogenic shift from reparative to pro-inflammatory macrophages [18]. MDMs mediate NLRP3 inflammasome activation, clear dead cells, and, produce several chemokines (CxCl1, Cxcl2, CCL2 and CCL9) and pro-inflammatory cytokines (IL-1β, TNFα) necessary to initiate wound-healing processes [18, 23]. Phenotypically heterogeneous embryonically-derived RMs (CCR2−) and infiltrating MDMs (CCR2+) have also been reported in the failing human hearts, the latter being directly associated with pro-inflammation and ischemic cardiomyopathy [24]. Although, ‘M1- and M2-like’ macrophages have also been described in the pro-inflammatory and resolution phases, respectively; due to controversies associated with this phenotype-based characterization, these are not discussed here.
Dendritic Cells
Dendritic cells (DCs) are the professional antigen-presenting cells and by modulating adaptive immune responses play a vital role in IC. Phenotypically, DCs are similar to macrophages and share ontogeny but are superior in regulating T-cells to induce either immunogenic or tolerogenic responses [25]. Broadly, DCs can be categorized as conventional DCs (cDC1 and cDC2) or plasmacytoid DCs (pDCs) and all of these subsets have been shown to reside in non-injured hearts mostly in close proximity to RMs [26]. Resident DCs process proteins released during the regular wear and tear of cardiomyocytes and maintain immune-homeostasis by activating regulatory T-cells to induce tolerogenic responses. However, during MI, resident DCs also escort cardiac-specific self-antigens to the LNs and activate effector CD4+ T-cells, thereby waning self-tolerance and inducing auto-immunity [27]. Studies have shown that at around 5d post-MI, cDC2s are maximal in the ischemic hearts and promote pro-inflammation to aid in the clearance of cellular debris. However, by day 7 post-MI, cDC2s are replaced by cDC1s to induce Treg-mediated tolerance, promote fibrotic scar-formation and initiate angiogenesis during the reparative phase [26]. Although, the exact function and role of tolerogenic pDCs is not clear but they also have been found to infiltrate the injured heart by 3d post-MI [26]. Due to their phenotypic diversity, it is not trivial to conclusively ascertain the function of each DC subset in MI and IC. However, it is clear that cDCs are important for tissue-healing as their depletion immediately after MI improves cardiac function, reduce myocyte hypertrophy and blunt IC by curbing pro-inflammation (cDC2 mediated), and T-cell infiltration during resolution (7d post-MI) [26]. Similarly, adoptive transfer of tolerogenic pDCs also subside pro-inflammatory responses by inducing immune-suppressive Tregs [28]. It, therefore, appears that DCs exert indirect effects on ischemic cardiomyopathy by altering T-cell mediated pro-inflammation and/or favoring Treg-mediated wound-resolution.
In summary, a distinct innate immune-cell landscape involving several players is activated during acute ischemic insult with the purpose of activating phasic responses to clear apoptotic cells and induce pro-inflammation followed by repair and immune resolution. However, sustained inflammation or eventual re-activation of these innate immune components fuel pathological cardiac responses leading to progressive cardiac dysfunction and maladaptive LV remodeling during IC [30].
Adaptive Immune Response
Helper T-cells
CD4+ helper (Th) T-lymphocytes play a critical role in mediating transition from the pro-inflammatory to immune-resolution phase. By reacting to a variety of tissue-cytokines, they can polarize into a spectrum of subsets, such as Th1, Th2, Th9, Th17, Th22, follicular helper (Tfh) or regulatory T-cells (Tregs) capable of exerting varied effects ranging from inflammation to fibrosis and angiogenesis [29]. T-cell activation is the most stringently controlled immune activation mechanism requiring strong interaction between the cognate T-cell receptors (TCRs), the antigens presented in conjunction with MHC molecules expressed on the APCs (RMs, MDMs and DCs) and other appropriate co-stimulatory signals [30]. Studies with reporter mice expressing mCherry protein under the cardiac-specific αMyHC, have clearly shown the migration of mCherry+ myeloid cells to the mediastinal lymph nodes (mLNs) and spleens of infarcted mice [31]. These findings suggest that intracellular cardiac-antigens or neo-antigens formed during MI are phagocytosed and processed by the APCs and are shuttled to the mLNs/spleens to effect T-cell activation and proliferation. Several independent groups have shown that T-cell activation follows innate immune activation and cardiac T-cell levels are maximal in 3-7 days post-MI, return to baseline levels by day 14 [32] and exhibit a 2nd wave of T-cell activation during IC [3]. Moreover, T-cell subsets exhibit phase-dependent plasticity and interconvert from pro-inflammatory (Th1 and Th17) to pro-fibrotic (Th2 and Treg) subsets during the transition from pro-inflammatory to resolution phase [33]. Ischemic and non-ischemic models of HF also show that T-cells can modulate fibroblast activity and collagen secretion as well. While defective scar formation with disarrayed collagen fibers has been observed in CD4−/− [34] and TCR−/− [35] mice during ischemic and non-ischemic injury, respectively; T-cell depletion during ischemic cardiomyopathy results in reduced fibrosis and LV remodeling [3]. These contradictory findings suggest that T-cells are indispensable for wound healing and immune resolution early after injury but during IC undergo a phenotypically pathological shift (presumably owing to persistent low-grade inflammation) that by accentuating pro-inflammation and fibrotic LV remodeling aggravate cardiac dysfunction [3]. Furthermore, this T-cell activation is directed against specific cardiac antigens such as MyHCA [36] as OT-II mice exhibit significant protection from T-cell induced autoimmune damage. Analogues to these preclinical studies, T-cells with distinct TCR repertoires [37] or specificity against cardiac antigens, such as αMyHC and troponin have also been reported in clinical studies [38].
Among all Th subsets, Tregs demand a specific mention due to their potent immune-suppressive potential. Tregs are critical to regulate immune responses, promote cardiac repair and initiate immune resolution [39]. Similar to effector CD4+ T-cells, Tregs also infiltrate the myocardium immediately after MI (day 1), reach maximal levels by day 3 and return to baseline by day 14 post-MI. These Tregs promote ‘M2-like’ macrophage phenotype characterized by arginase-1, IL-13, TGF-β and osteopontin expression thereby suggesting cardio-protective effects of Tregs during the acute phase [40]. This is also consistent with the fact that adoptive transfer of Tregs during MI subside inflammation and promote cardiac repair [41]. However due to chronic inflammation during IC, Tregs have also been shown to undergo a temporal phenotypic shift with expression of pro-inflammatory cytokines and loss of immunosuppressive potential [42]. Importantly, their partial depletion and subsequent reconstitution resets their phenotype and immunosuppressive potential leading to improved cardiac function, reduced LV remodeling and partial reversal of IC [42].
CD8+ cytotoxic T-cells are also significantly increased by day 1 with a peak observed by 5-7 days post MI [32]. Like Th cells, CD8+ T-cells are also known to be increased at 8 weeks post-MI during IC [3]. Increased incidence of cardiac rupture, compromised scar formation and exacerbated pro-inflammation at 7 days post-MI have been observed in the absence of functional CD8+ T-cells. Paradoxically, the cardiac function improves significantly in CD8 deficient mice also at 7d post-MI suggesting their protective role in adequate scar formation but an overall pathological effect on cardiac physiology [43] immediately after MI. In-vitro co-culture studies have shown that during late stages of MI (2-3 weeks post-infarction) CD8+ T-cells exert antigen-specific cytotoxic effects on cardiomyocytes and escalate their apoptotic death [44]. Despite this knowledge, the exact role of CD8+ T-cells in mediating LV remodeling during IC is not known and require further studies to adequately discern their spatio-temporal effects on cardiac pathophysiology.
γδT-cells
Inherent complexity of T-cell responses can also be appreciated from the fact that several unconventional T-cells that do not require MHC-restriction (and hence antigen-presenting cells) for their activation have also been identified in humans and rodents [45]. These T-cells i) recognize proteins that are outside the MHC locus such as MR1, and CD1a-d, ii) are apt for rapid responses (hour to days), iii) bridge innate and adaptive immune responses iv) do not recognize classical antigens, v) are not donor-specific and, last but not the least, vi) most individuals respond similarly as opposed to conventional T-cells where the degree and extent of activation can be different in different individuals [45]. Innate natural killer T-cells (iNKT), γδT-cells and mucosal-associated invariant T-cells (MAIT) are some important members of this class [45, 46]. To maintain brevity and focus only γδT-cells are discussed below and readers are recommended to explore other detailed reviews discussing the role of iNKT cells in cardiovascular diseases [47] and MAIT cells in autoimmune responses [48].
It is well-established that unconventional T-cells are poised to mount a rapid immune response during ischemic [49] and non-ischemic tissue injuries [46] as they directly recognize lipidic- and phosphatidic-antigens in the environment and do not need antigen-presentation by other cells. Indeed, studies have shown that γδT-cells quickly respond to ischemic cardiac injury, and using CCL20-CCR6 signaling infiltrate the myocardium within 1 day post-MI, peak levels being observed by 7 days [49]. Moreover, these T-cells are a major source of IL-17A which is known to alter endothelial cell activation, regulate neutrophil recruitment, mediate pro-inflammatory macrophage phenotype, facilitate myofibroblast activation and exert pro-apoptotic effects on cardiomyocytes [50]. Consequently, mice deficient in γδT-cells exhibit higher survival during the initial 7 days, and significantly reduced IC characterized by blunted LV remodeling, decreased fibrosis and improved cardiac function at 28 days post-MI [49]. Similar results have also been observed by direct IL-17A neutralization using selective antibodies [50]. Considering that IL-17A expression is regulated by IL-23 and IL-1β [51], it is possible that during acute MI these cytokines directly bolster IL-17A production in γδT-cells which then act on the cardiomyocytes, fibroblasts and innate immune cells to pathologically alter their function, and overall inflammatory/reparative milieu.
B-cells
B-lymphocytes mediate humoral immunity by generating antibodies against self or non-self antigens. Presence of auto-antibodies against cardiac antigens such as actin, myosin, troponin I, Na+K+ATPase, in humans [52] as well as in rodents [53], therefore, points to B-cell mediated adaptive responses in cardiac diseases. Naïve murine heart contains two different populations of B220+ B-cells characterized by IgMhighIgDlow (larger fraction) and IgMlowIgDhigh (smaller fraction) expression [54]. Although, the ontogeny of these populations is not known, they populate the heart by E13.5 and despite certain phenotypic differences reside in equilibrium with circulating and splenic B-cells [55]. During ischemic injury, B220+ and CD19+IgD+IgMloW cells are actively recruited into the infarcted myocardium, reach maximal levels by day 3-5 post-MI and return to baseline levels by day 14 [56]. It appears that B-cells exert a pathological role in cardiac wound healing post-ischemic injury as their anti-CD20 mediated depletion confer a protective response by decreasing LV remodeling and improving cardiac function by day 14 post-MI [56]. Since, anti-CD20 only depletes B2 and B1b cells without affecting B1a levels [57] it is possible that more complex subset-specific effects exist for different B-cell subtypes. Several mechanisms, such as reduction in cell apoptosis, ischemic damage, pro-inflammatory cytokines and Ly6Chigh monocyte recruitment, appears to play a role in this protective response. Moreover, these effects are CCL7 (a ligand for CCR2) dependent as B-cell specific CCL7 gene knockout results in negation of these effects [56]. Moreover, owing to the complex interactions of B-cells with other components of the innate and adaptive immune responses, and lack of appropriate pre-clinical and clinical studies, much remains to be investigated to better understand the role of B-cells in IC.
Conclusion
Considerable evidence suggests direct and indirect protective effects of immune activation during MI and overall pathological response during IC. Several players of the innate and adaptive immune systems form a complex interconnected network that can exert beneficial as well as detrimental effects depending upon the cues received from the tissue-milieu, the nature and extent of the injury and the previous history of cardiac insults. Although, highly specific pre-clinical models compel to appreciate the therapeutic benefits of immunomodulation during IC, direct evidence from clinical studies are scant and require carefully planned studies to dissect acute vs chronic effects to devise efficient immune cell based therapeutic strategies for cardiac repair.
Acknowledgments
Sources of Funding
This work was supported by the National Institutes of Health (NIH) R00 HL132123 grant to SSB, and NIH R01 HL127442 and AHA GIA 17GRNT33700188 to RJG. SN was supported (partly) by the funds provided by the ‘Drug Development Institute’ at the Ohio State University. Funding sources did not influence any part of this work.
Abbreviations:
- MI
Myocardial Infarction
- IC
Ischemic cardiomyopathy
- BM
Bone marrow
- CCL7
Chemokine (C-C motif) ligand 7
- CCR2/CCL2
C-C chemokine receptor type 2/Chemokine (C-C motif) ligand 2
- cDC
Conventional dendritic cells
- CX3CR1
CX3C chemokine receptor 1
- DC
Dendritic cells
- IL-1β
Interleukin 1β
- Ly6C
Lymphocyte antigen 6 complex
- MDM
Monocyte derived macrophages
- mLN
Mediastinal lymph nodes
- MyHCA
Myosin heavy chain alpha
- pDC
Plasmacytoid dendritic cell
- RM
Resident macrophages
- TCR
T-cell receptor
- TGF
Transforming growth factor
- TNFα
Tumor necrosis factor alpha
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest: None
Conflict of Interest: None
References:
- [1].Prabhu SD, Frangogiannis NG, The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis, Circ Res 119(1) (2016) 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Biesbroek PS, Beek AM, Germans T, Niessen HW, van Rossum AC, Diagnosis of myocarditis: Current state and future perspectives, Int J Cardiol 191 (2015) 211–9. [DOI] [PubMed] [Google Scholar]
- [3].Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD, Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure, Circ Heart Fail 10(3) (2017) e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Strassheim D, Dempsey EC, Gerasimovskaya E, Stenmark K, Karoor V, Role of Inflammatory Cell Subtypes in Heart Failure, J Immunol Res 2019 (2019) 2164017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, Lindner D, Inflammation and fibrosis in murine models of heart failure, Basic Res Cardiol 114(3) (2019) 19. [DOI] [PubMed] [Google Scholar]
- [6].Mann DL, Innate immunity and the failing heart: the cytokine hypothesis revisited, Circ Res 116(7) (2015) 1254–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, Quaife-Ryan GA, Al-Sharea A, Pernes G, Dragoljevic D, Lal H, Schroder K, Hanaoka BY, Raman C, Grant MB, Hudson JE, Smyth SS, Porrello ER, Murphy AJ, Nagareddy PR, Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction, Circulation 141(13) (2020) 1080–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Epelman S, Liu PP, Mann DL, Role of innate and adaptive immune mechanisms in cardiac injury and repair, Nat Rev Immunol 15(2) (2015) 117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meissner J, Irfan A, Twerenbold R, Mueller S, Reiter M, Haaf P, Reichlin T, Schaub N, Winkler K, Pfister O, Heinisch C, Mueller C, Use of neutrophil count in early diagnosis and risk stratification of AMI, Am J Med 124(6) (2011) 534–42. [DOI] [PubMed] [Google Scholar]
- [10].Daseke MJ 2nd, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML, Neutrophil proteome shifts over the myocardial infarction time continuum, Basic Res Cardiol 114(5) (2019) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S, Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype, Eur Heart J 38(3) (2017) 187–197. [DOI] [PubMed] [Google Scholar]
- [12].Soehnlein O, Steffens S, Hidalgo A, Weber C, Neutrophils as protagonists and targets in chronic inflammation, Nat Rev Immunol 17(4) (2017) 248–261. [DOI] [PubMed] [Google Scholar]
- [13].Auffray C, Sieweke MH, Geissmann F, Blood monocytes: development, heterogeneity, and relationship with dendritic cells, Annu Rev Immunol 27 (2009) 669–92. [DOI] [PubMed] [Google Scholar]
- [14].Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ, The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions, J Exp Med 204(12) (2007) 3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]; This was the first study to elegantly show phasic recruitment of Ly6Chigh (during pro-inflammatory phase) and Ly6Clow monocytes (during repair/resolution phase) into the ischemic myocardium. This is also one of the classical papers in cardiovascular immunology.
- [15].Crane MJ, Daley JM, van Houtte O, Brancato SK, Henry WL Jr., Albina JE, The monocyte to macrophage transition in the murine sterile wound, PLoS One 9(1) (2014) e86660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD, Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis, Circ Res 114(2) (2014) 266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that lymphoid tissues such as the spleen act as the extramedullary sources of immune-cells during ischemic cardiomyopathy and regulate left-venrtricular remodeling. By adoptive transfer of splenocytes, authors also provided hints about the tissue-speciifcity of immune responses.
- [17].Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney J, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, Thorp EB, Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction, Circ Res 113(8) (2013) 1004–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL, Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation, Immunity 40(1) (2014) 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper is of high-significance in cardiovascular immunology. Authors, for the first time, showed cardiac-specific resident macrophages that are derived from embryonic lineages and are maintained independent of other hematopoietically derived monocytes/macrophages during steady-state.
- [19].Frantz S, Nahrendorf M, Cardiac macrophages and their role in ischaemic heart disease, Cardiovasc Res 102(2) (2014) 240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M, Macrophages Facilitate Electrical Conduction in the Heart, Cell 169(3) (2017) 510–522 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper is highly recommended to the readers. Authors, very elegantly, showed that cardiac resident macrophages reside near the atrio-ventricular node and form gap-junctions with cardiomyocytes using connexin-43 channels. RMs synchronuously depolarize with cardiomyocytes and facilitate myocardial conduction in the heart. Moreover, depletion of RMs result in fulminant arrythmias.
- [21].Liu B, Zhang HG, Zhu Y, Jiang YH, Luo GP, Tang FQ, Jian Z, Xiao YB, Cardiac resident macrophages are involved in hypoxiainduced postnatal cardiomyocyte proliferation, Mol Med Rep 15(6) (2017) 3541–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN, Macrophages are required for neonatal heart regeneration, J Clin Invest 124(3) (2014) 1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]; It is well-known that injured rodent hearts can completely regenerate for upto P7. Authors showed that RMs play an important role in this. They promote neoangiogenesis and facilitate heart-regeneration in neonates as depletion of macrophages signifciantly impairs their ability to fully regenerate.
- [23].Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ, Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury, Circ Res 124(2) (2019) 263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG, Lavine KJ, The human heart contains distinct macrophage subsets with divergent origins and functions, Nat Med 24(8) (2018) 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]; This was the first study to show CCR2− RMs and CCR2+ hematopoietic monocyte-derived macrophages in the human hearts. Using gender mis-matched heart tissues, authors very elegantly showed that in human hearts also RMs are maintained independent of their hematopoeitic counterparts. This paper is highly recommended to readers.
- [25].Worbs T, Hammerschmidt SI, Forster R, Dendritic cell migration in health and disease, Nat Rev Immunol 17(1) (2017) 30–48. [DOI] [PubMed] [Google Scholar]
- [26].Lee JS, Jeong SJ, Kim S, Chalifour L, Yun TJ, Miah MA, Li B, Majdoubi A, Sabourin A, Keler T, Guimond JV, Haddad E, Choi EY, Epelman S, Choi JH, Thibodeau J, Oh GT, Cheong C, Conventional Dendritic Cells Impair Recovery after Myocardial Infarction, J Immunol 201(6) (2018) 1784–1798. [DOI] [PubMed] [Google Scholar]; This paper pointed to the complexity of cardiac immune cell niche by showing the presence of specific dendritic cell populations near the resident macrophages during steady-state. These dendritic cells exhibit complex interactions with other immune cells and different subtypes mediate disparate effects on cardiac (patho)physiology.
- [27].Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, Sonderegger I, Bachmaier K, Kopf M, Penninger JM, Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity, Nat Med 9(12) (2003) 1484–90. [DOI] [PubMed] [Google Scholar]
- [28].Choo EH, Lee JH, Park EH, Park HE, Jung NC, Kim TH, Koh YS, Kim E, Seung KB, Park C, Hong KS, Kang K, Song JY, Seo HG, Lim DS, Chang K, Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization, Circulation 135(15) (2017) 1444–1457. [DOI] [PubMed] [Google Scholar]
- [29].Mirlekar B, Co-expression of master transcription factors determines CD4(+) T cell plasticity and functions in auto-inflammatory diseases, Immunol Lett 222 (2020) 58–66. [DOI] [PubMed] [Google Scholar]
- [30].Zhan Y, Carrington EM, Zhang Y, Heinzel S, Lew AM, Life and Death of Activated T Cells: How Are They Different from Naive T Cells?, Front Immunol 8 (2017) 1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].DeBerge M, Yu S, Dehn S, Ifergan I, Yeap XY, Filipp M, Becker A, Luo X, Miller S, Thorp EB, Monocytes prime autoreactive T cells after myocardial infarction, Am J Physiol Heart Circ Physiol 318(1) (2020) H116–H123. [DOI] [PMC free article] [PubMed] [Google Scholar]; We found this paper to be of special significance as this was the first study to provide experimental proof for the trafficking of myeloid cells (loaded with cardiac-specific proteins) to the media-stinal lymph nodes post-MI and subsequent activation of auto-immune T-cells.
- [32].Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M, Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction, J Mol Cell Cardiol 62 (2013) 24–35. [DOI] [PubMed] [Google Scholar]
- [33].Hofmann U, Frantz S, Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction, Circ Res 116(2) (2015) 354–67. [DOI] [PubMed] [Google Scholar]
- [34].Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, Frantz S, Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice, Circulation 125(13) (2012) 1652–63. [DOI] [PubMed] [Google Scholar]
- [35].Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM, Alcaide P, Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure, J Exp Med 214(11) (2017) 3311–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rieckmann M, Delgobo M, Gaal C, Buchner L, Steinau P, Reshef D, Gil-Cruz C, Horst ENT, Kircher M, Reiter T, Heinze KG, Niessen HW, Krijnen PA, van der Laan AM, Piek JJ, Koch C, Wester HJ, Lapa C, Bauer WR, Ludewig B, Friedman N, Frantz S, Hofmann U, Ramos GC, Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses, J Clin Invest 129(11) (2019) 4922–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhong Z, Wu H, Zhang Q, Zhong W, Zhao P, Characteristics of T cell receptor repertoires of patients with acute myocardial infarction through high-throughput sequencing, J Transl Med 17(1) (2019) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, Matsui T, Rosenzweig A, Bronson RT, Smith R, Fletcher AL, Turley SJ, Wucherpfennig K, Kyewski B, Lipes MA, Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans, J Clin Invest 121(4) (2011) 1561–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Matsumoto K, Ogawa M, Suzuki J, Hirata Y, Nagai R, Isobe M, Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice, Int Heart J 52(6) (2011) 382–7. [DOI] [PubMed] [Google Scholar]
- [40].Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S, Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation, Circ Res 115(1) (2014) 55–67. [DOI] [PubMed] [Google Scholar]
- [41].Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, Frangogiannis NG, Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function, Am J Physiol Heart Circ Physiol 307(8) (2014) H1233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD, Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy, Circulation 139(2) (2019) 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]; Regulatory T-cells (Tregs) have long been considered as protective during ischemic injury and are essential to check overt immune activation. This paper for the first-time showed that during chronic inflammation Tregs undergo dynamic temporal phenotypic shifts during IC, lose their immunosuppressive potential and express pro-inflammatory cytokines. Moreover, Treg dysfunction is one of the critical events in mediating maladaptive LV remodeling as their reversible depletion promotes neoangiogenesis, improves cardiac function and ameliorates fibrosis.
- [43].Ilatovskaya DV, Pitts C, Clayton J, Domondon M, Troncoso M, Pippin S, DeLeon-Pennell KY, CD8(+) T-cells negatively regulate inflammation post-myocardial infarction, Am J Physiol Heart Circ Physiol 317(3) (2019) H581–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Varda-Bloom N, Leor J, Ohad DG, Hasin Y, Amar M, Fixler R, Battler A, Eldar M, Hasin D, Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro, J Mol Cell Cardiol 32(12) (2000) 2141–9. [DOI] [PubMed] [Google Scholar]
- [45].Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB, The burgeoning family of unconventional T cells, Nat Immunol 16(11) (2015) 1114–23. [DOI] [PubMed] [Google Scholar]
- [46].Toubal A, Nel I, Lotersztajn S, Lehuen A, Mucosal-associated invariant T cells and disease, Nat Rev Immunol 19(10) (2019) 643–657. [DOI] [PubMed] [Google Scholar]
- [47].van Puijvelde GHM, Kuiper J, NKT cells in cardiovascular diseases, Eur J Pharmacol 816 (2017) 47–57. [DOI] [PubMed] [Google Scholar]
- [48].Chiba A, Murayama G, Miyake S, Mucosal-Associated Invariant T Cells in Autoimmune Diseases, Front Immunol 9 (2018) 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yan X, Shichita T, Katsumata Y, Matsuhashi T, Ito H, Ito K, Anzai A, Endo J, Tamura Y, Kimura K, Fujita J, Shinmura K, Shen W, Yoshimura A, Fukuda K, Sano M, Deleterious effect of the IL-23/IL-17A axis and gammadeltaT cells on left ventricular remodeling after myocardial infarction, J Am Heart Assoc 1(5) (2012) e004408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, Yuan J, Jevallee H, Wei F, Shi GP, Cheng X, Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration, J Am Coll Cardiol 59(4) (2012) 420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH, Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity, Immunity 31(2) (2009) 331–41. [DOI] [PubMed] [Google Scholar]
- [52].De Scheerder I, Vandekerckhove J, Robbrecht J, Algoed L, De Buyzere M, De Langhe J, De Schrijver G, Clement D, Post-cardiac injury syndrome and an increased humoral immune response against the major contractile proteins (actin and myosin), Am J Cardiol 56(10) (1985) 631–3. [DOI] [PubMed] [Google Scholar]
- [53].Kaya Z, Leib C, Katus HA, Autoantibodies in heart failure and cardiac dysfunction, Circ Res 110(1) (2012) 145–58. [DOI] [PubMed] [Google Scholar]
- [54].Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, Friedrich M, Pinnecker J, Abesser M, Heinze KG, Schuh K, Beyersdorf N, Kerkau T, Demengeot J, Frantz S, Hofmann U, Myocardial aging as a T-cell-mediated phenomenon, Proc Natl Acad Sci U S A 114(12) (2017) E2420–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rocha-Resende C, Yang W, Adamo L, Myocardial associated B cells are a dynamic subgroup of circulating B cells, in equilibrium with splenic and circulating B cells, but with organ specific features, The Journal of Immunology 204(1 Supplement) (2020) 153.14–153.14. [Google Scholar]
- [56].Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, Vilar J, Caligiuri G, Tsiantoulas D, Laurans L, Dumeau E, Kotti S, Bruneval P, Charo IF, Binder CJ, Danchin N, Tedgui A, Tedder TF, Silvestre JS, Mallat Z, B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction, Nat Med 19(10) (2013) 1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Adamo L, Staloch LJ, Rocha-Resende C, Matkovich SJ, Jiang W, Bajpai G, Weinheimer CJ, Kovacs A, Schilling JD, Barger PM, Bhattacharya D, Mann DL, Modulation of subsets of cardiac B lymphocytes improves cardiac function after acute injury, JCI Insight 3(11) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]