

Abstract
Excitatory amino-acid transporters (EAATs) bind and transport glutamate, limiting spillover from synapses due to their dense perisynaptic expression primarily on astroglia. Converging evidence suggests that abnormalities in the astroglial glutamate transporter localization and function may underlie a disease mechanism with pathological glutamate spillover as well as alterations in the kinetics of perisynaptic glutamate buffering and uptake contributing to dysfunction of thalamo-cortical circuits in schizophrenia. We explored this hypothesis by performing cell- and region-level studies of EAAT1 and EAAT2 expression in the mediodorsal nucleus of the thalamus in an elderly cohort of subjects with schizophrenia. We found decreased protein expression for the typically astroglial-localized glutamate transporters in the mediodorsal and ventral tier nuclei. We next used laser-capture microdissection and quantitative polymerase chain reaction to assess cell-level expression of the transporters and their splice variants. In the mediodorsal nucleus, we found lower expression of transporter transcripts in a population of cells enriched for astrocytes, and higher expression of transporter transcripts in a population of cells enriched for relay neurons. We confirmed expression of transporter protein in neurons in schizophrenia using dual-label immunofluorescence. Finally, the pattern of transporter mRNA and protein expression in rodents treated for 9 months with antipsychotic medication suggests that our findings are not due to the effects of antipsychotic treatment. We found a compensatory increase in transporter expression in neurons that might be secondary to a loss of transporter expression in astrocytes. These changes suggest a profound abnormality in astrocyte functions that support, nourish and maintain neuronal fidelity and synaptic activity.
INTRODUCTION
The thalamus is comprised of discrete, modality-specific nuclei that facilitate the transmission of information to the neocortex.1,2 Auditory and visual sensory information is filtered and relayed through the geniculate body, while the ventral tier and other nuclei comprise the motor portion of the thalamus, which relay sensory information critical for normal motor movements.1–3 The limbic part of the thalamus includes the mediodorsal (MD) and anterior nuclei. These areas have an integrative role within the limbic system, illustrated by the diverse inputs they receive from the ventral pallidum, hippocampus, entorhinal cortex, insula, amygdala and temporal cortex.2,4 These nuclei also have dense reciprocal projections with the dorsal lateral prefrontal and anterior cingulate cortices and are believed to have a role in emotional and executive functions.4,5 The juxtaposition of the MD nucleus within limbic circuitry led to the hypothesis that alterations in this structure could account for the variety of symptoms observed in schizophrenia due to its central role in processing sensory information.6,7
The thalamus contains three main neuron types: excitatory relay cells, local inhibitory interneurons and gamma-aminobutryic acid (GABA)-ergic neurons in the reticular nucleus.8 Large glutamatergic relay cells account for 60–70% of the neurons in the dorsal thalamus, whereas inhibitory interneurons account for the remaining 30–40% of neurons in this structure.9
Converging lines of evidence implicate abnormalities of thalamocortical circuits in schizophrenia.10 Resting thalamocortical oscillations were reduced in subjects with schizophrenia in prefrontal, premotor and motor, but not parietal, cortices, whereas another study found reduced thalamic functional magnetic resonance imaging–blood-oxygen-level-dependent (fMRI BOLD) responses following cortical stimulation with single-pulse transcranial magnetic stimulation.11,12 In addition, there was reduced functional connectivity between the frontal cortex and the MD/anterior thalamus and increased connectivity between motor and somatosensory cortex and the thalamus in subjects with schizophrenia measured using resting-state fMRI.13 Taken together, these data suggest a marked abnormality of thalamocortical circuitry in schizophrenia.
The function of these excitatory circuits is dependent on the release and reuptake of glutamate, as well as the expression and localization of myriad receptors, enzymes and transporters. We have previously reported cell-specific decreases in ionotropic glutamate-receptor transcripts in thalamic relay neurons in schizophrenia.14 We have also found increased region-level expression of excitatory amino-acid transporter (EAAT)-1 and EAAT2 transcripts in the thalamus using in situ hybridization, suggesting a profound alteration in glutamate transmission within the thalamus in this illness.15 To further test this hypothesis, we performed cell- and region-level studies of glutamate transporter expression using western blot analyses, laser-capture microdissection (LCM) and quantitative polymerase chain reaction (QPCR) to assess the fidelity of the glutamate reuptake machinery in the limbic thalamus in schizophrenia. Several glutamate transporter splice variants are expressed in the brain, including exon 9 skipping variants that lack parts of the glutamate translocation site and a 3’ variant that contains a PDZ trafficking domain.16 As EAATs are expressed as trimers, expression of EAAT splice variants in disease states might profoundly affect glutamate uptake and biological processes associated with normal transporter functions. We also assessed expression of a negative modulator of EAAT2 activity, G-protein pathway suppressor-1 (GPS1), as well as a marker of GABAergic inhibitory neurons, glutamic acid decarboxylase 67 (GAD67). We specifically hypothesize that cell-level changes in expression of glutamate transporters occur in thalamic relay neurons in schizophrenia.
MATERIALS AND METHODS
Tissue acquisition and preparation
Post-mortem brain tissue from patients with schizophrenia and a nonpsychiatrically ill comparison group was obtained from the Mount Sinai NIH Brain and Tissue Repository. We performed four specific experiments using post-mortem brain: two mRNA studies using LCM and QPCR, and two experiments using western blot analysis that examined different thalamic nuclei. Five subjects with schizophrenia and 10 control subjects were common to all four experiments (Supplementary Table S1). The Taqman and SYBR-Green mRNA studies have 15 control and 9 subjects with schizophrenia in common. Samples were not pooled in any of the experiments. Brains were obtained after autopsy and cut coronally into 10 mm slabs and frozen until further dissection. Twenty-micrometer sections, cut from tissue blocks of the thalamus at a horizontal level ~ 6–9 mm dorsal to the interventricular plane (about 23.5–27.5 mm caudal to the anterior commissure), were mounted on 1×3 inch penfoil polymer or superfrost plus glass slides. After sectioning, the ventral tier and MD nuclei were grossly dissected from the remaining tissue blocks. The last section from the tissue block for each case was nissl stained and used as a guide for dissections. Thalamic nuclei were identified and anatomically matched using descriptions of thalamic architecture, detailed atlases and (previously published) in situ hybridization studies.15,17
We performed LCM, QPCR, western blot analyses, immunofluorescence and rodent antipsychotic studies as previously described.14,18,19 Full details of these methods and extensive quality control studies (Supplementary Figures S1–S4) are provided in the Supplementary Materials.
Quantification and statistical analysis
All data sets were analyzed for normal distribution. Data from the mRNA studies were probed for associations between age, postmortem interval (PMI), tissue pH or RNA integrity (RIN) and the dependent measures using Pearson’s product-moment correlation. Data from the protein studies were probed for associations between age, tissue pH or PMI and the dependent measures. For the mRNA and protein studies, outliers more than three standard deviations from the mean were not included in the analyses. Analysis of covariance was performed if significant correlations were found. If no correlations were present, data were analyzed with analysis of variance. Secondary analyses were performed to assess the effects of sex on the dependent measures. All data sets were analyzed with Statistica (Statsoft, Tulsa, OK, USA).
RESULTS
We examined the expression of EAAT1, EAAT2, EAAT3, GPS1 and GAD67 protein using western blot analysis in the MD nucleus and the ventral tier nuclei of the thalamus.20 There were no significant associations between expression of EAAT1–3, GPS1 or GAD67 protein and age, pH, or PMI in the MD or ventral tier nuclei. In the MD nucleus, we found decreases in EAAT1 (F(1,23) = 5.03, P<0.05) and EAAT2 (F(1,21) = 11.3, P<0.01), but not EAAT3, GPS1 or GAD67 protein levels in subjects with schizophrenia (Figure 1). In the ventral tier nuclei, we detected a decrease in EAAT2 (F(1,22) = 4.73, P<0.05), but not EAAT1, EAAT3, GPS1 or GAD67 (Figure 1). No significant differences in β-tubulin were detected in any of our protein studies (data not shown). No effect of sex on the expression of EAAT1–3, GPS1 or GAD67 proteins was detected in the MD or ventral tier nuclei (data not shown).
Figure 1.

Western blot analyses of excitatory amino-acid transporter 1 (EAAT1), EAAT2, EAAT3, G-protein pathway suppressor-1 (GPS1) and glutamic acid decarboxylase-67 (GAD67) protein in tissue homogenate from the mediodorsal (SCZ N = 13, Comp N = 16) and ventral (SCZ N = 13, Comp N = 15) nuclei of the thalamus in schizophrenia (SCZ) and comparison subjects (Comp). Data are normalized to β-tubulin protein levels assayed in the same lane on the same blot. Data are expressed as means ± s.e.m. *P<0.05.
To determine which cell types account for the changes in EAAT protein expression, we used LCM to harvest excitatory glutamatergic relay neurons (large cells) and astrocytes (small cells) from the MD nucleus of the thalamus at a horizontal level ~ 6–9 mm dorsal to the interventricular plane, about 17.5–23.5 mm caudal to the anterior commissure (Figure 2).20 Large cells were identified based on their size and distinctive morphology, whereas small cells were selected on the basis of size and the absence of distinctive structures such as an apical dendrite. Cells with intermediate size profiles or ambiguous morphology were not selected for harvest. The large cell population was enriched for vesicular glutamate transporter 2 (VGLUT2) mRNA, a marker of glutamatergic neurons expressed in thalamic relay neurons.21 Neuron-specific enolase (NSE) transcripts were slightly enriched in the large cell population, whereas glial fibrillary acid protein (GFAP) transcripts, a marker for astrocytes, were not enriched (Figure 2). The enriched population of astrocytes likely contains some GABAergic interneurons due to the presence of GAD67 transcripts in our small cell samples (Supplementary Figure S2).
Figure 2.
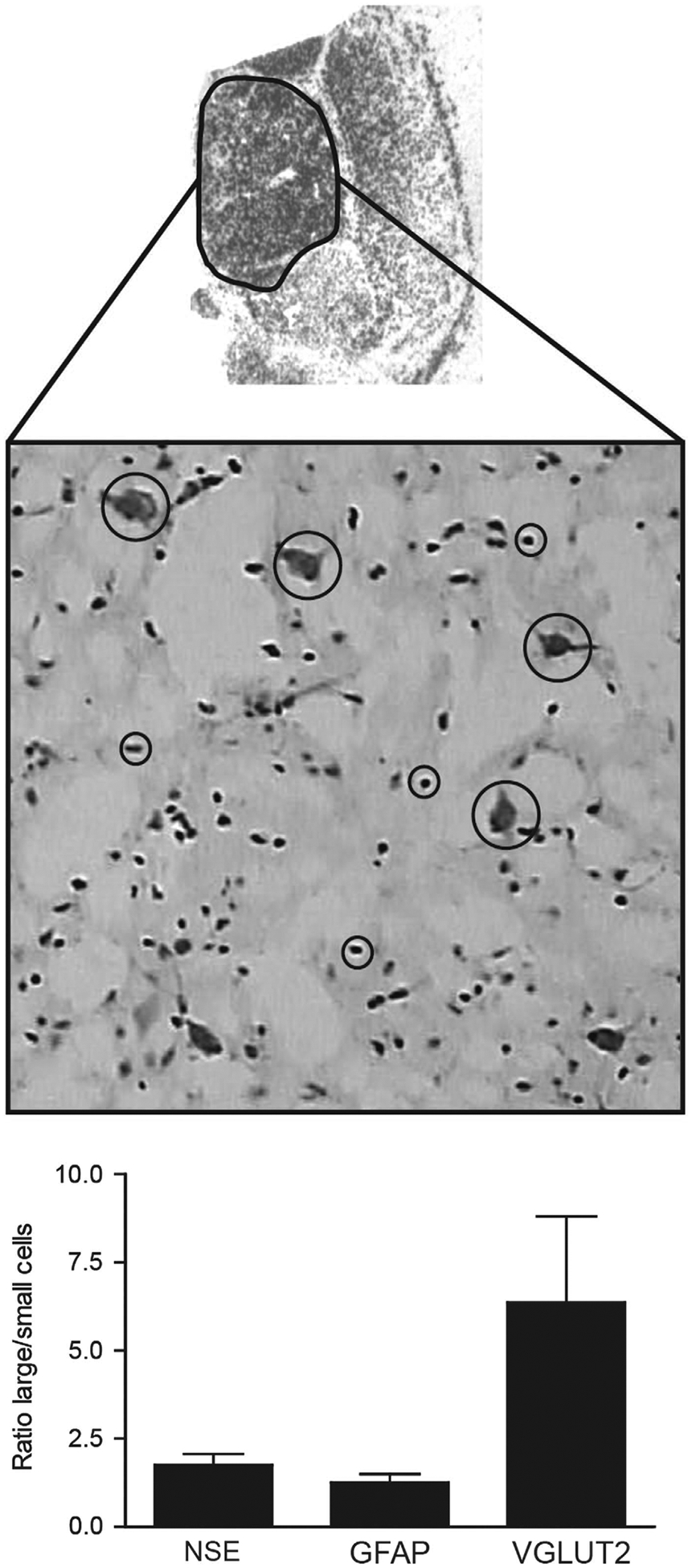
Top: Autoradiogram of in situ hybridization for EAAT2 mRNA in the human thalamus. The highlighted region is the mediodorsal (MD) nucleus of the thalamus. Middle: Nissl stain of a 20-μm tissue section from the MD nucleus, × 20 magnification. Relay neurons (large cells, big circles) and astrocytes (small cells, small circles) were identified based on morphology. Bottom: QPCR analysis of markers for relay neurons (VLGUT2, NSE) and astrocytes (GFAP) harvested using laser-capture microdissection. Data are expressed as means ± s.e.m. of the ratios of values for each subject for 500 large cells/500 small cells (n = 23). Large and small cells were captured from the same tissue section with different caps. EAAT2, excitatory amino-acid transporter 2; GFAP, glial fibrillary acidic protein; NSE, neuron-specific enolase; VGLUT2, vesicular glutamate transporter 2.
We measured expression of transcripts for EAAT1, EAAT2 and EAAT3 using Taqman QPCR assays that detect regions of the EAAT mRNAs common to most splice variants (Figure 3a). In small cells, we found a significant association between EAAT1 and EAAT2 expression and tissue pH (EAAT1: R = 0.63, P<0.05, EAAT2: R = 0.39, P<0.05), but not age or RIN. We also found a significant association between EAAT1 expression and PMI (R = 0.47, P<0.05). In large cells, we found a significant association between EAAT2 and age (R = 0.44, P<0.05), but not PMI, RIN or tissue pH. We did not detect any associations between EAAT3 expression and age, pH, PMI or RIN.
Figure 3.
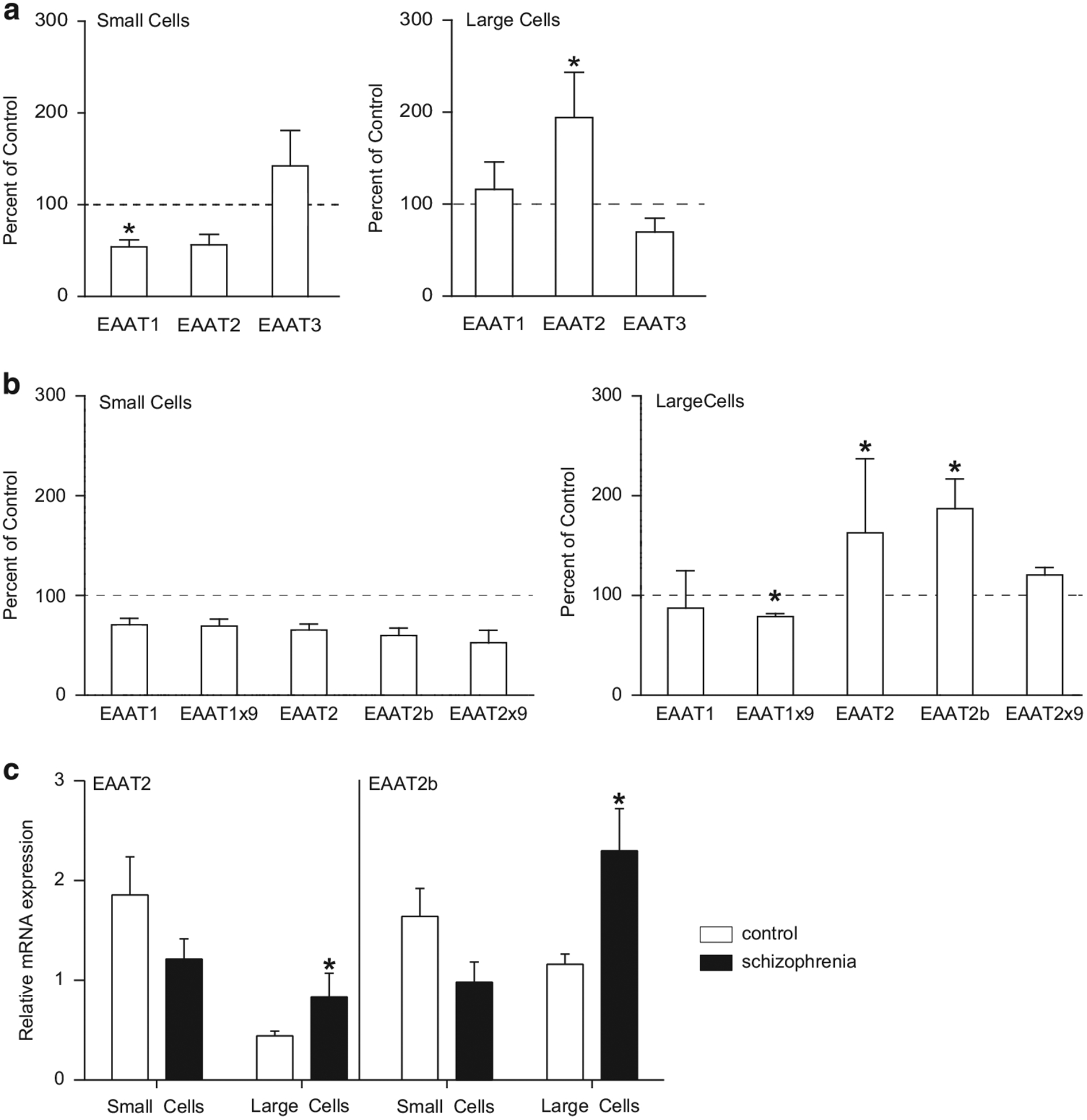
(a) Probe-based QPCR analysis of excitatory amino-acid transporters (EAATs) 1, 2 and 3 transcripts isolated from enriched populations of astrocytes (small cells) and relay neurons (large cells) from the mediodorsal (MD) nucleus of the thalamus obtained using laser-capture microdissection (LCM). N = 12 subjects with schizophrenia, N = 20 comparison subjects. (b) Dye-based QPCR analysis of native EAATs (EAAT1 and EAAT2) and the splice variant EAAT1 exon 9 skipping (EAAT1×9), EAAT2b and EAAT2 exon 9 skipping (EAAT2×9) transcripts isolated from enriched populations of astrocytes (small cells) and relay neurons (large cells) from the MD nucleus of the thalamus obtained using LCM. N = 9 subjects with schizophrenia, N = 15 comparison subjects. (c) Relative expression levels of EAAT2 and EAAT2b in large and small cells from the dye-based QPCR experiments. Large and small cell samples were processed separately, normalized to the geometric mean of three housekeeping genes, and analyzed with the standard curve method for relative quantification using a pooled calibrator sample for each population of cells. Data are expressed as means ± s.e.m. *P<0.05.
We found a decrease in EAAT1 (F(1, 26) = 6.15, P<0.05), but not EAAT2 or EAAT3, mRNA expression in small cells harvested from the MD nucleus in subjects with schizophrenia (Figure 3a). We found an increase in EAAT2 (F(1,23) = 6.23, P<0.05), but not EAAT1 or EAAT3, mRNA in the large cell population. No effect of sex on the expression of EAAT1–3 transcripts was detected in the small or large cell samples (data not shown).
In a separate experiment, we measured expression of EAAT1 and EAAT2 mRNAs and several EAAT splice variants using SYBR-Green QPCR assays in large and small cells harvested from the MD nucleus. In small and large cell populations, we did not find any significant associations between EAAT transcript expression and tissue pH, RIN, PMI or age of death. In small cells, we did not detect any changes in EAAT transcript expression in subjects with schizophrenia (Figure 3b). In the large cell sample, we detected increases in EAAT2 (F(1,17) = 6.46, P<0.05) and EAAT2b (F(1, 16) = 9.0, P<0.01) mRNAs, a decrease in EAAT1 exon 9 skipping (EAAT1×9) (F(1,19) = 15.1, P<0.01) mRNA, but no changes in EAAT1 or EAAT2 exon 9 skipping (EAAT2×9), (Figure 3b).
To graphically demonstrate the shift in expression of transporter splice variants in an astrocyte-enriched sample to a neuronal-enriched sample, we plotted the relative expression levels of EAAT2 and EAAT2b mRNAs in large and small cells from the dye-based QPCR experiments (Figure 3c). As the large and small cell populations were analyzed separately using a relative quantification approach, we only compared schizophrenia versus the control group for each transcript.
Other than EAAT2 mRNA expression in the small cell sample (females4males, F(1, 22) = 7.36, P<0.01), no effect of sex on the expression of EAATs was detected in the SYBR-Green PCR studies (data not shown).
As EAAT2 mRNA is reportedly expressed at low levels in neurons, we performed double-label immunofluorescence to determine if EAAT2 protein localizes to neurons in schizophrenia.22,23 EAAT2 protein was expressed in a diffuse, punctate manner consistent with the expression in astrocytic processes in the MD nucleus in subjects with schizophrenia (Figures 4a and d).24,25 EAAT2 was also expressed in small or intermediate size cells with the morphology of neurons (Figures 4a and d, small white arrows). EAAT2 expression colocalized with the neuronal marker NeuN (Figures 4c and e, arrows), but not GAD67 (data not shown) in these cells. A subset of larger cells with an excitatory relay neuron-like morphology also expressed NeuN and EAAT2 (Figure 4e). We did not detect a fluorescent signal in sections stained with control antibodies (Supplementary Figure S4).
Figure 4.
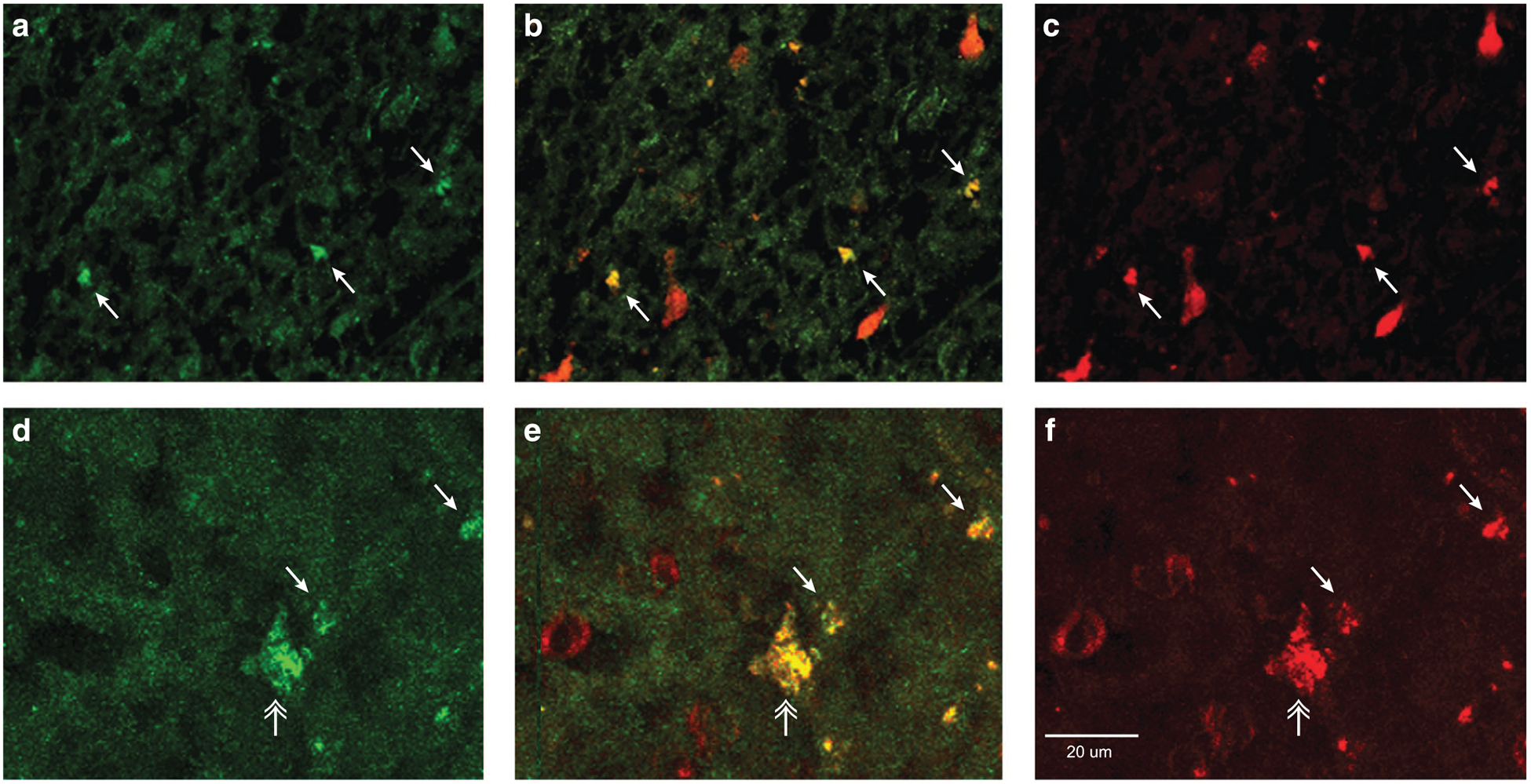
EAAT2 protein localization in the mediodorsal nucleus of the thalamus in subjects with schizophrenia. Labeling of cell profiles with (a and d) EAAT2 (green), (c and f) NeuN (red) and (b and e) merge of NeuN and EAAT2 (yellow). The top row (a–c) are images from a Mount Sinai NIH Brain and Tissue Repository subject, the bottom row (d–f) images are from an Alabama Brain Collection subject. The small white arrows indicate intermediate size cells double labeled for NeuN and EAAT2. The white double arrow indicates a larger cell with relay neuron-like morphology. Isotype and species-specific preimmune IgG control studies are shown in Supplementary Figure S4.
We assessed the effects of antipsychotic medication on the expression of EAATs in the thalamus in rats treated for 9 months with haloperidol decanoate. Haloperidol was used for these studies as most of the human subjects in our study were taking typical antipsychotics. Using SYBR-Green QPCR, we found increased expression of EAAT1 (F(1,15) = 15.9, P = 0.05), EAAT2 (F(1,15) = 19.2, P<0.01) and EAAT1×9 (F(1,13) = 4.6, P<0.01) transcripts (Figure 5a). We did not detect changes in EAAT2b mRNA. We were unable to measure EAAT2×9 mRNA because we could not validate an assay for this splice variant in the rodent. Using western blot analysis, we found an increase in EAAT2 protein (F(1,18) = 17.2, P<0.01), whereas changes in EAAT1 protein were not detected (Figure 5b).
Figure 5.
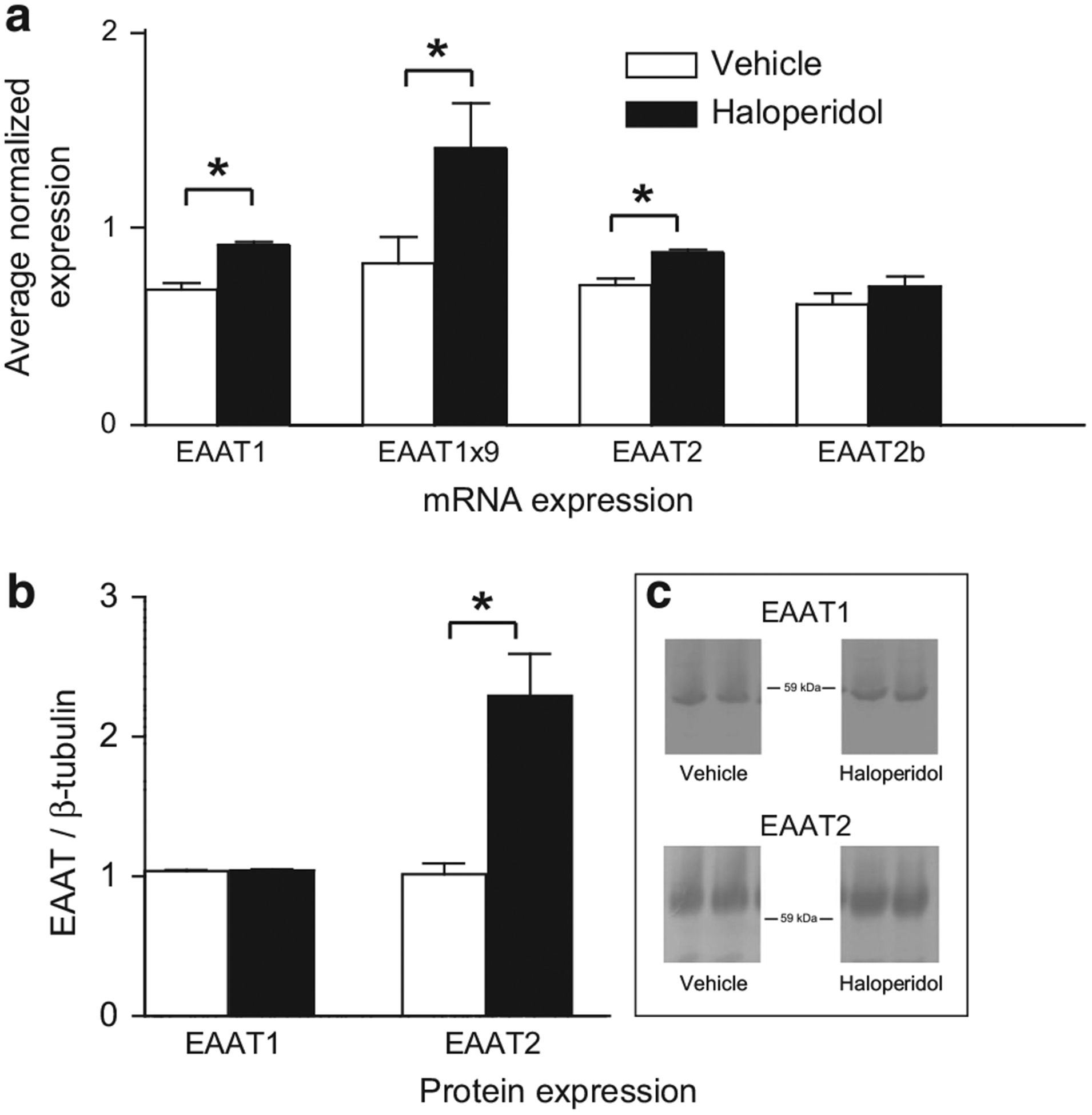
(a) Dye-based QPCR analysis of excitatory amino-acid transporter 1 (EAAT1), EAAT2, EAAT1 exon 9 skipping (EAAT1×9) and EAAT2b transcripts isolated from the thalamus of rats (N = 10 per group) treated with haloperidol decanoate (28.5 mg kg−1 every 3 weeks) or vehicle (sesame oil) for 9 months. (b) Western blot analyses of EAAT1 and EAAT2 protein in tissue homogenate from the thalamus in rats treated as described above for panel (a). Protein data are normalized to β-tubulin protein levels assayed in the same lane on the same blot. Data are expressed as means ± s.e.m. *P<0.05.
DISCUSSION
We have previously reported increased region-level expression of EAAT1 and EAAT2 transcripts in the thalamus in schizophrenia using in situ hybridization. From these results, we hypothesized that glutamate uptake capacity in astrocytes may be increased, based on the observations that (1) EAAT1 and EAAT2 are predominately expressed on astroglia26 and (2) these transporters account for the large majority (about 90%) of Na+-dependent glutamate reuptake in most brain regions.27,28 In the present study, we first measured protein levels of the transporters in the MD nucleus and ventral tier nuclei, and found significant decreases in EAAT1 and EAAT2 protein expression. This discrepancy with our previous mRNA studies may be explained by subsequent work demonstrating that EAAT1–2 may also be expressed in neurons in normal brain,22,23 and that cellular localization of glutamate transporters may be altered in diseases of the central nervous system.29–31 Thus, we tested the hypothesis that there are cell-level changes in transporter expression in the thalamus in schizophrenia. In the MD nucleus, using LCM–QPCR, we detected a shift in expression of transporter splice variants from a population of cells enriched for astrocytes (small cells), to a population enriched for excitatory relay neurons (large cells) in this illness. Using immunofluorescence, we confirmed the expression of EAAT2 protein in cells positive for the neuronal marker NeuN in a subset of subjects with schizophrenia. Changes in region-level EAAT1–2 expression in antipsychotic-treated rats, with the one exception of EAAT2 transcripts, were in the opposite direction of our human findings. Increased expression of glutamate transporters on neurons suggests a loss of astroglial reuptake capacity, and may be a pathophysiological sign of disrupted coupling of glutamate metabolism between neurons and astrocytes.
Localization of glutamate transporters to astrocytes facilitates Na+-dependent glutamate reuptake; transporters are co-expressed in perisynaptic regions with Na+/K+ ATPases, mitochondria and glycolytic enzymes.32–35 Functional coupling of these elements facilitates rapid glutamate reuptake, maintenance of the Na+/K+ gradients, and provides adenosine triphosphate (ATP), lactate, and other energy intermediates to maintain and regulate these processes.36 A shift in transporter expression from astrocytes to neurons may significantly impair critical reciprocal functions between these cells that facilitate normal brain function.
A cellular shift in the expression of EAAT2 has been previously characterized in amyotrophic lateral sclerosis (ALS).31 Cell-level immunohistochemical studies found decreased expression of EAAT2 in astrocytes, with an increase of the EAAT2 splice variant EAAT2b in motor cortex neurons.31 Diminished global EAAT2 protein expression in astrocytes likely accounts for the large deficits in glutamate reuptake present in ALS, and is the most consistently replicated finding amongst animal models of ALS, as well as postmortem studies of spontaneous and familial ALS cases.31 Some cases of ALS have as high as a 90% decrease in region-level expression of EAAT2 protein, while region-level decreases of EAAT2 protein expression in the superior temporal gyrus, hippocampus and thalamus in schizophrenia are more modest, in the 20–30% range.37
The authors of these ALS studies speculate that diminished glutamate reuptake increases inward calcium currents in post-synaptic neurons, initiating excitotoxic events including dissociation of mitochondrial proteins.31 Recent findings may support this hypothesis.
Dissociation of the enzyme hexokinase from mitochondrial voltage-dependent anion channel diminished EAAT2-mediated glutamate reuptake in rodent brain.35 EAAT2, hexokinase and voltage-dependent anion channel are components of a putative glutamate transport protein complex that is tightly linked spatially and functionally to mitochondria.19,38 Interestingly, we found an increase in the ratio of cytoplasmic:mitochondrial hexokinase protein expression in frontal cortex in schizophrenia, suggesting disruption of the glutamate transport protein complex in this illness.19
Increased expression of transcripts for EAAT2 splice variants was also found in neurons following hypoxia, spinal cord injury and traumatic brain injury.29,39–43 These data suggest a common pathological mechanism in diseased or injured brain, where increased neuronal expression of glutamate transporter isoforms may represent a compensatory response in the setting of diminished astroglial glutamate reuptake.44,45 This shift, a sort of ‘glialization’ of neurons, also appears to be present in schizophrenia, as we found a similar effect in the thalamus in this illness. Although the expression of EAAT2 splice variants is postulated to have a protective effect, the benefits of a shift of EAAT2-mediated glutamate reuptake may be limited, as overexpression of EAAT2 in neurons in hippocampal slices lowered the threshold for glutamate-mediated excitotoxicity.29,46
The predominant isoform of EAAT2 that we found increased in an enriched population of thalamic relay neurons is EAAT2b, a variant with a unique intronic coding sequence that retains the ability to transport glutamate and regulate extrasynaptic glutamate levels.16,44,47 EAAT2b has 11 unique amino acids at the C terminus containing a PDZ domain.48 PDZ domains facilitate protein–protein interactions by binding specific target domains on other proteins and are found on a large number of post-synaptic density (PSD) proteins.49
Interestingly, glutamate transporter-1b (GLT1b), the rodent variant of EAAT2b, likely has a direct protein–protein interaction with PSD95, a ubiquitous PSD marker found in post-synaptic neurons, as well as protein interacting with C-kinase-1 (PICK1), an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor interacting protein.48,50 These properties suggest that EAAT2b may be trafficked to PSDs, an idea supported by ultrastructural studies localizing EAAT2 to the PSD in normal human brain.23 Our immunohistochemistry studies (Figure 5) suggest a somatic distribution for EAAT2 protein expression in some neurons, which is consistent with the pattern observed in ALS.44 However, the present work lacks the resolution to determine if neuronal expression of EAAT2 protein is selectively increased in the PSD in schizophrenia.
We have previously reported an increase in EAAT2b protein levels in schizophrenia in a cytosolic fraction prepared from cortical tissue homogenate containing astroglia and neurons.19 In this previous study, although the PSD was not directly assessed, we did not detect changes in EAAT2b protein levels in the fractions containing PSDs. We have also found an increase in EAAT2b transcripts in an enriched population of pyramidal neurons harvested from the anterior cingulate cortex in subjects with schizophrenia.51 Taken together, these data suggest that neuronal EAAT2b expression is somatic and/or cytosolic in disease states, and might not be properly trafficked to the PSD or other domains where EAAT2 is expressed at low levels in neurons in normal brain.23 Trafficking of proteins to and from microdomains such as the PSD is tightly regulated, and accumulating evidence suggests that this process may be disrupted in schizophrenia.52–54
We found a decrease in EAAT1 protein in the MD nucleus, and a corresponding decrease in EAAT1 mRNA in cells enriched for astrocytes. We previously reported diminished EAAT1 protein levels in the frontal cortex and superior temporal gyrus in schizophrenia.37,55,56 EAAT1 and EAAT2 expression is generally found in nonoverlapping subsets of glia, suggesting that changes in EAAT1 expression are in a functionally discrete subset of cells that do not express EAAT2.57 Glutamate aspartate transporter (GLAST; rodent EAAT1) knockout mice have behavioral endophenotypes associated with schizophrenia, including deficits in working memory and acoustic startle, without a decrease in EAAT2 expression.58,59 These rodent studies suggest that diminished EAAT1-mediated glutamate reuptake alone is sufficient to contribute to the genesis of core signs and symptoms of this illness. Supporting this hypothesis, one subject with schizophrenia with a partial deletion of EAAT1 has been reported. Generation of a transgenic animal with this genetic lesion might provide an important model for studying the role of glutamate transport in this illness.60 Regardless of whether the loss of EAAT1 expression is genetic or acquired, decreases in EAAT1 protein expression across limbic regions may significantly contribute to the pathophysiology of this disease.
We also found decreased transcripts for the EAAT1×9 splice variant in an enriched population of relay neurons in schizophrenia. The EAAT1×9 variant does not transport glutamate, as it lacks the glutamate translocation domain within exon 9.61 In cell culture, coexpression of EAAT1×9 with native EAAT1 reduced insertion of EAAT1 complexes into the plasma membrane, exerting a dominant negative effect of EAAT1-mediated glutamate reuptake.62 The authors of this study speculate that this splice variant has a potent regulatory role in astroglia, as EAAT1×9 is widely distributed in normal brain.62 The effects of EAAT1×9 in neurons have not been investigated. EAAT1×9 expression has been reported in pyramidal cells in Alzheimer’s disease, and in neurons in hypoxia, where it may act as a marker of cells at risk for excitotoxic injury.30,63 Further work is needed to determine if EAAT1×9 transcripts are translated to protein in neurons and what role a change in expression of this variant would have on neuronal function.
In contrast to EAAT1×9, we did not find changes in thalamic EAAT2×9 expression in our study. We also did not find changes in EAAT2×9 transcripts in LCM-acquired pyramidal neurons from the frontal cortex,51 suggesting that cell-level changes in EAAT2 splice variant expression in neurons in schizophrenia are specific to EAAT2b.
Although the changes in glutamate transporter expression in thalamus could be compensatory, causative or both, we postulate that the region-level decrease in EAAT1 and EAAT2 protein expression reflects a loss of astrocytic glutamate reuptake and buffering in this structure. These changes might indicate a primary deficit in astrocytes, a selective loss of function, a decrease in astrocytes themselves, and/or a loss of astrocyte processes. Several studies have reported thinning of cortical gray matter, accompanied by decreased density of astrocytes, as well as a loss of neuropil.64 These findings suggest a marked abnormality in the ultrastructural elements that account for the large volume of gray matter not occupied by cell bodies or synapses. Regardless of the mechanism, diminished astrocyte-mediated glutamate reuptake could lead to pathological spillover of glutamate from excitatory synapses, activating extrasynaptic glutamate receptors that modulate diverse processes, including presynaptic neurotransmitter release as well as cell death pathways that regulate spine turnover.65–69 Such changes may lead to a compensatory response in neurons that includes increased expression of glutamate transporters. Interestingly, the kinetics of excitatory post-synaptic currents may be slowed by limiting surface diffusion of astrocytic transporters, suggesting that impairment of perisynaptic glutamate reuptake and buffering directly leads to alterations in neuroplasticity.70
Although more work is needed to assess the nature and degree of alterations in thalamic neurons, accumulating evidence from neuroimaging studies suggests dysfunction in thalamocortical networks in schizophrenia with the MD nucleus as the primary locus of dysconnectivity.6,11,13,71 These deficits may underlie abnormalities of cortical oscillations, as thalamo–cortico–thalamic circuit activation organizes and supports neuronal synchrony in the gamma band width, and may be essential during cognitive and sensory processing.72–76
In summary, in the MD nucleus we found cell-type-specific changes in EAAT mRNA expression in enriched populations of neurons and astrocytes, with a global decrease in EAAT2 protein expression. These data suggest that astrocyte functions that rely on glutamate uptake to support neuronal activity are compromised, contributing to well-characterized deficits of thalamocortical circuits in schizophrenia. Further work will be required to determine if this pathophysiological mechanism is specific to schizophrenia, or whether it is also found in other psychiatric illnesses, such as mood disorders, where derangements of glutamate transmission may also have a central role.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by MH53327 (JHM-W), MH88752 (JHM-W), MH094445 (REM), MH074016 (REM) and Doris Duke Clinical Scientist Award (REM). This work was also supported by the Lindsay Brinkmeyer Schizophrenia Research Fund.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
REFERENCES
- 1.Steriade M, Jones EG, McCormick DA (eds). The Thalamus. Elsevier: Oxford, 1997. [Google Scholar]
- 2.Alelu-Paz R, Gimenez-Amaya JM. The mediodorsal thalamic nucleus and schizophrenia. J Psychiatry Neurosci 2008; 33, 489–498. [PMC free article] [PubMed] [Google Scholar]
- 3.Briggs F, Usrey WM. Emerging views of corticothalamic function. Curr Opin Neurobiol 2008; 18: 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russchen FT, Amaral DG, Price JL. The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis. J Comp Neurol 1987; 256: 175–210. [DOI] [PubMed] [Google Scholar]
- 5.Cummings JL. Anatomic and behavioral aspects of frontalsubcortical circuits In: Grafman J, Holyoak KJ, Boller F (eds). Structures and Functions of the Human Prefrontal Cortex, vol. 769 New York Academy of Sciences: New York, 1995, pp 1–13. [DOI] [PubMed] [Google Scholar]
- 6.Andreasen NC, Arndt S, Swayze V 2nd, Cizadlo T, Flaum M, D O’Leary et al. Thalamic abnormalities in schizophrenia visualized through magnetic resonance image averaging. Science 1994; 266: 294–298. [DOI] [PubMed] [Google Scholar]
- 7.Jones EG. Cortical development and thalamic pathology in schizophrenia. Schizophr Bull 1997; 23: 483–501. [DOI] [PubMed] [Google Scholar]
- 8.Jones EG. A new view of specific and nonspecific thalamocortical connections. Adv Neurol 1998; 77: 49–71. [PubMed] [Google Scholar]
- 9.Dorph-Petersen KA, Pierri JN, Sun Z, Sampson AR, Lewis DA. Stereological analysis of the mediodorsal thalamic nucleus in schizophrenia: volume, neuron number, and cell types. J Comp Neurol 2004; 472: 449–462. [DOI] [PubMed] [Google Scholar]
- 10.Torrey EF, Peterson MR. Schizophrenia and the limbic system. Lancet 1974; 2: 942–946. [DOI] [PubMed] [Google Scholar]
- 11.Ferrarelli F, Sarasso S, Guller Y, Riedner BA, Peterson MJ, Bellesi M et al. Reduced natural oscillatory frequency of frontal thalamocortical circuits in schizophrenia. Arch Gen Psychiatry 2012; 69: 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guller Y, Ferrarelli F, Shackman AJ, Sarasso S, Peterson MJ, Langheim FJ et al. Probing thalamic integrity in schizophrenia using concurrent transcranial magnetic stimulation and functional magnetic resonance imaging. Arch Gen Psychiatry 2012; 69: 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodward ND, Karbasforoushan H, Heckers S. Thalamocortical dysconnectivity in schizophrenia. Am J Psychiatry 2012; 169: 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sodhi MS, Simmons M, McCullumsmith R, Haroutunian V, Meador- Woodruff JH. Glutamatergic gene expression is specifically reduced in thalamocortical projecting relay neurons in schizophrenia. Biol Psychiatry 2011; 70: 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Expression of excitatory amino acid transporter transcripts in the thalamus of subjects with schizophrenia. Am J Psychiatry 2001; 158: 1393–1399. [DOI] [PubMed] [Google Scholar]
- 16.Lauriat TL, McInnes LA. EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders. Mol Psychiatry 2007; 12: 1065–1078. [DOI] [PubMed] [Google Scholar]
- 17.Dekaban A. Human thalamus; an anatomical, developmental and pathological study. I. Division of the human adult thalamus into nuclei by use of the cyto-myelo-architectonic method. J Comp Neurol 1953; 99: 639–683. [DOI] [PubMed] [Google Scholar]
- 18.Shan D, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Expression of equilibrative nucleoside transporter type 1 protein in elderly patients with schizophrenia. Neuroreport 2012; 23: 224–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shan D, Mount D, Moore S, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal partitioning of hexokinase 1 suggests disruption of a glutamate transport protein complex in schizophrenia. Schizophr Res 2014; 154: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morel A, Magnin M, Jeanmonod D. Multiarchitectonic and stereotactic atlas of the human thalamus. J Comp Neurol 1997; 387: 588–630. [DOI] [PubMed] [Google Scholar]
- 21.Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Vesicular glutamate transporter transcript expression in the thalamus in schizophrenia. Neuroreport 2001; 12: 2885–2887. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R et al. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 2002; 22: 2142–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts RC, Roche JK, McCullumsmith RE. Localization of excitatory amino acid transporters EAAT1 and EAAT2 in human postmortem cortex: a light and electron microscopic study. Neuroscience 2014; 277: 522–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1995; 15: 711–720. [DOI] [PubMed] [Google Scholar]
- 25.Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 1995; 15: 1835–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65: 1–105. [DOI] [PubMed] [Google Scholar]
- 27.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996; 16: 675–686. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997; 276: 1699–1702. [DOI] [PubMed] [Google Scholar]
- 29.Pow DV, Naidoo T, Lingwood BE, Healy GN, Williams SM, Sullivan RK et al. Loss of glial glutamate transporters and induction of neuronal expression of GLT-1B in the hypoxic neonatal pig brain. Brain Res Dev Brain Res 2004; 153: 1–11. [DOI] [PubMed] [Google Scholar]
- 30.Sullivan SM, Macnab LT, Bjorkman ST, Colditz PB, Pow DV. GLAST1b, the exon-9 skipping form of the glutamate-aspartate transporter EAAT1 is a sensitive marker of neuronal dysfunction in the hypoxic brain. Neuroscience 2007; 149: 434–445. [DOI] [PubMed] [Google Scholar]
- 31.Philips T, Rothstein JD. Glial cells in amyotrophic lateral sclerosis. Exp Neurol 2014; 262: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rose EM, Koo JC, Antflick JE, Ahmed SM, Angers S, Hampson DR. Glutamate transporter coupling to Na,K-ATPase. J Neurosci 2009; 29: 8143–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC et al. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 2011; 31: 18275–18288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cholet N, Pellerin L, Magistretti PJ, Hamel E. Similar perisynaptic glial localization for the Na+,K+-ATPase alpha 2 subunit and the glutamate transporters GLAST and GLT-1 in the rat somatosensory cortex. Cereb Cortex 2002; 12: 515–525. [DOI] [PubMed] [Google Scholar]
- 35.Jackson JG, O’Donnell JC, Krizman E, Robinson MB. Displacing hexokinase from mitochondrial voltage-dependent anion channel impairs GLT-1-mediated glutamate uptake but does not disrupt interactions between GLT-1 and mitochondrial proteins. J Neurosci Res 2014; 93: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouzier-Sore AK, Pellerin L. Unraveling the complex metabolic nature of astrocytes. Front Cell Neurosci 2013; 7: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shan D, Lucas EK, Drummond JB, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal expression of glutamate transporters in temporal lobe areas in elderly patients with schizophrenia. Schizophr Res 2013; 144: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shan D, Yates S, Roberts RC, McCullumsmith RE. Update on the neurobiology of schizophrenia: a role for extracellular microdomains. Minerva Psichiatr 2012; 53: 233–249. [PMC free article] [PubMed] [Google Scholar]
- 39.Tawfik VL, Regan MR, Haenggeli C, Lacroix-Fralish ML, Nutile-McMenemy N, Perez N et al. Propentofylline-induced astrocyte modulation leads to alterations in glial glutamate promoter activation following spinal nerve transection. Neuroscience 2008; 152: 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J et al. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol 1997; 42: 335–348. [DOI] [PubMed] [Google Scholar]
- 41.Cimarosti H, Jones NM, O’Shea RD, Pow DV, Salbego C, Beart PM. Hypoxic preconditioning in neonatal rat brain involves regulation of excitatory amino acid transporter 2 and estrogen receptor alpha. Neurosci Lett 2005; 385: 52–57. [DOI] [PubMed] [Google Scholar]
- 42.Yi JH, Pow DV, Hazell AS. Early loss of the glutamate transporter splice-variant GLT-1v in rat cerebral cortex following lateral fluid-percussion injury. Glia 2005; 49: 121–133. [DOI] [PubMed] [Google Scholar]
- 43.Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma 2013; 30: 1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maragakis NJ, Dykes-Hoberg M, Rothstein JD. Altered expression of the glutamate transporter EAAT2b in neurological disease. Ann Neurol 2004; 55: 469–477. [DOI] [PubMed] [Google Scholar]
- 45.McCullumsmith RE. Evidence for schizophrenia as a disorder of neuroplasticity. Am J Psychiatry 2015; 172: 312–313. [DOI] [PubMed] [Google Scholar]
- 46.Selkirk JV, Stiefel TH, Stone IM, Naeve GS, Foster AC, Poulsen DJ. Over- expression of the human EAAT2 glutamate transporter within neurons of mouse organotypic hippocampal slice cultures leads to increased vulnerability of CA1 pyramidal cells. Eur J Neurosci 2005; 21: 2291–2296. [DOI] [PubMed] [Google Scholar]
- 47.Lee A, Pow DV. Astrocytes: glutamate transport and alternate splicing of transporters. Int J Biochem Cell Biol 2010; 42: 1901–1906. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez-Gonzalez IM, Garcia-Tardon N, Cubelos B, Gimenez C, Zafra F. The glutamate transporter GLT1b interacts with the scaffold protein PSD-95. J Neurochem 2008; 105: 1834–1848. [DOI] [PubMed] [Google Scholar]
- 49.Feng W, Zhang M. Organization and dynamics of PDZ-domain-related supra-modules in the postsynaptic density. Nat Rev Neurosci 2009; 10: 87–99. [DOI] [PubMed] [Google Scholar]
- 50.Bassan M, Liu H, Madsen KL, Armsen W, Zhou J, Desilva T et al. Interaction between the glutamate transporter GLT1b and the synaptic PDZ domain protein PICK1. Eur J Neurosci 2008; 27: 66–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Donovan SM, Hassefeld K, Bauer D, Simmons M, Roussos P, Haroutunian V et al. Glutamate transporter splice variant expression in an enriched pyramidal cell population in schizophrenia. Transl Psychiatry 2015; 5: e579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hammond JC, McCullumsmith RE, Funk AJ, Haroutunian V, Meador-Woodruff JH. Evidence for abnormal forward trafficking of AMPA receptors in frontal cortex of elderly patients with schizophrenia. Neuropsychopharmacology 2010; 35: 2110–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kristiansen LV, Bakir B, Haroutunian V, Meador-Woodruff JH. Expression of the NR2B-NMDA receptor trafficking complex in prefrontal cortex from a group of elderly patients with schizophrenia. Schizophr Res 2010; 119: 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kristiansen L, Patel S, Haroutunian V, Meador-Woodruff JH. Expression of the NR2B-NMDA receptor subunit and its Tbr-1/CINAP regulatory proteins in postmortem brain suggest altered receptor processing in schizophrenia. Synapse 2010; 64: 495–502. [DOI] [PubMed] [Google Scholar]
- 55.Bauer D, Gupta D, Harotunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia. Schizophr Res 2008; 104: 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bauer D, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal glycosylation of EAAT1 and EAAT2 in prefrontal cortex of elderly patients with schizophrenia. Schizophr Res 2010; 117: 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM et al. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci 2007; 27: 6607–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karlsson RM, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 2009; 34: 1578–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karlsson RM, Tanaka K, Heilig M, Holmes A. Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol Psychiatry 2008; 64: 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008; 320: 539–543. [DOI] [PubMed] [Google Scholar]
- 61.Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR. Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging 2011; 32: 553.e1–e11. [DOI] [PubMed] [Google Scholar]
- 62.Vallejo-Illarramendi A, Domercq M, Matute C. A novel alternative splicing form of excitatory amino acid transporter 1 is a negative regulator of glutamate uptake. J Neurochem 2005; 95: 341–348. [DOI] [PubMed] [Google Scholar]
- 63.Pow DV, Cook DG. Neuronal expression of splice variants of “glial” glutamate transporters in brains afflicted by Alzheimer’s disease: unmasking an intrinsic neuronal property. Neurochem Res 2009; 34: 1748–1757. [DOI] [PubMed] [Google Scholar]
- 64.Pakkenberg B, Scheel-Kruger J, Kristiansen LV. Schizophrenia; from structure to function with special focus on the mediodorsal thalamic prefrontal loop. Acta Psychiatr Scand 2009; 120: 345–354. [DOI] [PubMed] [Google Scholar]
- 65.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 2002; 5: 405–414. [DOI] [PubMed] [Google Scholar]
- 66.Lozovaya NA, Grebenyuk SE, Tsintsadze T, Feng B, Monaghan DT, Krishtal OA. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape ‘superslow’ afterburst EPSC in rat hippocampus. J Physiol 2004; 558: 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsvetkov E, Shin RM, Bolshakov VY. Glutamate uptake determines pathway specificity of long-term potentiation in the neural circuitry of fear conditioning. Neuron 2004; 41: 139–151. [DOI] [PubMed] [Google Scholar]
- 68.Marcaggi P, Attwell D. Short- and long-term depression of rat cerebellar parallel fibre synaptic transmission mediated by synaptic crosstalk. J Physiol 2007; 578: 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci 1998; 21: 8–14. [DOI] [PubMed] [Google Scholar]
- 70.Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J et al. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci 2015; 18: 219–226. [DOI] [PubMed] [Google Scholar]
- 71.Anticevic A, Cole MW, Repovs G, Murray JD, Brumbaugh MS, Winkler AM et al. Characterizing thalamo-cortical disturbances in schizophrenia and bipolar illness. Cereb Cortex 2014; 24: 3116–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fries P. Neuronal gamma-band synchronization as a fundamental process in cortical computation. Annu Rev Neurosci 2009; 32: 209–224. [DOI] [PubMed] [Google Scholar]
- 73.Ribary U. Dynamics of thalamo-cortical network oscillations and human perception. Prog Brain Res 2005; 150: 127–142. [DOI] [PubMed] [Google Scholar]
- 74.Roux F, Wibral M, Singer W, Aru J, Uhlhaas PJ. The phase of thalamic alpha activity modulates cortical gamma-band activity: evidence from resting-state MEG recordings. J Neurosci 2013; 33: 17827–17835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lisman J. Working memory: the importance of theta and gamma oscillations. Curr Biol 2010; 20: R490–R492. [DOI] [PubMed] [Google Scholar]
- 76.Lisman J, Buzsaki G. A neural coding scheme formed by the combined function of gamma and theta oscillations. Schizophr Bull 2008; 34: 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.