

Abstract
Intrinsic resistance of unknown mechanism impedes the clinical utility of inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6i) in malignancies other than breast cancer. Here we used melanoma patient-derived xenografts (PDXs) to study the mechanisms for CDK4/6i resistance in pre-clinical settings. We observed that melanoma PDXs resistant to CDK4/6i frequently displayed activation of phosphatidyl inositol 3 kinase (PI3K)-AKT pathway, and inhibition of this pathway improved CDK4/6i response in a p21-dependent manner. We showed that a target of p21, CDK2, was necessary for proliferation in CDK4/6i-treated cells. Upon treatment with CDK4/6i, melanoma cells up-regulated cyclin D1, which sequestered p21 and another cyclin-dependent kinase inhibitor, p27, leaving a shortage of p21 and p27 available to bind and inhibit CDK2. Therefore, we tested whether induction of p21 in resistant melanoma cells would render them responsive to CDK4/6i. Because p21 is transcriptionally driven by p53, we coadministrated CDK4/6i with a murine double minute (MDM2) antagonist to stabilize p53, allowing p21 accumulation. This resulted in improved anti-tumor activity in PDXs and in murine melanoma. Furthermore, co-administration of CDK4/6 and MDM2 antagonists with standard of care therapy caused tumor regression. Notably, the molecular features associated with response to CDK4/6 and MDM2 inhibitors in PDXs were recapitulated by an ex vivo organotypic slice culture assay, which could potentially be adopted in clinic for patient stratification. Our findings provide a rationale for co-targeting CDK4/6 and MDM2 in melanoma.
One Sentence Summary:
CDK4/6 inhibition becomes effective in preclinical models of melanoma when combined with MDM2 antagonism.
Introduction
Metastatic melanoma is an aggressive disease with high mortality rates. Immunotherapy is usually the first-line treatment for metastatic melanoma. However, nearly half of melanoma patients are unresponsive to immunotherapeutic agents (1). Although drugs targeting serine/threonine-protein kinase B-Raf which is a product of v-Raf murine sarcoma viral oncogene homolog B gene (BRAF) is an effective treatment option for patients with tumors driven by oncogenic BRAFV600E, and mitogen-activated protein kinase kinase (MEK) inhibitors are somewhat beneficial for patients whose tumors have NRAS mutations, these drugs are often ineffective for long-term disease control due to the prompt onset of acquired resistance (2-4). Therefore, there remains a large group of melanoma patients that are resistant to the standard of care treatments and urgently require new therapeutic approaches.
Cell cycle kinases CDK4 and CDK6 are often constitutively activated in melanoma tumors through deletions and mutations of CDKN2A (43% of melanoma cases, TCGA), which encodes a negative regulator of CDK4/6, p16, and a negative regulator of MDM2, p14 (5-7). Furthermore, about 8% of melanomas amplify the genetic loci encoding either CDK4 or CDK6, and 7% amplify the CCND1 gene encoding CDK4/6 activator cyclin D1 (TCGA provisional melanoma dataset, 6/14/18, https://www.cancer.gov/tcga). In response to growth factor stimulation and/or oncogenic signaling, cyclin D1 activates CDK4/6 to facilitate G1 to S cell cycle transition by phosphorylating and inactivating the tumor suppressor retinoblastoma protein (RB). Inactive RB is released from complexes with the transcription factor E2F1, which is then free to initiate expression of genes involved in DNA replication and mitosis (8). Preclinical studies have shown that CDK4/6 inhibitors can reduce melanoma cell proliferation (9, 10) and synergize with MEK inhibitors to block melanoma growth in murine models (10). This suggests that activation of CDK4/6 is a key mechanism of melanoma development and progression and that targeting these kinases may benefit melanoma patients (11).
There are several specific small molecule inhibitors of CDK4/6 (CDK4/6i) that have been evaluated in clinical trials (12). Three such inhibitors are currently FDA-approved for the treatment of ER+ metastatic breast cancer in combination with hormone therapy (12). A number of clinical trials are underway evaluating the therapeutic utility of CDK4/6i in other malignancies, including melanoma (NCT02974725, NCT03484923, NCT03454919, NCT02645149, NCT02465060, NCT02791334, NCT02857270, ClinicalTrials.gov). Surprisingly, despite the clear role of CDK4/6 in melanomagenesis, only limited efficacy of CDK4/6i against metastatic melanoma has been reported thus far. For example, in a multi-cancer study of CDK4/6i palbociclib, only 1 out of 6 patients with metastatic melanoma achieved stable disease (13). Similarly, in a study of another CDK4/6i, abemaciclib, only 1 out of the 26 patients within the melanoma cohort achieved RECIST partial response and 6 patients achieved stable disease, for a disease control rate of 27% (14). Greater therapeutic benefit can be achieved by combining CDK4/6i with other agents. For instance, combined treatment with the CDK4/6i ribociclib and a MEKi showed a 33% response rate (7 out of 21) in patients with NRAS-mutated melanomas (15). Taken together, clinical data indicate that a large proportion of melanomas are intrinsically resistant to CDK4/6i. This suggests that molecular pathways compensating for the loss of CDK4 and CDK6 are either rapidly induced or constitutively activated in melanoma cells. In this study, we used a patient-derived xenograft (PDX) model to elucidate the molecular pathways driving CDK4/6i resistance in melanoma with the goal of interrupting resistance pathways to improve therapeutic efficacy of CDK4/6i.
Results
Resistance to CDK4/6i is associated with activation of PI3K/AKT pathway
We first evaluated whether melanoma PDXs recapitulate intrinsic resistance of melanoma tumors to CDK4/6i observed in clinical studies (14, 15). Overall, the response to the CDK4/6i ribociclib (LEE011) was modest. None of the tested PDX tumors regressed on therapy. Six out of thirteen tested PDXs showed slowing of tumor growth after CDK4/6i treatment, whereas seven other PDXs were intrinsically resistant to CDK4/6i therapy (Fig. 1A, see data file S1 for tumor measurements). Of note, xenografts of A375 melanoma cells were also resistant to ribociclib treatment (Fig. 1A), and therefore were chosen for additional in vitro studies.
Figure 1. Melanoma PDXs are intrinsically resistant to CDK4/6 inhibition.

A. Tumor volume change over time was assessed in 13 distinct melanoma PDXs and A375 melanoma xenografts grown in BALB/c nude mice that were treated daily with 100 mg/kg of the CDK4/6i ribociclib or vehicle. B. Genetic mutations and copy number alterations in tumors that were used to generate PDXs shown in A. Paired targeted DNA seq analysis was performed on tumor tissue and corresponding blood sample. Tumor-specific genetic alterations are shown. The map was generated using OncoPrinter. C. RPPA analysis of protein lysates prepared from vehicle-treated PDXs. Statistical analysis was performed using multiple T-test without correction for multiple comparisons, with alpha=0.05%. Each row (protein marker) was analyzed individually, without assuming a consistent SD. Results from proteomics analysis of proteins differentially expressed in CDK4/6i-responsive and resistant PDXs are shown.
To gain insight into the potential mechanisms of intrinsic resistance to CDK4/6i, we compared somatic mutations and copy number alterations of 160 cancer-related genes in six CDK4/6i-resistant and six partially responsive PDXs. The frequencies of common melanoma oncogenic driver mutations NRASQ61 and BRAFV600E were similar in responsive and resistant PDXs (Fig. 1B, data files S2, S3). It has been previously suggested that CDK4 gene amplification or CDKN2A deletion or mutations may facilitate CDK4/6i sensitivity (16). However, we found no enrichment for CDKN2A mutations/loss or CDK4 amplifications in CDK4/6-responsive PDXs. We also observed that three out of six CDK4/6i-resistant PDXs had genetic alterations inactivating the tumor suppressor p53, including TP53 mutations and MDM2 amplification (Fig. 1B, black rectangle). Overall, none of the tested genetic alterations were statistically significantly associated with CDK4/6i response.
We next compared proteomic and phospho-proteomic profiles in CDK4/6i-sensitive and resistant PDXs. Reverse phase protein array (RPPA) analysis identified 8 phospho-proteins and 9 total proteins associated with CDK4/6i response, (Fig. 1C, data file S4). Notably, CDK4/6i-sensitive tumors showed evidence of MAPK signaling activation based on increased amounts of the phospho-MEK and RSK (both are critical MAPK pathway nodes(17)), increased BAD phosphorylation (downstream MAPK pathway target(17)), and increased inhibitory phosphorylation of PEA-15 (MAPK pathway inhibitor(18)). In contrast, CDK4/6i-resistant tumors exhibited evidence of PI3K pathway activation based on the increase of phosphorylated AKT, and phosphorylated 4E-BP1, which are key PI3K effectors(19), and down-regulation of AMPKa and WIPI1 which are implicated in mTOR inhibition (20, 21). These data indicate that the PI3K/AKT/mTOR pathway may play a role in CDK4/6i-resistance.
To assess functional relevance of the PI3K/AKT pathway in response to CDK4/6i, we inactivated it in melanoma cell lines A375 and WM115 using specific small molecule inhibitors targeting this pathway: pan-PI3K inhibitor (PI3Ki) buparlisib (BKM120) and the AKT inhibitor (AKTi) ipatasertib. EdU incorporation assays showed that DNA replication was the lowest in cells treated with combined CDK4/6i and PI3Ki or CDK4/6i and AKTi, suggesting an improved inhibition of cell proliferation as compared to either monotherapy (Fig. 2A). These findings indicate that PI3K/AKT signaling can facilitate CDK4/6i resistance. We next sought to identify the downstream effectors of the PI3K/AKT pathway that modulate CDK4/6i response. It has been shown that PI3K/AKT pathway promotes cancer cell proliferation by inactivating CDK inhibitors p21 and p27 (22). Western blot analysis in melanoma cells showed that PI3Ki induced p21 expression, whereas p27 was not significantly affected (Fig. 2B). We also tested whether PI3K inhibition inhibits the cell cycle by inducing down-regulation of cyclin D1, and found this not to be the case based on western blot analysis (Fig. 2B). In comparison to single agents and vehicle treatments, CDK4/6i and PI3Ki co-treated cells had greater reduction of the transcription factor FoxM1, which regulates genes involved in replication and mitosis (23) (Fig. 2B). Furthermore, combined CDK4/6i and PI3Ki treatment prominently reduced the amount of inactivating phosphorylation of RB, which is a key replication inhibitor and one of the main targets of CDK4/6 and CDK2 (24) (Fig. 2B). To test the functional relevance of p21 in CDK4/6i response, we used p21-specific siRNA (Fig. 2C). The inhibition of DNA replication by CDK4/6i combined with PI3Ki or with AKTi was compromised by p21 knockdown (Fig. 2D). Similar results were obtained using isogenic HCT116 cells with or without knockout of CDKN1A gene encoding p21 (Fig. 2E). These findings suggest that PI3K/AKT pathway can facilitate CDK4/6i resistance by inhibiting p21.
Figure 2. Inhibition of PI3K/AKT pathway enhances response to CDK4/6i by inducing p21.
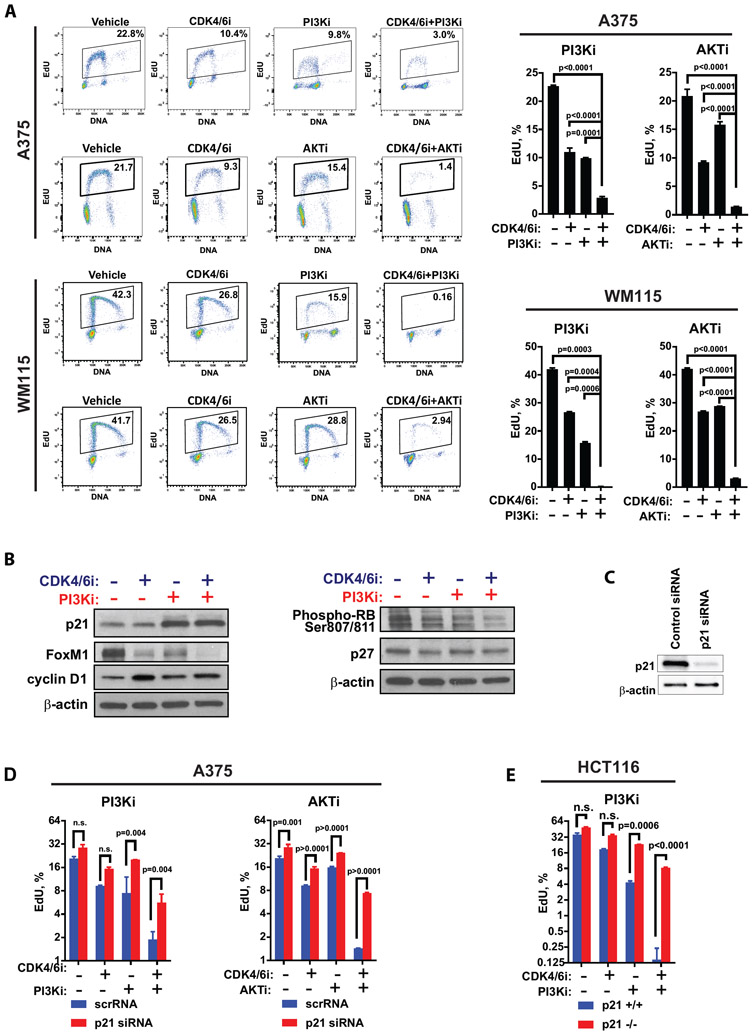
A. The percentages of A375 or WM115 cells replicating DNA after 3 days of treatment with DMSO (vehicle), CDK4/6i (ribociclib, 3 μM), PI3Ki (buparlisib, 5 μM), AKTi (ipatasertib, 10 μM), or indicated combinations of these drugs as determined by EdU incorporation assay. Representative flow cytometry plots are shown on the left. Graphs on the right summarize data from 3 biological replicates (mean ± SD) with statistical comparison of log transformed values performed using one-way ANOVA with Tukey's multiple comparisons test. B. Western blot analysis of indicated proteins in A375 cells treated for 3 days with DMSO (vehicle), CDK4/6i (ribociclib, 3 μM), PI3Ki (buparlisib, 5 μM), or combination of ribociclib and buparlisib. C. Western blot analysis of p21 expression in A375 cells transfected with control non-targeting or p21-targeting siRNA. D-E. Results of the EdU incorporation assay in A375 expressing control or p21 siRNA (D) and in isogenic HCT116 p21+/+ and p21−/− cells(E). Cells were treated with DMSO (vehicle), CDK4/6i (ribociclib, 3 μM), PI3Ki (buparlisib/BKM120, 5 μM), AKTi (ipatasertib, 10 μM), or indicated drug combinations for 2 days. Mean percentage of EdU-positive cells ± SD shown. Statistical analysis of the log-transformed percentages of the EdU+ cells was performed using 2-way ANOVA and corrected for multiple comparisons with Sidak's test.
Endogenous p21 and p27 are sequestered by cyclin D1, which is induced by CDK4/6i
Our data show that inhibition of p21 by active PI3K/AKT pathway can result in CDK4/6i resistance. However, not all of the CDK4/6i-resistant PDXs displayed markers of PI3K/AKT pathway activation. Further, in addition to p21, cells express another potent endogenous CDK inhibitor, p27, which has been implicated in CDK4/6i response previously (25). Of note, p27 was not induced by PI3K pathway inhibition, suggesting that there may be additional molecular mechanisms that inactivate endogenous CDK inhibitors p21 and p27 in melanoma cells.
Binding of p21 and p27 to CDK2 and CDK1 blocks their kinase activity, causing cell cycle arrest. In proliferating cells, p21 and p27 are disabled by binding and sequestration by the D type cyclins, which are increased in response to mitogenic stimulation. This sequestration enables CDK2 and CDK1 activity and cell cycle progression (26-29). Therefore, we first tested how cyclin D1 was affected by CDK4/6i. We found cyclin D1 to be up-regulated in CDK4/6i-treated melanoma and breast cancer cells (Fig. 3A), which is consistent with earlier reports showing cyclin D1 induction in response to CDK4/6 inhibitors in a variety of cancer cells (30-32). Cyclin D1 accumulation in response to CDK4/6i was also detected in PDX explants (PDXE) treated with CDK4/6i ex vivo (Fig. 3B) and in PDX tumors treated in vivo (Fig. 3C). No significant induction of cyclin E was detected (fig. S1A).
Figure 3. Cyclin D1 and CDK2 facilitate CDK4/6i resistance.

A. Western blot analysis of cyclin D1 in lysates from indicated cells treated with DMSO (Veh.), 3 μM ribociclib (Ribo), or 1 μM palbociclib (Palbo) for 3 days. B. Western blot analysis of cyclin D1 in lysates from PDX explants (PDXE) cultured with DMSO or 3 μM CDK4/6i ribociclib for 3 days. C. Cyclin D1 expression in PDX1688 tumors from mice treated daily with 100 mg/kg of ribociclib as shown in Fig. 1A. Statistical comparison performed with t test after log transformation. D. Co-immunoprecipitation assay in A375 cells treated with DMSO or 3 μM ribociclib for 3 days. E. The efficiency of cyclin D1 knockdown in A375 cells stably transfected with cyclin D1 shRNA or control non-targeting shRNA. F. EdU incorporation analysis of DNA replication in A375 cells transfected with control or cyclin D1-targeting shRNA and treated with DMSO (vehicle), 3 μM ribociclib (Ribo), or 1 μM palbociclib (Palbo) for 3 days. Two-way ANOVA with Sidak's multiple comparisons test was used after log transformation for statistical comparison of cyclin D1-deficient and control cells. G. The CDK1 and CDK2 shRNA knockdown efficiency in A375 cells treated with DMSO (veh.), 3 μM ribociclib (Ribo), or 1 μM palbociclib (Palbo) for 2 days. H. Colony formation assay. Indicated shRNA-expressing cells were plated at 1000 cells per well in 6-well plates and treated with DMSO (veh.), 3 μM ribociclib (Ribo), or 1 μM palbociclib (Palbo) for 7 days. Cells were visualized with crystal violet. Scale bar: 1 mm. I. DNA replication analysis for A375 cells transduced with CDK1 or CDK2 shRNA. Cells were treated as in G. The experiment was repeated three times, and mean ± SD from representative experiment is shown. Statistical comparison was performed using two-way ANOVA after log transformation. Sidak's test was used to correct for multiple comparisons. J. Results of the CellTiter-Blue viability assay in indicated cells treated with 3 μM ribociclib (CDK4/6i), 10 μM seliciclib (Pan-CDKi), the combination of both drugs (combo), or vehicle for 3 days. Graphs were produced in Prism. Statistical significance of cell viability differences between the “combo” and all other treatment groups was examined using one-way ANOVA after log transformation with Tukey's multiple comparisons test. K. The percentages of SK-Mel5 cells replicating DNA after 3 days of treatment with 3 μM ribociclib (CDK4/6i), 10 μM seliciclib (Pan-CDKi), combination of both drugs (combo), or vehicle measured by flow cytometry after EdU incorporation assay. One-way ANOVA after log transformation with Tukey's multiple comparisons test was performed to calculate statistical significance.
We next tested whether cyclin D1 is able to bind p21 and p27 in the presence of CDK4/6i. Indeed, we found that p21 and p27 were bound to cyclin D1 in melanoma cells. Furthermore, although the total protein expression of p21 and p27 was not increased after CDK4/6i treatment, more p21 and p27 proteins were bound to cyclin D1 (Fig. 3D). These data suggest that cyclin D1, induced by CDK4/6i, can sequester p21 and p27 in CDK4/6i treated cells. To test whether cyclin D1 upregulation and p21/p27 contribute to CDK4/6i resistance, we knocked cyclin D1 down with shRNA (Fig. 3E). This resulted in a decreased EdU-incorporation, indicative of G1 cell cycle arrest (Fig. 3F). Notably, the percentages of cells replicating DNA were the lowest in samples of cyclin D1-deficient cells treated with CDK4/6i. Similar results were obtained when cyclin D1 expression was down-regulated with a small molecule inhibitor of ERK (ERKi) (fig. S1B). Similar to the effect of cyclin D1 shRNA, ERKi enhanced CDK4/6i-mediated G1 arrest in melanoma cells (fig. S1C). In summary, these data suggest that compensatory induction of cyclin D1 after CDK4/6i treatment can contribute to CDK4/6i resistance by inhibiting p21 and p27.
CDK2 can facilitate CDK4/6i resistance
Our data indicated that p21 activation can overcome CDK4/6i resistance. The key function of p21 is to inhibit activities of CDK2 and CDK1 (33, 34), and genetic knockout studies demonstrated that cell cycle CDKs can compensate for each other’s activities (35-38). Therefore, we next tested whether CDK1 or CDK2 can facilitate CDK4/6i resistance by knocking them down using specific shRNAs (Fig. 3G). Only partial knockdown of CDK1 was achieved, consistent with CDK1 being essential for cell division (35). However, CDK4/6i treatment reduced the residual amount of CDK1 in CDK1 shRNA-expressing cells and also reduced the amount of CDK1 in CDK2 shRNA-expressing cells. These data could be interpreted to indicate a partial early phase cell cycle arrest (Fig. 3G). However, there was no dramatic reduction in cell growth in the colony formation assay (Fig. 3H), or in 5-Ethynyl-2′-deoxyuridine (EdU)EdU incorporation (Fig. 3I) after CDK4/6i in shCDK1 cells as compared to control GFP shRNA-expressing cells. These data suggest that CDK1 is not the main driver of CDK4/6i resistance. However, we cannot completely exclude its contribution because CDK1 knockdown was not complete. In contrast, knockdown of CDK2 was efficient (Fig. 3G). Loss of CDK2 had no prominent effect on cell proliferation, which is consistent with data from CDK2 knockout mice, which develop normally and exhibit functional mitosis (39, 40). Notably, treatment of CDK2 shRNA-expressing cells with CDK4/6i ribociclib or palbociclib produced greater anti-proliferative effect as compared to that in control cells, seen as slower growth in the colony formation assay (Fig. 3H) and reduced EdU incorporation (Fig. 3I). These data could be interpreted to indicate that CDK2 drives cell cycle progression through interphase in CDK4/6i-treated melanoma cells.
To further confirm that activities of other CDKs may contribute to CDK4/6i resistance, we used pan-CDK inhibitor seliciclib. This drug inhibits multiple cell cycle kinases including CDK1, CDK2, CDK5, CDK7, CDK9 and potentially other kinases (41). We found that addition of seleciclib to CDK4/6i treatment further reduced cell growth (Fig. 3J) and replication (Fig. 3K) in various melanoma cells. These data suggest that when CDK4/6 are inhibited by a specific inhibitor, other CDK/CDKs can facilitate cell cycle progression.
MDM2 antagonists sensitize melanoma cells to CDK4/6i
Next, we sought out a therapeutic strategy to overcome intrinsic resistance to CDK4/6i in melanoma. Our data showed that inactivation of CDK inhibitors p21 and p27 promotes CDK4/6i resistance. Therefore, we sought to pharmacologically induce p21 expression. We have previously demonstrated that MDM2 antagonists can induce p21 in melanoma tumors by stabilizing tumor suppressor p53 (42). Here, we investigated whether p21 induction by an MDM2 antagonist will augment CDK4/6i response. As expected, treatment with MDM2 inhibitor (MDM2i) nutlin-3a increased the amounts of p21 and p53 proteins (Fig. 4A). Co-targeting CDK4/6 and MDM2 resulted in de-phosphorylation of RB on Ser 807/811, indicative of RB-mediated cell cycle arrest (Fig. 4A). In wild type p53-expressing cells, the addition of MDM2i further reduced the percentages of viable cells, whereas cells expressing mutated non-functional p53 were resistant to MDM2i (Fig. 4B, C). Concurrent administration of CDK4/6i and MDM2i was most effective in blocking cell growth; however, sequential administration of these agents also inhibited cell growth, albeit with lower efficacy (fig. S2A, B). Furthermore, proliferation of CDK4/6i and MDM2i co-treated cells remained inhibited even after the treatment was paused for 7 days (fig. S2C, D). Notably, malignant melanoma cells were more sensitive to the combined CDK4/6i and MDM2i treatment compared to non-malignant human melanocytes, which were relatively insensitive (fig. S2E). This suggests that CDK4/6 and MDM2 co-inhibition preferentially inhibits rapidly dividing tumor cells but has relatively low toxicity to normal cells. We also found a greater decrease of EdU and BrdU incorporation in cells co-treated with CDK4/6i and MDM2i, indicating cell cycle arrest (Fig. 4D, fig. S2F). To confirm that p21 was the main effector of MDM2i-mediated improvement of CDK4/6i response, we compared CDK4/6i ribociclib and MDM2i CGM097 response in A375 melanoma cells transfected with control or p21-targeting siRNA and in isogenic HCT116 cells with and without p21 knockout. Treatment with MDM2i decreased EdU uptake in A375 cells expressing control siRNA (Vehicle 11.1±0.566, CGM097 6.375±0.064, p<0.0001 with 2-way ANOVA followed by Dunnett’s multiple comparison test). However, MDM2i-induced cell cycle arrest was abrogated in p21-deficient cells (Fig. 4E). Consequently, the anti-proliferative effect of combined CDK4/6i and MDM2i was reduced in p21-knockdown and knockout cells as compared to p21-expressing cells (Fig. 4E). Similarly, RB activation and down-regulation of proliferation markers FoxM1 and phospho-Ser10 H3 were augmented by MDM2i co-treatment only in p21-expressing and not in p21-deficient cells (Fig. 4F). The efficiencies of p21 knockdown and knockout are shown in Fig. 2C and 4F, respectively. These data suggest that p21 is a principal effector of MDM2i-mediated enhancement of CDK4/6i anti-proliferative effect.
Figure 4. MDM2 inhibition improves response to CDK4/6 inhibition via the p53-mediated induction of p21.

A. Western blot analysis of protein lysates from A375 cells treated with 2.5 μM of palbociclib (CDK4/6i) ± 2.5 μM of Nutlin-3A (MDM2i) or vehicle for 3 days. B. Representative photographs of plate wells and microscopy images of A375 and WM115 cells treated as described in A and stained with crystal violet. Scale bar: 100 μm. C. Relative numbers of indicated melanoma cells after 3 days of treatment with 1 μM CDK4/6i ribociclib, 0.5 μM MDM2i CGM097, 0.1 μM MDM2i HDM201, or indicated combinations of these drugs as determined by CellTiterBlue assay. D. Percentages of SK-Mel5 cells replicating DNA after treatment with 1 μM ribociclib, 0.5 μM CGM097, 0.1 μM HDM201, or indicated combinations of these drugs for 3 days was determined using EdU incorporation assay. C-D. For statistical analysis, we used one-way ANOVA after log transformation with Tukey’s post-test. E. EdU incorporation in A375 cells transfected with control or p21 siRNA and in WT and p21−/− HCT116 cells after 2 days of treatment with 3 μM of ribociclib (CDK4/6i), 0.5 μM of MDM2i CGM097 (for A375), 0.2 μM of MDM2i HDM201 (for HCT116), combination of both drugs, or vehicle. Mean ± SD is shown. Two-way ANOVA with Sidak’s multiple comparison test was performed on log-transformed values. F. Western blot analysis of indicated proteins in HCT116 cells treated as described in E.
MDM2i and CDK4/6i combination inhibits melanoma growth in vivo
We next evaluated the effect of combined treatment with CDK4/6i and MDM2i in vivo using eleven distinct melanoma PDXs, one human melanoma cell xenograft, and two immunocompetent murine melanoma models. All tumors expressed wt p53. We found that combined administration of CDK4/6i and MDM2 antagonists resulted in greater inhibition of tumor growth as compared to single agent treatments (Fig. 5A-F). Nine out of eleven PDX tumors (82%) showed inhibition of tumor growth after treatment with the combined therapy as compared to vehicle (Fig. 5B). Notably, CDK4/6 and MDM2 inhibitor combination inhibited growth of NRAS mutant PDX1826, which is in contrast to an FDA-approved MEKi, trametinib, that had no anti-tumor activity on this PDX (Fig. 5C). Similarly, the growth of BRAF-mutant PDX1351 was abrogated after CDK4/6i and MDM2i co-inhibition, whereas treatment with standard of care BRAFi/MEKi drug combination had limited efficacy (Fig. 5D). Combined CDK4/6i and MDM2i treatment was also effective in inhibiting growth of A375 melanoma xenografts (Fig. 5E) and NRAS-mutant mouse melanoma tumors (YUMM10.1) grown in immunocompetent mice (Fig. 5F). In a BRAF-mutant immunocompetent melanoma model (YUMM1.7), CDK4/6i and MDM2i combination alone had modest efficacy, however it was strongly effective when both inhibitors were co-administered with standard of care BRAF/MEK inhibitors (Fig. 5G), causing regression of 100% of tumors (Fig. 5H, I). Tumor measurements for all experiments in Figure 5 are provided in data file S5. The treatment did not induce severe toxicity in mice, as there was no significant loss of body weight in either C57B1/6 mice bearing YUMM10.1 tumors or nude mice bearing PDX1595 tumors (Fig. 5J, data file S6), or increase of serum concentrations of liver enzymes ALT and AST as compared to vehicle group in nude mice bearing PDX1826 (Fig. 5K).
Figure 5. Combined treatment with CDK4/6i and MDM2i is effective in PDXs and murine melanoma models.

A. The effect of CDK4/6i and MDMi on growth of melanoma PDXs. PDX tumors 1642, 1460, and 1688 were implanted into nude mice. Animals were treated daily with 100 mg/kg CDK4/6i ribociclib, 50 mg/kg MDM2i CGM097, the combination of both drugs, or vehicle (MC+HPMC). Tumor volume change over time is shown for each treatment group. B. Eleven melanoma PDXs with wt p53 were grown in nude mice treated as described in A. Final tumor volumes in treatment groups relative to vehicle group are shown. Statistical analysis was performed using linear mixed effects regression model. C. Tumor volume change over time in nude mice implanted with PDX1826 and treated as described in A or with MEKi (trametinib, 1 mg/kg) once a day. D. Tumor volume change over time in nude mice implanted with PDX1351 and treated as described in A or with MEKi+BRAFi (trametinib, 1 mg/kg + dabrafenib, 30 mg/kg) once a day. E. Growth of A375 tumors implanted into nude mice and treated as described in A. F. YUMM10.1 tumors were grown in C57Bl/6 mice. Animals were treated daily with 100 mg/kg CDK4/6i ribociclib, 20 mg/kg MDM2i HDM201, the combination of both drugs, or vehicle (MC+HPMC). G. YUMM1.7 tumors were grown in C57Bl/6 mice. Animals were treated daily with combination of 100 mg/kg CDK4/6i ribociclib and 20 mg/kg MDM2i HDM201, combination of MEKi+BRAFi (trametinib, 1 mg/kg + dabrafenib, 30 mg/kg), the four drugs combined, or vehicle (MC+HPMC) once a day. Tumor volume changes over time are shown. H. Waterfall plot of YUMM1.7 tumor volume changes over baseline after indicated treatment from experiment shown in G. I. Frequencies of distinct types of responses in experiment with YUMM1.7 tumors shown in G. J. Body weight changes during the course of treatment in C57Bl mice bearing YUMM10.1 tumors and in nude mice bearing PDX1595 tumors. K. Serum concentrations of AST and ALT in mice bearing PDX1826 tumors and treated as shown in C. Statistics calculated on log-transformed values with one-way ANOVA and Dunnett’s post-test.
The combined CDK4/6i and MDM2i therapy had a cytostatic effect in vivo, as shown by the down-regulation of the proliferation marker Ki67 in 3 out 7 tested PDXs that were responsive to combined CDK4/6i and MDM2i treatment (Fig. 6A, B; fig. S3). Of note, both PDXs that were resistant to combined CDK4/6i and MDM2i treatment (based on data in Figure 5B) exhibited Ki67 index above 25% (Fig. 6B). This indicates that highly proliferative melanoma tumors may require more aggressive therapeutic approach, such as, for example, addition of inhibitors of BRAF/MEK/ERK signaling, to effectively counteract tumor progression.
Figure 6. CDK4/6i and MDM2i combination reduces proliferation and induces expression of immune-related proteins and p53 in PDX tumors.

A. IHC staining of Ki67 in tumors shown in 5B. Scale bar: 0.1 mm. B. Quantification of the percentages of Ki67-positive cells in tumors shown in 5B. Statistical analysis performed using two-way ANOVA after log transformation with Tukey’s test for multiple comparisons. C. Heat map of proteins up- and down-regulated by ribociclib (CDK4/6i), CGM097 (MDM2i), and combination of both drugs (Combo) in PDX tumors 1642, 1688, 1179, 1826, and 1460 treated as shown in Figure 5B. Analysis by quantitative ELISA-based proteomics performed by Ray Biotech. D. Heat map of p53 protein expression in 5 indicated PDX tumors treated with ribociclib (CDK4/6i), CGM097 (MDM2i), and combination of both drugs (Combo) as shown in Fig. 5B.
To gain further insight into molecular mechanisms of anti-tumor activity of CDK4/6i and MDM2i in vivo, we performed a 660-biomarker proteomics assay in tumor lysates prepared from five distinct PDXs (1642, 1688, 1179, 1826, and 1460), each of which was treated with vehicle, CDK4/6i, MDM2i, or a combination of both. Tumor volumes for these tumors are shown in Fig. 5B. A number of immune mediators, cytokines, growth factors, and adhesion molecules were up-regulated in response to combined CDK4/6i and MDM2i treatment (Fig. 6C, data file S7). Similar expression changes have been reported in cells undergoing senescence (43). This is in agreement with our earlier data demonstrating CDK4/6i-mediated induction of senescence and pro-inflammatory gene expression signatures in melanoma cells (44). Gene set analysis of proteins modulated by CDK4/6i and MDM2i co-treatment identified an enrichment of GO annotations “cell adhesion”, “inflammatory response”, “negative regulation of cell proliferation”, and KEGG pathways “proteoglycans in cancer”, “PI3K-AKT signaling pathway”, “focal adhesion”, and “cytokine-cytokine receptor interaction”. Notably, “p53 signaling pathway” was among the top 10 enriched KEGG pathways in our set of proteins differentially regulated by CDK4/6 and MDM2 co-targeting (table S1). Furthermore, based on proteomics analysis, p53 was significantly (p=0.02, one-way ANOVA with Dunnett's multiple comparisons test) up-regulated by combined CDK4/6i and MDM2i in a set of five tested PDXs with wt p53 (Fig. 6D). These data suggest that induction of the p53 pathway is associated with response to combined CDK4/6i and MDM2i treatment.
We next evaluated the expression of p53 and its target p21 in individual PDX tumors treated with single agents or combined CDK4/6i and MDM2i. We also assessed the degree of inactivating Ser807/811 phosphorylation of RB and expression of proliferation marker phospho-Ser10 H3. p21 was consistently induced by CDK4/6i and MDM2i combination treatment (fig. S4A, B). The difference between p21 expression with combination therapy and with MDM2i alone was not statistically significant. This may reflect an adaptive resistance developing in tumors treated with MDM2i alone or the sample quality, because tumors treated with single agent MDM2 tend to be more necrotic (42). In addition, p21 was induced in PDX1129 that had wtp53 but was unresponsive to CDK4/6i and MDMi treatment (fig. S4C), indicating that unidentified alternative factors may be blocking a growth inhibitory response in this tumor. There was no significant p21 induction in PDX1214, which had a p53 mutation (fig. S4D). This suggests that p21 induction reflects the overall activity of p53, which is induced by MDM2i. The expression of mitotic marker phosphor-H3 was reduced in two tested PDXs indicating cell cycle arrest (fig. S4A, B). Other tested markers, such as p53 and phosphor-RB exhibited variability in expression between individual tumors (fig. S4A, B).
Tumor response to CDK4/6i and MDM2i treatment can be tested ex vivo
We next investigated whether CDK4/6i and MDM2i response biomarkers identified above could be evaluated ex vivo to guide treatment decision for individual patients. We used a simple organotypic culture model, where therapy-naive PDX tumors were freehand cut into slices with thickness of 0.25-1 mm (for subsequent FFPE and IHC) or small fragments 0.5-2 mm in diameter (for subsequent protein expression analysis). The size of these fragments is comparable to the size of a standard clinical tumor biopsy specimen. Slices and fragments were then cultured for 3 days in the presence of CDK4/6i and/or MDM2i using standard cell culture conditions (Fig. 7A). Notably, tumors that were responsive to CDK4/6i and MDM2i co-targeting in vivo showed down-regulation of the Ki67 index when treated with these drugs ex vivo, whereas tumors that were resistant in vivo had high basal Ki67 index which was not significantly affected by ex vivo drug treatment (Fig. 7B, C). Furthermore, we detected strong induction of p53 and p21 by MDM2i and combined MDM2i and CDK4/6i treatment in ex vivo cultured PDX explants (PDXEs) (Fig. 7D, E), indicating that ex vivo tumor cultures recapitulate treatment-associated molecular changes observed in vitro and in vivo. We also observed consistent downregulation of phosphorylated H3 and RB in PDXEs from PDXs that were sensitive to CDK4/6 and MDM2 co-targeting in vivo. These results show that a simple short-term ex vivo treatment assay of a viable tumor biopsy specimen may aid in identifying patients who are likely to gain benefit from the CDK4/6i and MDM2i drug combination.
Figure 7. Ex vivo slice culture model recapitulates tumor response to CDK4/6i and MDM2i.

A. Diagram depicting an experimental setup of organotypic PDX tumor explant (PDXE) culture and representative image of PDXE. B. Representative photomicrographs of Ki67 IHC staining in PDXEs treated ex vivo with 2.5 μM of ribociclib, 2.5 μM of CGM097, the combination of both drugs, or vehicle for 3 days. Scale bar: 0.1 mm. C. Summary of the Ki67 staining in indicated PDXEs treated as described in A. Statistical analysis with two-way ANOVA after log-transformation with Tukey’s multiple comparison test. D. Western Blot analysis of PDXEs treated as described in A. E. Proposed model of CDK4/6i resistance in melanoma. Pathways associated with therapeutic resistance are indicated by red connecting arrows and lines, whereas pathways facilitating response are marked by blue connections. CDKs are orange, and cyclins are yellow. All other proteins are shown in blue.
Discussion
Aberrant activation of CDK4 and CDK6 is common in melanoma as a result of frequent genetic alterations in CDKN2A and CDK4. This makes CDK4/6 an attractive therapeutic target. However clinical studies of CDK4/6i demonstrated only modest efficacy in melanoma (13, 14). To date the mechanisms of intrinsic resistance to CDK4/6i remained largely unknown (16). Our study uncovered molecular mechanisms of CDK4/6i resistance in melanoma associated with disabling of CDK inhibitors p21 and p27 by cyclin D1 and PI3K/AKT pathway, thus allowing cell cycle progression driven by CDK2. The finding that CDK2 can compensate for the inhibition of CDK4/6i in melanoma cells aligns well with results obtained using breast cancer cells (45, 46). Collectively these findings illustrate functional redundancy between cell cycle CDKs, such that when one of them is inactivated, the remaining CDKs can phosphorylate the targets of inactivated CDK. This was demonstrated by cell cycle studies using yeast, as well as CDK4/CDK6, CDK4/CDK2, and CDK2/CDK4/CDK6 knockout mice (36, 37, 47, 48). Our study revealed that melanomas can exploit these basic adaptation mechanisms to overcome anti-proliferative effects of CDK4/6 inhibition.
The implication of cyclin D1 in CDK4/6i resistance is of particular interest. Originally, cyclin D1 was considered to be a CDK4/6i sensitizer based on in vitro studies in breast cancer cell lines (49). However, in the Phase II Paloma 1 study, patients with cyclin D1 amplification had essentially the same outcome as an unselected patient population (50). Furthermore, a recent study of 560 cancer cell lines showed that cyclin D1 amplifications were associated with resistance to CDK4/6i (51). Notably, all of the previously reported CDK4/6i sensitizers, including endocrine therapy in ER+ breast cancer (52), EGFR inhibitors in HER2+ breast cancer (53), and RAF-MEK-ERK pathway inhibitors in melanoma and other cancers (10, 30, 54, 55) limit expression of D-type cyclins (16). Our data suggest that cyclin D1 facilitate CDK4/6i resistance by sequestering CDK inhibitors p21 and p27. Nevertheless, we cannot fully exclude the possibility that cyclin D1 promotes CDK4/6i resistance by acting through residual CDK4/6 activity remaining after CDK4/6i treatment. Furthermore, cyclin D1 can also contribute to CDK4/6i resistance by directly activating cell cycle CDKs other than CDK4 and CDK6, because it has been previously shown that in mice lacking CDK4 and CDK6, cyclin D can bind and activate other cell cycle CDKs (36, 37, 56, 57).
We found that therapeutic resistance to CDK4/6i can be counteracted using MDM2 antagonists, which induce p21 by stabilizing its transcriptional activator p53 (58). Of note, genetic alterations targeting p53 (and, presumably, p21) are associated with poor response to CDK4/6i in cancer cell lines (51) and in breast cancer patients (14). Similarly, we found that three out of six CDK4/6-resistant PDXs had p53-inactivating gene alterations. We also found that p21 induction was responsible for enhanced CDK4/6i response by PI3Ki co-treatment. These data explain the synergistic anti-tumor activity of CDK4/6i and PI3Ki that was reported by a number of pre-clinical studies (32, 59-62), as well as poor response to CDK4/6i in tumors with activating PIK3CAE545K mutation (63). This further underscores the clinical potential of MDM2 antagonists, which induce p21 to counteract CDK4/6i resistance. Of note, a recent study described a different approach to overcome CDK4/6i resistance by stabilizing p27 using peptide that blocks its tyrosine phosphorylation (25). This suggests that activation of either p21 or p27 can be a viable approach to improve CDK4/6i responses.
Addition of MDM2i to CDK4/6i treatment is likely to benefit patients with p53 and RB wild-type melanoma. Notably, p53 wild-type melanomas exhibit oncogenic addiction to MDM2-mediated inactivation of p53 (64) and respond well to MDM2i (65). Furthermore, MDM2 inhibitors are effective in combination with current and emerging melanoma therapeutics including MEK, BRAFV600E, iASPP, and Aurora kinase inhibitors (65-68). Of note, the majority of melanoma tumors retain the wild-type TP53 gene (83% in TCGAA dataset) and thus may benefit from MDM2i and CDK4/6i combination. In addition, the combination of MDM2 and CDK4/6 inhibitors is effective in a model of liposarcoma, where co-amplifications of MDM2 and CDK4 genes are common (69). For more precise selection of potentially responsive tumors, an ex vivo assessment of pharmacodynamic biomarkers can be performed. This can be done by simply culturing the leftover tumor resection/biopsy tissue material in the presence of CDK4/6i and MDM2i, followed by detection of drug-induced expression changes such as decrease of Ki67 index, loss of RB and H3 phosphorylation, and induction of p53 and p21, as demonstrated by this study.
There are limitations of our study. For instance, the analysis of protein markers in melanoma tumors should be interpreted with caution because at the time of tissue collection some of the tumors were large (especially in the control group) with regions of necrosis. This and potential other factors not directly related to therapy may influence the expression pattern of proteins within the tumors, which would account for some variability between biological replicates. Another limitation is that complete knockdown or knockout of CDK1 is not feasible because this kinase is essential for mammalian cell division, and therefore it is difficult to test its contribution to CDK4/6i resistance.
In summary, our study identified molecular mechanisms of CDK4/6i intrinsic resistance in melanoma, which was associated with inactivation of p21. On the basis of these findings, we present a promising strategy to counteract CDK4/6i resistance by increasing p21 expression using MDM2 antagonists. We also identified markers of response to these agents that allow ex vivo prescreening of potential responders. These findings may be of great value for further clinical development of CDK4/6i in melanoma and other malignancies.
Materials and methods
Study design.
Experiments were designed to have at least 6 mice per treatment group bearing two tumors each. Due to technical challenges with the PDX model (failed tumor engraftment, accidental death), the actual sample size in some treatment groups was 5 mice for PDXs 1214, 1530, 1595,1642, 1662, 1688, 4 for PDX 1460, and 3 for PDX 1904 (data file S5). Calculations of sample size for in vivo experiments were based on simple regression model requiring larger sample sizes than what would be expected compared to the mixed model approach used in this study. For the comparisons of tumor size between treatments, we chose all experiments to have at least 80% power to detect standardized effect sizes of 2 or greater; such effect sizes in our preliminary data ranged from 2.5 to 4.9. Especially considering the gain in information from longitudinal measurements, samples sizes for these experiments provided sufficient power, to detect biologically meaningful differences among treatments. Animals were allocated to different treatment groups randomly. Analysis of the immunohistochemical staining of tumors was performed by a dermatopathologist who was blinded to the study design and expected results.
Cell lines and reagents.
A375, SK-Mel5, MCF7 cells were purchased from ATCC. YUMM10.1 and YUMM1.7 cells were provided by Marcus Bosenberg (Yale University). Cells were cultured in DMEM/F12 medium supplemented with 10% FBS and 1% penicillin/streptomycin (all Gibco). HDM201, CGM097, ribociclib, and buparlisib were provided by Novartis. GDC-0994 and ipatasertib were purchased from Medkoo. Palbociclib and roscovitine were purchased from LC laboratories. Nutlin-3a was synthesized as described previously (70). Working concentrations of CDK4/6 inhibitors ribociclib and palbociclib used for in vitro experiments were chosen based on previously reported human pharmacokinetics data and kinase-specificity assays (71-74). Antibodies used in this study are indicated in table S2.
Mouse studies.
Animal studies were approved by the Vanderbilt IACUC. Balb/C nude mice, C57BL/6, and NSG mice were purchased from Jackson Labs. PDX tumors were established and propagated as described previously (42). All drugs were administered five days a week by oral gavage in a total volume of 100-200 μl. Ribociclib and HDM201 were prepared in 0.5% methylcellulose, and CGM097 was prepared in 0.5% hydroxypropyl methyl cellulose. Mouse body weight was assessed once a week, and tumor measurements were taken twice a week with micro-calipers. Tumor volume was estimated as 0.5*length*width* width. Treatment began when tumors reached 50-150 mm3 volume on average and continued until tumors exceeded 15 mm in diameter or became perforated.
Targeted DNA sequencing.
DNA from tumor and peripheral blood derived from PDX donor was submitted to VANTAGE core for QC and DNAseq using Human Comprehensive Cancer Panel (Qiagen). Identification of tumor-associated somatic mutations and copy number alterations was performed using QIAGEN NGS Data Analysis Web Portal. Color-coded map of genetic alterations in PDXs was constructed using OncoPrinter available through cBioPortal (75).
Proteomics.
Tumor tissue was submerged in RIPA buffer and processed on Precellys Homogenizer. Lysates were submitted to MD Anderson Cancer Center RPPA core for RPPA analysis or to RayBiotech for 660 biomarker Quantibody Multiplex ELISA. Clustering and construction of heat maps was performed using MORPHEUS web tool (Broad Institute). Gene ontology and KEGG pathway enrichment analyses were performed using DAVID Bioinformatics Resources 6.8 (76). RPPA protein expression was compared between CDK4/6i-responsive and resistant PDX tumors using multiple T-test computed in Prism 7.03 (Graphpad). Each row was analyzed individually, without assuming a consistent SD. Statistical significance was determined without correction for multiple comparisons, with α=0.05%.
Organotypic culture.
Immediately after removal from mice, PDX tumors were submerged in PBS and sliced into 0.5 mm slices using surgical scalpel. Slices were placed into cell culture-treated 6 well plates containing 2 ml of medium supplemented with 10% FBS, 1% antibiotics/antimycotics (all Gibco), and experimental drugs. Tissue explants were incubated for 3 days using regular cell culture conditions followed by either fixation in formalin for subsequent paraffin embedding and Ki67 IHC or homogenization in RIPA lysis buffer for subsequent western blot analysis.
DNA replication analysis.
EdU and BrdU incorporation was measured using Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (ThermoFisher) and BrdU Flow Kit (BD Pharmigen), respectively, according to manufacturers’ recommendations.
Viability Assay.
CellTiter-Blue Cell Viability Assay (Promega) was performed according to the manufacturer’s recommendations. To convert fluorescent assay readout units into cell percentages, a standard curve was plotted for each of the tested cell lines based on the measurement of titrated cells plated with known cell density ranging from 10-100%. The fluorescence measurements obtained from unknown drug-treated specimens of the corresponding cell line were interpolated from standard curve using Prism 7 (Graphpad). The percentages of viable cells were presented relative to untreated control, which was considered to have 100% viable cells. Crystal violet staining was performed with commercial 1% solution (Sigma-Aldrich) on cells that were fixed with 100% methanol for 10 min.
Western blot and co-immunoprecipitation (co-IP).
RIPA buffer (Sigma-Aldrich) and nondenaturing buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 1% Nonidet P-40 (NP-40), 2 mM EDTA) were used to prepare lysates for western blot and co-IP, respectively. Protein concentration was measured using Bradford Protein Assay reagent (BioRad). Co-immunoprecipitation was performed using Dynabeads immunoprecipitation kit (ThermoFisher) according to manufacturer’s recommendations. For western blots, primary antibodies were used at 1:500 dilutions, except antibodies recognizing housekeeping proteins (HSP90, β-actin), which were used at 1:2000. Blots were incubated with primary antibodies overnight. See table S2 for primary antibodies used in this study. Secondary antibodies (Jackson ImmunoResearch) were used at 1:10000 dilution for 2-3 hours at room temperature. Densitometry analysis of western blot results was performed using ImageStudio or ImageJ software.
Pathology/histology assessment.
AST and ALT analysis, histology, and IHC were performed by Vanderbilt’s Translational Pathology Shared Resource (TPSR). Immunohistochemistry (IHC) slides were scanned at the Digital Pathology Shared resource. The automated quantification of the percentages of the Ki67-positive cells was performed by Leica Biosystems’ Digital Image Hub.
Statistical analysis.
Standard T-test and analysis of variances (ANOVA) were used for analysis comparing two samples and multiple samples, respectively. GraphPad’s Prism 7.03 software and R language (version 3.3.0.) were used for the statistical analysis. For in vivo experiments, we compared the progression of tumor area (mm2) over time among groups of mice receiving different therapy with a linear mixed effects regression model. To meet the normality assumptions for these parametric methods, a square root or a natural log transformation was implemented to ameliorate the heterogeneity evident in the data. Mixed models estimate corrected variance estimates in the presence of correlated measurements taken in the same mouse (such as left and right flank) and for measures on the same tumor over time. The Akaike information criterion was used to select among competing correlation structures. Standard residual analysis and goodness-of-fit statistics were evaluated. All tests of statistical significance were two-sided.
Supplementary Material
Figure S1. Cyclins play role in CDK4/6i resistance.
Figure S2. Combined CDK4/6i and MDM2i treatment inhibits growth and DNA replication in melanoma cells.
Figure S3. Combined administration of CDK4/6i and MDM2i inhibits proliferation of tumor cells in vivo.
Figure S4. Several protein markers are induced in melanoma PDXs after CDK4/6i and MDM2i treatment.
Table S1. Pathway enrichment analysis of genes significantly altered by combined CDK4/6i and MDM2i treatment.
Table S2. Antibodies used in this study.
Data file S3. Raw data for the copy number alterations genetic analysis shown in Figure 1B.
Data file S1. Tumor measurements for experiments shown in Figure 1A.
Data file S4. Raw data for the RPPA analysis shown in Figure 1C.
Data file S6. Mouse weight measurements shown in Figure 5J.
Data file S2. Raw data for the single nucleotide variation (SNVs) genetic analysis shown in Figure 1B.
Data file S5. Tumor measurements for all mouse experiments shown in Figure 5.
Data file S7. Raw data for the analysis of 660 protein biomarker shown in Figure 6C.
Acknowledgements:
We thank Marcus Bosenberg for providing YUMM10.1 and YUMM1.7 cells, and Mary Hooks for providing melanoma surgical material.
Funding: This work was supported by grants from Harry J. Lloyd Charitable Trust award (019720-001, AEV), BCRF (IIDRP-16-001, AEV), NCI SPORE in Breast Cancer (P50CA098131) pilot award (AEV), NIH R37 CA233770-01 (AEV), the Department of Veterans Affairs (5101BX000196-04, AR), NIH (CA116021 (AR), CA116021-S1 (AR)), Senior Research Career Scientist Award to AR, and Vanderbilt Clinical Oncology Career Development program (AEV) funded through NIH K12 training grant (CA90625). Support for Core Facilities utilized in this study was provided by Vanderbilt Ingram Cancer Center (P30 CA68485).
Footnotes
Competing interests: Ensar Halilovic is an employee of Novartis and an inventor on patent application (US20170143723A1) held by Novartis AG that covers a pharmaceutical combination of an MDM2/4 inhibitor and a CDK4/6 inhibitor. Douglas Johnson serves on scientific advisory boards for Array Biopharma, BMS, Incyte, Merck, and Novartis and received research funding from BMS and Incyte. Ann Richmond is on the Advisory Board for Polyphor. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or Supplementary Materials. Ribociclib, CGM097, and HDM201 were obtained from Novartis under an MTA.
References and notes
- 1.Gide TN, Wilmott JS, Scolyer RA, Long GV, Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res 24, 1260–1270 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D, Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372, 30–39 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, Puzanov I, Lewis KD, Amaravadi RK, Chmielowski B, Lawrence HJ, Shyr Y, Ye F, Li J, Nolop KB, Lee RJ, Joe AK, Ribas A, Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366, 707–714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DB, Puzanov I, Treatment of NRAS-mutant melanoma. Curr. Treat. Options Oncol 16, 15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, Do HD, Mitchell C, Cullinane C, Liu W, Fox SB, Dutton-Regester K, Hayward NK, Jene N, Dobrovic A, Pearson RB, Christensen JG, Randolph S, McArthur GA, Sheppard KE, Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigm Cell Melanoma R 27, (2014). [DOI] [PubMed] [Google Scholar]
- 6.Zhao R, Choi BY, Lee MH, Bode AM, Dong Z, Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 8, 30–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheppard KE, McArthur GA, The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res 19, 5320–5328 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, Morgan DO, Tsai LH, Wolgemuth DJ, Cyclin-dependent kinases: a family portrait. Nat. Cell Biol 11, 1275–1276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, Sicinski P, A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20, 620–634 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, Jakubosky D, Genovese G, Muller FL, Jeong JH, Bender RP, Chu GC, Flaherty KT, Wargo JA, Collins JJ, Chin L, Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nature medicine 18, 1503–1510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller DM, Flaherty KT, Cyclin-dependent kinases as therapeutic targets in melanoma. Pigment Cell Melanoma Res 27, 351–365 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Sherr CJ, Beach D, Shapiro GI, Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 6, 353–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O'Dwyer PJ, Schwartz GK, Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res 18, 568–576 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, Papadopoulos KP, Beeram M, Rasco DW, Hilton JF, Nasir A, Beckmann RP, Schade AE, Fulford AD, Nguyen TS, Martinez R, Kulanthaivel P, Li LQ, Frenzel M, Cronier DM, Chan EM, Flaherty KT, Wen PY, Shapiro GI, Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 6, 740–753 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Sosman JA, Kittaneh M, Lolkema MPJK, Postow MA, Schwartz G, Franklin C, Matano A, Bhansali S, Parasuraman S, Kim K, A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: Early encouraging clinical activity. J. Clin. Oncol 32, (2014). [Google Scholar]
- 16.Knudsen ES, Witkiewicz AK, The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 3, 39–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison DK, MAP kinase pathways. Cold Spring Harb Perspect Biol 4, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh JC, Canton B, Nguyen XT, Barnier JV, Camonis J, Ginsberg MH, Chneiweiss H, PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell 1, 239–250 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Hemmings BA, Restuccia DF, The PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 7, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho H, Kapadia R, Al-Tahan S, Ahmad S, Ganesan AK, WIPI1 coordinates melanogenic gene transcription and melanosome formation via TORC1 inhibition. J Biol Chem 286, 12509–12523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laplante M, Sabatini DM, mTOR Signaling. Cold Spring Harb Perspect Biol 4, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szymonowicz K, Oeck S, Malewicz NM, Jendrossek V, New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers (Basel) 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalin TV, Ustiyan V, Kalinichenko VV, Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle 10, 396–405 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams PD, Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim Biophys Acta 1471, M123–133 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Patel P, Tsiperson V, Gottesman SRS, Somma J, Blain SW, Dual Inhibition of CDK4 and CDK2 via Targeting p27 Tyrosine Phosphorylation Induces a Potent and Durable Response in Breast Cancer Cells. Mol Cancer Res 16, 361–377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto T, Sicinski P, Cell cycle proteins as promising targets in cancer therapy. Nature Reviews Cancer 17, 93–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng M, Sexl V, Sherr CJ, Roussel MF, Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc Natl Acad Sci U S A 95, 1091–1096 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H, Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J 18, 5310–5320 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geng Y, Yu Q, Sicinska E, Das M, Bronson RT, Sicinski P, Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proc Natl Acad Sci U S A 98, 194–199 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen SH, Gong X, Zhang Y, Van Horn RD, Yin T, Huber L, Burke TF, Manro J, Iversen PW, Wu W, Bhagwat SV, Beckmann RP, Tiu RV, Buchanan SG, Peng SB, RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 37, 821–832 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Wong CH, Ma BBY, Hui CWC, Lo KW, Hui EP, Chan ATC, Preclinical evaluation of ribociclib and its synergistic effect in combination with alpelisib in non-keratinizing nasopharyngeal carcinoma. Sci. Rep 8, 8010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, Bellet M, Cortes J, Elliott R, Pancholi S, Baselga J, Dowsett M, Martin LA, Turner NC, Serra V, Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 76, 2301–2313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satyanarayana A, Hilton MB, Kaldis P, p21 inhibits Cdk1 in the absence of Cdk2 to maintain the G1/S phase DNA damage checkpoint. Mol. Biol. Cell 19, 65–77 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charrier-Savournin FB, Chateau MT, Gire V, Sedivy J, Piette J, Dulic V, p21-mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Mol. Biol. Cell 15, 3965–3976 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, Kaldis P, Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci U S A 109, 3826–3831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M, Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M, Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Barriere C, Santamaria D, Cerqueira A, Galan J, Martin A, Ortega S, Malumbres M, Dubus P, Barbacid M, Mice thrive without Cdk4 and Cdk2. Mol Oncol 1, 72–83 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M, Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35, 25–31 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P, Cdk2 knockout mice are viable. Curr Biol 13, 1775–1785 (2003). [DOI] [PubMed] [Google Scholar]
- 41.McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, Mackenzie M, Melville J, Stewart K, Wang S, Zhelev N, Zheleva D, Lane DP, In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int J Cancer 102, 463–468 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, Turner DC, Essaka DC, Stewart CF, Sosman JA, Kelley MC, Ecsedy JA, Johnston JN, Richmond A, Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res 75, 181–193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodier F, Campisi J, Four faces of cellular senescence. J. Cell Biol. 192, 547–556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vilgelm AE, Johnson CA, Prasad N, Yang J, Chen SC, Ayers GD, Pawlikowski JS, Raman D, Sosman JA, Kelley M, Ecsedy JA, Shyr Y, Levy SE, Richmond A, Connecting the Dots: Therapy-Induced Senescence and a Tumor-Suppressive Immune Microenvironment. Journal of the National Cancer Institute 108, djv406 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES, Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 29, 4018–4032 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, Bellet M, Cortes J, Elliott R, Pancholi S, Baselga J, Dowsett M, Martin LA, Turner NC, Serra V, Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res 76, 2301–2313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malumbres M, Cyclin-dependent kinases. Genome Biol. 15, 122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berthet C, Klarmann KD, Hilton MB, Suh HC, Keller JR, Kiyokawa H, Kaldis P, Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev Cell 10, 563–573 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ, PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast cancer research : BCR 11, R77 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, Fowst C, Huang X, Kim ST, Randolph S, Slamon DJ, The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 16, 25–35 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, Dowless M, Dempsey J, Zeng Y, Torres R, Boehnke K, Mur C, Marugan C, Baquero C, Yu C, Bray SM, Wulur IH, Bi C, Chu S, Qian HR, Iversen PW, Merzoug FF, Ye XS, Reinhard C, De Dios A, Du J, Caldwell CW, Lallena MJ, Beckmann RP, Buchanan SG, Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 32, 761–776 e766 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Deng Y, Ma G, Li W, Wang T, Zhao Y, Wu Q, CDK4/6 Inhibitors in Combination With Hormone Therapy for HR(+)/HER2(−) Advanced Breast Cancer: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Clin Breast Cancer 18, e943–e953 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, Ramm S, Palmer AC, Yuzugullu H, Varadan V, Tuck D, Harris LN, Wong KK, Liu XS, Sicinski P, Winer EP, Krop IE, Zhao JJ, Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer cell 29, 255–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ziemke EK, Dosch JS, Maust JD, Shettigar A, Sen A, Welling TH, Hardiman KM, Sebolt-Leopold JS, Sensitivity of KRAS-Mutant Colorectal Cancers to Combination Therapy That Cotargets MEK and CDK4/6. Clin. Cancer Res. 22, 405–414 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG, Chan EM, Starling JJ, Beckmann RP, Peng SB, The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol. Cancer Ther. 13, 2253–2263 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Padmakumar VC, Aleem E, Berthet C, Hilton MB, Kaldis P, Cdk2 and Cdk4 activities are dispensable for tumorigenesis caused by the loss of p53. Mol Cell Biol 29, 2582–2593 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, Geng Y, Zagozdzon A, Jecrois M, Young RA, Liu XS, Cepko CL, Gygi SP, Sicinski P, Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature 463, 374–378 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vilgelm AE, Cobb P, Malikayil K, Flaherty D, Andrew Johnson C, Raman D, Saleh N, Higgins B, Vara BA, Johnston JN, Johnson DB, Kelley MC, Chen SC, Ayers GD, Richmond A, MDM2 Antagonists Counteract Drug-Induced DNA Damage. EBioMedicine 24, 43–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, Lehar J, Wiesmann M, Wartmann M, Chen Y, Cao ZA, Pinzon-Ortiz M, Kim S, Schlegel R, Huang A, Engelman JA, CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26, 136–149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teo ZL, Versaci S, Dushyanthen S, Caramia F, Savas P, Mintoff CP, Zethoven M, Virassamy B, Luen SJ, McArthur GA, Phillips WA, Darcy PK, Loi S, Combined CDK4/6 and PI3Kalpha Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 77, 6340–6352 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Asghar US, Barr AR, Cutts R, Beaney M, Babina I, Sampath D, Giltnane J, Lacap JA, Crocker L, Young A, Pearson A, Herrera-Abreu MT, Bakal C, Turner NC, Single-Cell Dynamics Determines Response to CDK4/6 Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 23, 5561–5572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teh JLF, Cheng PF, Purwin TJ, Nikbakht N, Patel P, Chervoneva I, Ertel A, Fortina PM, Kleiber I, HooKim K, Davies MA, Kwong LN, Levesque MP, Dummer R, Aplin AE, In Vivo E2F Reporting Reveals Efficacious Schedules of MEK1/2-CDK4/6 Targeting and mTOR-S6 Resistance Mechanisms. Cancer Discov. 8, 568–581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romano G, Chen PL, Song P, McQuade JL, Liang RJ, Liu M, Roh W, Duose DY, Carapeto FCL, Li J, Teh JLF, Aplin AE, Chen M, Zhang J, Lazar AJ, Davies MA, Futreal PA, Amaria RN, Zhang DY, Wargo JA, Kwong LN, A Preexisting Rare PIK3CA(E545K) Subpopulation Confers Clinical Resistance to MEK plus CDK4/6 Inhibition in NRAS Melanoma and Is Dependent on S6K1 Signaling. Cancer Discov 8, 556–567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verhaegen M, Checinska A, Riblett MB, Wang S, Soengas MS, E2F1-dependent oncogenic addiction of melanoma cells to MDM2. Oncogene 31, 828–841 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ji Z, Njauw CN, Taylor M, Neel V, Flaherty KT, Tsao H, p53 rescue through HDM2 antagonism suppresses melanoma growth and potentiates MEK inhibition. J. Invest. Dermatol. 132, 356–364 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, Knapp S, Kessler BM, Middleton MR, Siebold C, Jones EY, Sviderskaya EV, Cebon J, John T, Caballero OL, Goding CR, Lu X, Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer cell 23, 618–633 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Vilgelm AE, Mdm2 and Aurora A inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res., (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vilgelm A, Richmond A, Combined therapies that induce senescence and stabilize p53 block melanoma growth and prompt antitumor immune responses. Oncoimmunology 4, e1009299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laroche-Clary A, Chaire V, Algeo MP, Derieppe MA, Loarer FL, Italiano A, Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davis TA, Vilgelm AE, Richmond A, Johnston JN, Preparation of (−)-Nutlin-3 using enantioselective organocatalysis at decagram scale. J Org Chem 78, 10605–10616 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poratti M, Marzaro G, Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib. Eur J Med Chem 172, 143–153 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, Bergqvist S, Solowiej J, Diehl W, He YA, Yu X, Nagata A, VanArsdale T, Murray BW, Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol Cancer Ther 15, 2273–2281 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, Chakraborty A, Matano A, Dobson JR, Crystal AS, Parasuraman S, Shapiro GI, A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin Cancer Res 22, 5696–5705 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL, Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3, 1427–1438 (2004). [PubMed] [Google Scholar]
- 75.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6, pl 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cyclins play role in CDK4/6i resistance.
Figure S2. Combined CDK4/6i and MDM2i treatment inhibits growth and DNA replication in melanoma cells.
Figure S3. Combined administration of CDK4/6i and MDM2i inhibits proliferation of tumor cells in vivo.
Figure S4. Several protein markers are induced in melanoma PDXs after CDK4/6i and MDM2i treatment.
Table S1. Pathway enrichment analysis of genes significantly altered by combined CDK4/6i and MDM2i treatment.
Table S2. Antibodies used in this study.
Data file S3. Raw data for the copy number alterations genetic analysis shown in Figure 1B.
Data file S1. Tumor measurements for experiments shown in Figure 1A.
Data file S4. Raw data for the RPPA analysis shown in Figure 1C.
Data file S6. Mouse weight measurements shown in Figure 5J.
Data file S2. Raw data for the single nucleotide variation (SNVs) genetic analysis shown in Figure 1B.
Data file S5. Tumor measurements for all mouse experiments shown in Figure 5.
Data file S7. Raw data for the analysis of 660 protein biomarker shown in Figure 6C.