

Abstract
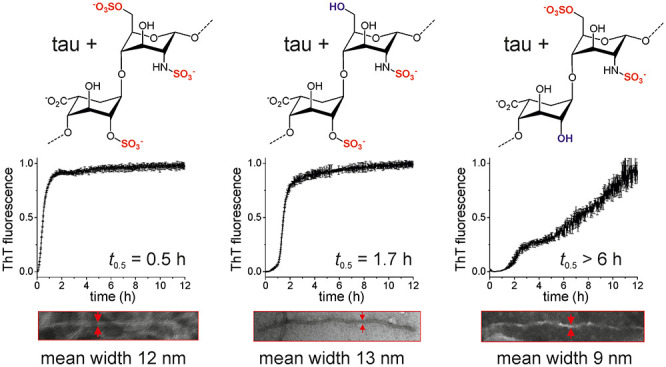
A pathological signature of Alzheimer’s disease (AD) is the formation of neurofibrillary tangles comprising filamentous aggregates of the microtubule associated protein tau. Tau self-assembly is accelerated by polyanions including heparin, an analogue of heparan sulfate. Tau filaments colocalize with heparan sulfate proteoglycans (HSPGs) in vivo, and HSPGs may also assist the transcellular propagation of tau aggregates. Here, we investigate the role of the sulfate moieties of heparin in the aggregation of a recombinant tau fragment Δtau187, comprising residues 255–441 of the C-terminal microtubule-binding domain. The effects that the selective removal of the N-, 2-O-, and 6-O-sulfate groups from heparin have on the kinetics of tau aggregation, aggregate morphology, and protein structure and dynamics were examined. Aggregation kinetics monitored by thioflavin T (ThT) fluorescence revealed that aggregation is considerably slower in the presence of 2-O-desulfated heparin than with N- or 6-O-desulfated heparin. Transmission electron microscopy revealed that tau filaments induced by 2-O-desulfated heparin were more slender than filaments formed in the presence of intact heparin or 6-O-desulfated heparin. The 2-O-desulfated heparin-induced filaments had more extensive regions of flexibility than the other filaments, according to circular dichroism and solid-state NMR spectroscopy. These results indicate that the sulfation pattern of heparin regulates tau aggregation, not purely though electrostatic forces but also through conformational perturbations of heparin when the 2-O-sulfate is removed. These findings may have implications for the progression of AD, as the sulfation pattern of GAGs is known to change during the aging process, which is the main risk factor for the disease.
The assembly of microtubule-associated protein tau (MAPT, or tau; UniProtKB P10636) into filamentous aggregates is a pathological hallmark of neurodegenerative diseases, including Alzheimer’s disease (AD) and Pick’s disease, collectively known as tauopathies.1,2 Tau is a water-soluble, intrinsically disordered protein under normal physiological conditions, and its function is to assist the assembly and stabilization of microtubules and other neuronal cytoskeletal elements. Six isoforms of tau occur in the adult human brain;3 the largest (441-aa) isoform consists of two N-terminal inserts (N1, N2) and four repeat units (R1–R4) in the microtubule-binding C-terminal region. The shorter isoforms lack one or both of the N1 and N2 units and/or the R2 unit. Neurofibrillary tangles of abnormally phosphorylated, aggregation-prone tau occur in the brains of patients affected by AD, and these consist predominantly of insoluble paired helical filaments (PHF) and straight filaments (SF).4,5 The filaments consist of an ordered β-sheet amyloid core flanked by a fuzzy, unstructured coat of around 200 residues that is invisible to transmission electron microscopy (TEM).6 Recent visualization by electron cryo-microscopy, supported by earlier solid-state NMR studies,7,8 has revealed that the nanoscale polymorphism of tau PHF and SF originates from disease-specific molecular conformations of tau within the fibers,5,9 raising the possibility that neuropathological strains may propagate in a prion-like mechanism. The molecular processes that lead to the formation and propagation of these strains in vivo are far from clear, and our understanding has until recently depended upon mechanistic studies in vitro.
Studies of the aggregation mechanisms of intact or truncated tau proteins in vitro require the addition of arachidonic acid or polyanionic cofactors such as RNA and, most commonly, heparin to initiate aggregation.10,11 Recent results from tissue-derived filaments pose the question of whether the heparin-induced tau aggregation is pathologically relevant.12,13 Heparin-induced filaments are heterogeneous,13 possibly reflecting the heterogeneity and polydispersity of the heparin cofactor, and the protein molecular conformation differs somewhat from the structures associated with AD or Pick’s. It is worth noting, however, that there is insufficient information about the structural variability of filaments obtained from different AD or Pick’s brains, given the limited current data and challenges involved. Further, heparin is closely related to heparan sulfate, which was found early on to accumulate with neurofibrillary tangles in the AD brain14 and, when conjugated to proteoglycans, assist the cellular uptake and possible propagation of tau via a prion-like seeding mechanism.15
It has often been assumed that heparin–tau interactions are driven largely by nonspecific ionic interactions between the protein and the sulfate and carboxylate groups of heparin,16 but this notion has been challenged by recent results showing that the sulfation pattern of HS influences the cellular internalization of tau, with the 6-O-sulfate being critical for this process and for tau binding.17−19 Here, using selectively desulfated heparin molecules, we show that the sulfation pattern of heparin also influences the rate of aggregation of a truncated tau construct and modulates the mass, nanoscale morphology, and atomic structure of the filaments formed. All experiments were performed with the Δtau187 construct20 comprising residues 255–441 of the tau N2R4 isoform, spanning repeat units R1–R4 and encompassing the amyloid core region from N256 to F378 (Scheme 1A). Porcine mucosal low-molecular weight heparin (LMWH; approximately 6 kDa, dp18) and high molecular weight heparin (HMWH; approximately 16 kDa, dp24–32) were chemically modified to remove the 2-O-sulfate groups and/or the 6-O-sulfate groups and/or replace the N-sulfates with acetyl groups, yielding 10 derivatives (Scheme 1B). The residual sulfate levels of the 6-O- and 2-O- desulfated LMW heparin derivatives are 61 and 70% and of the HMW heparin derivatives are 50 and 42% of the sulfate levels of the respective unmodified heparin, as measured by enzymatic digestion followed by high-performance anion exchange chromatography (HPAEC).
Scheme 1. (A) Sequence of the Four Microtubule Binding Repeats (R1–R4) and β-Strand Regions of the Heparin-Induced 2N4R Tau Polymorphs (Orange, Snake; Blue, Twisted; Green, Jagged) Identified by Cryo-EM and (B) Generic Structure of the Heparin Disaccharide Unit and List of the Desulfated Heparin Derivatives Prepared.

Experimental Methods
Tau Expression
The tau construct comprised residues 255–441 of human 4R tau (cDNA clone htau46) with the aggregation impeding N terminus removed, leaving the second and third repeat microtubule binding (MTB) units, including the highly amyloidogenic sequences VQIINK and VQIVYK, respectively. The protein was expressed and purified as previously described.20
Preparation of Desulfated Heparin
Unmodified LMWH (approximately dp 18) and high molecular weight heparin (approximately 36 dp) were purchased from Iduron (Manchester, U.K.). Modified heparin derivatives were prepared through appropriate combinations of solvolytic desulfation,21,22 involving the removal of iduronate 2-O-sulfation via 2,3-epoxide formation and subsequent nucleophilic ring opening23 and N-acetylation, followed by purification. Purification involved, for the 6-de-O-sulfated derivatives, a precipitation step in ethanol, followed by dialysis (3.5 kDa cutoff, 2 L × 3), gel permeation chromatography (Sephadex G-25), cation exchange (Dowex-50 Na+ form), and then freeze-drying. For the 2-O-desulfated derivatives, purification involved a dialysis step (3.5 kDa cutoff, 2L × 3) followed by GPC (Sephadex G-25) and cation exchange (Dowex-50 Na+ form) prior to freeze-drying. The compounds were characterized by 1H and 13C NMR spectroscopy in D2O, essentially as described,24 to establish the purity and extent of modification by reference to established chemical shift positions. The extent of modification was measured by analysis of the HPAEC (Propac PA-1 column 2 × 250 mm) profiles (monitoring the double bond formed by lyase treatment at 232 nm) of complete digestion products following bacterial (Flavobacterium heparinium) heparinase digestion (hI, hII, and hIII) in lyase buffer (50 mM sodium acetate, 2.5 mM calcium acetate) and reference to standard disaccharides (Dextra Laboratories, U.K.) to provide the proportions of each constituent disaccharide to be established and the extent of particular substitutions to be calculated with reference to the values obtained for the relevant (LMW or HMW) heparin starting material.
Aggregation Kinetics of Tau
The formation of amyloid was measured with the amyloid specific dye Thioflavin T (ThT). Tau with heparin (20 and 5 μM respectively) was incubated with Tris (30 mM) and DTT (1 mM) at pH 7.5 in the presence of 20 μM Thioflavin at 37 °C. Fluorescence measurements, with excitation at 450 nm and emission at 482 nm, were taken from triplicate samples on a Molecular Devices Flexstation 3 Microplate Reader (Molecular Devices) every 2 min for up to 24 h with shaking for 10 s prior to each read.
Circular Dichroism
Tau (20 μM) was incubated with Tris (30 mM) and DTT (1 mM) at pH 7.5 with heparin derivates (5 μM) at 37 °C with agitation. Spectra were acquired hourly during the first 8 h, followed by acquisition of a final spectrum after 24 h. Spectra were acquired on a Chirascan Plus CD spectrometer between 180 and 260 nm with a bandwidth of 1 nm using a path-length of 0.1 mm. Background signals of buffer and the relevant heparin derivative were removed from the spectra. Measurements were collected from triplicate samples, and the averaged spectra were fitted with the BestSel algorithm between 190 and 250 nm, and percentages of helical, β-sheet, turn, and unordered components were calculated.25
Transmission Electron Microscopy
Tau and heparin (20 and 5 μM, respectively) were incubated with Tris (30 mM) and DTT (1 mM) at pH 7.5 at 37 °C for 24 h. A 10 μL suspension was spotted onto carbon coated formar grids. After 5 min, the excess liquid was removed via blotting. For negative staining, 10 μL of 2% phosphotungstic acid was spotted onto the loaded grids and left for 3 min before blotting the excess. Grids were viewed on a Jeol JEM-1010 electron microscope, and images were captured that were representative of the entire grid. Fibril widths were measured using ImageJ software, and the averages from 50 measurements were calculated.
Solid-State NMR experiments
Following 24 h of agitated growth of tau (20 μM) at 37 °C in the presence of the heparin derivatives (5 mM), tau aggregates were pelleted by centrifugation at 50 000g for 1 h in an ultracentrifuge (Beckman Coulter), and the supernatant was removed. The fibrils were sedimented by a second centrifugation step. The pellet was separated from the supernatant and packed in its hydrated state by ultracentrifugation into a 3.2 mm zirconia MAS rotor without further treatment for analysis by SSNMR.
Two-dimensional 13C–13C SSNMR spectra were recorded at a magnetic field of 16.3 T on a Bruker Avance III 700 spectrometer with a 3.2 mm HXY probe operating in double resonance mode. The magic-angle frequency was 13 kHz. The operating temperature was 4 °C. Hartmann–Hahn cross-polarization was achieved with a 2 ms contact time and 100 kHz proton decoupling with SPINAL-64 was applied during signal acquisition. Spectra were recorded with either 10 ms, 50 or 200 ms mixing times during which the proton nutation frequency was adjusted to the MAS frequency of 14 kHz to meet the dipolar-assisted rotational resonance (DARR) condition.26 Typically, 480 increments were acquired in the indirect (t1) dimension with 400–600 transients per increment, and the total measurement time varied from 2 to 7 days depending on the efficiency of rotor packing. Phase-sensitive detection in the indirect dimensions was achieved using the States-TPPI method. Chemical shifts are expressed relative to tetramethylsilane.
The simulated 13C–13C spectrum (Supporting Information, Figure S1) was calculated from chemical shifts predicted from the cryo-EM structure of heparin induced 2N4R tau snake filaments12 using a C program written specifically for this purpose.
Results
Desulfation of Heparin Influences Δtau187 Aggregation Kinetics
Incubation of Δtau187 (20 μM) with unmodified LMWH in the presence of the amyloid-sensitive dye thioflavin T (ThT) indicates that tau aggregation reaches completion within 5 h and the aggregation kinetics follow a sigmoidal trajectory consistent with a lag phase, filament growth, and maturation with a half-time t0.5 of 0.5 h (Figure 1A). Removal of only the 6-O-sulfates of LMWH or selective replacement of the N-sulfates with COCH3 (Figure 1B) has a measurable effect on the aggregation kinetics, increasing t0.5 to 1.4 h in both cases. A considerably slower rate of aggregation is seen in the presence of selectively 2-O-desulfated heparin (t0.5> 6 h). Removal of the 6-O-sulfate groups combined with N-acetylation increases t0.5 to 2.4 h, but aggregation is much slower when 2-O-desulfation is combined with either 6-O-desulfation or N-acetylation (t0.5 = 4.5 h) (Figure 1C). Aggregation kinetics in the presence of HMWH are also dependent on the sites of heparin desulfation, with Δtau187 aggregation again being considerably slower in the presence of 2-O-desulfated heparin than with the other derivatives (Figure 1D). In summary, the overall rate of heparin-induced Δtau187 aggregation to completion is impeded to a greater extent by removing the 2-O-sulfates of heparin than it is by removing the 6-O- or N-sulfates. Although the data for Δtau187 are normalized to the final fluorescence emission that was measured, the curve is clearly continuing in an upward trajectory, indicating that aggregation had not reached its conclusion even after 12 h, and t0.5 is likely to be considerably longer than 6 h. The different effect of the selectively desulfated heparin derivatives argues against the tau–heparin interaction being mediated only by nonspecific charge interactions. Other chemical and/or conformational properties must be considered.
Figure 1.

Aggregation kinetics of Δtau187 (20 μM) in the presence of unmodified heparin (5 μM) monitored by ThT fluorescence. (A) Unmodified and fully desulfated LMWH. (B) LMWH desulfated at a single site per disaccharide unit. (C) LMWH desulfated at two sites per disaccharide unit. (D) Unmodified HMWH and HMWH desulfated at one site per disaccharide. Means and error bars are shown for triplicate measurements.
Concentration Dependence of Desulfated Heparin-Induced Δtau187 Aggregation
To further investigate the effect of the sulfate groups of heparin on Δtau187 aggregation, the kinetics were followed (by ThT) at different concentrations of LMWH and the 6-O, N-acetylated, and 2-O-desulfated derivatives (Figure 2). The time to completion of aggregation (quantified by t0.5) is sensitive to heparin concentration, as is the ThT fluorescence at the end point of aggregation, suggesting that the mass of filaments is heparin concentration dependent. Curves calculated for the apparent rate constants for primary nucleation (kn), elongation (k+), and secondary nucleation (k2)27,28 agree well with the experimental data (Figure 2 and Table 1). The aggregation rate in the presence of unmodified LMWH remains constant up to 10 μM heparin (t0.5 ∼ 0.5 h), with the final fibril mass scaling approximately with heparin concentration. The end-point ThT fluorescence also increases with heparin concentration up to 10 μM. This trend has been attributed to an increase in the fibrillar mass with increasing heparin concentration, which gives rise to enhanced ThT fluorescence.29 Although it is known that many other environmental factors or conformational differences can influence ThT fluorescence, it is likely that here the ThT curves do indeed reflect the fibril mass, as the heparin concentration (which is not ThT reactive) is the only controlled variable. At an equimolar (20 μM) LMWH and Δtau187 concentration, the aggregation rate decreases markedly (t0.5 = 1.7 h), and no further increase in ThT fluorescence (or end-point fibril mass) is observed (Figure 2, top left). The reduced rate can be attributed to a reduction in both the apparent kn and k+ values (Table 1). The retarding effect of heparin at higher concentrations is known and has been attributed to a screening effect of high heparin concentrations because of increased ionic strength.30 It has also been proposed that high heparin concentrations promote the formation of dead-end, off-pathway tau–heparin complexes.31 A further explanation, which will be explored in the next section, is that higher heparin concentrations increase the number of 1:1 tau:heparin complexes that are below the critical mass necessary to nucleate filament growth.
Figure 2.

Aggregation kinetics of Δtau187 (20 μM) in the presence of LMWH or derivatives (desulfated at one site per disaccharide) at four concentrations, measured by thioflavin T fluorescence. Means and standard errors are shown from triplicate measurements. Bold lines are the best fitting curves corresponding to the rate constants given in Table 1. Means and error bars are shown for triplicate measurements.
Table 1. Summary of the Apparent Rate Constants for Tau Aggregation in the Presence of Unmodified Heparin and Desulfated Derivatives, Obtained from the Lines of Best Fit to the Data in Figure 2a.
| cofactor | conc/μM | kn/10–6 M–1 s–1 | k+/106 M–1 s–1 | k2/M–2 s–1 | t0.5/h |
|---|---|---|---|---|---|
| LMWH | 2.5 | 32 | 17 | 10 | 0.51 (0.10) |
| 5.0 | 35 | 19 | 10 | 0.46 (0.10) | |
| 10.0 | 33 | 18 | 70 | 0.45 (0.10) | |
| 20.0 | 6 | 4 | 1 | 1.70 (0.13) | |
| LMW-6OH | 2.5 | 4 | 7 | 13 | 1.47 (0.24) |
| 5.0 | 6 | 9 | 12 | 1.12 (0.14) | |
| 10.0 | 9 | 15 | 16 | 0.72 (0.12) | |
| 20.0 | 6 | 11 | 16 | 0.95 (0.06) | |
| LMW-NAc | 2.5 | 6 | 4 | 10 | 1.4 (0.13) |
| 5.0 | 5 | 6 | 10 | 1.4 (0.15) | |
| 10.0 | 5 | 8 | 11 | 0.9 (0.10) | |
| 20.0 | 3 | 4 | 10 | 1.7 (0.10) | |
| LWM-2OH | 2.5 | 1 | 2 | 1 | 6.5 (0.25) |
| 5 | 2 | 2 | 1 | 5.5 (0.30) | |
| 10 | 2 | 3 | 1 | 4.4 (0.30) | |
| 20 | 2 | 2 | 0 | ||
| HWMH | 2.5 | 36 | 19 | 10 | 0.5 (0.03) |
| 5 | 28 | 15 | 9 | 0.5 (0.09) | |
| 10 | 6 | 4 | 6 | 2.0 (0.05) | |
| HMW-6OH | 2.5 | 11 | 7 | 1 | 1.7 (0.09) |
| 5 | 10 | 7 | 1 | 1.6 (0.1) | |
| HMW-2OH | 2.5 | >6 | |||
| 2.5 |
The calculations were also based on fixed values for the fibril dissociation rate constant (km = 1.0 M–1 s–1) and the reaction orders of primary nucleation (nc = 2) and secondary nucleation (n2 = 2).27,28 The reaction orders do not necessarily correspond to the size of the nuclei, but heparin has been shown to interact with two tau molecules, forming a dimer that nucleates fibril growth.31 Dashes indicate where constants could not be calculated because of poor fits to the experimental data. Errors in t0.5 are given in parentheses.
When Δtau187 is incubated with 6-O-desulfated or N-acetylated LMWH, a 2–3-fold overall decrease in the rate of aggregation is seen relative to unmodified LMWH with t0.5 being 0.9–1.7 h (Figure 2, top right and bottom left; Table 1). Aggregation again follows a sigmoidal profile, and the reduced rate appears to originate chiefly from a reduction in kn and k+. Fluorescence at the conclusion of aggregation has a nonlinear relationship with heparin concentrations up to 20 μM heparin. In the presence of 2-O-desulfated LMWH, aggregation is substantially slower at all heparin concentrations with t0.5 in excess of 4 h, and the growth curves are consistent with an apparent reduction of kn, k+, and k2 (Figure 2, bottom right; Table 1) compared to the rate constant values in the presence of the other cofactors. Interestingly, the final aggregate mass decreases by 50% on increasing the 2-O-desulfated LMWH concentration from 10 to 20 μM.
Heparin 6-O-Desulfation, but Not 2-O-Desulfation, Reduces the Affinity for Δtau187
The total residual sulfation levels of the 6-O- and 2-O-desulfated LMWH molecules are very similar (as are 6-O- and 2-O-desulfated HMWH), and so the large differences in the behavior of these desulfated heparins cannot be attributed entirely to their reduced charge density. Indeed, the ThT data reveal a complex relationship between the sulfation patterns and concentration of heparin, Δtau187 aggregation rates, and filament mass that cannot be explained fully by the apparent rate constants in Table 1. These rate constants describe a cofactor-independent aggregation mechanism represented by the equations27,28
| 1 |
and
| 2 |
Here, M(t) and P(t) are the filament mass and number, respectively, at time t, and m(t) is the monomer concentration. The term knm(t)nc represents the formation of primary nuclei from monomers (m) with reaction order nc (typically assigned a value of 2). The term k2m(t)n2M(t) describes secondary nucleation in a reaction of order n2 (typically given a value of 2 or 3) with respect to the mass of monomer.
The term 2k+m(t)P(t) represents fibril elongation by the addition of monomers to the two growing ends of the fibers. We considered minor modifications to these equations that might account for the critical involvement of the heparin cofactor, h, in the Δtau187 nucleation and elongation processes, and which may also explain the influence of heparin on both aggregation rate and filament mass (Scheme 2). The trends observed can be rationalized if it is assumed that Δtau187 monomers and heparin molecules rapidly establish an equilibrium state 1:1 complex (mh). Neglecting depletion of monomer and heparin concentrations by other interaction pathways at this initial condition, the equilibrium concentration of the complex [mh] is, according to the standard relationship, given by
| 3 |
where [m]T and [h]T are the total concentrations of free protein monomer and free heparin and Kd is the dissociation constant. The equilibrium is perturbed as mh recruits further monomers to form a nucleating species. Aggregation of the 4-repeat domain tau construct in the presence of heparin has been shown to be nucleation dependent, with a single heparin molecule binding tau forming an aggregation-promoting dimer, which then serves as a building block for further fibril growth.31 We therefore assume that the smallest primary nucleating species N consists of two tau molecules bound to one heparin molecule, m2h. The rate of formation of primary nuclei N thus depends on collisions between free monomeric protein and the mh complex such that
| 4 |
where nc = 1. Eqs 3 and 4 thus take into account the role of heparin in the initiation of tau aggregation. Eq 1 becomes
| 5 |
These equations likely oversimplify the tau–heparin interaction but serve as a working hypothesis until the details of the model are further refined with experimental input. The simulated curves share similarities with the experimental data in Figure 2; the simulations do not replicate exactly the behavior of the ThT curves, and so quantitative analysis using global fitting of eqs 4 and 5 to the curves was not attempted. However, qualitative inspection of numerically simulated curves based on these adaptations suggests that t0.5 for aggregation and filament mass are both sensitive to heparin concentration and Kd (Figure 3).
Scheme 2. Basic Pathway for Heparin-Induced Tau Assembly into Filaments.
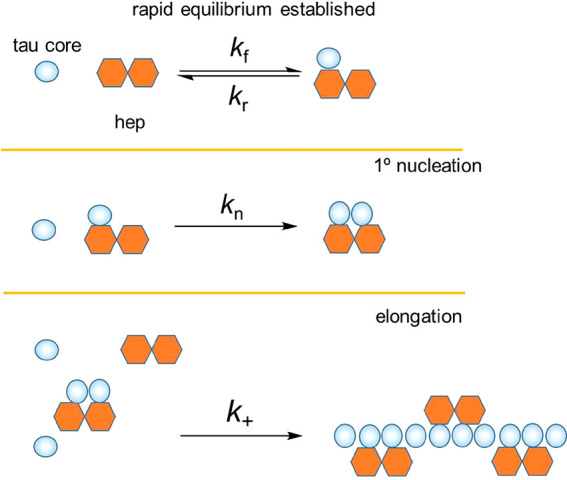
Figure 3.
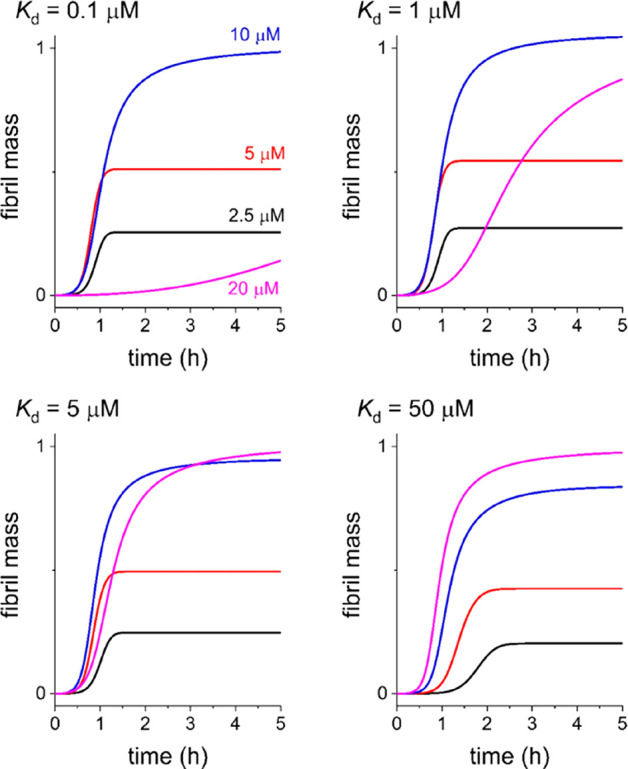
Calculated fibril mass at four heparin concentrations based on the proposed model. The value of Kd for the initial tau–heparin complex is the only variable in the calculations. Using the terminology of Knowles and colleagues, values of the remaining constants are kn = 5 × 10–4 M–1 s–1, k+ = 8 × 106 M–1 s–1. No fibril fragmentation of secondary nucleation (i.e., k2 = 0) was assumed for simplification.
An equilibrium association constant exceeding 106 M–1 (i.e., Kd < 1 μM) for tau and heparin has been reported.30 According to eq 5, when Kd = 0.1 μM, t0.5 for filament growth does not change markedly at heparin concentrations up to 10 μM, and the filament mass increases with heparin concentration up to 10 μM (Figure 3, top left panel). At a heparin concentration of 20 μM, a considerable increase in t0.5 is predicted, and the end-point filament mass decreases significantly. This apparent inhibitory effect at higher heparin concentrations can be rationalized as the high-affinity heparin–tau interaction, removing free monomer from solution, leaving little remaining heparin to form nuclei and/or elongate the filaments. These trends in fibril mass and t0.5 are reminiscent of the ThT fluorescence curves for Δtau187 with LMWH and LMW-2OH (Figure 2 and Table 1), although the latter curve is consistent with much slower aggregation than that represented by the simulation. Hence, removal of the 2-O-sulfate group does not appear to reduce the affinity of heparin for Δtau187 according to these curves. The slower rate of tau aggregation in the presence of LMW-2OH, compared to the rates in the presence of the other heparin derivatives, is reflected in the lower rate constants kn, k+, and k2 calculated by curve fitting (Figure 2 and Table 1), which indicates that 2-O-sulfate group is critical for tau primary and secondary nucleation and filament elongation.
For progressively weaker tau–heparin interactions, the calculated fibril mass in the presence of 20 μM heparin increases as Kd increases (Figure 3). At Kd = 50 μM, a progressive decrease in t0.5 and increase in fibril mass is predicted as the heparin concentration is increased. These trends are reminiscent of the ThT curves for tau in the presence of LMW-6OH (Figure 2, top right) and support the previous reports that 6-O-desulfation of heparin reduces its affinity for tau.19 Similarly, at Kd = 5 μM, the curves are reminiscent of the ThT profiles for tau in the presence of LMW-NAc, although there are also some important differences that indicate that our model is incomplete.
Heparin 2-O-Desulfation, But Not 6-O-Desulfation, Affects the Structure and Morphology of Δtau187 Aggregates
We next used circular dichroism (CD) spectroscopy to follow the structural transformation accompanying Δtau187 aggregation in the presence of HMWH, HMW-6OH, and HMW-2OH. CD has indicated that tau aggregation accompanies a transition from an unfolded structure to a partially folded structure with approximately ∼36% β-sheet.32 Here, we obtained spectra from freshly prepared Δtau187–heparin solutions and again after incubation at 37 °C for 8 h. In the absence of heparin, the spectrum changes little over 8 h (Figure 4A), with a minimum at 200 nm consistent with the protein remaining unfolded, and a secondary structure content of 3.7 (±0.8)% α-helices, 25 (±0.4)% β-sheet, 18 (±0.1)% turns, and 54 (±1.2)% unordered. In the presence of unmodified heparin, Δtau187 undergoes a structural transition over 8 h (Figure 4B, left). The spectrum at the end-point of aggregation with intact heparin is consistent with 0.7 (±1.3)% α-helices, 46 (±11)% β-sheet, 14 (±1.2)% turns, 39 (±9.3)% unordered content, and an isodichroic point at around 208 nm. A similar trend is seen in the CD spectra of Δtau187 in the presence of 6-O-desulfated HMWH (Figure 4B, middle) with an increase in β-sheet content to 37 (±3.6)% and no further change, indicating that aggregation has reached completion after 8 h with the structural content being similar to aggregates formed with unmodified heparin. By contrast, the spectrum of Δtau187 in the presence of 2-O-desulfated HMWH (Figure 4B, right) changes little over 8 h and is consistent with 5.3 (±0.1)% α-helices, 27 (±2.5)% β-sheet, 17 (±0.5)% turns, and 51 (±1.8)% unordered residues, closely resembling the structure content of monomeric Δtau187 after 8 h. Furthermore, there is no clear isodichroic point, as seen in the spectra of tau with HMWH or LMW-6OH. Hence, the CD spectra concur with ThT measurements that the rate of Δtau187 aggregation is slower in the presence of 2-O-desulfated heparin than with the other heparin derivatives.
Figure 4.

Secondary structure and morphology of Δtau187 aggregates. (A) CD spectra of Δtau187 (20 μM) at 0 and 8 h. (B) CD spectra of Δtau187 in the presence of 5 μM HMWH, 6-O-desulfated HMWH, or 2-O-desulfated HMWH at 0 and 8 h. The samples were incubated at 37 °C without agitation. All CD spectra are shown as the average of triplicate repeats. (C) TEM images of aggregates formed from Δtau187 (20 μM) after incubating with 5 μM of HMWH or the desulfated derivatives without agitation for 24 h. Scale bar = 200 nm. (D) Distribution of filament widths measured from the TEM images.
The morphology of aggregates formed after incubation with heparin or each of the desulfated analogues was visualized by negative-stain TEM. Four tau morphologies formed in the presence of heparin, termed snake, twister, hose, and jagged, have been observed by cryo-EM.12 Here, in the presence of unmodified HMWH, Δtau187 forms a dense network of fibrillar aggregates, and upon closer inspection, the filaments display a sinusoidal curvature pattern with a mean width of 11.8 (±5.2) nm (Figure 4C and D, left). The intertwined filaments that are characteristic of PHFs, or their crossover points, cannot be discerned here, but the width of the filaments is similar to that typically seen for paired filaments (10–25 nm).5,32−34 Aggregates formed in the presence of 6-O-desulfated heparin display a similar morphology and a mean width of 12.7 (±4.6) (Figure 4C and D, middle), although their sparse deposition is consistent with a lower yield of filaments. Filaments formed in the presence of HMW-2OH (Figure 4C, right) are also sparsely distributed and display a sinusoidal curvature pattern. These filaments are noticeably more slender than the filaments formed with unmodified heparin, with mean width of 8.5 (±2.6) nm (Figure 4D, right). The width of these filaments is more typical of the single filaments that are the minor population of tau aggregates associated with AD.33,34 To summarize, heparin 2-O-desulfation has a much more profound effect on tau aggregation rate, filament mass, β-sheet content, and filament morphology than either 6-O-desulfation or N-acetylation.
Tau Filaments Formed with 2-O-Desulfated Heparin Have a Reduced β-Sheet Core
Finally, we used magic-angle spinning (MAS) solid-state NMR (SSNMR) to compare the structure and dynamics of uniformly 13C-labeled Δtau187 fibrils formed in the presence of HWMH and HMW-2OH. Previous SSNMR analyses of heparin-induced aggregates of truncated tau constructs indicated that amino acids 306–324 form the rigid β-sheet core and adopt two distinct conformations7,8 consistent with the polymorphism of tau filaments visualized by cryo-EM.12 Unlike electron microscopy, the flexible N- and C-terminal sequences constituting the “fuzzy” regions may be observed by MAS SSNMR using INEPT-based methods that correlate nuclear spins through J-couplings, while the ordered core is observed selectively using Hartmann–Hahn cross-polarization combined with nuclear dipolar recoupling methods.32,35
Here, two-dimensional 13C–13C dipolar correlation NMR spectra were obtained from fibrils formed by Δtau187 (20 μM) in the presence of 5 μM HMWH or HMW-2OH. By using a short dipolar mixing time (10 ms), the spectra report on the motionally restrained residues forming the core of the fibrils. The more flexible outer residues give rise to weaker cross-peaks or are not observed. The peaks are quite broad, which probably reflects the known structural heterogeneity of tau filaments formed in the presence of heparin.13 No attempt was made to sequentially assign the spectra owing to severe crowding and overlap, as the tau construct we studied is much larger than the constructs previously studied by SSNMR. Sequential assignment and structural determination could in principle be achieved with a combination of 2D and 3D NMR experiments and selective isotope labeling, but this was beyond the scope of the present work. The experimental spectrum of tau filaments formed with HMWH is overlaid with a simulated spectrum (Supporting Information, Figure S1) generated from 13C chemical shifts predicted from the cryo-EM structure of heparin-induced filaments (Table S1),12 which are predominantly β-sheet/hairpin folds. Good agreement between the simulated and experimental spectra suggests that the observed signals originate from the β-sheet core of the fibrils. A striking reduction in cross-peak intensities, particularly for cross-peaks representing longer-range couplings, is seen in the spectrum of the fibrils induced by HMW-2OH, as compared to the spectrum of fibrils induced by HMWH (Figure 5A and B, top panels). The reduction in intensity is consistent with a higher degree of overall flexibility within the fibrils induced by HMW-2OH. In contrast to the 13C–13C spectra, 1H–13C INEPT spectra of the exhibit many more peaks than are seen than in the spectrum of HMWH-induced filaments (Figure 5A and B, bottom panels), confirming that the HMW-2OH-induced fibrils contain a greater number of flexible residues than do fibrils induced by HMWH. In the INEPT spectrum of HMW-2OH-induced filaments, much stronger signals are observed from methyl bearing side chains, but also in the Cα region, indicating that more residues are flexible in the protein backbone. Taken together, these observations are consistent with the HMW-2OH-induced filaments having a smaller β-sheet core, flanked by a greater number of mobile residues, compared to the filaments formed in the presence of unmodified heparin.
Figure 5.

Solid-state magic-angle spinning 13C NMR spectra of uniformly 13C-labeled Δtau187 aggregates formed in the presence of HMWH or 6-O- or 2-O-desulfated HMWH derivatives. (A) Top: region of a 13C–13C dipolar correlation NMR spectrum of [U–13C] Δtau187 aggregates formed in the presence of HMWH. Dipolar interactions were reintroduced by a 5 ms DARR mixing period. Bottom: A 1H–13C INEPT spectrum of the sample. (B) The 13C–13C dipolar correlation NMR spectrum and INEPT spectrum of tau filaments formed in the presence of HMW-2OH. The MAS frequency was 13 kHz in all experiments.
Discussion
Heparin is a convenient experimental aid to induce the rapid formation of tau filaments/fibrils in vitro for mechanistic and structural investigations. It is a close analogue of the GAG heparan sulfate, and heparan sulfate proteoglycans (HSPGs) such as agrin are commonly associated with Aβ plaques and NFTs in AD. The sulfation patterns of GAGs are known to change with aging,36,37 the principal risk factor for AD, and so there is biological incentive to investigate how GAG sulfation affects tau aggregation, using heparin as a representative GAG.
Experimental evidence gained over the past decade suggests that the progression of neurodegeneration in AD is driven by the transcellular propagation of tau aggregates, which seed protein aggregation in the recipient cells in a prion-like manner.38,39 This process may not necessarily be limited to tau aggregates, as monomeric tau can also be internalized and seed the aggregation of endogenous tau.40 The mechanism of tau release from neurons into the extracellular space is not currently known, but the cellular uptake of tau fibrils can occur via binding to HSPGs.15 It is conceivable that HSPGs also promote the fibrillization of tau monomers and oligomers in the extracellular space, just as heparin (and HS) do in vitro, in addition to facilitating the cellular uptake of fibrils.
Here, we report that selectively altering the sulfation pattern of heparin affects the kinetics of heparin-induced Δtau187 aggregation and the mass of filaments formed and can also modify the molecular structure of the filaments. The rate of aggregation is slower in the presence of all the desulfated heparins than in the presence of native heparin, which may be attributed in part to the lower negative charge of the derivatives. All the heparins desulfated at a single position have approximately the same overall sulfation levels, however, and the differences in the behavior of tau in the presence of 6-O-desulfated, 2-O-desulfated, and N-acetylated heparins suggest that the derivatives have distinctive chemical properties that modulate their interactions with tau.
We propose a basic mechanism for heparin-induced tau aggregation, which goes some way toward explaining the aggregation kinetics in the presence of the different heparin derivatives, but significant differences between the experimental ThT curves and calculated curves indicate that the model is incomplete. A more robust analysis may need to take into account the formation of oligomers, polymerization via multiple pathways and complex secondary nucleation processes involving protein fibrillar and heparin surfaces. As it stands, the basic model is consistent with the 6-O-desulfated and, to a lesser extent, N-desulfated heparin having lower affinity for tau than intact heparin. This interpretation agrees with previous work: using a heparin-immobilized chip, surface plasmon resonance was used to show that N-desulfation and 2-O-desulfation had no effect on heparin binding to a tau construct, whereas 6-O-desulfation severely reduced binding.19 In addition, cellular studies have shown that heparin interacts with heparan sulfate binding sites in tau and prevents binding to cell-surface HSPGs,15 but removal of the 6-O-sulfates from heparin abolishes the inhibitory effect, whereas 2-O-desulfated heparin remains strongly inhibitory.17 Further studies concur that 6-O-sulfation is critical for tau–heparan sulfate interactions and that this modification regulates uptake in human cell lines and mouse brain slice culture.18 Interestingly, the 6-OS and NS glucosamine sulfates, but not the 2-OS iduronate sulfate, of heparin is required for binding to Aβ fibrils,41 suggesting a common function for this moiety in amyloid binding.
The most interesting finding here is that 2-O-desulfation of heparin results in a markedly greater reduction of the Δtau187 aggregation rate than does desulfation at the other two sites, and templates tau assembly into a different filament structure and morphology. These differences may be rationalized by considering the effects of removing sulfate groups on the conformation of heparin. In addition to losing a charge in the form of a sulfate group, the conformation of heparin derivatives is sensitive to the substitution pattern in several ways. The iduronate residue is known to be in an equilibrium of 1C4 and 2S0 chair and skew boat forms in heparin (67:33 1C4:2S0)42 but, following de-O-sulfation at position-2 of the iduronate residue, this equilibrium is disturbed, moving in favor of the 1C4 chair form (85:15).43 In addition, removal of 2-O-sulfate groups alters the 1JCH NMR coupling constants (C-1 to H-1 and C-4 to H-4) around the glycosidic linkages, consistent with additional changes in conformation;44,45 for example, the 1JCH values change at A-1 (C-1 to H-1 of glucosamine) from 172 to 176 Hz and at I-1 (C-1 to H-1 of the iduronate residue) from 147 to 156 Hz.46 The removal of 6-O-sulfate groups from glucosamine, on the other hand, while altering chemical shift values principally adjacent to the site of the sulfation as expected,24 has less effect elsewhere in the heparin chain, consistent with relatively little perturbation of the overall conformation.47 We propose that the unique conformational perturbations resulting from 2-O-desulfation must therefore modify the tau–heparin interaction in such a way that the conformation of tau is less amenable to self-assembly. One explanation, which is supported by the NMR data, is that the core β-sheet region of tau is reduced and the flanking fuzzy coat region extends further into the core. Why a different filament structure and morphology occurs in the presence of 2-O-desulfated heparin can only be speculated upon at this stage. Alterations in the global and local conformation of heparin after 2-O-desulfation may reduce or otherwise change the charged surface that interacts with tau monomers. A distinct primary nucleating species may be formed that directs propagation along a structurally and kinetically altered pathway involving interactions between fewer core residues.
There is ample evidence that heparin sulfates accelerate the aggregation of monomeric tau in vitro.48 Further work will be necessary to understand fully the nature of the tau species that interact with HSPGs during transcellular propagation and to ascertain whether GAGs play a role in filament formation. It should be noted, that protein aggregates in the Alzheimer’s brain principally contains phosphorylated tau (P-tau), rather than normal tau, and as such the higher negative charge density would likely repel polyanionic GAGs. The interplay among tau, P-tau, and GAGs must therefore be considered in future investigations. The key message of this work is that to understand the role of GAG-protein interactions in the formation of amyloid fibrils, it is necessary to look beyond electrostatics and to consider the chemical and conformational effects conferred on GAGs by their sulfation patterns.
Acknowledgments
Funding was provided by BBSRC (U.K.) Grant BB/K015958/1.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00443.
Regions of NMR spectra and a table of predicted 13C chemical shifts (PDF)
Accession Codes
Full length human MAPT: UniProtKB P10636.
The authors declare no competing financial interest.
Supplementary Material
References
- Gibbons G. S.; Lee V. M. Y.; Trojanowski J. Q. (2019) Mechanisms of Cell-to-Cell Transmission of Pathological Tau a Review. Jama Neurology 76, 101–108. 10.1001/jamaneurol.2018.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T.; Noble W.; Hanger D. P. (2017) Roles of Tau Protein in Health and Disease. Acta Neuropathol. 133, 665–704. 10.1007/s00401-017-1707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G.; Jakes R.; Rutherford D.; Crowther R. A. (1989) Multiple Isoforms of Human Microtubule-Associated Protein-Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 3, 519–526. 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Iqbal K.; Alonso A. D. C.; Chen S.; Chohan M. O.; El-Akkad E.; Gong C. X.; Khatoon S.; Li B.; Liu F.; Rahman A.; Tanimukai H.; Grundke-Iqbal I. (2005) Tau Pathology in Alzheimer Disease and Other Tauopathies. Biochim. Biophys. Acta, Mol. Basis Dis. 1739, 198–210. 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P.; Falcon B.; He S.; Murzin A. G.; Murshudov G.; Garringer H. J.; Crowther R. A.; Ghetti B.; Goedert M.; Scheres S. H. W. (2017) Cryo-Em Structures of Tau Filaments from Alzheimer’s Disease. Nature 547, 185–190. 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S.; Medalsy I. D.; Mandelkow E.; Muller D. J. (2013) The Fuzzy Coat of Pathological Human Tau Fibrils Is a Two-Layered Polyelectrolyte Brush. Proc. Natl. Acad. Sci. U. S. A. 110, E313–E321. 10.1073/pnas.1212100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daebel V.; Chinnathambi S.; Biernat J.; Schwalbe M.; Habenstein B.; Loquet A.; Akoury E.; Tepper K.; Muller H.; Baldus M.; Griesinger C.; Zweckstetter M.; Mandelkow E.; Vijayan V.; Lange A. (2012) Beta-Sheet Core of Tau Paired Helical Filaments Revealed by Solid-State Nmr. J. Am. Chem. Soc. 134, 13982–13989. 10.1021/ja305470p. [DOI] [PubMed] [Google Scholar]
- Andronesi O. C.; von Bergen M.; Biernat J.; Seidel K.; Griesinger C.; Mandelkow E.; Baldus M. (2008) Characterization of Alzheimer’s-Like Paired Helical Filaments from the Core Domain of Tau Protein Using Solid-State Nmr Spectroscopy. J. Am. Chem. Soc. 130, 5922–5928. 10.1021/ja7100517. [DOI] [PubMed] [Google Scholar]
- Falcon B.; Zhang W. J.; Murzin A. G.; Murshudov G.; Garringer H. J.; Vidal R.; Crowther R. A.; Ghetti B.; Scheres S. H. W.; Goedert M. (2018) Structures of Filaments from Pick’s Disease Reveal a Novel Tau Protein Fold. Nature 561, 137–140. 10.1038/s41586-018-0454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichou Y.; Lin Y. X.; Rauch J. N.; Vigers M.; Zeng Z. K.; Srivastava M.; Keller T. J.; Freed J. H.; Kosik K. S.; Han S. I. (2018) Cofactors Are Essential Constituents of Stable and Seeding-Active Tau Fibrils. Proc. Natl. Acad. Sci. U. S. A. 115, 13234–13239. 10.1073/pnas.1810058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M.; Valpuesta J. M.; Medina M.; Montejo de Garcini E.; Avila J. (1996) Polymerization of Tau into Filaments in the Presence of Heparin: The Minimal Sequence Required for Tau-Tau Interaction. J. Neurochem. 67, 1183–1190. 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- Zhang W. J.; Falcon B.; Murzin A. G.; Fan J.; Crowther R. A.; Goedert M.; Scheres S. H. W. (2019) Heparin-Induced Tau Filaments Are Polymorphic and Differ from Those in Alzheimer’s and Pick’s Diseases. eLife 8, e43584 10.7554/eLife.43584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichou Y.; Vigers M.; Goring A. K.; Eschmann N. A.; Han S. I. (2018) Heparin-Induced Tau Filaments Are Structurally Heterogeneous and Differ from Alzheimer’s Disease Filaments. Chem. Commun. 54, 4573–4576. 10.1039/C8CC01355A. [DOI] [PubMed] [Google Scholar]
- Snow A. D.; Mar H.; Nochlin D.; Sekiguchi R. T.; Kimata K.; Koike Y.; Wight T. N. (1990) Early Accumulation of Heparan-Sulfate in Neurons and in the Beta-Amyloid Protein-Containing Lesions of Alzheimer’s Disease and Down’s Syndrome. Am. J. Pathol. 137, 1253–1270. [PMC free article] [PubMed] [Google Scholar]
- Holmes B. B.; DeVos S. L.; Kfoury N.; Li M.; Jacks R.; Yanamandra K.; Ouidja M. O.; Brodsky F. M.; Marasa J.; Bagchi D. P.; Kotzbauer P. T.; Miller T. M.; Papy-Garcia D.; Diamond M. I. (2013) Heparan Sulfate Proteoglycans Mediate Internalization and Propagation of Specific Proteopathic Seeds. Proc. Natl. Acad. Sci. U. S. A. 110, E3138–E3147. 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capila I.; Linhardt R. J. (2002) Heparin-Protein Interactions. Angew. Chem., Int. Ed. 41, 390–412. . [DOI] [PubMed] [Google Scholar]
- Stopschinski B. E.; Holmes B. B.; Miller G. M.; Manon V. A.; Vaquer-Alicea J.; Prueitt W. L.; Hsieh-Wilson L. C.; Diamond M. I. (2018) Specific Glycosaminoglycan Chain Length and Sulfation Patterns Are Required for Cell Uptake of Tau Versus Alpha-Synuclein and Beta-Amyloid Aggregates. J. Biol. Chem. 293, 10826–10840. 10.1074/jbc.RA117.000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J. N.; Chen J. J.; Sorum A. W.; Miller G. M.; Sharf T.; See S. K.; Hsieh-Wilson L. C.; Kampmann M.; Kosik K. S. (2018) Tau Internalization Is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 8, 6382. 10.1038/s41598-018-24904-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Huvent I.; Lippens G.; Eliezer D.; Zhang A. Q.; Li Q. H.; Tessier P.; Linhardt R. J.; Zhang F. M.; Wang C. Y. (2017) Glycan Determinants of Heparin-Tau Interaction. Biophys. J. 112, 921–932. 10.1016/j.bpj.2017.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M.; Smith M. J.; Goedert M. (1998) Tau Proteins with Ftdp-17 Mutations Have a Reduced Ability to Promote Microtubule Assembly. FEBS Lett. 437, 207–210. 10.1016/S0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- Inoue Y.; Nagasawa K. (1976) Selective N-Desulfation of Heparin with Dimethyl Sulfoxide Containing Water or Methanol. Carbohydr. Res. 46, 87–95. 10.1016/S0008-6215(00)83533-8. [DOI] [PubMed] [Google Scholar]
- Nagasawa K.; Inoue Y.; Kamata T. (1977) Solvolytic Desulfation of Glycosaminoglycuronan Sulfates with Dimethyl-Sulfoxide Containing Water or Methanol. Carbohydr. Res. 58, 47–58. 10.1016/S0008-6215(00)83402-3. [DOI] [PubMed] [Google Scholar]
- Jaseja M.; Rej R.; Sauriol F.; Perlin A. (1989) Novel Regioselective and Stereoselective Modifications of Heparin in Alkaline Solution - Nuclear Magnetic Resonance Spectroscopic Evidence. Can. J. Chem. 67, 1449–1456. 10.1139/v89-221. [DOI] [Google Scholar]
- Yates E. A.; Santini F.; Guerrini M.; Naggi A.; Torri G.; Casu B. (1996) H-1 and C-13 Nmr Spectral Assignments of the Major Sequences of Twelve Systematically Modified Heparin Derivatives. Carbohydr. Res. 294, 15–27. 10.1016/S0008-6215(96)90611-4. [DOI] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Bulyáki É.; Kun J.; Moussong É.; Lee Y.-H.; Goto Y.; Réfrégiers M.; Kardos J. (2018) Bestsel: A Web Server for Accurate Protein Secondary Structure Prediction and Fold Recognisiton from the Circular Dichorism Spectra. Nucleic Acids Res. 46 (W1), W315–W322. 10.1093/nar/gky497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takegoshi K.; Nakamura S.; Terao T. (2001) C-13-H-1 Dipolar-Assisted Rotational Resonance in Magic-Angle Spinning Nmr. Chem. Phys. Lett. 344, 631–637. 10.1016/S0009-2614(01)00791-6. [DOI] [Google Scholar]
- Cohen S. I. A.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. (2013) Proliferation of Amyloid-Beta 42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. U. S. A. 110, 9758–9763. 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J.; Waudby C. A.; Devlin G. L.; Cohen S. I. A.; Aguzzi A.; Vendruscolo M.; Terentjev E. M.; Welland M. E.; Dobson C. M. (2009) An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 326, 1533–1537. 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- Friedhoff P.; Schneider A.; Mandelkow E. M.; Mandelkow E. (1998) Rapid Assembly of Alzheimer-Like Paired Helical Filaments from Microtubule-Associated Protein Tau Monitored by Fluorescence in Solution. Biochemistry 37, 10223–10230. 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- Zhu H. L.; Fernandez C.; Fan J. B.; Shewmaker F.; Chen J.; Minton A. P.; Liang Y. (2010) Quantitative Characterization of Heparin Binding to Tau Protein: Implication for Inducer-Mediated Tau Filament Formation. J. Biol. Chem. 285, 3592–3599. 10.1074/jbc.M109.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G.; Udgaonkar J. B. (2011) Understanding the Kinetic Roles of the Inducer Heparin and of Rod-Like Protofibrils During Amyloid Fibril Formation by Tau Protein. J. Biol. Chem. 286, 38948–38959. 10.1074/jbc.M111.271874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S.; Davies P.; Mandelkow E. (2004) Tau Paired Helical Filaments from Alzheimer’s Disease Brain and Assembled in Vitro Are Based on Beta-Structure in the Core Domain. Biochemistry 43, 1694–1703. 10.1021/bi0357006. [DOI] [PubMed] [Google Scholar]
- Ksiezak-Reding H.; Wall J. S. (2005) Characterization of Paired Helical Filaments by Scanning Transmission Electron Microscopy. Microsc. Res. Tech. 67, 126–140. 10.1002/jemt.20188. [DOI] [PubMed] [Google Scholar]
- Nizynski B.; Dzwolak W.; Nieznanski K. (2017) Amyloidogenesis of Tau Protein. Protein Sci. 26, 2126–2150. 10.1002/pro.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronesi O. C.; Becker S.; Seidel K.; Heise H.; Young H. S.; Baldus M. (2005) Determination of Membrane Protein Structure and Dynamics by Magic-Angle-Spinning Solid-State Nmr Spectroscopy. J. Am. Chem. Soc. 127, 12965–12974. 10.1021/ja0530164. [DOI] [PubMed] [Google Scholar]
- Foscarin S.; Raha-Chowdhury R.; Fawcett J. W.; Kwok J. C. F. (2017) Brain Ageing Changes Proteoglycan Sulfation, Rendering Perineuronal Nets More Inhibitory. Aging 9, 1607–1622. 10.18632/aging.101256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyzi E.; Saldeen T.; Larsson E.; Lindahl U.; Salmivirta M. (1998) Age-Dependent Modulation of Heparan Sulfate Structure and Function. J. Biol. Chem. 273, 13395–13398. 10.1074/jbc.273.22.13395. [DOI] [PubMed] [Google Scholar]
- Kfoury N.; Holmes B. B.; Jiang H.; Holtzman D. M.; Diamond M. I. (2012) Trans-Cellular Propagation of Tau Aggregation by Fibrillar Species. J. Biol. Chem. 287, 19440–19451. 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M.; Guo J. L.; McBride J. D.; Zhang B.; Trojanowski J. Q.; Lee V. M. Y. (2013) Synthetic Tau Fibrils Mediate Transmission of Neurofibrillary Tangles in a Transgenic Mouse Model of Alzheimer’s-Like Tauopathy. J. Neurosci. 33, 1024–1037. 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel C. H.; Kumar S.; Pinotsi D.; Tunnacliffe A.; St. George-Hyslop P.; Mandelkow E.; Mandelkow E.-M.; Kaminski C. F.; Kaminski Schierle G. S. (2014) Extracellular Monomeric Tau Protein Is Sufficient to Initiate the Spread of Tau Protein Pathology. J. Biol. Chem. 289, 956–967. 10.1074/jbc.M113.515445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart K. L.; Hughes E.; Yates E. A.; Middleton D. A.; Radford S. E. (2017) Molecular Origins of the Compatibility between Glycosaminoglycans and a Beta 40 Amyloid Fibrils. J. Mol. Biol. 429, 2449–2462. 10.1016/j.jmb.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hricovini M.; Hricovini M. (2018) Solution Conformation of Heparin Tetrasaccharide. Dft Analysis of Structure and Spin-Spin Coupling Constants. Molecules 23, 3042. 10.3390/molecules23113042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd T. R.; Guimond S. E.; Skidmore M. A.; Duchesne L.; Guerrini M.; Torri G.; Cosentino C.; Brown A.; Clarke D. T.; Turnbull J. E.; Fernig D. G.; Yates E. A. (2007) Influence of Substitution Pattern and Cation Binding on Conformation and Activity in Heparin Derivatives. Glycobiology 17, 983–993. 10.1093/glycob/cwm062. [DOI] [PubMed] [Google Scholar]
- Hricovini M.; Tvaroska I. (1990) Conformation Dependence of Th Eone-Bond Carbon Proton Coupling-Constants in Oligosaccharides. Magn. Reson. Chem. 28, 862–866. 10.1002/mrc.1260281005. [DOI] [Google Scholar]
- Tvaroska I.; Taravel F. R. (1992) One-Bond Carbon Proton Coupling Constants - Angular Dependence in Beta-Linked Oligosaccharides. J. Biomol. NMR 2, 421–430. 10.1007/BF02192806. [DOI] [PubMed] [Google Scholar]
- Yates E. A.; Santini F.; De Cristofano B.; Payre N.; Cosentino C.; Guerrini M.; Naggi A.; Torri G.; Hricovini M. (2000) Effect of Substitution Pattern on H-1, C-13 Nmr Chemical Shifts and (1)J(Ch) Coupling Constants in Heparin Derivatives. Carbohydr. Res. 329, 239–247. 10.1016/S0008-6215(00)00144-0. [DOI] [PubMed] [Google Scholar]
- Rudd T. R.; Yates E. A. (2010) Conformational Degeneracy Restricts the Effective Information Content of Heparan Sulfate. Mol. BioSyst. 6, 902–908. 10.1039/b923519a. [DOI] [PubMed] [Google Scholar]
- Maiza A.; Chantepie S.; Vera C.; Fifre A.; Huynh M. B.; Stettler O.; Ouidja M. O.; Papy-Garcia D. (2018) The Role of Heparan Sulfates in Protein Aggregation and Their Potential Impact on Neurodegeneration. FEBS Lett. 592, 3806–3818. 10.1002/1873-3468.13082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.