

Abstract

The calculation of
electron correlation is vital for the description
of atomistic phenomena in physics, chemistry, and biology. However,
accurate wavefunction-based methods exhibit steep scaling and often
sluggish convergence with respect to the basis set at hand. Because
of their delocalization and ease of extrapolation to the basis-set
limit, plane waves would be ideally suited for the calculation of
basis-set limit correlation energies. However, the routine use of
correlated wavefunction approaches in a plane-wave basis set is hampered
by prohibitive scaling due to a large number of virtual continuum
states and has not been feasible for all but the smallest systems,
even if substantial computational resources are available and methods
with comparably beneficial scaling, such as the Møller–Plesset
perturbation theory to second order (MP2), are used. Here, we introduce
a stochastic sampling of the MP2 integrand based on Monte Carlo summation
over continuum orbitals, which allows for speedups of up to a factor
of 1000. Given a fixed number of sampling points, the resulting algorithm
is dominated by a flat scaling of 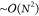 . Absolute correlation energies are accurate
to <0.1 kcal/mol with respect to conventional calculations for
several hundreds of electrons. This allows for the calculation of
unbiased basis-set limit correlation energies for systems containing
hundreds of electrons with unprecedented efficiency gains based on
a straightforward treatment of continuum contributions.
. Absolute correlation energies are accurate
to <0.1 kcal/mol with respect to conventional calculations for
several hundreds of electrons. This allows for the calculation of
unbiased basis-set limit correlation energies for systems containing
hundreds of electrons with unprecedented efficiency gains based on
a straightforward treatment of continuum contributions.
1. Introduction
Electron correlation lies at the heart of a wide range of fundamental physical and chemical phenomena, which range from the structural diversity and dynamics of water1 over the dissociation of liquid hydrogen at high pressure2 and the stability and mobility of point defects in semiconductors3 to the barrier height of chemical reactions. Wavefunction-based methods allow for a conceptually simple and convenient treatment of electron correlation4−6 and have found widespread and long-lasting use in theoretical chemistry. Correlated wavefunction methods have been widely applied as a benchmarking tool7,8 in the development of computationally more expedient methods such as the Kohn–Sham density functional theory (KS-DFT).9,10 More recently, their scope has been enlarged by rigorous hybridization schemes that combine KS-DFT with correlated wavefunction approaches,11−15 giving rise to some of the most accurate density functional approximations available to date.12,13,16−19 In particular, while it has been pointed out that many recently developed density functional approximations fail to yield correct densities and energies at the same time,20 double hybrid (DH) functionals have been shown to be able to overcome this fundamental problem.21 Recently, modern machine learning techniques have considerably increased timescales and system sizes that can be sampled on conventional infrastructure, but the generation of reliable input data for the training of such methods still relies on the computational feasibility of reference calculations of sufficient accuracy. In this perspective, the importance and scope of wavefunction-based first-principles techniques applied to condensed matter systems can therefore only be expected to grow further, both as a standalone method and in combination with DFT.
To this day, wavefunction-based correlation methods are hampered by a scaling that is polynomial at best and that is associated with a considerable prefactor. This implies that for larger systems, trade-offs have to be made between the accuracy of the basis set employed and the number of electrons that can be treated with reasonable computational resources. Moreover, correlated wavefunction approaches have only scarcely been applied in the condensed phase, which is due to additional difficulties encountered in periodic systems.22−40 These difficulties can be further exacerbated by the large basis sets needed to obtain basis-set limit reference energies.41 This precludes the routine use of wavefunction-based methods for large condensed phase systems; at the same time, benchmarking possibilities, for example, against newly developed density functionals, remain restricted to comparably few atoms and small supercells27,31,34,35,38 or have to be based on basis sets which are far from completeness. In benchmarking, this can be particularly problematic in combination with the erroneous convergence behavior of certain density functionals, which obfuscates any comparison that is not explicitly made at the complete basis-set limit.42,43 The availability of basis-set limit results is therefore necessary not only for formal reasons, but is also of great importance for the assessment of the physical accuracy of existing models and approximations, representing an important guideline in the development of new techniques and approximations that are able to reach far beyond current system sizes and limitations.
In principle, plane wave (PW) basis sets would constitute an ideal choice for the calculation of basis-set limit correlation energies since they do not introduce any localization bias and allow for a controlled, simple, and well-defined extrapolation to the complete basis-set limit44 without the linear dependency issues commonly encountered in large atom-centered bases.28,35,45,46 In particular, since a single PW is the solution of the Schrödinger equation of a free electron, its use enables the description of continuum states, which have been shown to play a crucial role in the complete description of electron correlation.47 In the following, we will refer to continuum states in finite systems as those virtual states that resemble a free electron; it is also this resemblance that lies at the heart of simple extrapolation to basis-set limit values.44 By virtue of their very nature, conventional atom-centered bases such as Gaussian functions or combined Gaussian/PW (GPW)48 bases are unable to describe such continuum states, which also account for the absence of physical models that would allow for a simple extrapolation to the basis-set limit. Instead, they usually rely on specifically constructed basis sets that allow for certain extrapolation models to be applied; this, however, does not commonly hold for density functionals.42,49 PWs have not been reported to suffer from this drawback.43 In addition, PWs are equally suitable for the treatment of both periodic and nonperiodic systems with either wavefunction-based methods, density functional techniques, or hybridizations thereof. These advantages, however, come at a price: The presence of a large number of continuum states in PW setups exacerbates the steep scaling of correlated wavefunction methods, making them computationally intractable for all but the smallest systems, which on their own will already require substantial resources on conventional high-performance compute clusters. In the following, we will show that PW-based correlated wavefunction calculations can be sped up by a factor of up to 1000 by stochastically sampling continuum state contributions via Monte Carlo summation. The error introduced by this stochastic approach remains below 0.01 kcal/mol per electron. This enables correlated wavefunction calculations in PWs for unprecedented system sizes on conventional computational infrastructure, making unbiased basis-set limit values routinely accessible for systems with up to hundreds of electrons. The same reflections hold for hybrid wavefunction/DFT methods, such as DH11 density functionals.
1.1. Møller–Plesset Perturbation Theory
Among
the correlated wavefunction methods, second-order Møller–Plesset
perturbation theory (MP2)50 exhibits a
comparably flat scaling of  with
the number of electrons or basis functions N, making
it one of the flattest scaling correlated, wavefunction-based
approaches available, second only to the random phase approximation
(RPA) and the direct RPA (dRPA), respectively.51,52 In general, MP2 has been found to provide a good first estimate
of the dynamic correlation energy.6,53 Conceptually
simple, the MP2 correlation energy EcMP2 is obtained
by a perturbative treatment that includes up to doubly excited determinants
and summation over pairs of all Nocc occupied
and Nvir virtual orbitals. For the spin-restricted
case,4
with
the number of electrons or basis functions N, making
it one of the flattest scaling correlated, wavefunction-based
approaches available, second only to the random phase approximation
(RPA) and the direct RPA (dRPA), respectively.51,52 In general, MP2 has been found to provide a good first estimate
of the dynamic correlation energy.6,53 Conceptually
simple, the MP2 correlation energy EcMP2 is obtained
by a perturbative treatment that includes up to doubly excited determinants
and summation over pairs of all Nocc occupied
and Nvir virtual orbitals. For the spin-restricted
case,4
| 1 |
where i, j and a, b denote spatial occupied and virtual orbitals ψ, respectively, that are eigenstates of the Hartree–Fock operator with eigenvalues ε. The MP2 matrix element e̅(i,j,a,b) is expressed in terms of four-electron Coulomb energies, which can be cast into a positive-definite form
| 2 |
where ⟨ij|ab⟩ are two-electron matrix elements. In DH density functionals, the second-order integrals in eq 1 are evaluated using a ground-state KS determinant, rather than the Hartree–Fock solution,11 and will only contribute to a fraction of the total correlation energy, the remainder being treated by pure density functional methods. If the advantages of DHs are to be made routinely available in condensed matter applications, they too require an efficient treatment of the terms in eq 1, and any improvements in the calculation of the MP2 term will directly benefit DH calculations.
Historically, EcMP2 has been evaluated using atom-centered basis sets or mixed GPW54 implementations. In particular, in periodic setups, use of localized basis functions has been reported to be susceptible to basis-set convergence issues,28,35,45,46 whereas problematic basis-set superposition effects and possible linear dependencies are absent in a PW representation. Applications in solid-state physics have also been scarce, which is in part due to the divergence of the MP2 integrand in zero-band gap systems, but a Thomas–Fermi screening of the MP2 amplitudes can resolve this issue.55
The presence of continuum (or continuum-like) states allows for simple extrapolation to the complete basis-set limit: Alavi and co-workers have demonstrated that in a PW basis at large Nvir, EcMP2 decays as ∝εa–3/2.44 PW-based methods therefore offer the unique advantage of a simple evaluation of basis-set limit values in both periodic and isolated systems, making them a potentially invaluable tool for basis-set bias-free calculations. PW MP2 calculations are few and have only recently been reported.34,35,56,57 This is in part due to their extensive memory requirements. The presence of a number of virtual states close to the number of PWs themselves (up to 109) can further complicate the calculations. However, given sufficient memory, the integrals of eq 2 are easily evaluated in the reciprocal space: Exchange-like matrix elements are easily obtained by solving the Poisson equation for a set of pair densities ρia(r) = ψi(r)ψa(r) in reciprocal space, where the Coulomb operator is diagonal. The real-space pair densities can simply be subjected to a fast Fourier transform (FFT), ρia(G) = FFT[ρia(r)]. Then, at the Γ-point, the reciprocal-space equivalent of a matrix element reads58
| 3 |
where † stands for the complex conjugate and index permutation. Ω is the volume of the system, G are reciprocal-space vectors, and Φ(G) is a suitably generalized form of the reciprocal-space Coulomb potential.59,60 Attempts to reduce the overall cost of the method have included mapping the virtual states onto a localized subspace,37,61−65 the use of stochastic orbitals66−69 and (real-space70,71 or graph-based57,72) sampling approaches, respectively, as well as exploiting Laplace transforms53,56,73−76 to enhance parallel efficiency. In an alternative strategy, one seeks to accelerate convergence of the correlation energy by improving the description of the electron–electron cusp. To this end, explicitly correlated basis sets can be used. In an ansatz commonly referred to as the F12 theory,77,78 the description of the electron–electron cusp is improved by combining a conventional, atom-centered Gaussian basis with a set of strongly orthogonal geminals. This approach has recently been extended to PWs.36
However, localization procedures fail for continuum states; previously reported stochastic techniques either introduce system-dependent errors of up to 2 kcal/mol per occupied orbital69 or carry a prefactor too large for practical applications,70 and approaches based on the Laplace transform56 require repeated calculations at different quadrature points, increasing the overall operation count for the sake of parallel efficiency, with modest reported speedups of around 4 to 5. A combined stochastic/Laplace-transform approach that results in appreciable computational speedups leads to errors of at least 20%.57 Similarly, due to the presence of nonfactorizable many-electron integrals, the cost of evaluating the MP2 integrand in PW-F12 is higher than for a pure PW basis set,36 and the efficiency of a graph-based approach72 was hindered by the absence of an optimized weighting scheme for graph generation. In a more general scope, a recent diagrammatic decomposition of the coupled cluster pair correlation function has allowed for the introduction of a basis-set correction in PWs that results in speedups of 2 orders of magnitude.79 Alternatively, in the context of Full Configuration Interaction Quantum Monte Carlo calculations,80 use of an effective, transcorrelated Hamiltonian81 has been shown to substantially improve convergence of the correlation energy of the uniform electron gas. In the GW theory,82 stochastic sampling schemes have successfully been applied with competitive accuracy and favorable timings with respect to deterministic calculations.83
In the following, we shall demonstrate how the presence of continuum states can be exploited to drastically reduce the computational cost of MP2 calculations without impacting their accuracy. The approach is based on a simple stochastic sampling of the integrands of eq 1 and can be implemented with little effort, representing a sleek and clean approach to tackle the issues arising in the continuum. This opens the path to routine applications of PW MP2-based approaches in both isolated and periodic systems with up to hundreds of occupied orbitals, making it possible to obtain basis-set limit DH or MP2 correlation energies on conventional computational infrastructure within a reasonable time.
2. Distribution of Continuum States and Stochastic Sampling
The possibility of introducing stochastic sampling is rooted in the behavior of the integrand at large εa, where continuum states arise. In this regime, EcMP2 grows as εa–3/2,44 and the overlap elements ⟨ij|ab⟩ must therefore be of low magnitude. It is obvious from eq 3 that overlaps will only be non-negligible whenever the symmetries of the continuum states match, but explicit symmetry determination for all states would be prohibitively expensive. Instead, given two high-lying virtual states, the statistical distribution of non-negligible overlaps is expected to be similar between truncations of eq 1 at a and some subsequent a + δ with arbitrary δ. For a spatially infinite supercell at infinite PW cutoff, one can define a cutoff energy εc with orbital index c, from where on all orbitals a ≥ c are part of the continuum. Then, the correlation energy can be separated
| 4 |
with the continuum contribution ηc(a) accounting for the incremental change in correlation energy when adding an additional continuum orbital to the system:
| 5 |
where ∑b′ is due to pairs of continuum (a) and noncontinuum (b′) virtual orbitals and we have used that, at the Γ-point, ψa = ψa*. Note that the last term in eq 4 contains only one explicit sum over virtual orbitals Nvir, with the orbital pairs themselves being formed by the triple sum in eq 5.
The simplest possible stochastic treatment of eq 5 is given by a uniform sampling of the summand, but this calls for a regular distribution of the overlap values obtained over all tuples ijab, which are the arguments of ηc(a); that is, the high-lying virtual orbitals of a finite system at finite PW cutoff need to reasonably approximate free, continuum electrons. Figure 1 shows the distribution of these arguments for a finite periodic box with εc – εHOMO = 50 eV and a total of Nvir = 3000 virtual orbitals. With the positive-definite definition of e̅ adopted in eq 2, the resulting distribution is indeed regular and smooth, indicating that ηc(a) could be predestinated to be treated by uniform Monte Carlo sampling. This is the approach we will privilege in the following. Note that in the limit of an infinite system, this is equal to Monte Carlo integration over the continuum; such integration techniques have been used as early as 1957 to calculate the high-density correlation energy of the uniform electron gas.84
Figure 1.

Figure showing the absolute occurrence P of orders of magnitude of the summand e̅(i,j,a,b) of ηc(a) of eq 5 from a = c to a = 3000 for εc – εHOMO = 50 eV. The order of magnitude of the matrix elements is small, and its distribution is smooth.
Let ps ∈ [0, 1) be a predefined sampling probability (i.e., ps is larger than or equal to zero but always smaller than 1). We define the function p as
| 6 |
Symmetry at the Γ-point, which gives rise to a factor of 2 in continuum/continuum and mixed continuum/noncontinuum terms of eq 5, can be accounted for by introducing a Kronecker delta. Then, the sums in ηc(a) can be simplified, and the stochastic expression for ηc(a) is
 |
7 |
where xijab ∼ U([0, 1)) is a sequence of random
numbers drawn from a uniform distribution U and b covers both b and b′
of eq 5. In the limit
of a continuum, the error of the sampling is expected to decrease
as  , with NMC being
the number of tuples ijab that are explicitly sampled.
The combination of eqs 4 and 7 amounts to a stochastic summation over
all orbital pairs that contribute to an increase in EcMP2, once
a continuum orbital a is added to a system already
containing a – 1 virtual orbitals.
, with NMC being
the number of tuples ijab that are explicitly sampled.
The combination of eqs 4 and 7 amounts to a stochastic summation over
all orbital pairs that contribute to an increase in EcMP2, once
a continuum orbital a is added to a system already
containing a – 1 virtual orbitals.
For any sampling probability ps < 1, only elements of ηc(a) with p(xijab, ps) = 1 have to be evaluated. In a finite cell with a finite PW cutoff and a sufficiently large number of virtual orbitals Nvir, estimates for ηc(a) are then obtained for every given continuum orbital a ∈ [c, Nvir] as follows: rather than randomly generating a tuple of indices for a predetermined number of Monte Carlo moves, a random number p(xijab, ps) is drawn for every element of the summand in eq 7, determining whether a particular tuple ijab will enter into the estimator of ηc(a). ps therefore defines a target value NMC ∝ ps of tuples that are expected to be sampled for every continuum orbital a. The advantages of such an algorithm are twofold: On the one hand, it avoids under- or oversampling of the subspace associated with orbital a; on the other hand, it enables efficient extrapolation to the basis-set limit in one single calculation, directly providing EcMP2 as a function of the highest virtual orbital included in eq 4.
In the following, we will adopt an orbital-dependent sampling probability ps(a) = NMC/Ncard(a), where Ncard(a) = (2a – 1)Nocc(Nocc + 1)/2 is the product of the cardinalities of the sums in eq 7. Ncard(a) therefore explicitly depends on the virtual orbital a that is added to eq 4. With this choice of a continuum-orbital-dependent ps(a), the density of the sampling decreases as a increases. This allows for NMC to remain a fixed, system-independent input quantity. We shall later show that conservative estimates for NMC and εc can be regarded as system-independent.
For orbitals that are part of the continuum, the resulting algorithm
scales formally as 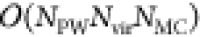 , where NPW is
the number of PWs in the basis set. This follows from eqs 4 and 7 since
the cost of evaluation of the triple sum over Nocc2Nvir in eq 7 is reduced to elements
with p(xijab, ps(a)) = 1,
which in turn is proportionate to NMC.
For virtual orbitals with εa <
εc, the conventional
, where NPW is
the number of PWs in the basis set. This follows from eqs 4 and 7 since
the cost of evaluation of the triple sum over Nocc2Nvir in eq 7 is reduced to elements
with p(xijab, ps(a)) = 1,
which in turn is proportionate to NMC.
For virtual orbitals with εa <
εc, the conventional  scaling applies (cf. eqs 1 and 3). Further on
in the text, we will show that in practice, the number of terms due
to eigenvalues εa < εc does not dominate scaling. Once NMC can be made both independent of the orbital
index a and the system at hand, the scaling of the
resulting method reduces to
scaling applies (cf. eqs 1 and 3). Further on
in the text, we will show that in practice, the number of terms due
to eigenvalues εa < εc does not dominate scaling. Once NMC can be made both independent of the orbital
index a and the system at hand, the scaling of the
resulting method reduces to 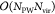 integral evaluations for all orbitals with
eigenvalue ε > εc.
integral evaluations for all orbitals with
eigenvalue ε > εc.
3. Computational Methods
3.1. General Setup
Hard pseudopotentials of the Goedecker–Teter–Hutter (GTH) form85 parametrized for Hartree–Fock calculations31 have been used for all calculations. PW MP2 and stochastic MPs2 energies were calculated using a modified version of the CPMD code.86 The convergence threshold on the residual gradient on occupied orbitals was set to 10–7 a.u., whereas a threshold of 10–5 a.u. was used for the virtual space. For isolated systems, periodic images were decoupled using the Poisson solver by Martyna and Tuckerman.87 The wavefunction cutoff energies Ecutψ were set to 150 Ry for all systems but for the ethylene crystal, where a value of 140 Ry was used. A density cutoff Ecutρ = 4Ecutψ was adopted for all systems, while cutoff energies for MP2 pair densities were set to Ecutψ without impacting accuracy. The calculation of the MP2 term is based on straightforward evaluation of eq 3 as in ref (58). Since no derivatives with respect to Ec have to be evaluated, reciprocal-space orbital pairs ρia(G) are stored in memory, allowing for substantial speedups with respect to an on-the-fly evaluation of every pair density. The size of the periodic super- or unit-cells, the number of electronic states as well as molecular geometries, and PW energy cutoffs are given in the Supporting Information.
3.2. Extrapolation of Correlation Energies
Correlation energies are obtained from single-point extrapolation using the εNvir–3/2 dependency (equivalent to 1/Nvir) as described in ref (44). Such a leading-order approximation requires a sufficient number of high-energy orbitals in order to gather sufficient statistics and reliable extrapolated values. In this work, we adopt a fitting scheme with various windows (ranges of points to fit) moving along the curve. Those vary in size with respect to the number of orbitals included, with a shift of ∼200 orbitals between them. The maximum window size is taken as the one that retains a fitting error comparable to smaller windows when finishing fitted ranges at Nvirmax, the highest orbital available from a Davidson diagonalization. The smallest window is given by the smallest possible window size that provides a stable fit.
MP(s)2 curves are fitted according to
| 8 |
The series of α(Nvir), obtained from all windows along the curves that terminate at Nvir up to Nvirmax, converge to an estimate of the MP(s)2 energies at the basis-set limit, Ec(εNvir → ∞) Averaging over different windows allows to account for sensitivity and variance of extrapolated values, which are given in the Supporting Information along with figures that illustrate the extrapolation procedure and error determination.
4. Results and Discussion
4.1. Accuracy of Stochastic Summation
Our stochastic sampling scheme was tested both on isolated systems as well as in periodic, condensed-phase setups. Test systems in the condensed phase include solid-state ethylene and benzene molecular crystals as well as a liquid consisting of a hydronium ion solvated in 32 water molecules. Isolated (gas-phase) systems are represented by a benzene monomer and a dimer in sandwich configuration. All calculations were carried out with a modified version of the CPMD code86 using GTH pseudopotentials generated for Hartree–Fock calculations.31,85
We first investigate the dependency of the accuracy of our stochastic sampling of eqs 4 and 7, called MPs2, on the number of orbitals in the occupied subspace and the choice of the continuum cutoff value εcgap. This quantity is given with respect to the highest occupied molecular orbital (HOMO) in order to be independent of the system setup or reference frame: εc = εc – εHOMO. Figure 2 shows the absolute difference between MP2 and MPs2 calculations for a benzene crystal and a benzene monomer for εcgap ranging from 20 to 180 eV. Differences rapidly decrease by increasing εc. Errors averaged over independent stochastic runs remain <0.01% for continuum cutoffs ≥120 eV. Reducing this value by half to 60 eV results in doubling of the relative error, which can still be acceptable. Further lowering of εcgap, however, leads to rapidly increasing errors. The standard deviation of the sampling error depends much more strongly on εc than on Nocc. From εcgap = 120 eV on, standard deviations become negligible and virtually identical for both systems. Notably, relative sampling errors are lower for the crystal at Nocc = 60 than for the monomer (graphs are provided in the Supporting Information). This strongly supports that accuracy is mainly influenced by εc, whereas at constant NMC, the error is independent of the partitioning of occupied and virtual states in eq 7.
Figure 2.

Absolute differences between stochastic and nonstochastic MP2 correlation energies, ΔEMPs2MP2, as a function of the continuum cutoff energy εc for a benzene crystal (Nocc = 60, four molecules per unit cell) and a benzene monomer (Nocc = 15) at NMC = 12,000. Error bars were obtained by averaging over six independent runs.
In the following and in line with the values of Figure 2, we will adopt NMC = 12,000 and εcgap = 120 eV to investigate the accuracy of stochastic sampling for different systems. Figure 3 shows the correlation energy as a function of the eigenvalue of the highest virtual orbital included (εNvir) for two exemplary systems: one periodic (ethylene crystal) and one isolated (benzene monomer) setup, calculated both with a full summation according to eq 1 as well as with our stochastic sampling. The corresponding extrapolated basis-set limit values are shown in Table 1. The largest errors of the extrapolated, absolute MP2 correlation energies lie between 0.02 and 0.1 kcal/mol. Errors in the binding energy of the benzene sandwich are of comparable magnitude, which is far below chemical accuracy. Differences per electron, ΔE/e–, do not exceed 0.1 meV. These values compare well to an expected stochastic error of ≤0.01%. Table 1 also shows the number of matrix element evaluations for all setups. With our system-independent choice of NMC, stochastic sampling reduces this number by 1–2 orders of magnitude compared to conventional calculations.
Figure 3.

EcMP2 as a function of the highest eigenvalue εNvir in the sum of eq 1 for both conventional and stochastic MP2 (MPs2) for an ethylene crystal (Nvir = 11,158) and an isolated benzene monomer (Nvirmax = 14,985). Domains without stochastic sampling are colored in gray. Differences between the curves, ΔEMPs2, are plotted on a secondary y-axis. Extrapolated basis-set limit values for all systems described here are found in Table 1.
Table 1. MP2 Correlation Energies EcMP2 Obtained from a Conventional MP2 Calculation and the Stochastic Approach, MPs2a.
| system | EcMP2 [a.u.] | EcMPs2 [a.u.] | ΔEMPs2MP2 [a.u.] | ΔE/e– [a.u.] | Nijab | psNijab | |
|---|---|---|---|---|---|---|---|
| ethylene | crystal | –0.78054 | –0.78056 | 2 × 10–5 | 8 × 10–7 | 5.06 × 109 | 3.26 × 108 |
| benzene | crystal | –4.69164 | –4.69149 | –1.5 × 10–4 | –1 × 10–6 | 1.30 × 1011 | 3.50 × 109 |
| monomer | –1.05681 | –1.05695 | 1.4 × 10–4 | 5 × 10–6 | 8.62 × 109 | 6.26 × 108 | |
| dimer | –2.12780 | –2.12777 | –3 × 10–5 | –5 × 10–7 | 3.33 × 1010 | 8.78 × 109 | |
| binding | –0.01417 | –0.01387 | –3 × 10–4 | –5 × 10–6 | |||
ΔEMPs2MP2 and ΔE/e– denote absolute and per-electron energy differences between stochastic sampling and conventional calculations, respectively. Nijab denotes the number of matrix elements (ijab-tuples) sampled in a conventional calculation, and psNijab is the number of effectively sampled matrix elements in a stochastic MPs2 calculation, with both numbers rounded to three digits. The threshold for stochastic sampling expressed with respect to the HOMO was identical for all systems, εc = εc – εHOMO = 120 eV. The same holds for NMC = 12,000, the number of terms sampled per virtual contribution ηc(a), as described in eq 7. For details on the systems used, cf. the Supporting Information.
For both absolute energies and energy differences, the observed deviations are several orders of magnitude lower than the reported error of other stochastic or Laplace transform-based schemes.57,66−72 For comparison, values of correlation energies obtained with atom-centered all-electron88 and GPW codes89 are listed in the Supporting Information; all values are consistently higher than those reported for our PW calculations. In particular, we note that differences between different basis sets tend to be substantially larger than the stochastic sampling error, further confirming the viability of uniform, stochastic sampling of the continuum space.
4.2. Performance and Speedups
With the
error of the stochastic sampling scheme being considerably lower than
the errors documented for other PW implementations, effective speedups
remain to be determined. Figure 4 shows the resulting cumulative execution times and
speedups of the stochastic sampling compared to the direct implementation
of eqs 1 and 3 for the ethylene crystal. Timings are reported as
a function of the highest orbital index included in the expansion, Nvir. For the ethylene crystal, at Nvir = 10,000, speedups of up to 957 can be reached with
stochastic summation, making the calculation about 3 orders of magnitude
faster compared to a conventional implementation. All the while, the
error introduced by uniform stochastic summation with respect to a
full calculation is only about 10–2 kcal/mol for
this system (cf. Table 1). This has to be compared to maximum speedups of about 5 documented
for Laplace transform-based schemes that allow for similar accuracy
to be retained56 and 20% errors in correlation
energies for algorithms that allow for larger speedups57 versus errors around 0.01% reported here. Figure 4 also includes a
fit of the CPU time of our stochastic scheme to a function O(Nvir) = a0 + a1Nvir, demonstrating that the scaling is described well by  , assuming that NMC =
cnst.
, assuming that NMC =
cnst. 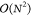 constitutes a formal improvement over the
scaling achieved with a stochastic graph-based approach in atom-centered
basis sets,72 which was reported to be
of
constitutes a formal improvement over the
scaling achieved with a stochastic graph-based approach in atom-centered
basis sets,72 which was reported to be
of  .
.
Figure 4.

Cumulative execution times (left) and speedups (right) compared between conventional algorithm and stochastic sampling for an ethylene crystal as a function of the highest virtual orbital index, Nvir, included in eq 4. O(Nvir) = a0 + a1Nvir denotes a least-squares fit on Nvir ∈ [5000, 10,000] with a0 = −4.76586 s, a1 = 0.010515 s. For sufficiently large Nvir > 2000, formal and practical scaling show excellent agreement. At Nvir = 10,000, the stochastic approach with NMC = 12,000 is 3 orders of magnitude faster. Timings were obtained by dividing the computational load over 5 OMP (Open Multi-Processing) threads and 24 MPI (Message Passing Interface) tasks.
Together with the high accuracy of the method, this considerable gain in efficiency allows for the treatment of systems that would be intractable when treated with conventional algorithms. One typical usage example of accurate wavefunction-based theories or computationally demanding high-quality DFT methods such as DH functionals lies in postprocessing of simulation data, for example, in a posteriori calculations of potential energy surfaces or reaction paths generated using lower-level methods in the context of molecular dynamics or Monte Carlo simulations. Recent developments have even made it possible to directly sample90 high-quality potential energy surfaces by virtue of multiple time step (MTS) propagators,91 which allow for an important decrease in computational cost and substantial improvements in tractability by permitting less-frequent evaluation of the full, high-quality Hamiltonian during a first-principles molecular dynamics run.
A liquid constituted of 1 hydronium ion solvated in 32 water molecules (264 electrons) will serve as an example of the performance of stochastic sampling in a typical setup encountered in first-principles (MTS-)molecular dynamics. Using the stochastic summation scheme reported here, calculating the basis-set limit correlation energy of this system, shown in Figure 5, is feasible in about 15 h on 25 16-core compute nodes with 128 GB of RAM, using the same, conservative estimates for εcgap reported in Table 1, which yielded accurate results for all test systems considered so far. Additionally, calculations using εc = 60 eV and εcgap = 90 eV have been carried out for the sake of comparison. Already at εc = 90 eV, the execution time is almost halved to about 8 h and can be further reduced by using εcgap = 60 eV, at which the basis-set limit correlation energy can be calculated in a mere 5 h. In particular, this drastic reduction in execution time is not accompanied by a considerable loss of accuracy. Extrapolated correlation energies are given in Table 2. It should be noted that postprocessing protocols can be applied when training machine learning algorithms with high-quality data, based on a coarse sampling of configuration space with a lower-level method (cf., e.g. ref (1)). Speedups in the calculation of correlation energies with MP2 or DH functionals will therefore be directly reflected in less time-consuming training procedures, thus considerably increasing throughput.
Figure 5.

Hydronium ion solvated in 32 water molecules (264 electrons explicitly accounted for). Calculation of EcMPs2 takes between 5 and 15 h on 25 16-core compute nodes. Molecules within the periodically repeated supercell are highlighted. Using εc = 120 eV and NMC = 12,000 yields an extrapolated EcMPs2 = −10.22952 a.u.
Table 2. Stochastic MP2 Correlation Energies EcMPs2 for a Hydronium Ion Solvated in 32 Water Molecules Using Different Thresholds for Continuum Energies εca.
| EcMPs2 [a.u.] | NMC | εcgap [eV] | t |
|---|---|---|---|
| –10.22771 | 12,000 | 60 | 4 h 55 |
| –10.23190 | 12,000 | 90 | 8 h 10 |
| –10.22952 | 12,000 | 120 | 15 h 25 |
Timings t, rounded to 5 min, are given for the execution time of the MP2 routine on 25 16-core nodes in a hybrid setup (50 MPI tasks, 8 OMP threads).
4.3. Generalization to RPA
The promising performance of the stochastic summation scheme described here also opens the possibility of its application to similar approaches in which continuum states can play a role. In DFT, the exact-exchange plus RPA approach has emerged as a promising method capable of more accurately predicting van der Waals binding energies, adsorption energies on surfaces, or lattice constants in molecular solids.44,54,92−94 Based on the similarity of the MP2 energy expression and the RPA, one can expect transferability of the stochastic sampling approach to the evaluation of the RPA. The reciprocal-space form of the RPA correlation energy EcRPA is51,52
| 9 |
where χ̃G,G′(q,iω) are elements of the full density response function, including the Coulomb interaction. At the Γ-point with q = 0, the diagonal elements in a stochastically sampled RPA scheme become
| 10 |
with
| 11 |
where the variables c, xjb, and ps are used analogously to eq 7. In this limit, we investigate the RPA integrand of eq 9 for an ethylene crystal at different values of ω. Figure 6 shows the values of EcRPA at varying iω for Nvir = 11,040 as well as an estimate of the overall RPA correlation energy based on trapezoidal integration. Stochastic sampling with ps = 1/3 introduces a maximum error of about 1% in the integrand. Overall, the stochastic sampling introduces a final error of less than 0.03 kcal/mol, which is comparable to the error obtained in MP2 calculations for the same system. These results demonstrate that the stochastic sampling of continuum states can also be applied for methods other than MP2 that include a substantial continuum-state contribution.
Figure 6.

Value of the integrand of eq 9, abbreviated as Tr{···}, for an ethylene crystal at q = 0 and Nvir = 11,040 as a function of iω for both conventional and stochastic calculations. The difference between the curves, ΔTr{···}, rapidly decreases as iω is increased; differences are plotted on the secondary y-axis and do not exceed 1.5%. Simple trapezoidal integration leads to EcRPA = −0.03365 a.u. for a full calculation and Ec = −0.03360 a.u. for stochastic sampling.
5. Conclusions and Outlook
Continuum states have been shown to be an important contributor to the overall electron correlation energy.47 Among the basis sets commonly used in solid-state physics, quantum chemical calculations, and first-principles molecular dynamics, PWs stand out as the only choice that can effectively account for continuum contributions, which are also the base of a simple basis-set limit extrapolation technique.44 Here, we have introduced a uniform stochastic sampling approach to treat continuum states arising in correlation energy calculations, where contributions due to states with orbital eigenvalues beyond some threshold εc are added by stochastic summation. This algorithm has been applied to the calculation of second-order perturbation energies which occur in both MP2 and DH density functionals. We have shown that stochastic summation over the continuum orbitals allows for the calculation of MP2 correlation energies with speedups of up to 3 orders of magnitude at remarkably low errors. This significant increase in efficiency enables calculations with several hundreds of electrons at a relatively low computational cost, making it possible to standardly apply MP2 and DH methods in a PW basis, which has so far been intractably expensive even on high-performance compute clusters. Importantly, the results presented here also enable straightforward DH calculations in the condensed phase, thus extending the availability of one of the most accurate density functional methods available to date for condensed matter systems. We have also shown that stochastic sampling of the continuum orbitals can easily be extended to other approaches, demonstrating the generality of the ansatz employed here. Calculations carried out within a stochastic RPA suggest errors comparable to the stochastic MP2 scheme. The stochastic sampling scheme itself is straightforward, easy to implement, and based on simple physical concepts.
Stochastic sampling of continuum states permits to easily obtain basis-set limit values, be it for periodic or isolated systems, with maximum errors of only 0.1 meV per electron. This accuracy makes basis-set limit values for correlation energies available using reasonable computational resources and execution times. This will allow for thorough benchmarking of new computational methods without basis-set bias, for routine postprocessing of potential energy landscapes generated using lower-level methods, as well as on-the-fly generation of high-accuracy first-principles molecular dynamics trajectories using multiple time stepping schemes. This data, in turn, can be used to feed high-throughput methods based on artificial intelligence. Overall, the techniques presented in this text pave the road to routinely apply accurate MP2 and DH calculations at the basis-set limit in condensed matter systems, ultimately extending the use of a method well-established for isolated systems to the condensed phase.
Acknowledgments
M.P.B. acknowledges a postdoctoral fellowship from the Swiss National Science Foundation (project 184500). U.R. acknowledges funding form the Swiss National Science Foundation via the NCCR MUST and individual grant no. 200020_185092. M.P.B. is indebted to Christoph Dellago for hosting him and thanks Clemens Moritz and Andreas Singraber for useful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.0c00724.
System setups, fitting errors for all basis-set limit values reported in this manuscript, graphs of EcMP(s)2(Nvir) for all systems, Ec obtained in Gaussian and Gaussian/PW basis sets for comparison, and relative sampling errors and their standard deviation as a function of εcgap for the benzene monomer and crystal (PDF)
Author Contributions
§ M.P.B. and J.V. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Morawietz T.; Singraber A.; Dellago C.; Behler J. How van der Waals interactions determine the unique properties of water. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 8368–8373. 10.1073/pnas.1602375113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzola G.; Yunoki S.; Sorella S. Unexpectedly high pressure for molecular dissociation in liquid hydrogen by electronic simulation. Nat. Commun. 2014, 5, 3487. 10.1038/ncomms4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W.; Tkatchenko A. Electronic Structure and van der Waals Interactions in the Stability and Mobility of Point Defects in Semiconductors. Phys. Rev. Lett. 2013, 111, 045501. 10.1103/physrevlett.111.045501. [DOI] [PubMed] [Google Scholar]
- Szabo A.; Ostlund N. S.. Modern Quantum Chemistry; Dover Publications, 1996. [Google Scholar]
- Helgaker T.; Jørgensen P.; Olsen J.. Molecular Electronic-Structure Theory; John Wiley & Sons, 2000. [Google Scholar]
- Jensen F.Introduction to Computational Chemistry, 3rd ed.; John Wiley & Sons, 2017. [Google Scholar]
- Peverati R.; Truhlar D. G. Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc., A 2014, 372, 20120476. 10.1098/rsta.2012.0476. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Hansen A.; Bauer C.; Ehrlich S.; Najibi A.; Grimme S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. 10.1039/c7cp04913g. [DOI] [PubMed] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. 10.1103/physrev.136.b864. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. 10.1103/physrev.140.a1133. [DOI] [Google Scholar]
- Zhao Y.; Lynch B. J.; Truhlar D. G. Doubly hybrid meta DFT: New multi-coefficient correlation and density functional methods for thermochemistry and thermochemical kinetics. J. Phys. Chem. A 2004, 108, 4786–4791. 10.1021/jp049253v. [DOI] [Google Scholar]
- Bartlett R. J.; Grabowski I.; Hirata S.; Ivanov S. The exchange-correlation potential in ab initio density functional theory. J. Chem. Phys. 2005, 122, 034104. 10.1063/1.1809605. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Peverati R.; Head-Gordon M. Orbital optimized double-hybrid density functionals. J. Chem. Phys. 2013, 139, 024110. 10.1063/1.4812689. [DOI] [PubMed] [Google Scholar]
- Su N. Q.; Xu X. Construction of a parameter-free doubly hybrid density functional from adiabatic connection. J. Chem. Phys. 2014, 140, 18A512. 10.1063/1.4866457. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Assessment of model chemistries for noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 1009–1018. 10.1021/ct060044j. [DOI] [PubMed] [Google Scholar]
- Schwabe T.; Grimme S. Towards chemical accuracy for the thermodynamics of large molecules: New hybrid density functionals including non-local correlation effects. Phys. Chem. Chem. Phys. 2006, 8, 4398–4401. 10.1039/b608478h. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Zhao Y.; Truhlar D. G. The DBH24/08 database and its use to assess electronic structure model chemistries for chemical reaction barrier heights. J. Chem. Theory Comput. 2009, 5, 808–821. 10.1021/ct800568m. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. Efficient and accurate double-hybrid-meta-GGA density functionals- evaluation with the extended GMTKN30 database for general main group thermochemistry, kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2011, 7, 291–309. 10.1021/ct100466k. [DOI] [PubMed] [Google Scholar]
- Medvedev M. G.; Bushmarinov I. S.; Sun J.; Perdew J. P.; Lyssenko K. A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. 10.1126/science.aah5975. [DOI] [PubMed] [Google Scholar]
- Su N. Q.; Zhu Z.; Xu X. Doubly hybrid density functionals that correctly describe both density and energy for atoms. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 2287–2292. 10.1073/pnas.1713047115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala P. Y.; Scuseria G. E. Electron Correlation in Large Molecular Systems Using the Atomic Orbital Formalism. the Case of Intermolecular Interactions in Crystalline Urea as an Example. J. Comput. Chem. 2000, 21, 1524–1531. . [DOI] [Google Scholar]
- Ayala P. Y.; Konstantin K. N.; Kudin N.; Scuseria G. E. Atomic orbital laplace-transformed second-order møller-plesset theory for periodic systems. J. Chem. Phys. 2001, 115, 9698–9707. 10.1063/1.1414369. [DOI] [Google Scholar]
- Pisani C.; Busso M.; Capecchi G.; Casassa S.; Dovesi R.; Maschio L.; Zicovich-Wilson C.; Schütz M. Local-MP2 electron correlation method for nonconducting crystals. J. Chem. Phys. 2005, 122, 094113. 10.1063/1.1857479. [DOI] [PubMed] [Google Scholar]
- Casassa S.; Halo M.; Maschio L.; Roetti C.; Pisani C. Beyond a Hartree-Fock description of crystalline solids: The case of lithium hydride. Theor. Chem. Acc. 2007, 117, 781–791. 10.1007/s00214-006-0198-x. [DOI] [Google Scholar]
- Pisani C.; Maschio L.; Casassa S.; Halo M.; Schütz M.; Usvyat D. Periodic local MP2 method for the study of electronic correlation in crystals: Theory and preliminary applications. J. Comput. Chem. 2008, 29, 2113–2124. 10.1002/jcc.20975. [DOI] [PubMed] [Google Scholar]
- Pisani C.; Schütz M.; Casassa S.; Usvyat D.; Maschio L.; Lorenz M.; Erba A. Cryscor: A program for the post-Hartree-Fock treatment of periodic systems. Phys. Chem. Chem. Phys. 2012, 14, 7615–7628. 10.1039/c2cp23927b. [DOI] [PubMed] [Google Scholar]
- Paulus B. The method of increments—A wavefunction-based ab initio correlation method for solids. Phys. Rep. 2006, 428, 1–52. 10.1016/j.physrep.2006.01.003. [DOI] [Google Scholar]
- Manby F. R.; Alfè D.; Gillan M. J. Extension of molecular electronic structure methods to the solid state: Computation of the cohesive energy of lithium hydride. Phys. Chem. Chem. Phys. 2006, 8, 5178–5180. 10.1039/b613676a. [DOI] [PubMed] [Google Scholar]
- Gillan M. J.; Alfè D.; de Gironcoli S.; Manby F. R. High-precision calculation of Hartree-Fock energy of crystals. J. Comput. Chem. 2008, 29, 2098–2106. 10.1002/jcc.21033. [DOI] [PubMed] [Google Scholar]
- Del Ben M.; Hutter J.; Vandevondele J. Second-order møller-plesset perturbation theory in the condensed phase: An efficient and massively parallel gaussian and plane waves approach. J. Chem. Theory Comput. 2012, 8, 4177–4188. 10.1021/ct300531w. [DOI] [PubMed] [Google Scholar]
- Del Ben M.; Hutter J.; Vandevondele J. Electron correlation in the condensed phase from a resolution of identity approach based on the gaussian and plane waves scheme. J. Chem. Theory Comput. 2013, 9, 2654–2671. 10.1021/ct4002202. [DOI] [PubMed] [Google Scholar]
- Hutter J.; Wilhelm J.; Rybkin V. V.; Ben M. D.; VandeVondele J.. MP2- and RPA-Based Ab Initio Molecular Dynamics and Monte Carlo Sampling. Handbook of Materials Modeling; Springer, 2018; pp 1–21. [Google Scholar]
- Marsman M.; Grüneis A.; Paier J.; Kresse G. Second-order Møller-Plesset perturbation theory applied to extended systems. I. Within the projector-augmented-wave formalism using a plane wave basis set. J. Chem. Phys. 2009, 130, 184103. 10.1063/1.3126249. [DOI] [PubMed] [Google Scholar]
- Grüneis A.; Marsman M.; Kresse G. Second-order Møller-Plesset perturbation theory applied to extended systems. II. Structural and energetic properties. J. Chem. Phys. 2010, 133, 074107. 10.1063/1.3466765. [DOI] [PubMed] [Google Scholar]
- Grüneis A.; Shepherd J. J.; Alavi A.; Tew D. P.; Booth G. H. Explicitly correlated plane waves: Accelerating convergence in periodic wavefunction expansions. J. Chem. Phys. 2013, 139, 084112. 10.1063/1.4818753. [DOI] [PubMed] [Google Scholar]
- Grüneis A. Efficient Explicitly Correlated Many-Electron Perturbation Theory for Solids: Application to the Schottky Defect in MgO. Phys. Rev. Lett. 2015, 115, 066402. 10.1103/physrevlett.115.066402. [DOI] [PubMed] [Google Scholar]
- Grüneis A.Coupled Cluster and Quantum Chemistry Schemes for Solids. Handbook of Materials Modeling; Springer, 2018; pp 1–16. [Google Scholar]
- Gruber T.; Liao K.; Tsatsoulis T.; Hummel F.; Grüneis A. Applying the Coupled-Cluster Ansatz to Solids and Surfaces in the Thermodynamic Limit. Phys. Rev. X 2018, 8, 21043. 10.1103/physrevx.8.021043. [DOI] [Google Scholar]
- Sharkas K.; Toulouse J.; Maschio L.; Civalleri B. Double-hybrid density-functional theory applied to molecular crystals. J. Chem. Phys. 2014, 141, 044105. 10.1063/1.4890439. [DOI] [PubMed] [Google Scholar]
- Loos P.-F.; Pradines B.; Scemama A.; Toulouse J.; Giner E. A Density-Based Basis-Set Correction for Wave Function Theory. Phys. Chem. Lett. 2019, 10, 2931–2937. 10.1021/acs.jpclett.9b01176. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Characterizing and Understanding the Remarkably Slow Basis Set Convergence of Several Minnesota Density Functionals for Intermolecular Interaction Energies. J. Chem. Theory Comput. 2013, 9, 4453–4461. 10.1021/ct400660j. [DOI] [PubMed] [Google Scholar]
- Bircher M. P.; López-Tarifa P.; Rothlisberger U. Shedding Light on the Basis Set Dependence of the Minnesota Functionals: Differences Between Plane Waves, Slater Functions, and Gaussians. J. Chem. Theory Comput. 2019, 15, 557–571. 10.1021/acs.jctc.8b00897. [DOI] [PubMed] [Google Scholar]
- Shepherd J. J.; Grüneis A.; Booth G. H.; Kresse G.; Alavi A. Convergence of many-body wave-function expansions using a plane-wave basis: From homogeneous electron gas to solid state systems. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 86, 035111. 10.1103/physrevb.86.035111. [DOI] [Google Scholar]
- Halo M.; Casassa S.; Maschio L.; Pisani C. Periodic local-MP2 computational study of crystalline neon. Phys. Chem. Chem. Phys. 2009, 11, 586–592. 10.1039/b812870g. [DOI] [PubMed] [Google Scholar]
- Del Ben M.; Hutter J.; Vandevondele J. Second-order møller-plesset perturbation theory in the condensed phase: An efficient and massively parallel gaussian and plane waves approach. J. Chem. Theory Comput. 2012, 8, 4177–4188. 10.1021/ct300531w. [DOI] [PubMed] [Google Scholar]
- Kelly H. P. Correlation effects in atoms. Phys. Rev. 1963, 131, 684–699. 10.1103/physrev.131.684. [DOI] [Google Scholar]
- VandeVondele J.; Krack M.; Mohamed F.; Parrinello M.; Chassaing T.; Hutter J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. 10.1016/j.cpc.2004.12.014. [DOI] [Google Scholar]
- Jensen F. How Large is the Elephant in the Density Functional Theory Room?. J. Phys. Chem. A 2017, 121, 6104–6107. 10.1021/acs.jpca.7b04760. [DOI] [PubMed] [Google Scholar]
- Møller C.; Plesset M. S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. 10.1103/PhysRev.46.618. [DOI] [Google Scholar]
- Adler S. L. Quantum Theory of the Dielectric Constant in Real Solids. Phys. Rev. 1962, 126, 413–420. 10.1103/physrev.126.413. [DOI] [Google Scholar]
- Wiser N. Dielectric Constant with Local Field Effects Included. Phys. Rev. 1963, 129, 62–69. 10.1103/physrev.129.62. [DOI] [Google Scholar]
- Cremer D. Møller-Plesset perturbation theory: from small molecule methods to methods for thousands of atoms. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 509–530. 10.1002/wcms.58. [DOI] [Google Scholar]
- Del Ben M.; Hutter J.; VandeVondele J. Electron Correlation in the Condensed Phase from a Resolution of Identity Approach Based on the Gaussian and Plane Waves Scheme. J. Chem. Theory Comput. 2013, 9, 2654–2671. 10.1021/ct4002202. [DOI] [PubMed] [Google Scholar]
- Shepherd J. J.; Grüneis A. Many-Body Quantum Chemistry for the Electron Gas: Convergent Perturbative Theories. Phys. Rev. Lett. 2013, 110, 226401. 10.1103/physrevlett.110.226401. [DOI] [PubMed] [Google Scholar]
- Schäfer T.; Ramberger B.; Kresse G. Quartic scaling MP2 for solids: A highly parallelized algorithm in the plane wave basis. J. Chem. Phys. 2017, 146, 104101. 10.1063/1.4976937. [DOI] [PubMed] [Google Scholar]
- Schäfer T.; Ramberger B.; Kresse G. Laplace transformed MP2 for three dimensional periodic materials using stochastic orbitals in the plane wave basis and correlated sampling. J. Chem. Phys. 2018, 148, 064103. 10.1063/1.5016100. [DOI] [PubMed] [Google Scholar]
- Chawla S.; Voth G. A. Exact exchange in ab initio molecular dynamics: An efficient plane-wave based algorithm. J. Chem. Phys. 1998, 108, 4697–4700. 10.1063/1.476307. [DOI] [Google Scholar]
- Gygi F.; Baldereschi A. Self-consistent Hartree-Fock and screened-exchange calculations in solids: Application to silicon. Phys. Rev. B: Condens. Matter Mater. Phys. 1986, 34, 4405–4408. 10.1103/physrevb.34.4405. [DOI] [PubMed] [Google Scholar]
- Broqvist P.; Alkauskas A.; Pasquarello A. Hybrid-functional calculations with plane-wave basis sets: Effect of singularity correction on total energies, energy eigenvalues, and defect energy levels. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 80, 085114. 10.1103/physrevb.80.085114. [DOI] [Google Scholar]
- Huzinaga S.; Arnau C. Virtual Orbitals in the Hartree-Fock Theory. Phys. Rev. A 1969, 1, 1285–1288. 10.1103/physreva.1.1285. [DOI] [Google Scholar]
- Huzinaga S.; Arnau C. Virtual orbitals in hartree-fock theory. II. J. Chem. Phys. 1971, 54, 1948–1951. 10.1063/1.1675123. [DOI] [Google Scholar]
- Csepes Z.; Kozmutza C. Many-Body Perturbation Theory Based on Localized Orbitals. Croat. Chem. Acta 1984, 57, 855–864. [Google Scholar]
- Palmieri P.; Tarroni R.; Rettrup S. Hartree-Fock operators to improve virtual orbitals and configuration interaction energies. J. Chem. Phys. 1994, 100, 5849–5856. 10.1063/1.467148. [DOI] [Google Scholar]
- Neogrády P.; PitoňáK M.; Urban M. Optimized virtual orbitals for correlated calculations: An alternative approach. Mol. Phys. 2005, 103, 2141–2157. 10.1080/00268970500096251. [DOI] [Google Scholar]
- Neuhauser D.; Rabani E.; Baer R. Expeditious stochastic approach for MP2 energies in large electronic systems. J. Chem. Theory Comput. 2013, 9, 24–27. 10.1021/ct300946j. [DOI] [PubMed] [Google Scholar]
- Ge Q.; Gao Y.; Baer R.; Rabani E.; Neuhauser D. A guided stochastic energy-domain formulation of the second order Møller-Plesset perturbation theory. J. Phys. Chem. Lett. 2014, 5, 185–189. 10.1021/jz402206m. [DOI] [PubMed] [Google Scholar]
- Neuhauser D.; Baer R.; Zgid D. Stochastic Self-Consistent Second-Order Green’s Function Method for Correlation Energies of Large Electronic Systems. J. Chem. Theory Comput. 2017, 13, 5396–5403. 10.1021/acs.jctc.7b00792. [DOI] [PubMed] [Google Scholar]
- Takeshita T. Y.; De Jong W. A.; Neuhauser D.; Baer R.; Rabani E. Stochastic Formulation of the Resolution of Identity: Application to Second Order Møller-Plesset Perturbation Theory. J. Chem. Theory Comput. 2017, 13, 4605–4610. 10.1021/acs.jctc.7b00343. [DOI] [PubMed] [Google Scholar]
- Willow S. Y.; Kim K. S.; Hirata S. Stochastic evaluation of second-order many-body perturbation energies. J. Chem. Phys. 2012, 137, 204122. 10.1063/1.4768697. [DOI] [PubMed] [Google Scholar]
- Willow S. Y.; Kim K. S.; Hirata S. Brueckner-Goldstone quantum Monte Carlo for correlation energies and quasiparticle energy bands of one-dimensional solids. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 90, 201110. 10.1103/physrevb.90.201110. [DOI] [Google Scholar]
- Thom A. J. W.; Alavi A. Stochastic Perturbation Theory: A Low-Scaling Approach to Correlated Electronic Energies. Phys. Rev. Lett. 2007, 99, 143001. 10.1103/physrevlett.99.143001. [DOI] [PubMed] [Google Scholar]
- Almlöf J. Elimination of energy denominators in Møller-Plesset perturbation theory by a Laplace transform approach. Chem. Phys. Lett. 1991, 181, 319–320. 10.1016/0009-2614(91)80078-c. [DOI] [Google Scholar]
- Häser M.; Almlöf J. Laplace transform techniques in Møller-Plesset perturbation theory. J. Chem. Phys. 1992, 96, 489–494. 10.1063/1.462485. [DOI] [Google Scholar]
- Wilson A. K.; Almlöf J. Møller-Plesset correlation energies in a localized orbital basis using a Laplace transform technique. Theor. Chim. Acta 1997, 95, 49–62. 10.1007/s002140050182. [DOI] [Google Scholar]
- Kats D.; Usvyat D.; Schütz M. On the use of the Laplace transform in local correlation methods. Phys. Chem. Chem. Phys. 2008, 10, 3430. 10.1039/b802993h. [DOI] [PubMed] [Google Scholar]
- Kutzelnigg W. r12-Dependent terms in the wave function as closed sums of partial wave amplitudes for large l. Theor. Chim. Acta 1985, 68, 445–469. 10.1007/bf00527669. [DOI] [Google Scholar]
- Kutzelnigg W.; Klopper W. Wave functions with terms linear in the interelectronic coordinates to take care of the correlation cusp. I. General theory. J. Chem. Phys. 1991, 94, 1985–2001. 10.1063/1.459921. [DOI] [Google Scholar]
- Irmler A.; Gallo A.; Hummel F.; Grüneis A. Duality of Ring and Ladder Diagrams and Its Importance for Many-Electron Perturbation Theories. Phys. Rev. Lett. 2019, 123, 156401. 10.1103/physrevlett.123.156401. [DOI] [PubMed] [Google Scholar]
- Booth G. H.; Thom A. J. W.; Alavi A. Fermion Monte Carlo without fixed nodes: A game of life, death, and annihilation in Slater determinant space. J. Chem. Phys. 2009, 131, 054106. 10.1063/1.3193710. [DOI] [PubMed] [Google Scholar]
- Luo H.; Alavi A. Combining the Transcorrelated Method with Full Configuration Interaction Quantum Monte Carlo: Application to the Homogeneous Electron Gas. J. Chem. Theory Comput. 2018, 14, 1403–1411. 10.1021/acs.jctc.7b01257. [DOI] [PubMed] [Google Scholar]
- Hedin L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796–A823. 10.1103/physrev.139.a796. [DOI] [Google Scholar]
- Vlček V. Stochastic Vertex Corrections: Linear Scaling Methods for Accurate Quasiparticle Energies. J. Chem. Theory Comput. 2019, 15, 6254–6266. 10.1021/acs.jctc.9b00317. [DOI] [PubMed] [Google Scholar]
- Gell-Mann M.; Brueckner K. A. Correlation Energy of an Electron Gas at High Density. Phys. Rev. 1957, 106, 364–368. 10.1103/physrev.106.364. [DOI] [Google Scholar]
- Goedecker S.; Teter M.; Hutter J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 1703–1710. 10.1103/physrevb.54.1703. [DOI] [PubMed] [Google Scholar]
- www.cpmd.org, CPMD. Copyright IBM Corp 1990-2008, Copyright MPI für Festkörperforschung Stuttgart 1997-2001.
- Martyna G. J.; Tuckerman M. E. A reciprocal space based method for treating long range interactions in ab initio and force-field-based calculations in clusters. J. Chem. Phys. 1999, 110, 2810–2821. 10.1063/1.477923. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, CT, 2016.
- VandeVondele J.; Krack M.; Mohamed F.; Parrinello M.; Chassaing T.; Hutter J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. 10.1016/j.cpc.2004.12.014. [DOI] [Google Scholar]
- Liberatore E.; Meli R.; Rothlisberger U. A Versatile Multiple Time Step Scheme for Efficient ab Initio Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 2834–2842. 10.1021/acs.jctc.7b01189. [DOI] [PubMed] [Google Scholar]
- Tuckerman M. E.; Martyna G. J.; Berne B. J. Molecular dynamics algorithm for condensed systems with multiple time scales. J. Chem. Phys. 1990, 93, 1287–1291. 10.1063/1.459140. [DOI] [Google Scholar]
- Langreth D. C.; Perdew J. P. Exchange-correlation energy of a metallic surface: Wave-vector analysis. Phys. Rev. B: Solid State 1977, 15, 2884–2901. 10.1103/physrevb.15.2884. [DOI] [Google Scholar]
- Olsen T.; Thygesen K. S. Random phase approximation applied to solids, molecules, and graphene-metal interfaces: From van der Waals to covalent bonding. Phys. Rev. B: Condens. Matter Mater. Phys. 2013, 87, 075111. 10.1103/physrevb.87.075111. [DOI] [Google Scholar]
- Nguyen N. L.; Colonna N.; de Gironcoli S. Ab initio self-consistent total-energy calculations within the EXX/RPA formalism. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 90, 045138. 10.1103/physrevb.90.045138. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.