

Abstract
Colorectal cancer is the third leading cause of cancer related-death in the United States. Search for new alternatives to treat this type of cancer is necessary. In a previous report, auransterol from Penicillium aurantiacobrunneum showed cytotoxicity in HT-29 cancer cells. Thus, the goal of this study was to examine the potential cytotoxic mechanism of auransterol in HT-29 cells. Real-time cytotoxicity of auransterol was determined in HT-29 colon cancer cells, using the SRB assay. Loss of MTP, overproduction of ROS, cell cycle, cell migration, and caspase activity were analyzed. Western blot analysis was used to evaluate protein expression. Auransterol reduced cell proliferation rate in a time and concentration-dependent manner, with an IC50 value > 100, 49.1 and 23.8 μM at 24, 48 and 72 h of treatment, respectively. After 24 h of treatment, 50 μM of auransterol induced loss of MTP, overproduction of ROS, increased caspase activity, induced cell cycle G1-phase accumulation and inhibition of migration in HT-29 cells compared to control. These results were supported by protein upregulation of Cyt c, BAX, PARP-1, p21 and procaspase-3, and downregulation of Bcl-2 with no modifications in procaspase-7 and p53. The cytotoxic effect of auransterol in HT-29 colon cancer cells is mediated by mitochondrial apoptosis independent of p53 activation, cell cycle G1 phase arrest, and inhibition of cell migration. This work encourages further preclinical and clinical studies of auransterol and suggests auransterol as a good candidate for colorectal cancer treatment.
Keywords: Auransterol, HT-29 cells, intrinsic apoptosis, toxicity
1. Introduction
Colorectal cancer is the third leading cause of cancer related-death in the United States. In 2019, in men and women deaths due to colorectal cancer were estimated as 27 640 and 23 380, respectively, and, a similar number of deaths are estimated for 2020 [1]. It is expected around 150 000 new cases of this cancer for 2020 in the United States [2]. In patients with colon cancer under last stages (III-IV), colonoscopy and colectomy are not enough to delay cancer development; thus, chemotherapy is commonly used as adjunct to surgical interventions to increase in patients chances of survival. Among drugs commonly used are: 5-fluorouracil, capecitabine, irinotecan, oxaliplatin, trifluridine and tipiracil [2]. However, cellular abnormalities in apoptosis (a form of programed cell death) of colorectal cancer contributes to chemoresistance development, which is an important factor that limit the efficacy of anticancer drugs [3]. Nowadays, it is necessary to discover new drugs that help to overcome and improve the survival of patients with colorectal cancer.
There are two major pathways that induce apoptosis and activate a group of cysteine-aspartic proteases called caspases [4]. The first one, known as extrinsic apoptosis is activated by an external stimuli and act primarily in the death receptors family. The second one is also called mitochondrial apoptosis; it is triggered and regulated by the B cell lymphoma (BCL-2) family [5]. The pro- or anti-apoptotic protein members of this family are activated by an internal stimulus in response to cellular stress. The activation of pro-apoptotic members, like BAX and BAK, provokes the releasing of cytochrome c (Cyt c) to the cytosol, then, apoptosome formation started with Cyt c binding to Apaf-1 and procaspase-9, resulting in the activation of caspase-9, to finally activate caspase effectors (caspase-3 and-7) and get cell death process [5,6]. In cancer cells, it has been observed an upregulation of anti-apoptotic BCL-2 proteins like Bcl-2, Bcl-W, Bcl-xL, Bfl-1 and Mcl-1, and downregulation of pro-apoptotic members [7]. Probably, intrinsic apoptosis is the most common mode to induce programmed cell death. Thus, a strategy to induce apoptosis in cancer cells is the regulation of BCL-2 family.
Nowadays, natural products, especially fungi derivatives continue to be useful source for development of new anticancer lead drugs. Auransterol, a natural compound isolated from Penicillium aurantiacobrunneum, belongs to the sterol family, which many members have been demonstrated their anticancer activity [8, 9]. In the present study, the antiproliferative effect and a preliminary analysis of the mode of action of auransterol was investigated in HT-29 colorectal cancer cells.
2. Materials and methods
2.1. Reagents
Paclitaxel, dimethylsulfoxide (DMSO), cycloheximide, trichloroacetic acid, sulforhodamine B (SRB), propidium iodide (PI), and 2´,7´-dichlorofluorescein-diacetate (DCFH-DA) were purchased from Sigma-Aldrich. The mitochondrial transmembrane potential (MTP) assay kit was purchased from Cayman Chemical Company. RNAse was purchased from Invitrogen. Caspase-Glo 3/7® kit was purchased from Promega corporation. Roswell Park Memorial Institute Media (RPMI-1640), fetal bovine serum (FBS) and antibiotic-antimycotic were purchased from Gibco. PhosphoSafe™ was purchased from Novagen. Pierce™ BCA Protein assay kit was obtained from Thermo Fisher Scientific. Primary antibodies: anti-procaspase-3 (9662), anti-procaspase-7 (9492), anti-Bcl-2 (15071), anti-PARP-1 (5625), anti-cytochrome c (11940), anti-BAX (2772) anti-p21 Waf1/Cip1 (12D1) and anti-p53 (9282) were purchased from Cell Signaling Technologies. Anti-rabbit or anti-mouse horseradish peroxidase (HRP)-conjugated antibody and anti-β-actin (sc-130656) were purchased from Santa Cruz Biotechnology, Inc.
2.2. Sample and preparation
Auransterol was obtained according to a previously reported procedure [10] (Fig. 1A). Auransterol and controls were dissolved in DMSO 100% at 4 or 10 mg/mL. Stocks were stored at 4 °C until use. Auransterol was tested at 0, 12.5, 25 and 50 μM, and paclitaxel was used as positive control at 0.01 μM. The final DMSO concentration in treated cells was 0.2 %.
Fig. 1.
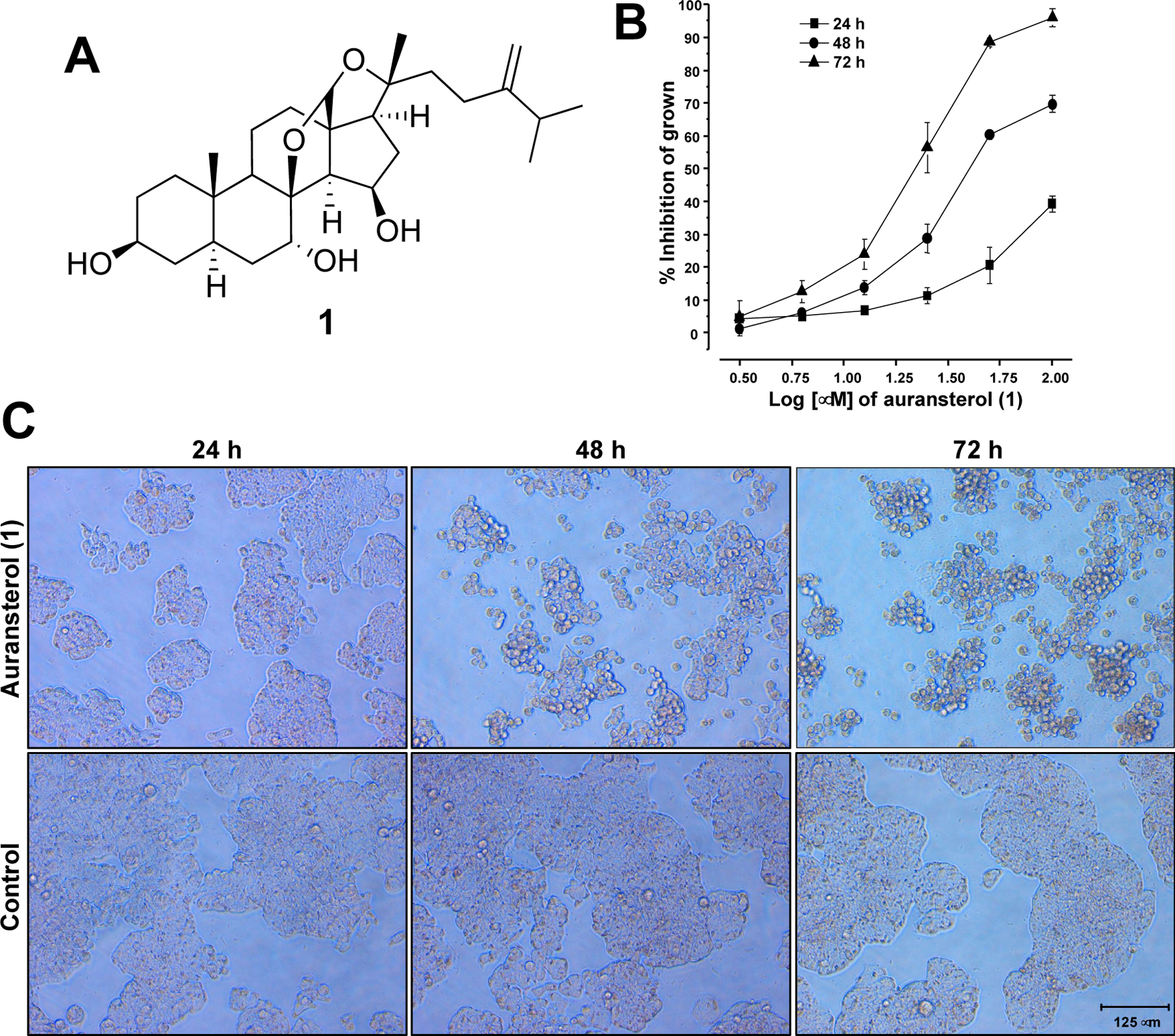
(A) Structure of auransterol. (B) Cytotoxic activity of auransterol in HT-29 colon cancer cells after 24, 48 and 72 h of treatment using the SRB assay. (C) Representative microscopic view (magnification 20x) of cells after 24, 48 and 72 h of auransterol treatment at 25 μM. Each data is expressed as the mean ± SEM (n = 9) of three independent experiments with three repetitions each one.
2.3. Cell culture
The HT-29 human colorectal cancer cells were obtained from the American Type Culture Collection (Manassas, Virginia, USA). Cells were cultured in RPMI-1640 containing 10% FBS and 1% antibiotic-antimycotic. The cells were grown as a monolayer in T75 tissue culture flasks and kept at 37°C and in an atmosphere with 5% of CO2.
2.4. Cytotoxicity assay
To determine cytotoxicity of auransterol, the sulforhodamine B (SRB) assay was used [11]. Briefly, HT-29 cells were seeded and incubated overnight in a 96-well plate at 5 x 104 cells/mL. To obtain the IC50 (50% inhibitory concentration) value, cells were treated with auransterol or paclitaxel at different concentrations for 24, 48 or 72 h at 37°C and 5% CO2. Then, cells were fixed with trichloroacetic acid (20%), washed, and stained with SRB (0.4%). After removing excess of dye and drying, Tris base (10 mM) was added and, absorbance (515 nm) was read in FLUOstar Optima plate reader (BMG Labtech Inc., Durham, NC, USA). IC50 values were calculated by non-linear regression analysis, using TableCurve 2Dv4 (System Software Inc., San Jose, CA, USA).
2.5. Mitochondrial transmembrane potential assay
The outer mitochondrial membrane potential (ΔΨm) was assessed using JC-1 (5,5´,6,6´-tetrachloro-1,1´,3,3´-tetraethyl-benzimidazoylcarbocyanine iodide) Mitochondrial Membrane Potential Assay Kit. HT-29 cells were seeded and treated 24 h as described above. After treatment, fluorescent dye JC-1 (10 μL) was administrated and incubated. Then, fluorescence values were determined in a FLUOstar Optima plate reader. Healthy cells were detected at an excitation and emission wavelength of 535/595 nm. Unhealthy cells were detected at an excitation and emission wavelength of 485/535 nm. Data is presented as the ratio of healthy/unhealthy cells. Paclitaxel was used as positive control at 0.01 μM.
2.6. Reactive oxygen species assay
The reactive oxygen species (ROS) overproduction was evaluated using the fluorescent 2´,7´-dichlorofluorescein-diacetate (DCFH-DA) probe in HT-29 colon cancer cells. After 24 h of treatment with auransterol (0–50 μM) or paclitaxel (0.01 μM) or vitamin c (50 μM), as above described, cells were incubated with DCFH-DA (10 μM). Fluorescence was measured using a FLUOstar Optima plate reader with an excitation and emission wavelength of 485 and 530 nm, respectively. All treatments were performed in triplicate and data are representative of two independent experiments [11].
2.7. Western blot analysis
To determine protein expression, western blot analysis was performed. Briefly, after 24 h of treatment with auransterol (0–50 μM) or paclitaxel (0.01 μM), cells were lysed with 70 μL of Phosphosafe™, then, protein concentration (20 μg) was calculated by BCA protein analysis. Electrophoresis was performed using Nu-PAGE 10% SDS-PAGE Bis-Tris gel in SDS-PAGE buffer. Polyvinylidene fluoride membrane (PVDF) was used for transfer. Next, membrane was blocked with bovine serum albumin (3%). Afterwards, membranes were washed with tris buffered saline containing tween 20 (TBST) and incubated overnight with primary antibody (procaspase-3, procaspase-7, Bcl-2, PARP-1, cytochrome c, BAX, p21 Waf1/Cip1 (12D1), p53 and β-actin) diluted 1:1000. After that, membrane was washed three times with TBST and secondary antibody (1:1000) was added to be incubated for 1 h and washed with TBST. Then, band intensities were detected using chemiluminescent substrate Supersignal Femto kit and band densities were analyzed using ImageJ 1.52a program (Bethesda, Maryland, USA ) [11].
2.8. Caspase-3 and −7 activity
Activity of caspase-3 and −7 was measured using the Caspase-Glo 3/7® kit. HT-29 cells were seeded in a white 96 well plate and treated for 24 h with auransterol (0–50 μM) or paclitaxel (0.01 μM) used as a positive control. Then, equal volume of Caspase-Glo 3/7® reagent was added and incubated for 2 h. Afterwards, luminescence was measured using a FLUOstar Optima plate reader.
2.9. Cell cycle analysis
Cell cycle analysis was carried out using the fluorescent intercalating agent propidium iodide (PI) method. Briefly, HT-29 cells were seeded on 6-cm plates and treated with auransterol (0–50 μM) or paclitaxel (0.01 μM) for 24 h. Cells washed with phosphate-buffered saline (PBS) were treated with trypsin, and re-suspended in cold water. Then, cells were fixed with ethanol (70%), washed, and re-suspended in PBS. The RNAse (20 μg/mL) was then added to cells and incubated for 1 h to add PI (1.0 μg/mL) prior reading. The analysis was made using a BD FACS Calibur cytometer at an excitation wavelength of 488 nm and an emission wavelength of 617 nm. The cellular DNA profile was analyzed using the software WinMDI version 2.8 (BD, USA) [11].
2.10. Cell migration assay
Cell migration was measured using the wound-healing assay. In brief, cells were seeded in 24 well plate and using a sterile 200 μL pipette tip a scratch wound was shaped. HT-29 cells were treated 48 h with auransterol (0–50 μM) or paclitaxel (0.01 μM) in RPMI-1640. At 24 or 48 h post treatment images of wound recovery were taken with Axiovert 40 CFL Zeiss microscope (magnifier Zeiss CP-Achromat 5x/0.12) and ProgRes C10 plus camera. Wound recovery percent was calculated using the ImageJ 1.52a program.
2.11. Statistical Analysis
All data are presented as the mean ± standard error of the mean (SEM). Each assay was carried out in triplicate of two or three independent experiments. TableCurve 2D 4v (System Software Inc., San Jose, CA, USA) was used for statistical evaluations to obtain IC50 values. Statistical significance values were determined by an analysis of variance (ANOVA) followed by a post-hoc Dunett’s test. These analyses were made using GraphPad Prism 5.0 software (San Diego, California, USA). A value of *p < 0.05, **p < 0.01, ***p < 0.001 indicated significant differences between groups against the controls.
3. Results
3.1. Auransterol suppressed HT-29 cell proliferation
To investigate the antiproliferative effect of auransterol (Fig. 1A) in HT-29 colon cancer cells, the SRB assay was performed. Antiproliferative effect was recorded at 24, 48 and 72 h posttreatment, as shown in Fig. 1B. Auransterol suppressed proliferation of HT-29 cells in a concentration-dependent manner. After 24 h of auransterol treatment at 100 μM, the growth inhibition was 39.4% (IC50 > 100 μM); after 48 h the IC50 value was 49.1 ± 3.2 μM, and after 72 h the IC50 value was 23.8 ± 0.98 μM. The IC50 of paclitaxel used as positive control was 0.001 μM (Table 1). Fig. 1C shows that auransterol decreased the number and proliferation of the cells. Also, these effects are accompanied by morphological changes such as shrinkage of the cytoplasm and cell rounding. These observations suggest an apoptotic effect of auransterol in HT-29 cells.
Table 1.
Cytotoxicity of auransterol against HT-29 colon cells.
| Compound | HT-29a |
|---|---|
| Auransterol (24 h) | ≥ 100 |
| Auransterol (48 h) | 49.1 ± 3.2 |
| Auransterol (72 h) | 23.8 ± 0.98 |
| Paclitaxelb (72 h) | 0.001 ± 0.01 |
IC50 values are the concentration (μM) required for 50% inhibition of cell viability after treatment. Data represent three independent experiments with three repetitions each one.
Positive control.
3.2. Auransterol induced loss of mitochondrial transmembrane potential (MTP) in HT-29 cells
Next, to examine the effects of auransterol on intrinsic apoptosis, the mitochondrial transmembrane potential was evaluated using the JC-1 staining assay. The results in Fig. 2A showed, that after 24 h of treatment with 50 μM of auransterol (P < 0.05), HT-29 cells loss MTP as the positive control paclitaxel (0.01 μM, P < 0.001), when compared with the control. These results suggest that intrinsic apoptotic pathway may be involved in the antiproliferative effect of auransterol in HT-29 cells.
Fig. 2.
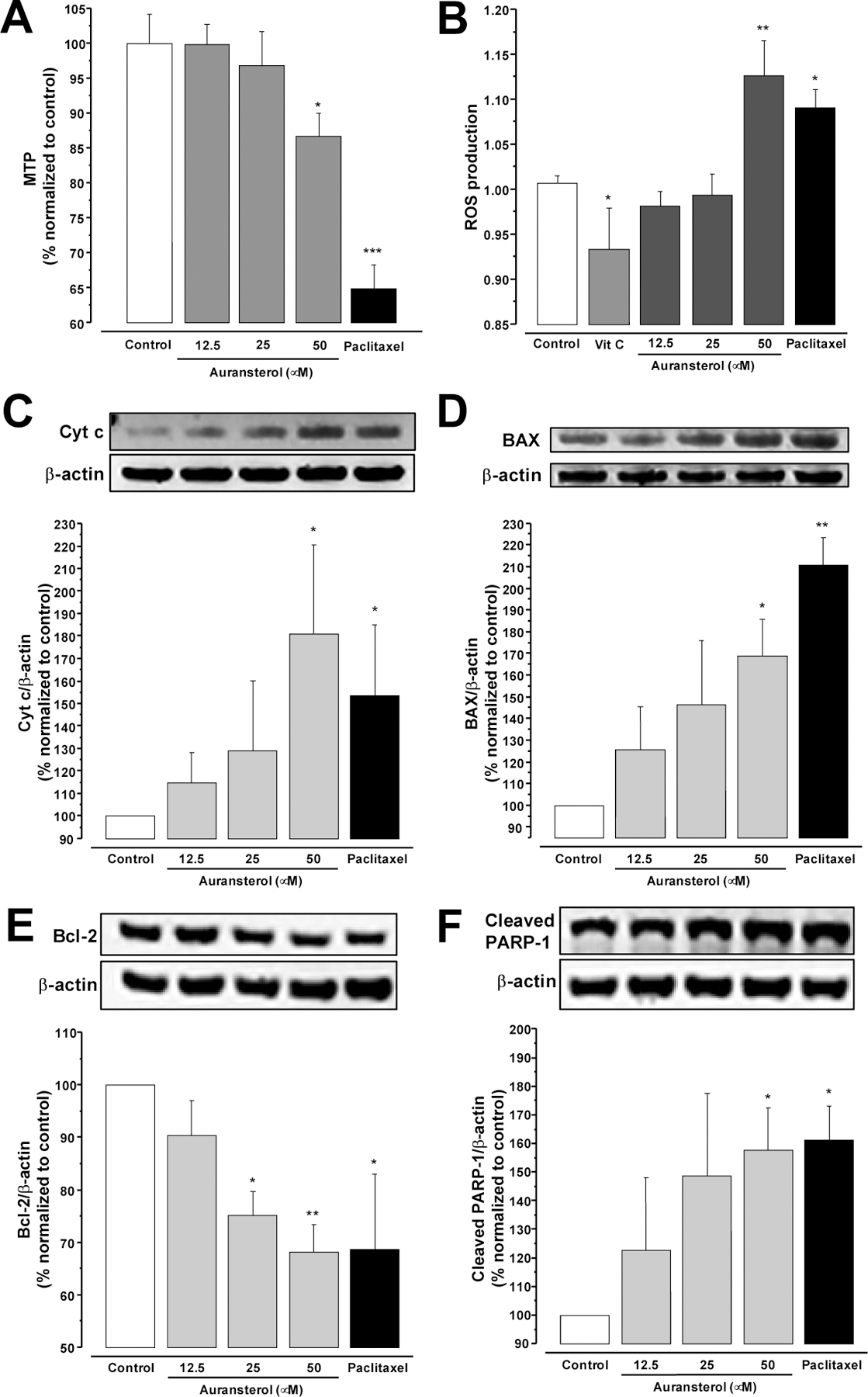
Effects of auransterol on intrinsic-apoptosis pathway in HT-29 colon cancer cells, (A) Effects on mitochondrial transmembrane potential (MTP). (B) Effects on overproduction of reactive oxygen species (ROS). (C) Cyt c, (D) BAX, (E) Bcl-2, and (D) cleaved PARP-1 protein levels. Cells were treated with auransterol (0–50 μM) for 24 h and protein concentration was determined for Western blot analysis. Paclitaxel was used as positive control (0.01 μM). Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (*P < 0.05, **P < 0.01, ***P < 0.01) of three independent assays.
3.3. Auransterol induced overproduction of reactive oxygen species (ROS) in HT-29 cells
In order to determinate whether auransterol exerts its cytotoxic effect by regulation of overproduction of ROS in HT-29 cells, the fluorescent DCFH-DA assay was used. Fig. 2B shows that, after 24 h of treatment with 50 μM of auransterol or paclitaxel (0.01 μM, positive control), a significance in overgeneration of ROS was recorded in HT-29 cells compared to the control (P < 0.01 and P < 0.05, respectively). However, vitamin C (50 μM) used as negative control, reduced ROS production (P < 0.05). These results indicate that overproduction of ROS with increasing concentrations of auransterol could have contributed to the death of HT-29 cells.
3.4. Auransterol regulated expression of some intrinsic apoptotic proteins in HT-29 cells
Because auransterol induced loss of MTP and overproduction of ROS, next experiments were focused to confirm the role of auransterol in the intrinsic-apoptosis pathway, by using western blot analysis in HT-29 cells. Results in Fig. 2C, D and F exhibited significantly elevated cytochrome c (Cyt c), Bcl-2 associated X (BAX) and cleaved poly (ADP-ribose) polymerase 1 (PARP-1) protein expression levels (P < 0.05) in HT-29 cells, after 24 h treatment with auransterol at 50 μM and paclitaxel at 0.01 μM. Moreover, by contrast Fig. 2E shows that auransterol at 25 and 50 μM and paclitaxel (0.01 μM) induced a downregulation of Bcl-2 (P ≤ 0.05). Taken together these outcomes support that auransterol induced intrinsic apoptosis by regulation of a number of proteins of BCL-2 family in HT-29 cells.
3.5. Auransterol modulated activity and expression of caspase executors in HT-29 cells
To further evaluate whether auransterol is involved in the activation or regulation of caspases, a luminescence assay and western blot analysis were employed. After 24 h of treatment in HT-29 cells, auransterol (50 μM) induced significant caspase-3 and −7 activity (Fig. 3A, P < 0.01), when compared with the control. Same activity was observed with paclitaxel (0.01 μM) that was used as positive control (P < 0.001). These results agree with the immunoblot experiments. Fig. 3B showed that 50 μM of auransterol and paclitaxel (0.01 μM) upregulated protein expression of procaspase-3 (P ≤ 0.01), but no modifications on procaspase-7 were detected (Fig. 3C). Thus, these results suggest that auransterol modulates caspase activity and protein synthesis in HT-29 colon cells.
Fig. 3.
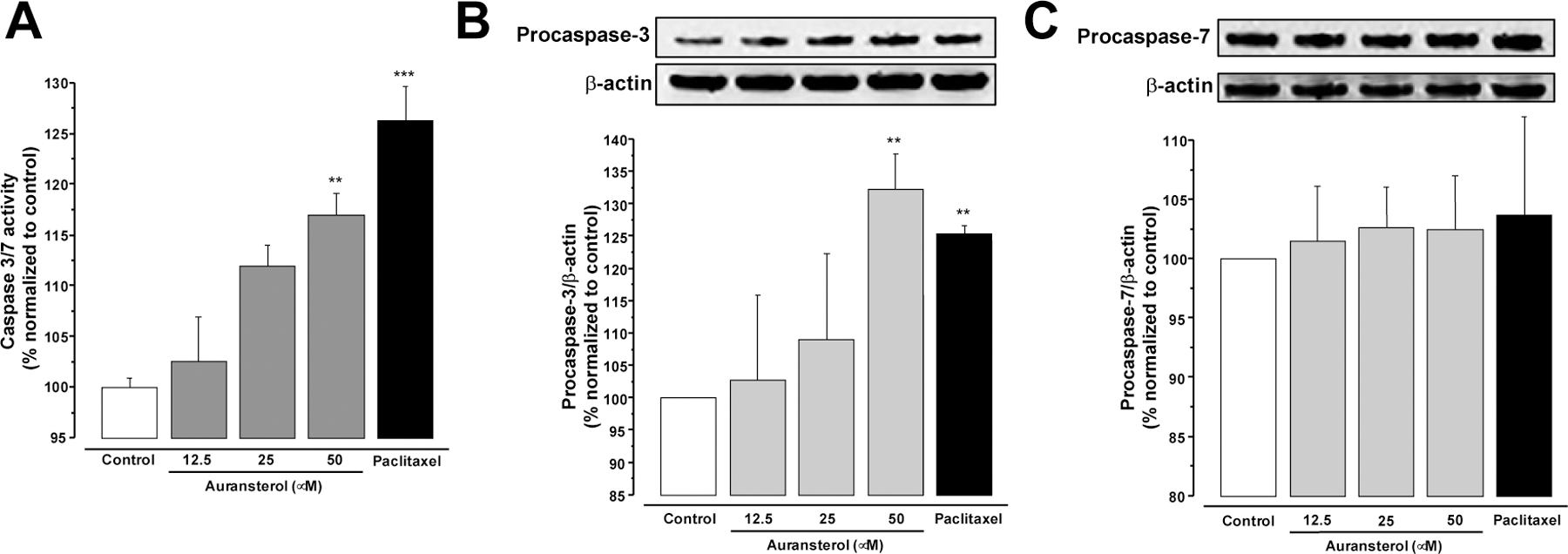
Effects of auransterol on caspase activity in HT-29 colon cancer cells. (A) Effects of auransterol (0–50 μM) on activity of caspase 3 and 7 after 24 h of treatment. (B) Protein expression of procaspase-3 and (C) procaspase-7. Paclitaxel was used as positive control (0.01 μM). Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (**P < 0.01, ***P < 0.01) of three independent assays.
3.6. Auransterol induced G1 cell cycle arrest in HT-29 cells
To explore whether auransterol regulates the cell cycle progression of HT-29 colon cancer cells, flow cytometry analysis was performed. After 24 h incubation of untreated cells, the percent phase distribution in sub-G1, G1, S and G2 was 12.6 ± 4.7, 43.5 ± 2.4, 13.19 ±1.6 and 30.59 ± 2.6, respectively. The percentage of the cell population accumulated with 25 and 50 μM of auransterol were 5.9 ± 2.5, 52.5 ± 3, 13 ± 1.7, 28.4 ± 1.1 and 2.4 ± 1, 55.6 ± 1.4, 12.9 ± 1.1, 29 ± 2.1, respectively. In case of paclitaxel (0.01 μM) used as positive control, the distribution was 23.3 ± 2.2, 29.3 ± 0.7, 12.6 ± 1.4 and 34.6 ± 1, respectively. The results indicate that auransterol induced G1 phase arrest, compared to the control (Fig. 4A, P ≤ 0.05).
Fig. 4.
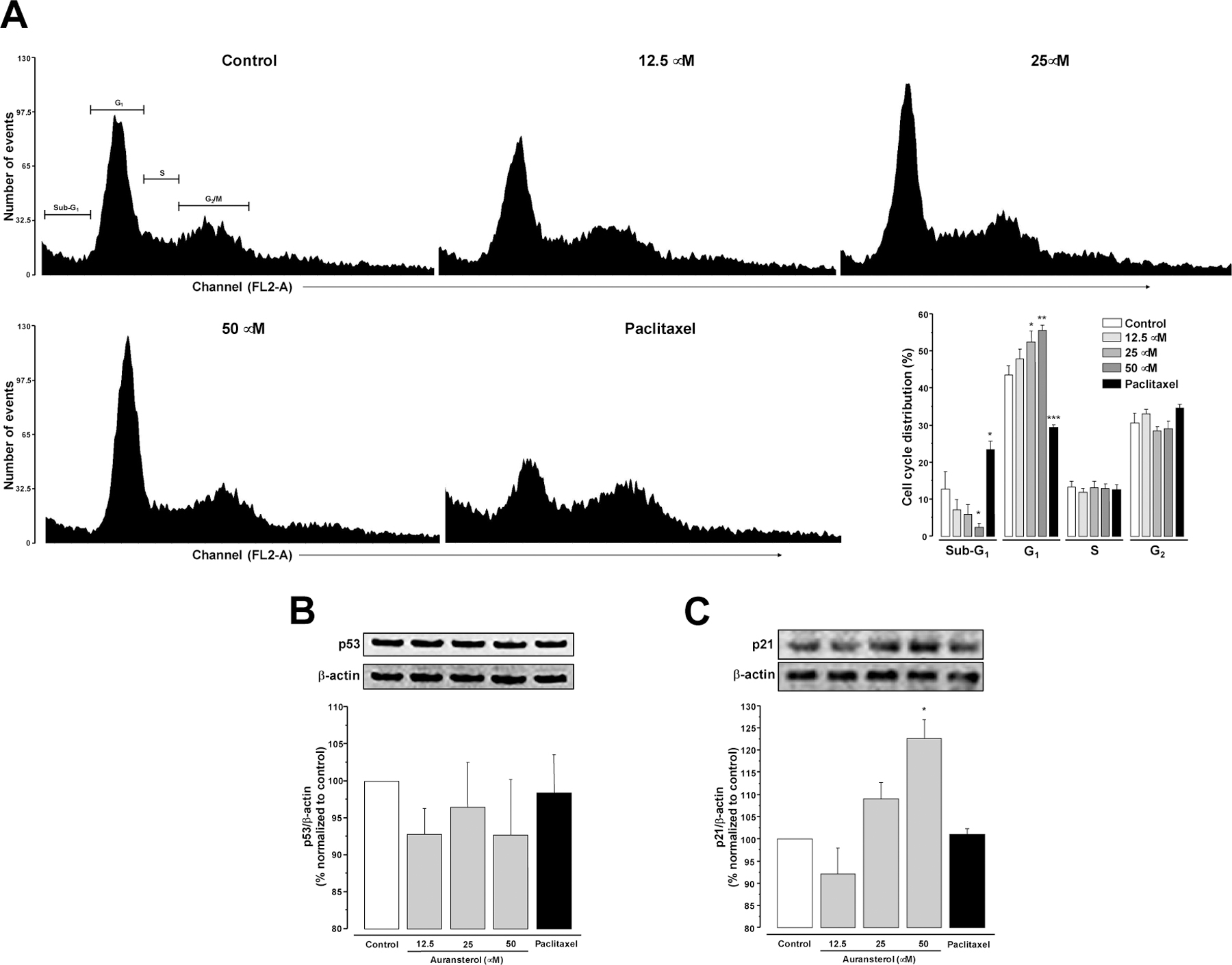
(A) Effect of auransterol on cell cycle distribution in HT-29 colon cancer cells. Cells were treated for 24 h with auransterol (0–50 μM). Data represent percentage of cell distribution in each cell cycle phase. (B) Protein expression of p53 and (C) p21. Paclitaxel (0.01 μM) was used as positive control. Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (*P < 0.05, **P < 0.01) of three independent assays.
3.7. Auransterol upregulated p21 in HT-29 cells
To further assess the effect of auransterol on p53 and p21 protein expression, western blot analysis was carried out. Fig. 4B shows that auransterol and paclitaxel do not modify the p53 tumor suppressor protein levels. But, the analysis of p21 protein expression indicated that auransterol (50 μM) upregulated this cyclin-dependent kinase inhibitor (P < 0.05). These outcomes suggest that auransterol induce cell cycle G1-phase arrest by activation of p21 independent of p53.
3.8. Auransterol inhibited migration of HT-29 cells
Since cell migration is one of the first steps involved in tumor metastasis [12]. The effect of auransterol in migration of HT-29 cells was investigated, by pursuing the wound healing assay. As shown in Fig. 5A, auransterol suppressed cell migration of HT-29 cells. The significant inhibitory effects on wound recovery of auransterol (25 and 50 μM, P ≤ 0.01) and paclitaxel (0.01 μM, P < 0.001) were observed after 24 h and compared to the control (Fig. 5B). At 48 h of treatment, only 50 μM of auransterol treated cells showed significant inhibition of cell migration (P < 0.001) as paclitaxel (0.01 μM, P < 0.001). These results suggest that auransterol may prevent the metastatic process in HT-29 cells.
Fig. 5.
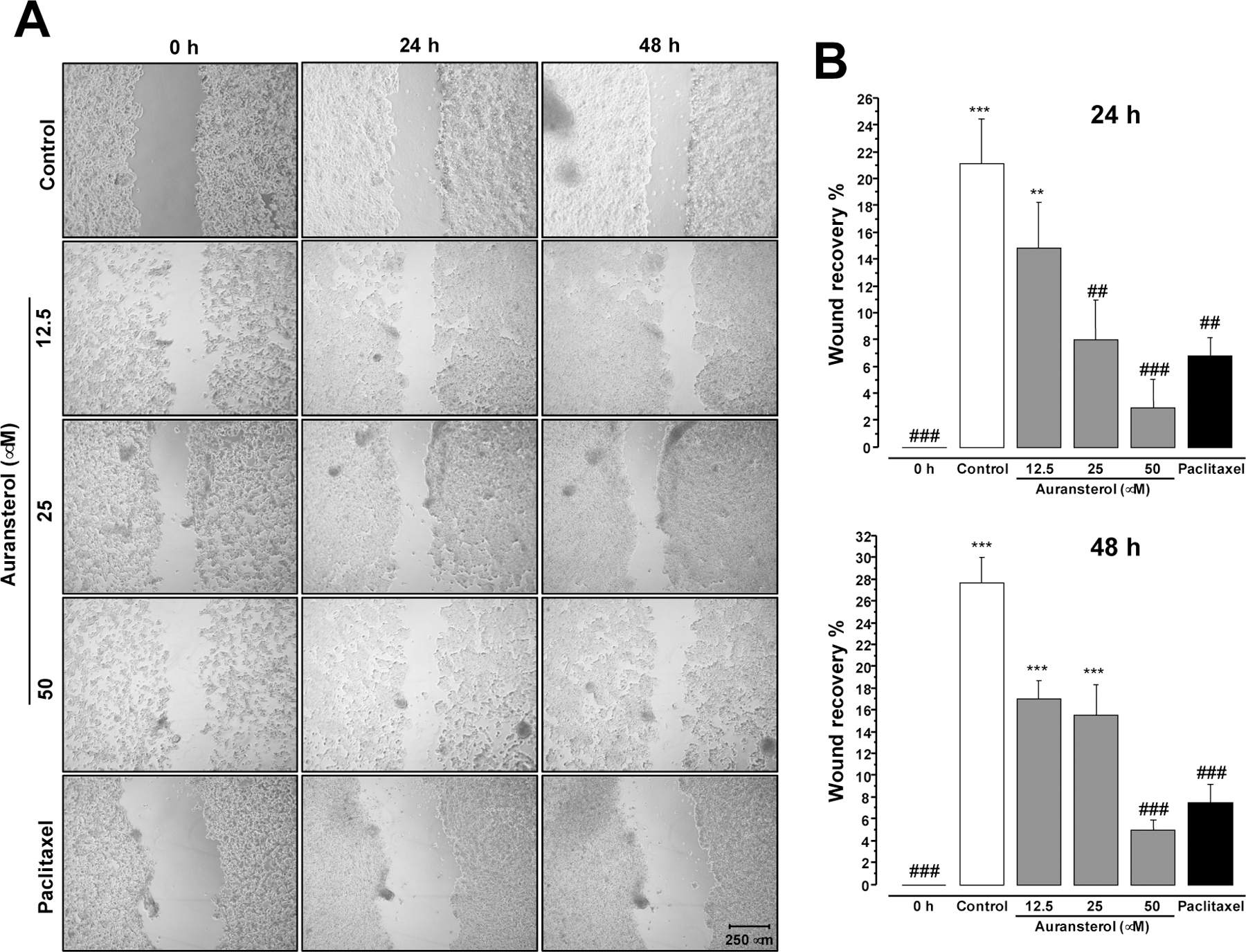
Effect of auransterol on HT-29 colon cells migration. (A) Microscopic view (magnifier Zeiss CP-Achromat 5x) of cells were treated 48 h with auransterol (0–50 μM) or paclitaxel (0.01 μM) on wound healing assay. Cell migration was monitored with 5 x magnification. (B) Quantitative analysis of cell migration into the scratch wound at 24 or 48 h post-scratch. Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (*P < 0.05, **P < 0.01, ***P < 0.01 compared to the 0 h; ##P < 0.01, ###P < 0.001 compared to the control) of three independent assays.
4. Discussion
In our previous study, the cytotoxicity of four compounds isolated from Penicillium aurantiacobrunneum of the lichen Niebla homalea were evaluated in a panel of cancer cells. One of these compounds, auransterol, showed cytotoxicity in HT-29 colon cancer cells [10]. In the present work, a preliminary elucidation of the cellular mechanism triggered by auransterol was explored in HT-29 cells. The results provide strong evidence that the cytotoxic effect of auransterol is mediated by activation of mitochondrial apoptosis. HT-29 cells treated with auransterol showed loss of MTP, overproduction of ROS, increased in caspase activity and upregulation of BAX, Cyt c, PARP-1 and procaspase-3 as well as downregulation of Bcl-2 and no modification in procaspase-7 protein. Additionally, auransterol inhibited cell migration and blocked cell cycle G1-phase with upregulation of p21, but no quantifiable change in p53 protein expression (Fig. 7).
Fig. 7.
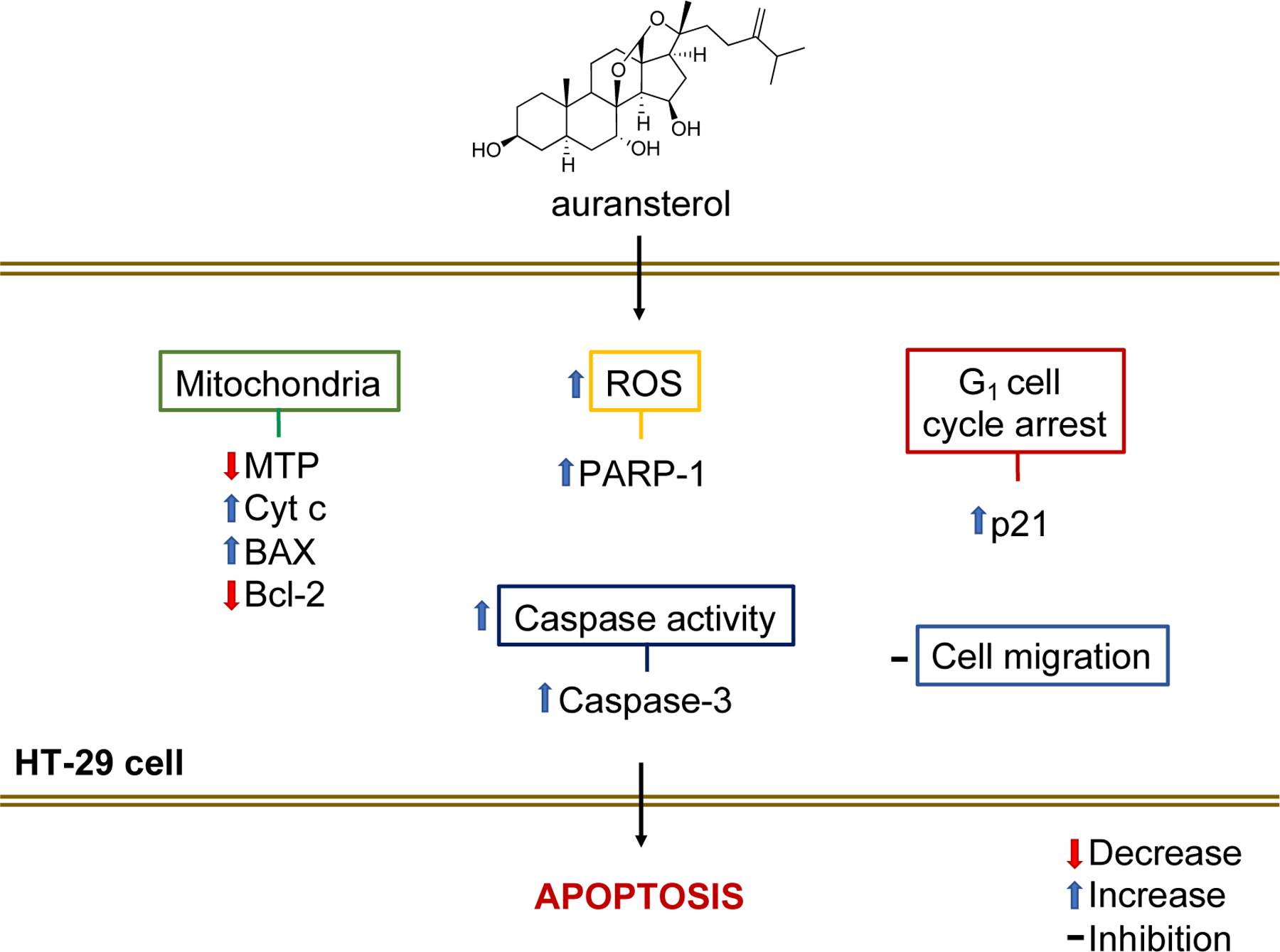
Hypothetic apoptosis-pathway induced by auransterol in HT-29 colon cells.
Secondary metabolites of Penicillium genus have demonstrated a broad range of biological activities including anticancer properties, with the most known compounds including the antifungal: griseofulvin, brefeldin A, 3-O-methylfunicone, chloctanspirone A, xanthocillin X mycophenolic acid, wortmannin, the antihyperlipidemic: lovastatin, compactin, emodin and dicatenarin, among others [13]. Despite the large number of fungal compounds with potential anticancer activity, few have been studied in depth and their mechanism of action is still unknown. Auransterol, is a compound of sterol family, of which many members have demonstrated anticancer activity [14,15], including the phytosterol rinoxiaB wich induced apoptotic cell death in colon cancer cells by targeting BAX/Bcl-2 pathway [16]. The most common sterols in plants with reported anticancer activity are stigmasterol, campesterol and β-sitosterol [17]. The polyvalent activity of β-sitosterol as an anticancer agent has been related to several cancer pathways [18]. In fungi, the most common sterols found with antiproliferative effect are ergosterol and ergosterol peroxide [19]. It has been found that ergosterol peroxide induce apoptosis by damage in the mitochondria when using A549 alveolar basal epithelial cancer cells [20]. To date, derivatives of natural products from fungi are generating greater interest in research as anticancer agents.
In this study, the results support that auransterol induced the activation of intrinsic apoptosis pathway in HT-29 colon cells. It is well known that stimuli independent to receptors activation like; DNA damage, stress in endoplasmic reticulum and overproduction of ROS, can start the induction of mitochondrial apoptosis [21]. Auransterol induced loss of mitochondrial transmembrane potential (MTP) and overproduction of ROS. In the cell, mitochondria and NADPH oxidases are the principal producers of ROS and, excess of ROS has been associated with damage of macromolecules and organelles like mitochondria itself [22]. Dysregulation in mitochondrial function can disrupt mitochondria outer membrane permeabilization (MOMP), which is mainly under control of BCL-2 protein family [23]. BCL-2 contains proapoptotic and antiapoptotic members. Many antiapoptotic proteins like Bcl-2, Bcl-xL and Mcl-1 are upregulated in colon cancer cells [24]. In brief, in cancer cells these anti-apoptotic proteins avoid MOMP by mutual inhibition and sequestration of BH3-only sensitizer (For example: BAD and NOXA) and BH3-only activators (BIM and BID) and pore formers BAX and BAK. Under many types of perturbations, including cellular stress or DNA damage, it induces the upregulation of BIM; this protein can activate BAX and inhibits anti-apoptotic members like Bcl-2. The pores formed by homo oligomerization of BAX allows the release of Cyt c [25]. In the cytoplasm, Cyt c interacts with apoptotic protease-activating factor 1 (APAF-1) to form the apoptosome, activating the initiator caspase-9, which in turn activate the effector caspase-3 [26]. Caspase effectors (caspase-3 and −7) are known to activate the execution phase and hallmarks of apoptosis, including typical morphological features [27]. Results in the present study demonstrated that auransterol upregulated BAX and Cyt c, increased the activity of caspase-3 and −7 and upregulated procaspase-3 protein. All together, these results suggested a possible mechanism of action for auransterol by induction of the intrinsic apoptotic pathway.
The ability to repair damage in cancer cells is one of the challenges to overcome in cancer treatment. The polymerization of nucleotides after DNA damage is performed by Poly (ADP-ribose) polymerase-1 (PARP-1). Thus, activation of PARP-1 helps cellular survival and induces apoptotic resistance [28]. Interestingly, the compound under investigation has shown an up regulation of PARP-1 in HT-29 cells, this effect could be due to the cellular response towards an increase in oxidative stress, displayed by loss of MTP and overproduction of ROS, also induced by auransterol. Further experiments are necessary to explore activity of PARP-1 after auransterol treatment.
A typical feature in cancer cells is the dysfunction of cell cycle machinery, which contributes to cellular overproliferation. Each phase in the cell cycle is regulated by cyclin-dependent kinases (CDKs) [29]. However, upregulation of cyclin-dependent kinase inhibitor p21 induce inhibition of the CDKs complex, thus prompting G1 phase arrest. Activation of p21 can be mediated by p53 or p53-independent pathway [30]. Data gathered from this study indicated that auransterol induced cell cycle arrest in G1 phase and upregulation of p21, with no modifications in p53 expression. Human HT-29 colorectal cancer cells have been described as p53 mutant [31], but in cancer cells, p21 activity is rarely affected [32], suggesting that activation of p21 by auransterol is p53-independent.
In cancer, the ability of cancer cells to migrate and invade surrounding tissues is closely related to cancer metastasis. Metastasis is the principal cause of patient death for those having cancer remission loss. Anticancer agents with inhibition of cell migration can help to avoid the proliferation of a secondary tumor and metastasis [12]. In this study, auransterol inhibited cell migration in HT-29 colon cancer cells; thus, there is a need for additional studies to analyze this effect further.
5. Conclusions
In conclusion, the present study demonstrated that the potential mechanism of cytotoxic effect induced by auransterol in HT-29 cells is mediated by activation of intrinsic apoptosis pathway, cell cycle arrest in G1 phase independent of p53, and also inhibition of cell migration. Thus, these results suggest that auransterol could be useful as potential new lead for colorectal cancer treatment.
Acknowledgement
We are grateful with Mr. Nathan Ezzone, OSU College of Pharmacy for helping in performing in vitro assays.
Funding
Isolation of auransterol was supported by a supplement of a program project grant from the National Cancer Institute (3P01 CA125066-10S1). The biological studies were supported by the main parent grant P01 CA125066.
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019, C.A. Cancer J. Clin 69 (1) (2019) 7–34. [DOI] [PubMed] [Google Scholar]
- [2].Treating colorectal cancer, American Cancer Society. https://www.cancer.org/cancer/colon-rectal-cancer/treating.html, 2019. (accessed 4 october 2019).
- [3].Wang D, Yang L, Yu W, Wu Q, Lian J, Li F, Liu S, Li A, He Z, Liu J, Sun Z, Yuan W, Zhang Y, Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-κB signaling, J. Immunother. Cancer 7 (1) (2019) 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sharma A, Boise LH, Shanmugam M, Cancer metabolism and the evasion of apoptotic cell death, Cancers. 11 (8) (2019) 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Singh R, Letai A, Sarosiek K, Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins, Nat. Rev. Mol. Cell. Biol 20 (3) (2019) 175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Letai A, Apoptosis and cancer, Annu. Rev. Cancer Biol 1 (2017) 275–294. [Google Scholar]
- [7].Campbell KJ, Tait SWG, Targeting BCL-2 regulated apoptosis in cancer, Open Biol 8 (5) (2018) 180002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koul M, Singh S, Penicillium spp.: prolific producer for harnessing cytotoxic secondary metabolites, Anticancer Drugs 28 (1) (2017) 11–30. [DOI] [PubMed] [Google Scholar]
- [9].Anaya-Eugenio GD, Ali T, Rakotondraibe LH, Carcache de Blanco E, Cytotoxic constituents from Penicillium concentricum, an endophytic fungus from Trichocolea tomentella, Anticancer Drugs 30 (4) (2019) 323–329. [DOI] [PubMed] [Google Scholar]
- [10].Tan CY, Wang F, Anaya-Eugenio GD, Gallucci JC, Goughenour KD, Rappleye CA, Spjut RW, Carcache de Blanco EJ, Kinghorn AD, Rakotondraibe LH, α-Pyrone and sterol constituents of Penicillium aurantiacobrunneum, a fungal associate of the lichen Niebla homalea, J. Nat. Prod 82 (9) (2019) 2529–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Anaya-Eugenio GD, Addo EM, Ezzone N, Henkin JM, Ninh TN, Ren Y, Soejarto DD, Kinghorn AD, Carcache de Blanco EJ, Caspase-dependent apoptosis in prostate cancer cells and zebrafish by corchorusoside C from Streptocaulon juventas, J. Nat. Prod 82 (6) (2019) 1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bravo-Cordero JJ, Hodgson L, Condeelis J, Directed cell invasion and migration during metastasis, Curr. Opin. Cell Biol 24 (2) (2012) 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bladt TT, Frisvad JC, Knudsen PB, Larsen TO, Anticancer and antifungal compounds from Aspergillus, Penicillium and other filamentous fungi, Molecules 18 (9) (2013) 11338–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roussi S, Winter A, Gosse F, Werner D, Zhang X, Marchioni E, Geoffroy P, Miesch M, Raul F, Different apoptotic mechanisms are involved in the antiproliferative effects of 7β-hydroxysitosterol and 7β-hydroxycholesterol in human colon cancer cells, Cell Death Differ 12 (2005) 128–135. [DOI] [PubMed] [Google Scholar]
- [15].Kloudova A, Guengerich FP, Soucek P, The role of oxysterols in human cancer, Trends Endocrinol. Metab 28 (7) (2017) 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gajendran B, Durai P, Madhu VK, Chinnasamy A, A novel phytosterol isolated from Datura inoxia, RinoxiaB is a potential cure colon cancer agent by targeting BAX/Bcl2 pathway, Bioorg. Med. Chem 28 (2) (2020) 115242. [DOI] [PubMed] [Google Scholar]
- [17].Shahzad N, Khan W, Ali A, Saluja SS, Sharma S, Al-Allaf FA, Abduljaleel Z, Ibrahim IAA, Abdel-Wahab AF, Afify MA, Al-Ghamdi SS, Phytosterols as a natural anticancer agent: Current status and future perspective, Biomed. Pharmacother 88 (2017) 786–794. [DOI] [PubMed] [Google Scholar]
- [18].Bin Sayeed MS, Ameen SS, Beta-Sitosterol: A Promising but Orphan Nutraceutical to Fight Against Cancer, Nutr. Cancer. 67 (8) (2015) 1214–1220. [DOI] [PubMed] [Google Scholar]
- [19].Dos Santos ACD, Couzinet-Mossion A, Ruiz N, Lakhdar F, Etahiri S, Bertrand S, Ory L, Roussakis C, Pouchus YF, Nazih E-H, Wielgosz-Collin G, Steroids from marine-derived fungi: Evaluation of antiproliferative and antimicrobial activities of eburicol, Mar Drugs 17 (6) (2019) 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wu H-Y, Yang F-L, Li L-H, Rao YK, Ju T-C, Wong W-T, Hsieh C-Y, Pivkin MV, Hua K-F, Wu S-H, Ergosterol peroxide from marine fungus Phoma sp. induces ROS-dependent apoptosis and autophagy in human lung adenocarcinoma cells, Sci. Rep 8 (1) (2018) 17956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Galluzi L, Vitale I, et al. , Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ 25 (3) (2018) 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Redza-Dutordoir M, Averill-Bates DA, Activation of apoptosis signalling pathways by reactive oxygen species, Biochim. Biophys. Acta 1863 (12) (2016) 2977–2992. [DOI] [PubMed] [Google Scholar]
- [23].Kalkavan H, Green DR, MOMP, cell suicide as a BCL-2 family business, Cell Death Differ 25 (1) (2018) 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Scherr A-L, Gdynia G, Salou M, Radhakrishnan P, Duglova K, Heller A, Keim S, Kautz N, Jassowicz A, Elssner C, He YW, Jaeger D, Heikenwalder M, Schneider M, Weber A, Roth W, Schulze-Bergkamen H, Koehler BC, Bcl-xL is an oncogenic driver in colorectal cancer, Cell Death Dis 7 (8) (2016) e2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kale J, Osterlund EJ, Andrews DW, BCL-2 family proteins: changing partners in the dance towards death, Cell Death Differ 25 (1) (2018) 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dorstyn L, Akey CW, Kumar S, New insights into apoptosome structure and function, Cell Death Differ 25 (7) (2018) 1194–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Heckmann BL, Tummers B, Green DR, Crashing the computer: apoptosis vs. necroptosis in neuroinflammation, Cell Death. Differ 26 (1) (2019) 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hunter JE, Willmore E, Irving JA, Hostomsky Z, Veuger SJ, Durkacz BW, NF-κB mediates radio-sensitization by the PARP-1 inhibitor, AG-014699, Oncogene 31 (2) (2012) 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shamloo B, Usluer S, p21 in Cancer Research, Cancers (Basel) 11(8) (2019) 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Karimian A, Ahmadi Y, Yousefi B, Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage, DNA Repair (Amst) 42 (2016) 63–71. [DOI] [PubMed] [Google Scholar]
- [31].Liu K, Chen W, Lei S, Xiong L, Zhao H, Liang D, Lei Z, Zhou N, Yao H, Liang Y, Wild-type and mutant p53 differentially modulate miR-124/iASPP feedback following pohotodynamic therapy in human colon cancer cell line, Cell Death Dis 8 (10) (2017) e3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M, Canovas B, Igea A, Ahuja AK, Zellweger R, Havaki S, Kanavakis E, Kletsas D, Roninson IB, Garbis SD, Lopes M, Nebreda A, Thanos D, Blow JJ, Townsend P, Sørensen CS, Bartek J, Gorgoulis VG, Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing, Nat. Cell Biol 18 (7) (2016) 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]