

Abstract
T cell ageing has a pivotal role in rendering older individuals vulnerable to infections and cancer and in impairing responses to vaccinations. Easy accessibility to peripheral human T cells as well as an expanding array of tools to examine T cell biology have provided opportunities to examine major ageing pathways and their consequences for T cell function. Here, we review emerging concepts of how the body attempts to maintain a functional T cell compartment with advancing age, focusing on three fundamental domains of the ageing process, namely self-renewal, control of cellular quiescence and cellular senescence. Understanding these critical elements in successful T cell ageing will allow the design of interventions to prevent or reverse ageing-related T cell failure.
Introduction
The adaptive immune system has been a prime area for researching the ageing process and its implications1, 2. Ageing-associated changes to the immune system are clinically important, leaving older individuals more vulnerable to new infections and to reactivation of latent viruses. Aggravating this problem is the fact that many of the current vaccine strategies only induce incomplete protection in older populations3. Improving vaccine responses is paramount for healthy ageing. This goal is achievable, as recently exemplified by the development of an adjuvanted varicella zoster virus (VZV) vaccine that is effective irrespective of age4. However, further progress will require approaches that are tailored to the ageing immune system and therefore a better knowledge of the specific immune defects. Strategies in young individuals cannot be simply translated to the older population, as shown by a recent meta-analysis of influenza virus vaccination studies5. In this analysis, biomarkers that were predictive of a superior vaccine response in the young were no longer informative in older individuals and an inflammatory signature had a positive effect in young individuals but was harmful in older adults5.
In addition to the implications for immune system function, studies on T cell ageing provide a unique opportunity to explore the fundamental mechanisms that drive the ageing process in general6. The T cell system has unique mechanisms of replenishment, with the production of new T cells entirely dependent on thymic activity, which rapidly declines during adolescence and early adulthood7. In the absence of thymic output, naive T cells essentially function as their own stem cells. The T cell system is also an excellent model to study the influence of ageing on cell population dynamics8. Immune competence is determined by the frequencies of T cells that recognize one particular antigenic peptide. Therefore, the population has to establish a balance between maintaining a highly diverse set of T cell specificities in sufficient frequencies to be able to respond and increasing the clonal size of the T cell specificities that are needed to control acute, chronic and latent infections over the life time of the individual. Finally, T cells are a model system enabling studies of cellular states that are relevant for ageing, including cellular quiescence, senescence and exhaustion9, 10, 11, 12.
Here, we review T cell ageing with respect to these mechanistic phases of the ageing process, focusing mainly on data available from human studies. By analogy to the stem cell theory, which postulates that the ageing process results from the inability of stem cells to replenish a tissue with functionally competent cells, we discuss whether and how the T cell population is maintained with age. Moreover, we discuss whether T cell ageing reflects cellular senescence or the failure to maintain quiescence and instead undergo differentiation. We highlight how the T cell ageing process is influenced by the accumulation of DNA damage and programmed pathways, in particular those that drive cell differentiation or senescence.
T cell replenishment in immune ageing
Naive T cell generation by peripheral T cell self-renewal.
One hallmark of ageing is the decline in homeostatic and regenerative capacity that is common to all tissues and organs and generally related to stem cell ageing6, 13, 14, 15. T cell replenishment in adult humans is special in that it is at least in part uncoupled from stem cells, relying less on thymic activity and more on homeostatic self-renewal of naive T cells. The generation of nascent T cells is entirely dependent on the thymus, where progenitor cells differentiate and are positively and negatively selected to generate the repertoire of self-restricted, self-tolerant and functional T cells. However, unlike any other organ, the thymus undergoes involution during childhood and adolescence, leading to reduced numbers of thymocytes and thymic epithelial cells and disruption of the tissue architecture7, 16. Thymic export rates decrease rapidly from the ages of one year to eight years and decrease at a more gradual rate in subsequent years17.
The consequences of reduced thymic activity for maintaining a naive T cell compartment are remarkably species specific. In young mice, thymic output is at least 2 to 4 times higher than the daily production by peripheral T cell proliferation18. As a consequence, naive T cell numbers cannot be maintained with declining thymic activity. Moreover, molecular defects in aged haematopoietic stem cells (HSCs) in mice mirror those in aged peripheral T cells, and T cell function in mice can be improved by reversing HSC defects such as by inhibiting CDC42 activity in aged HSCs19. By contrast, even in young adult humans the majority of T cells derives from peripheral T cell proliferation and not from the thymus (Figure 1). The contribution of the thymus to T cell generation is estimated to decline from only ~16% to <1% over the adult lifetime20, 21, a decline that can be compensated by minimal adjustments in homeostatic proliferation. This is consistent with the observation that turnover of peripheral naive T cells does not noticeably increase with age, possibly with the exception of the very elderly22, 23. Moreover, in contrast to mice, so far there is no evidence that defects in HSCs translate to T cell defects in humans. For example, mutations of the epigenetic modifiers TET2 and DNMT3A that have been associated with HSC clonality are found frequently in the elderly24 and could be detrimental to T cell diversity, but they have not been reported in peripheral T cells, possibly because low thymic activity prevents the generation of such mutant T cells.
Figure 1 |. Successful T cell replenishment by self-renewal.
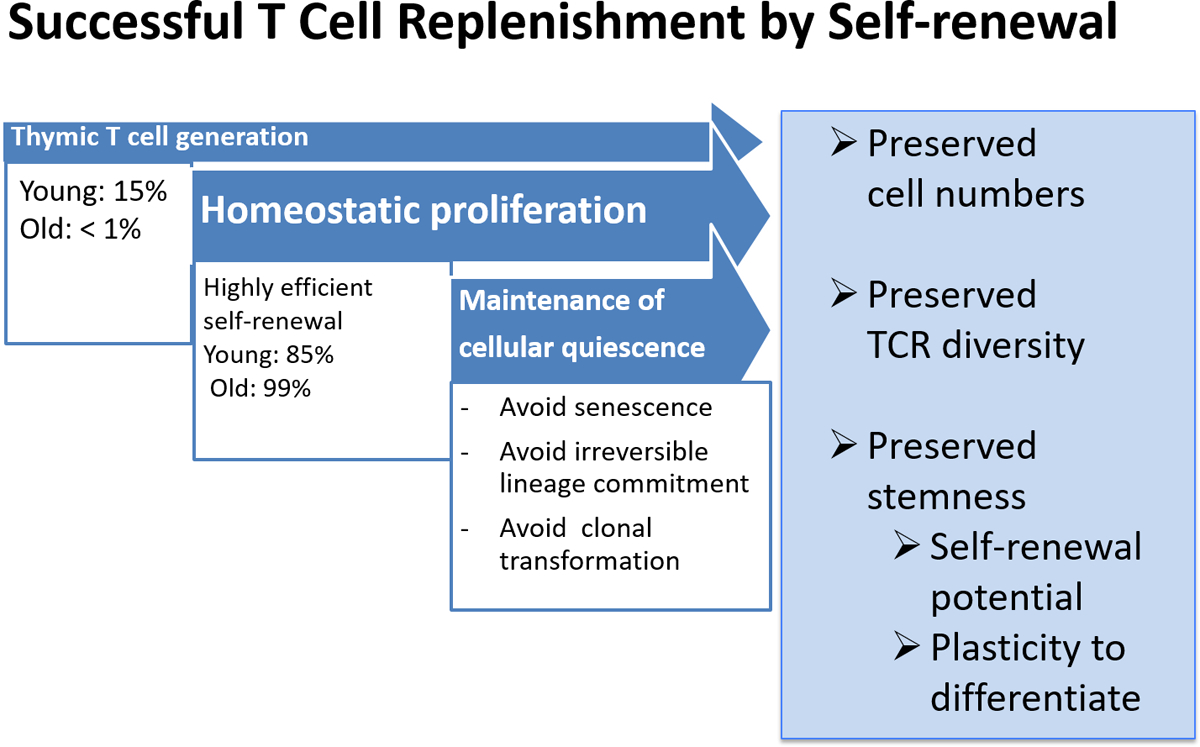
Throughout adult life, most T cell replenishment occurs by self-renewal of peripheral T cells rather than de novo generation by thymic activity. In healthy ageing, homeostatic proliferation is efficient to maintain a large compartment of naive T cells and is sufficiently stochastic to preserve a highly diverse T cell receptor (TCR) repertoire. Signals driving homeostatic proliferation should be below the threshold that interferes with cellular quiescence and initiates differentiation.
CD8+ T cells: the Achilles’ heel of T cell homeostasis.
Homeostatic proliferation in humans is sufficient to maintain a sizable naive CD4+ T cell compartment (Figure 1). By contrast, loss of circulating naive CD8+ T cells with age is more severe, in terms of both relative and absolute cell numbers25, 26, 27. In fact, reduced numbers of naive circulating CD8+ T cells is the most consistent and prominent marker of immune ageing in healthy older adults, independent of comorbidities and chronic latent infections28. The diminution of naive CD8+ T cells may even be underestimated, because CD8+ central memory T cells lose CD45RO expression and regain CD45RA expression after resolved viral infections, thereby masquerading as naive T cells29 (Figure 2). Consistent with this finding, a subset of naive CD8+ T cells is able to produce effector cytokines upon stimulation, indicating that this subset is primed30. It is not clear why there is reduced resilience of the naive CD8+ T cell compartment to age. It can be assumed that naive CD8+ T cells receive more consistent survival signals than naive CD4+ T cells, based on the facts that MHC class I molecules are ubiquitously expressed and CD8+ T cells are responsive to IL-15 in addition to IL-7. It is possible that IL-2 supports better homeostatic survival of naive CD4+ T cells, as CD4+ T cells but not CD8+ T cells gain expression of CD25 (the α-subunit of the high affinity IL-2 receptor) with age31. Because they are more easily activated, naive CD8+ T cells may also be depleted through apoptosis or assume a memory phenotype. Such cytokine-activated CD8+ T cells — known as virtual memory T cells — accumulate in mice with age and have been shown to develop features of cellular senescence32, 33, 34. Mouse naive CD8+ T cells are more susceptible to this homeostatic proliferation-induced differentiation into virtual memory T cells than naive CD4+ T cells. It is therefore possible that the generation of virtual memory T cells accounts for the preferential erosion of the naive CD8+ T cell compartment. A cell population in humans that has similarities to virtual memory T cells in mice has been described35, however, it is not known whether virtual memory T cells also accumulate in humans with age.
Figure 2 |. Mechanisms compromising T cell homeostasis.
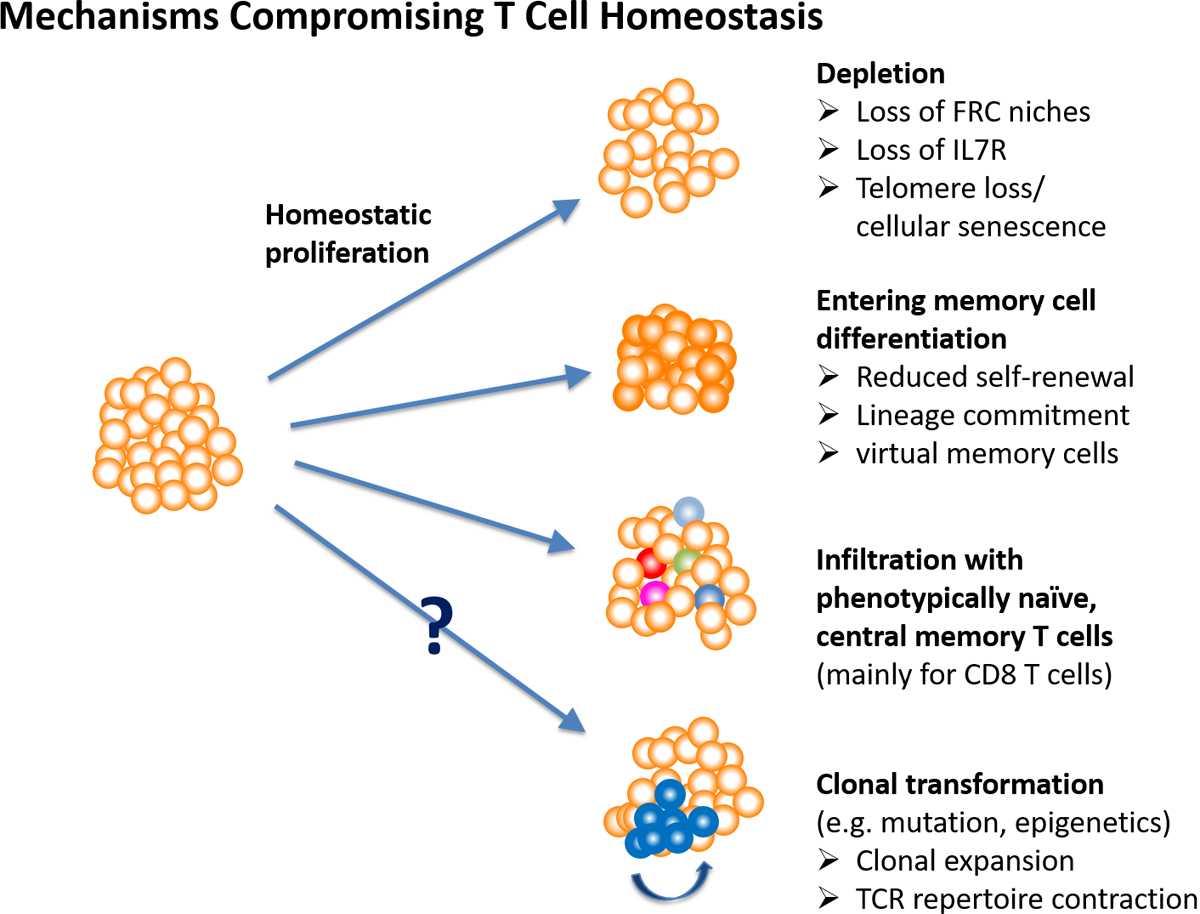
Maintenance of a large, diverse and functional naive T cell compartment can be compromised by several mechanisms. Homeostatic proliferation can be impaired through T cell-intrinsic defects or failing niches formed by fibroblast reticular cell (FRC) networks in the T cell zones of lymph nodes, leading to a reduction in compartment sizes. For unknown reasons, CD8+ naive T cells are more prone for homeostasis failure. If quiescence is not maintained, naive T cells undergo differentiation towards memory T cells. Whether differentiation proceeds as far in humans as seen with virtual memory cells in mice is unclear, but markers of early differentiation states are frequently observed, even in healthy old adults. Again, aged CD8+ T cells appear more susceptible to differentiation. Moreover, the naive T cell compartment can be infiltrated by memory T cells that masquerade as naive T cells and compete for niches. Phenotypic switches regularly occur in CD8+ central memory T cells, but are uncommon for CD4+ T cells. Finally, clonal expansion of selected T cells may occur because of changes in growth behaviour. Such transformations are common for haematopoietic stem cells, but have not been widely studied for T cells. Computer modelling predicts that they could result in rapid contraction of repertoire diversity. TCR, T cell receptor.
The tissue niche for homeostatic proliferation.
Peripheral homeostasis of T cells depends on their recruitment to secondary lymphoid organs where they encounter IL-7 produced by fibroblastic reticular cells (FRCs) and low-level T cell receptor (TCR) stimulation through antigen-presenting cells displaying self-antigens36, 37. Impaired access to or changes in the architecture of secondary lymphoid organs with age could therefore have a negative impact on the maintenance of the naive T cell compartment (Figure 2). This has indeed been found for aged mice, interestingly, independent of the production of IL-7 by FRCs38, 39 The importance of FRC networks for T cell homeostasis has also been demonstrated in humans, although not yet in the context of ageing. Initial studies showed that the inflammation associated with HIV infection leads to fibrosis of lymph nodes and contributes to peripheral T cell depletion40. In subsequent studies, this phenomenon was also seen in human populations that have a high incidence of chronic or recurrent infections other than HIV41. Lymph node fibrosis inversely correlated with peripheral naive T cell counts as well as with immune responses to yellow fever vaccination, indicating the functional relevance of this process.
In addition to failing FRC networks, diminished IL-7R expression on T cells contributes to a failure in T cell homeostasis with age. In genome-wide studies of ageing-associated epigenetic changes, reduced chromatin accessibility at the IL7R locus in CD8+ T cells was identified as an ageing hallmark42. In addition, microRNA networks in CD8+ T cells from older individuals are altered, reducing the activity of forkhead box protein O1 (FOXO1), the key transcription factor regulating IL7R gene expression43. Interestingly, compared with CD8+ T cells, CD4+ T cells show less epigenetic alteration with age, which may explain their higher resilience42.
Maintaining T cell diversity: how much is enough?
T cell homeostasis is unique in that it needs to maintain not only population size but also high diversity of TCR specificities (Figure 1). The notion that T cell specificities are lost with age, resulting in a contraction of the TCR repertoire, has been an attractive explanation for defective immune responses. However, there is little evidence that ‘holes’ in the repertoire occur in humans without extreme reduction in population sizes. In computer simulations of human T cell homeostasis, only minimal contraction in diversity was observed over 50 years of homeostatic proliferation despite the absence of thymic production; a marked reduction in diversity was not even observed when the size of the naive T cell compartment shrunk by >50%, as is regularly the case for CD8+ T cells44. Clonal extinction is more likely if the initial clonal size is very small, which does not appear to be the case in humans. Moreover, peripheral T cell selection implies that homeostatic proliferation occurs in TCR specificity-specific niches, where clones compete for the recognition of self-antigen in the context of self-MHC. Experimental evidence for the existence of such niches is lacking21.
Given the enormous diversity of the human naive TCR repertoire, clonal size estimates are difficult. Theoretical estimates have been in the order of 10 cells per clone45, which is likely at the lower end for humans, in particular for those clones that are generated early in life and have space to expand. Frequencies of naive T cells containing TCR excision circles in newborns suggest clonal sizes of 100, which would make complete extinction of a clone over a lifetime an unlikely event46. Several studies have used next-generation sequencing to estimate repertoire contraction with age. Due to undersampling and contamination with memory cells, estimates of naive T cell diversity are frequently misjudged47, 48, 49, 50. In part circumventing these limitations by analysing replicate samples of purified T cell subsets, we estimated there to be up to 108 unique TCR β-chain sequences in young adults51. TCR β-chain diversity declined with age, but only by a factor of 3 to 5 in healthy elderly, which still leaves a very diverse repertoire. Whether this decline is of functional significance is unclear.
Clonally expanded T cells with a fitness advantage challenge diversity.
In silico simulation of T cell homeostasis provided evidence that, under certain conditions, selection of peripheral T cell clones on the basis of “fitness” compromises repertoire diversity44. Rapid contraction in TCR diversity was observed if single clones changed their growth behaviour and gained the ability to rapidly expand (Figure 2). Acquired mutations conferring clonal growth advantage have been identified in HSCs with age, with TET2 and DNMT3A being frequent drivers of clonal haematopoiesis24, 52. As discussed above, thymic T cell generation at older ages is very low, preventing the differentiation of such HSCs into T cells. However, similar to HSCs, aged T cells have an extensive replicative history due to homeostatic proliferation and antigen-induced clonal expansion. It has been recently estimated that there are three mutations for every cell division53. It is therefore likely that T cells, unrelated to HSCs, accumulate mutations that could lead to the generation of T cell specificities with a growth advantage and rapid contraction in repertoire diversity44. Indeed, somatic mutations were discovered in clonally expanded CD8+ T cells in patients with rheumatoid arthritis54.
Oligoclonality is most prevalent in the subset of the T effector memory CD45RA cells (TEMRA cells), which are populations of T cells associated with memory inflation following certain viral infections55. Clonally expanded TEMRA cells are enriched in the bone marrow56 and therefore do not seem to compete with naive T cells for lymph node niches. In studies comparing memory T cells from the blood, spleen, bone marrow, lymph nodes and lungs, those in the lymph nodes were found to be less differentiated than those in other compartments57. It is therefore unlikely that TEMRA cells outcompete normal T cell self-renewal in the lymph nodes.
Memory T cell homeostasis: a surprisingly dynamic process to maintain long-term memory.
Regulation of memory T cell populations is highly complex and more information on the dynamics of the memory T cell compartment is required, particularly in humans, to fully understand how it is affected by age. In the most simplified model, memory T cells that are maintained in the absence of antigen have to be distinguished from those that re-encounter antigen. Antigen re-encounter may be periodic, for example in the setting of reinfection or reactivation of latent viruses, or chronic, for example by antigens expressed during viral latency or chronic active viral infection.
Studies in settings in which antigen re-exposure was excluded concluded that immune memory lasts for 40–60 years in the absence of antigen and therefore may be lifelong58. Frequencies of antigen-specific memory T cells after small pox vaccination decreased by 50% every 8–15 years59. However, the lifetime of individual memory T cells is much shorter; in humans, it is generally ~6 months as determined by deuterium labelling studies60. Thus, maintenance of immunological memory is a dynamic process that is not only determined by the survival of individual memory T cells, but also by homeostatic proliferation of these cells. Recent studies of antigen-specific CD8+ T cell kinetics after yellow fever vaccination have shown that by 30 days the cells have entered a quiescent state, which is characterized by a long intermitotic phase and division rate of once every 485 days29. The division rate of this antigen-specific CD8+ T cell population was lower than previous estimates for unfractionated CD4+ or CD8+ memory T cells (both ~180 days); however, it should be noted that the yellow fever-specific CD8+ T cells no longer express the classical memory cell markers, but are contained within the compartment of cells expressing naive T cell phenotypic markers and therefore resemble the subset of cells that have recently been coined stem cell-like memory T cells61, 62, 63. Recent studies of total stem cell-like memory CD4+ and CD8+ T cell populations yielded self-renewal estimates of ~430 days64, which is similar to estimates for yellow-fever-specific CD8+ T cell self-renewal64.
In summary, T cell memory appears to be maintained less by longevity of individual T cells than by homeostatic proliferation, which occurs at a slow rate, but is still faster than self-renewal of naive T cells. Based on data from mice, this proliferation is cytokine driven and has less requirement for tonic TCR signalling65, 66, 67. Limited kinetic data from in vivo deuterium labelling of healthy young and old individuals suggest similar turnover rates of central memory and effector memory populations, suggesting that this process is intact in older individuals, again with the caveat that only healthy individuals were studied23, 68. Moreover, TCR repertoire studies of memory cells only showed a small contraction in diversity51.
However, analysis of turnover of total T cell populations does not take into account possible heterogeneity in population dynamics. Indeed, analytical approaches to deuterium labelling data performed better using multi-exponential rather than single-exponential models indicating kinetic heterogeneity that is not reflected in conventional phenotypic markers69. Such heterogeneity is typical for memory T cells but not for naive T cells. One possible and intuitively appealing explanation is temporal kinetic heterogeneity — that is, a temporally limited increase in proliferative rate of a subpopulation due to antigenic re-exposure in the absence of clinical symptoms. Such proliferative burst could lead to the extinction of unrelated memory T cells and therefore the loss of T cell memory with age. Surprisingly, the computational analysis of available data is more consistent with the alternative model of kinetic heterogeneity — that is, the existence of memory cell subpopulations with stable distinct homeostatic proliferation kinetics60. Such population heterogeneity may in part challenge the concept that immune memory is maintained by the division of short-lived memory cells rather than the persistence of long-lived cells. Kinetic heterogeneity has also been described among stem cell-like memory T cells. Although most stem cell-like memory T cells are frequently dividing, a small subpopulation of these cells showed self-renewal dynamics that are consistent with the decline in memory64.
Recent studies of mice that were kept in a clean environment and were not infected indicated an even higher complexity of the kinetic structure of the CD4+ memory T cell compartment70. These studies revealed two central memory and two effector memory CD4+ T cell subpopulations of equal sizes, but with vastly different kinetics; one dividing and dying every three days, the other dividing every 170 days (of note, the average turnover rates of memory T cells in mice are tenfold higher than in humans). These studies also documented a constant influx of naive T cells into the memory cell compartment, with a surprisingly high replacement of up to 10% of cells per week in young mice in the absence of any obvious antigenic challenges. The replacement rate declined with age, which may reflect declining rates of memory T cell differentiation due to reduced numbers of naive T cells or higher resistance to replacement by memory T cells. Computational modelling could not clearly distinguish between these two models, but favoured the resistance model. These high replacement rates, as well as the existence of a highly proliferative memory T cell subset, represent major challenges to preserving memory.
It will be important to determine whether humans also have a pool of memory T cells that is rapidly and constantly replaced, whether and how this pool contributes to immune memory and, perhaps most importantly for immune health in older individuals, the nature of those memory T cells that appear to resist replacement. Longevity of T cell memory can differ depending on viral infection or modes of vaccination, which is currently not well understood. The most marked example in the context of ageing is the difference in immune memory to latent herpes viruses. Frequencies of VZV-specific T cells decline with age, resulting in frequent VZV reactivation presenting as shingles71, 72, whereas the T cell memory to cytomegalovirus (CMV), and in part to Epstein–Barr virus, tends to inflate, contributing to the age-associated increase of effector memory T cells and TEMRA cells26, 73. Further insights into the dynamics of the memory T cell population are needed to better understand what determines the longevity of memory.
T cell homeostasis: vulnerable to failure but generally robust.
In summary, CD4+ T cell homeostatic mechanisms can be surprisingly resilient to withstand the challenges of ageing in healthy individuals, such as thymic involution, peripheral selection, changing environments and recurring antigenic stimulation. The notion that T cell memory is maintained by unbiased homeostatic proliferation of memory T cells appears to be an oversimplification as the dynamics in the memory compartment are more complex than previously thought. The influence of age on memory cell dynamics and the implications for interventions to prolong immune memory have not been determined.
Most of the more mechanistic studies done so far have focused on healthy individuals, concluding that the ageing process does not necessarily induce major distortions. How far these findings can be generalized to less healthy individuals is currently unclear. While there is an association between frailty and a state of chronic activation of the innate immune system known as inflamm-ageing, it is unclear whether a similar relationship exists for adaptive immune defects. Poorer vaccine responses and higher risk of latent virus reactivation in frail individuals have been reported by some but not all studies74, 75, 76, 77. Mechanistic studies of T cell homeostasis in populations including frail elderly are needed. Larger population studies may also capture more infrequent and stochastic events, such as mutations that drive clonal expansion or cause massive contraction in diversity. Moreover, disease states are likely to have a major influence over T cell homeostasis, as shown by the effects of chronic or recurring inflammation on FRC niches.
Failed quiescence in ageing
T cells are quiescent but poised.
Naive and memory T cells exist in a quiescent state, poised to proliferate and differentiate upon antigen stimulation. Maintaining quiescence is vital to retain self-renewal potential and differentiation plasticity throughout life (Figure 1). In quiescence, cell division and growth are coordinately downregulated, cells are arrested in their cell cycle, and they have low metabolic and mammalian target of rapamycin complex (mTORC) activity resulting in reduced ribosome biogenesis and protein synthesis78. Disruption of quiescence has been implicated in stem cell ageing15. For example, activation of transcription factor networks including basic leucine zipper transcriptional factor ATF-like (BATF) induce clonality in lymphoid precursors and a preference for myeloid lineage differentiation with age79. As observed for FRC networks in the aged lymph node, stem cell niches in young mice generally promote stem cell proliferation more efficiently than those from old mice. However, the aged niche can also become more stimulatory, driving cells out of quiescence. For example, elevated fibroblast growth factor 2 (FGF2) signalling from the aged niche has been associated with depletion of the resident stem cell population in the muscle and diminished muscle regenerative capacity80. This model may also apply to HSCs that have been found to be more activated in the aged niche81, 82. Similarly, the favourable quiescent state of T cells is continuously challenged by environmental stimuli, including those that drive homeostatic proliferation, in particular under conditions of lymphopenia.
Differentiation of aged T cells due to failed quiescence.
Human T cell ageing shares many features with T cell differentiation, presumably due to a loss of quiescence (Figure 3). The clearest evidence for this is the accumulation of virtual memory CD8+ T cells in aged mice (Figure 2). These cells develop in response to cytokine stimulation in the absence of cognate antigen encounter and they express the same marker profile as cells undergoing lymphopenia-induced homeostatic proliferation83. The generation of virtual memory T cells has been considered to be beneficial, because these cells show innate properties and provide bystander protective immunity in response to secreted cytokines84, 85. However, being more-differentiated memory T cells, they have lost proliferative potential and therefore may account for the decline in primary CD8+ T cell responses with age34. How far human virtual memory T cells accumulate with age is not firmly established. If virtual memory T cells constitute a major proportion of the memory T cell compartment, TCR sequence diversity in the memory T cell population should increase with age, which is not the case. In fact, although virtual memory T cells are mostly CD8+ T cells, TCR diversity is 5–10 fold less for CD8+ memory T cells than for CD4+ memory T cells51.
Figure 3 |. Activation of differentiation pathways in T cell ageing.
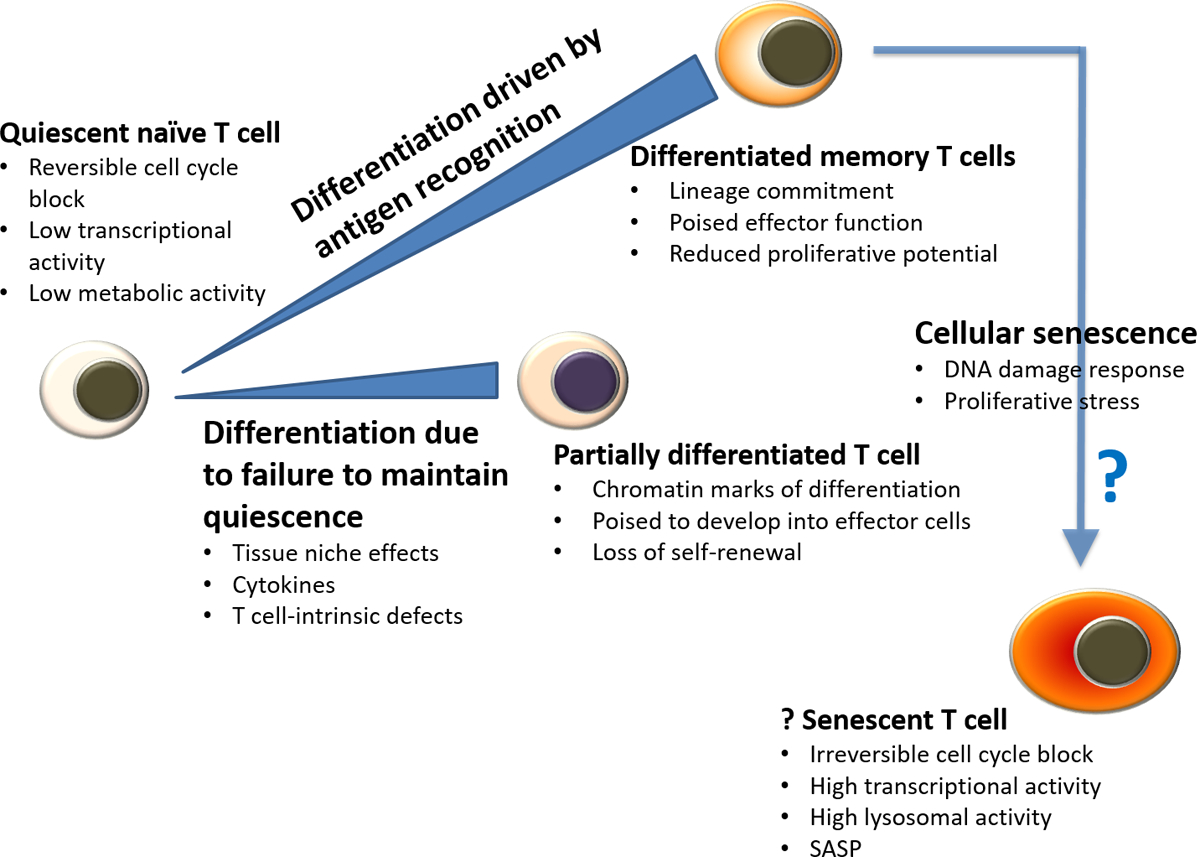
Many findings in T cell ageing are related to differentiation. Cumulative antigenic experience leads to the accumulation of differentiated cells. Even in the absence of antigen stimulation, T cells can leave their usual quiescent state and accumulate as partially differentiated cells. These cells, as well as functionally differentiated memory T cells, regain quiescence. Cellular senescence is fundamentally different from quiescence. SASP, senescence associated secretory phenotype.
Epigenetic evidence for loss of stemness in aged T cells.
Epigenetic studies have provided evidence that the quiescent state is not fully maintained in human T cells with ageing86, 87. Age-related changes in T cell chromatin accessibility are very similar to those that are seen with T cell activation and differentiation (Figure 3). These changes are much more prominent in CD8+ T cells than in CD4+ T cells. Sites of increased accessibility in naïve CD8+ T cells include motifs for basic leucine zipper domain-containing transcription factors, such as BATF and AP-1, but not motifs of the T-box or RUNX transcription factors, which is consistent with incomplete differentiation. Progression towards a more differentiated state with age is also observed for CD8+ memory T cells and, to some extent, for CD4+ naive T cells. At the single cell level, older T cells have increased population heterogeneity in histone acetylation, indicating diversity in activation and differentiation states88. Differentiation of naive T cells and/or phenotypic conversion of memory T cells could explain this finding.
Functional evidence for differentiation of aged naive T cells.
The naive T cell compartment is also increasingly recognized as being more heterogeneous than previously thought89. Gene expression studies of naive T cells suggest that T cell ageing, at least in part, involves the same pathways that are seen in T cell activation and differentiation31, 90. Single cell RNA-sequencing studies have shown that ageing increases the cell-cell variability in gene expression during early activation of mouse CD4+ T cells, possibly due to increased heterogeneity in differentiation states91. Age-associated changes in microRNA networks in part resemble those of differentiation43, 92, 93. For some key microRNAs, such as miR-146a, which increases with ageing and differentiation, and miRNA-181a, which decreases with ageing and differentiation, flow cytometry studies have shown that the microRNA expression levels follow a Gaussian distribution, supporting the idea that the entire population is affected and the observed changes are not due to subset contamination43. Transition to a more differentiated state is sufficient to change cell behaviour, without causing typical senescence (Figure 3). Most interesting is a bias in the differentiation potential of older T cells94. For example, increased expression of miR-21 in naive CD4+ T cells from older individuals targets for degradation negative regulators of several signalling pathways including PTEN, SPRY1 and PDCD4. The ensuing sustained activation of these signalling pathways favours the generation of inflammatory effector T cells over that of T follicular helper cells and memory precursor cells94. Also, transcription factor networks in aged memory CD4+ T cells are poised to favour effector states and expression of the ectoATPase CD3995, which has been associated with defective vaccine responses and effector states including T cell exhaustion95, 96.
Entry into a more differentiated state may also imply a bias in lineage commitment and therefore a loss in plasticity to respond. For example, a decrease in variation of CD38 expression with age among naïve CD4+ T cells was observed, and it was proposed that the increased heterogeneity in young T cells endows more plasticity in response patterns97. Similarly, we observed increased responsiveness to transforming growth factor-β in the presence of transcription factor such as BATF and IRF4 that favoured the generation of T helper 9 cells in older individuals98.
Cellular senescence in T cell ageing
Mechanism of cellular senescence.
Cellular senescence is generally implicated as a major mechanism of ageing-associated dysfunction6, 99. It is a form of irreversible growth arrest induced in response to telomere shortening or a variety of stress responses, mostly involving DNA damage100, 101. To avoid chromosomal instability and to safeguard genomic stability, DNA damage responses are triggered leading to permanent cell cycle arrest102. T cells are exposed to short-term and long-term stressors that could induce senescence103. Telomere shortening in naive T cells is generally thought to be a consequence of homeostatic proliferation104. Consistent with their increased replicative history, memory T cells have shorter telomeres than naive T cells105. Moreover, the DNA damage response is activated in T cells proliferating in response to antigen recognition106, 107 and is predictive for less successful vaccine responses in older individuals108.
Lack of typical cellular senescence features in aged naive and memory T cells.
Is cellular senescence functionally relevant for immune ageing in humans? Expression of the DNA damage response component p16INK4A (also known as CDKN2A) in human peripheral T cells has been described as a biomarker of ageing109. However, p16INK4A is also expressed during normal differentiation into effector T cells and is therefore not in itself sufficient to indicate senescence. Chromatin accessibility mapping has shown increased accessibility at the INK4A-ARF locus in memory and effector T cells compared with CD8+ naive T cells, but this is only slightly more so in older individuals90. Also, ageing-associated changes in heterochromatin accessibility (similar to changes occurring in senescent cells110, 111, 112) are subtle and occurred in CD8+ T cells but not in CD4+ T cells42. Moreover, most aged naive and memory T cells do not display the phenotypic hallmarks of senescent cells78, such as a secretory phenotype, increased size, increased lysosomal content and function, and vacuolated and granular morphology. Most importantly, the vast majority of aged naive and central memory T cells are able to proliferate, when appropriately activated. In fact, aged naive CD4+ T cells from patients with rheumatoid arthritis divide even faster although they show accelerated ageing (based on increased telomere attrition and DNA damage levels) compared with T cells from age-matched healthy controls107, 113, 114, 115.
TEMRA cells: effector T cells or senescent T cells?
Although most aged T cells do not exhibit functional defects due to cellular senescence, rare senescent T cells may still contribute to immune defects. In non-immune aged tissues, senescent cells only represent a small fraction of cells. Whether and how such infrequent cells have an impact on tissue function has been the basis for debate. It is now generally accepted that senescent cells, triggered by DNA damage, secrete a range of mediators, such as pro-inflammatory cytokines, that have potent paracrine and endocrine effects, a property known as senescence-associated secretory phenotype (SASP)116, 117, 118, 119, 120. This function of senescent cells can be beneficial, as long as it is of limited duration and senescent cells are rapidly cleared by the immune system. Long-term persistence of SASP can result in chronic inflammation and disease.
TEMRA cells meet several of the criteria for cellular senescence121 (Figure 4). These cells have short telomeres, exhibit cell cycle arrest, express DNA damage foci and have a secretome reminiscent of SASP. In common with exhausted T cells, TEMRA cells develop in the setting of chronic viral infection and exhibit cell cycle arrest, however unlike exhausted T cells, they maintain high effector functionality and actually increase their production of inflammatory mediators12. How these T cells accumulate in chronic viral infection is not entirely clear, but it may be due to failure in the attrition of effector T cells owing to increased expression of survival factors or reduced clearance by innate immune cells. Consistent with the concept that senescent cells exert systemic detrimental effects, TEMRA cells have been implicated in several chronic disease states as well as poor vaccine responses122, 123, 124. However, expanded TEMRA cells have been shown to have both positive and negative effects on life expectancy125.
Figure 4 |. Relationship between T cell differentiation and cellular senescence in T cell ageing.
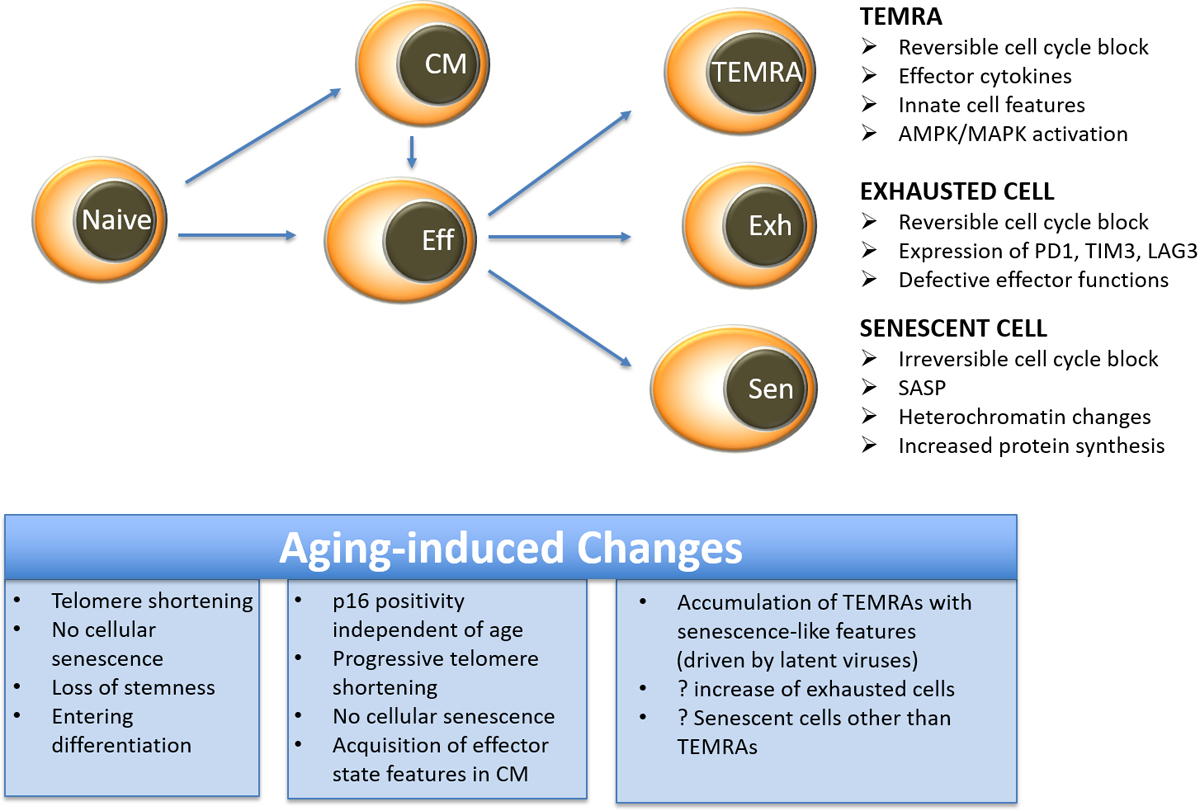
Following activation and differentiation, naive and memory T cells activate pathways that are also involved in developing cellular senescence, such as telomeric erosion, activation of DNA damage responses and expression of cell cycle inhibitors. However, true cellular senescence is not observed in most naive and memory T cells. T effector memory CD45RA cells (TEMRA cells) and exhausted T cells exhibit cell cycle blocks that are still, at least in part, reversible, distinguishing them from truly senescent cells. The excessive cytokine production by TEMRA cells is reminiscent of the senescence associated secretory phenotype (SASP), whereas exhausted T cells lack effector functions, setting them apart from TEMRA cells and senescent cells.
Nevertheless, it should be noted that TEMRA cells exhibit clear differences to true senescent cells. Most importantly, their cell cycle arrest is reversible126. In support of the notion that they are a form of terminal differentiation, they gain not only secretory features, but also the expression of regulatory cell surface receptors such as leukocyte immunoglobulin-like receptors and killer immunoglobulin-like receptors127, 128, 129. Their function depends on several biological pathways, including a reduced expression of NAD-dependent protein deacetylase sirtuin 1 that leads to increased lysosomal degradation of FOXO1 and metabolic reprograming that favours glycolytic activity and granzyme B production130. A characteristic hallmark of these cells is the activation of p38 mitogen-activated protein kinase (MAPK) downstream of AMPK and TAB1 and not downstream of TCR signalling131, 132, uncoupling T cell activation from antigen recognition (Figure 5). Inhibition of p38 MAPK restores telomerase activity and proliferative potential of TEMRA cells. More recent studies have shown that in old TEMRA JNK and ERK are activated downstream of sestrins and this activation was independent of regulators of the upstream MAPK cascade131, 133.
Figure 5 |. Regulation of cytokine transcription in effector T cells, TEMRA cells and senescent cells.
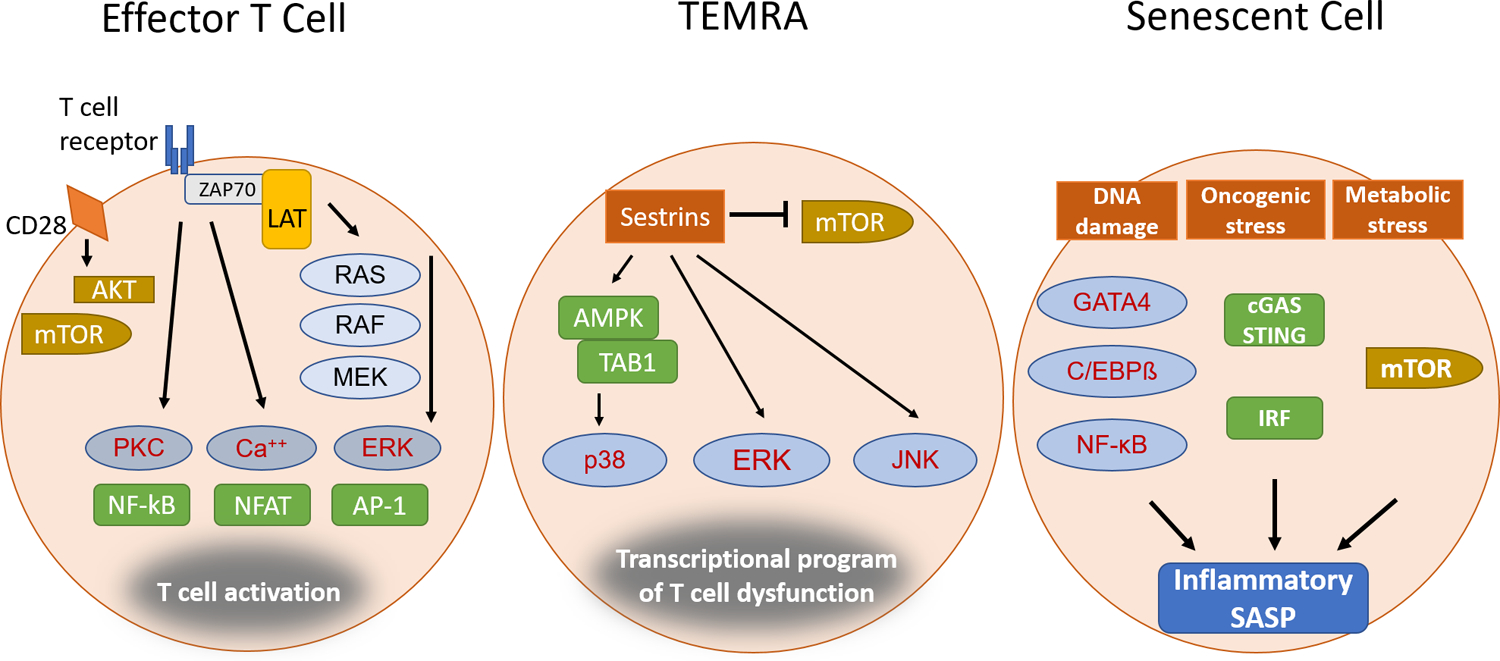
Effector T cells generated during normal immune responses, terminally differentiated T effector memory CD45RA cells (TEMRA cells), which are mostly responsive to latent viruses, and senescent T cells are all characterized by their excessive production of pro-inflammatory mediators. However, the signalling and transcriptional control mechanisms in the different cell states differ. For effector T cells, ligation of T cell receptor (TCR) and CD28 induces a signalling cascade that activates the mammalian target of rapamycin complex 1 (mTORC1) pathway as well as several transcriptional activators. For TEMRA cells, sestrins provide a scaffold for broad mitogen-activated protein kinase (MAPK) activation, in particular activation of p38 MAPK by the AMPK–TAB1 complex, whilemTORC1 is inhibited. For senescent T cells, DNA damage and cellular stress activate signalling pathways, with nuclear factor-κB (NF-κB), p38, C/EBPβ and mTORC1 playing critical roles in regulating the senescence associated secretory phenotype (SASP).
Targeting senescent T cells.
The idea that even infrequent senescent immune cells could have harmful system-wide effects and that clearance of senescent cells may be ineffective in ageing has led to the development of therapeutic approaches to eliminate senescent cells134. Several compounds — known as senolytics — have been developed that exhibit various degrees of selectivity in inducing death of senescent cells, by targeting apoptosis, chaperones and histone modifications99, 134. The best-examined senolytics are a combination of dasatinib and quercetin, which has shown benefits in model systems of cardiovascular disease and osteoarthritis and increases in lifespan135, 136, 137. Senolytics have not been explored to improve immune function; and it is unclear whether they would deplete TEMRA cells and, if they do so, whether they would be harmful or beneficial. As TEMRA cells maintain effector functionality and are specific for latent viruses, their depletion may enable viral reactivation. In fact, the memory inflation by TEMRA cells may account for the successful control of latent CMV infection in the elderly. In line with this, infection with VZV, which is also a herpes virus, does not induce memory inflation of TEMRA cells and shows frequent reactivation, presenting as shingles138.
An alternative approach to interfere with the harmful functions of senescent cells is to inhibit the production or activity of pro-inflammatory mediators (Figure 5)139. Treatment of senescent preadipocytes with a pan-JAK inhibitor reduced the production of inflammatory mediators in vivo, possibly by interfering with a positive feedback loop from secreted cytokines. Moreover, treatment of aged mice with a JAK1/JAK2 inhibitor alleviated SASP and frailty. However, T cell homeostasis depends on JAK-STAT5 signalling, and other JAK-STAT pathways are involved in T cell differentiation and function, indicating the limitation of this intervention. Indeed, the risk of developing shingles due to VZV reactivation was increased in patients with rheumatoid arthritis who were treated with the JAK1/JAK3 inhibitor tofacitinib140, 141. A similar increase in incidence of shingles was seen in patients treated with the JAK1/JAK2 inhibitor baricitinib142.
SASP inhibitors mostly target the nuclear factor-κB (NF-κB), mTORC and p38 MAPK pathways that are active in senescent cells. In in vitro studies, low dose inhibition of both mTORC1 and mTORC2 using the mTOR-specific ATP mimetic AZD8055 reversed major phenotypes of senescence in near-senescent fibroblasts143. In a recent Phase 2a study, a cohort of old individuals was treated for 6 weeks with a combination of a catalytic and an allosteric mTOR inhibitor in low doses to selectively inhibit mTORC1144. Treated individuals had an improved response to influenza virus vaccination, without any major side effects including no reactivation of latent viruses. It is possible that mTORC1 inhibition only modified the vaccine response without targeting senescent cells. In animal experiments, inhibition of the mTORC pathway has been shown to favour CD8+ T cell memory over effector T cell generation145. Moreover, mTORC inhibitors could counteract the more sustained signalling in CD4+ T cell responses from older individuals that is due to repression of the negative regulators PTEN and SPRY1, thereby favouring transcription factor networks involved in T follicular helper cell and memory T cell differentiation94. However, treated individuals also had significantly fewer infections over the subsequent year; this extended benefit following a short treatment period is surprising and indicated a sustained effect, possibly by eliminating senescent cells.
Other recent approaches are based on AMPK-dependent MAPK activation being central for senescence in TEMRA cells131. This implies that inhibition of mTORC1, which is in many aspects antagonistic to AMPK, could be harmful; the more appropriate treatment approaches would be the transcriptional repression of sestrins or inhibition of downstream p38 activation. Inhibition of p38 MAPK has been shown to partially restore proliferative capacity in TEMRA cells by inducing telomerase activity146.
Conclusions and future perspectives
Pathways implicated in the ageing process, and in particular in stem cell ageing, are highly relevant for the T cell system. T cells depend on well regulated replenishment and homeostasis; they need to maintain quiescence while being poised to respond; and, as highly proliferative cells, they are at risk of developing cellular senescence. However, studies in healthy elderly have shown that ageing-associated differences are not extreme and are frequently within the range of general population variation; accordingly, many healthy older individuals are immunocompetent. A recent extensive immune profiling study in a population selected for absence of major comorbidities and low frailty indices identified only a few immune markers that correlated with age28. Major immune defects therefore do not appear to be an inevitable consequence of ageing.
Studies into T cell ageing therefore provide an opportunity to understand how challenges can be overcome and healthy T cell ageing can be supported. The central theme that emerges is to promote cellular quiescence — for example, by reducing infectious burden or by lessening endogenous and exogenous inflammatory stimuli. Many of the T cell-intrinsic defects compromising T cell activation and differentiation in ageing arise from the activation of normal differentiation pathways rather than from the induction of irreversible cellular senescence. If the overall T cell compartment structure is largely intact, as appears to be the case in many healthy older individuals who have reduced but sufficient T cell numbers and diversity, these molecular defects represent druggable targets to improve the efficacy of vaccinations. However, we have a dearth of data on whether this also applies to T cells in less healthy old individuals. It is possible that frail individuals have major immune defects, such as a major diminution of cell numbers or contraction of diversity, or have many small changes that might act together to cause a clinical immune defect. Studies into systems immunology aimed at understanding the synergistic impact of small effects such as naturally occurring variation in immune parameters on immune responses, are still in their infancy, but eventually will provide insight into how to interpret and target ageing-associated changes.
Acknowledgements
This work was supported by the US National Institutes of Health (R01 AR042527, R01 HL117913, R01 AI108906, R01 HL142068, and P01 HL129941 to C.M.W. and R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191, and I01 BX001669 to J.J.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
GLOSSARY
- Homeostatic proliferation
A process of activation and proliferation of lymphocytes in the lymphopenic environment. T cell homeostatic proliferation is driven by T cell receptor interactions with self-peptide–MHC complexes and T cell responses to cytokines such as interleukin-7 (IL-7), IL-15 and possibly IL-21.
- Quiescence
A phase in the cell cycle in which the cell is not dividing or preparing to divide but still has the ability to do so in the presence of an appropriate signal. Quiescent cells have low metabolic activity and reduced protein synthesis.
- Senescence
A cellular state in which an irreversible growth arrest programme has been initiated that limits the lifespan of the cell and prevents unlimited cell proliferation. It can be caused by replication-induced telomere shortening or DNA damage. In contrast to quiescent cells, senescent cells have a secretory phenotype and upregulated protein synthesis.
- Exhaustion
Refers to an impaired ability of effector T cells to carry out their functions such as cytotoxicity and cytokine secretion owing to chronic stimulation by antigen.
- T effector memory CD45RA cells
(TEMRA cells). Terminally differentiated antigen-specific memory T cells that re-express CD45RA. These cells have been identified in both CD4+ and CD8+ T cell compartments, have short telomeres, exhibit cell cycle arrest, express DNA damage foci and have a secretome reminiscent of senescent cells.
- Memory inflation
The gradual accumulation of peptide-specific CD8+ T cells with an effector memory phenotype that occurs after the resolution of certain acute viral infections during viral latency (for example, cytomegalovirus infection). Induction in the setting of chronic antigen persistence suggests the clonal expansion is antigen-driven and not mutation-driven.
- Fibroblastic reticular cells
(FRCs). Specialized reticular fibroblasts located in the T cell areas of lymph nodes and other secondary lymphoid organs. They provide IL7 for T cell survival and produce collagen-rich reticular fibres and form stromal networks and conduits that are important for the trafficking of immune cells.
- Virtual memory T cells
Antigen-inexperienced memory-phenotype T cells, which may be induced by T cell receptor cross reactivity, low-affinity peptide and/or MHC ligands and certain cytokines.
- TCR excision circles
(TRECs). Small, stable circles of DNA excised during T cell receptor (TCR) gene rearrangement in the thymus.
- Stem cell-like memory T cells
Subset of memory T cells that has naïve-like features and phenotypes including enhanced self-renewal and multifunctional capacity.
- DNA damage responses
A cell response triggered by DNA damage, such as single or double strand breaks. The DNA damage response stops cell cycle progression to enable repair before the damage is transmitted to progeny cells. Checkpoints in the mammalian DNA damage response are controlled by the PI3K-related kinases ATM and ATR.
- Senolytics
Pharmacological compounds that preferentially deplete senescent cells.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Nikolich-Zugich J The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol 19, 10–19 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Goronzy JJ & Weyand CM Successful and Maladaptive T Cell Aging. Immunity 46, 364–378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Del Giudice G et al. Fighting against a protean enemy: immunosenescence, vaccines, and healthy aging. NPJ Aging Mech Dis 4, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lal H et al. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 372, 2087–2096 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Team H-CSP & Consortium H-I Multicohort analysis reveals baseline transcriptional predictors of influenza vaccination responses. Sci Immunol 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer DB The effect of age on thymic function. Front Immunol 4, 316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yanes RE, Gustafson CE, Weyand CM & Goronzy JJ Lymphocyte generation and population homeostasis throughout life. Semin Hematol 54, 33–38 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton SE & Jameson SC CD8 T cell quiescence revisited. Trends Immunol 33, 224–230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newton RH et al. Maintenance of CD4 T cell fitness through regulation of Foxo1. Nat Immunol 19, 838–848 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akbar AN & Henson SM Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol 11, 289–295 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Kirkwood TB Understanding the odd science of aging. Cell 120, 437–447 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Rando TA Stem cells, ageing and the quest for immortality. Nature 441, 1080–1086 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Ermolaeva M, Neri F, Ori A & Rudolph KL Cellular and epigenetic drivers of stem cell ageing. Nat Rev Mol Cell Biol 19, 594–610 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Dixit VD Impact of immune-metabolic interactions on age-related thymic demise and T cell senescence. Semin Immunol 24, 321–330 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Bains I, Thiebaut R, Yates AJ & Callard R Quantifying thymic export: combining models of naive T cell proliferation and TCR excision circle dynamics gives an explicit measure of thymic output. J Immunol 183, 4329–4336 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Hogan T, Gossel G, Yates AJ & Seddon B Temporal fate mapping reveals age-linked heterogeneity in naive T lymphocytes in mice. Proc Natl Acad Sci U S A 112, E6917–6926 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leins H et al. Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood 132, 565–576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.den Braber I et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 36, 288–297 (2012). [DOI] [PubMed] [Google Scholar]; This study provides evidence for species-specific differences in T cell replenishment of mice and humans and delineates the implications for T cell homeostasis with age.
- 21.Seddon B & Yates AJ The natural history of naive T cells from birth to maturity. Immunol Rev 285, 218–232 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Naylor K et al. The influence of age on T cell generation and TCR diversity. J Immunol 174, 7446–7452 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Westera L et al. Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover. Aging Cell 14, 219–227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busque L, Buscarlet M, Mollica L & Levine RL Concise Review: Age-Related Clonal Hematopoiesis: Stem Cells Tempting the Devil. Stem Cells 36, 1287–1294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czesnikiewicz-Guzik M et al. T cell subset-specific susceptibility to aging. Clin Immunol 127, 107–118 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wertheimer AM et al. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J Immunol 192, 2143–2155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thome JJ et al. Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci Immunol 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whiting CC et al. Large-Scale and Comprehensive Immune Profiling and Functional Analysis of Normal Human Aging. PLoS One 10, e0133627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Extensive immune profiling in a well-characterized cohort of healthy adults.
- 29.Akondy RS et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pulko V et al. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat Immunol 17, 966–975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Geest KS et al. Low-affinity TCR engagement drives IL-2-dependent post-thymic maintenance of naive CD4+ T cells in aged humans. Aging Cell 14, 744–753 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sprent J & Surh CD Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol 12, 478–484 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiu BC, Martin BE, Stolberg VR & Chensue SW Cutting edge: Central memory CD8 T cells in aged mice are virtual memory cells. J Immunol 191, 5793–5796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study shows that naïve CD8 T cells in mice fail to maintain quiescence and differentiate into virtual memory cells with age.
- 34.Quinn KM et al. Age-Related Decline in Primary CD8(+) T Cell Responses Is Associated with the Development of Senescence in Virtual Memory CD8(+) T Cells. Cell Rep 23, 3512–3524 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Jacomet F et al. Evidence for eomesodermin-expressing innate-like CD8(+) KIR/NKG2A(+) T cells in human adults and cord blood samples. Eur J Immunol 45, 1926–1933 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Link A et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol 8, 1255–1265 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Surh CD & Sprent J Homeostasis of naive and memory T cells. Immunity 29, 848–862 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Becklund BR et al. The aged lymphoid tissue environment fails to support naive T cell homeostasis. Sci Rep 6, 30842 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson HL et al. Lymph nodes as barriers to T-cell rejuvenation in aging mice and nonhuman primates. Aging Cell, e12865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The studies show in mice that ageing of the lymph node niche impairs T cell homeostasis.
- 40.Zeng M et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest 121, 998–1008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kityo C et al. Lymphoid tissue fibrosis is associated with impaired vaccine responses. J Clin Invest 128, 2763–2773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The manuscript provides evidence in humans that inflammation impairs the T cell niche in lymph nodes with negative implications for T cell homeostasis and T cell responses.
- 42.Ucar D et al. The chromatin accessibility signature of human immune aging stems from CD8(+) T cells. J Exp Med 214, 3123–3144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The studies show that age-associated epigenetic changes affect CD8 more than CD4 T cells.
- 43.Gustafson CE, Cavanagh MM, Jin J, Weyand CM & Goronzy JJ Functional pathways regulated by microRNA networks in CD8 T-cell aging. Aging Cell, e12879 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson PL, Yates AJ, Goronzy JJ & Antia R Peripheral selection rather than thymic involution explains sudden contraction in naive CD4 T-cell diversity with age. Proc Natl Acad Sci U S A 109, 21432–21437 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lythe G, Callard RE, Hoare RL & Molina-Paris C How many TCR clonotypes does a body maintain? J Theor Biol 389, 214–224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schonland SO et al. Homeostatic control of T-cell generation in neonates. Blood 102, 1428–1434 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Goronzy JJ, Qi Q, Olshen RA & Weyand CM High-throughput sequencing insights into T-cell receptor repertoire diversity in aging. Genome Med 7, 117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Britanova OV et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol 192, 2689–2698 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Warren RL et al. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res 21, 790–797 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laydon DJ, Bangham CR & Asquith B Estimating T-cell repertoire diversity: limitations of classical estimators and a new approach. Philos Trans R Soc Lond B Biol Sci 370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qi Q et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A 111, 13139–13144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study provides quantitative estimates of the decrease in T cell receptor richness and increase in clonality in human adults.
- 52.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tomasetti C, Li L & Vogelstein B Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Savola P et al. Somatic mutations in clonally expanded cytotoxic T lymphocytes in patients with newly diagnosed rheumatoid arthritis. Nat Commun 8, 15869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klenerman P The (gradual) rise of memory inflation. Immunol Rev 283, 99–112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pangrazzi L et al. Increased IL-15 Production and Accumulation of Highly Differentiated CD8(+) Effector/Memory T Cells in the Bone Marrow of Persons with Cytomegalovirus. Front Immunol 8, 715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miron M et al. Human Lymph Nodes Maintain TCF-1(hi) Memory T Cells with High Functional Potential and Clonal Diversity throughout Life. J Immunol 201, 2132–2140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crotty S & Ahmed R Immunological memory in humans. Semin Immunol 16, 197–203 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Hammarlund E et al. Duration of antiviral immunity after smallpox vaccination. Nat Med 9, 1131–1137 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Borghans JAM, Tesselaar K & de Boer RJ Current best estimates for the average lifespans of mouse and human leukocytes: reviewing two decades of deuterium-labeling experiments. Immunol Rev 285, 233–248 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Gattinoni L et al. A human memory T cell subset with stem cell-like properties. Nat Med 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahmed R et al. Human Stem Cell-like Memory T Cells Are Maintained in a State of Dynamic Flux. Cell Rep 17, 2811–2818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Benedetto S et al. Impact of age, sex and CMV-infection on peripheral T cell phenotypes: results from the Berlin BASE-II Study. Biogerontology 16, 631–643 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Costa Del Amo P et al. Human TSCM cell dynamics in vivo are compatible with long-lived immunological memory and stemness. PLoS Biol 16, e2005523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schluns KS & Lefrancois L Cytokine control of memory T-cell development and survival. Nat Rev Immunol 3, 269–279 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Lees JR & Farber DL Generation, persistence and plasticity of CD4 T-cell memories. Immunology 130, 463–470 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kedzierska K, Valkenburg SA, Doherty PC, Davenport MP & Venturi V Use it or lose it: establishment and persistence of T cell memory. Front Immunol 3, 357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wallace DL et al. Direct measurement of T cell subset kinetics in vivo in elderly men and women. J Immunol 173, 1787–1794 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Westera L et al. Closing the gap between T-cell life span estimates from stable isotope-labeling studies in mice and humans. Blood 122, 2205–2212 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Gossel G, Hogan T, Cownden D, Seddon B & Yates AJ Memory CD4 T cell subsets are kinetically heterogeneous and replenished from naive T cells at high levels. Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Asanuma H, Sharp M, Maecker HT, Maino VC & Arvin AM Frequencies of memory T cells specific for varicella-zoster virus, herpes simplex virus, and cytomegalovirus by intracellular detection of cytokine expression. J Infect Dis 181, 859–866 (2000). [DOI] [PubMed] [Google Scholar]
- 72.Levin MJ et al. Decline in varicella-zoster virus (VZV)-specific cell-mediated immunity with increasing age and boosting with a high-dose VZV vaccine. J Infect Dis 188, 1336–1344 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Klenerman P & Oxenius A T cell responses to cytomegalovirus. Nat Rev Immunol 16, 367–377 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Yao X et al. Frailty is associated with impairment of vaccine-induced antibody response and increase in post-vaccination influenza infection in community-dwelling older adults. Vaccine 29, 5015–5021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moehling KK et al. The effect of frailty on HAI response to influenza vaccine among community-dwelling adults >/= 50 years of age. Hum Vaccin Immunother 14, 361–367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Epps P et al. Preexisting Immunity, Not Frailty Phenotype, Predicts Influenza Postvaccination Titers among Older Veterans. Clin Vaccine Immunol 24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thomasini RL et al. Aged-associated cytomegalovirus and Epstein-Barr virus reactivation and cytomegalovirus relationship with the frailty syndrome in older women. PLoS One 12, e0180841 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Polymenis M & Kennedy BK Unbalanced growth, senescence and aging In: Gotta M & Meraldi P (eds). Cell Division Machinery and Disease, vol. 1002 Springer International Publishing AG: Switzerland, 2017, pp 189–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang J et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 148, 1001–1014 (2012). [DOI] [PubMed] [Google Scholar]
- 80.Chakkalakal JV, Jones KM, Basson MA & Brack AS The aged niche disrupts muscle stem cell quiescence. Nature 490, 355–360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morrison SJ, Wandycz AM, Akashi K, Globerson A & Weissman IL The aging of hematopoietic stem cells. Nat Med 2, 1011–1016 (1996). [DOI] [PubMed] [Google Scholar]
- 82.Sudo K, Ema H, Morita Y & Nakauchi H Age-associated characteristics of murine hematopoietic stem cells. J Exp Med 192, 1273–1280 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haluszczak C et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med 206, 435–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White JT et al. Virtual memory T cells develop and mediate bystander protective immunity in an IL-15-dependent manner. Nat Commun 7, 11291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee JY, Hamilton SE, Akue AD, Hogquist KA & Jameson SC Virtual memory CD8 T cells display unique functional properties. Proc Natl Acad Sci U S A 110, 13498–13503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goronzy JJ, Hu B, Kim C, Jadhav RR & Weyand CM Epigenetics of T cell aging. J Leukoc Biol 104, 691–699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Keenan CR & Allan RS Epigenomic drivers of immune dysfunction in aging. Aging Cell, e12878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheung P et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 173, 1385–1397 e1314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van den Broek T, Borghans JAM & van Wijk F The full spectrum of human naive T cells. Nat Rev Immunol 18, 363–373 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Moskowitz DM et al. Epigenomics of human CD8 T cell differentiation and aging. Sci Immunol 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study shows similarities in the epigenetic changes that occur with differentiation and ageing of human CD8 T cells.
- 91.Martinez-Jimenez CP et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355, 1433–1436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study shows that response patterns of mouse CD4 T cells to stimulation exhibit increasing heterogeneity with age.
- 92.Li G et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med 18, 1518–1524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ye Z et al. Regulation of miR-181a expression in T cell aging. Nat Commun 9, 3060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim C et al. Activation of miR-21-Regulated Pathways in Immune Aging Selects against Signatures Characteristic of Memory T Cells. Cell Rep 25, 2148–2162 e2145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fang F et al. Expression of CD39 on Activated T Cells Impairs their Survival in Older Individuals. Cell Rep 14, 1218–1231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gupta PK et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog 11, e1005177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu Y et al. Systematic Analysis of Cell-to-Cell Expression Variation of T Lymphocytes in a Human Cohort Identifies Aging and Genetic Associations. Immunity 45, 1162–1175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hu B et al. Transcription factor networks in aged CD4 naïve T cells bias lineage differentiation. Aging Cell In press (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.von Kobbe C Cellular senescence: a view throughout organismal life. Cell Mol Life Sci 75, 3553–3567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Campisi J Aging, cellular senescence, and cancer. Annu Rev Physiol 75, 685–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fumagalli M et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14, 355–365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Munoz-Espin D & Serrano M Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15, 482–496 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Chou JP & Effros RB T cell replicative senescence in human aging. Curr Pharm Des 19, 1680–1698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rufer N et al. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J Exp Med 190, 157–167 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weng NP, Levine BL, June CH & Hodes RJ Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc Natl Acad Sci U S A 92, 11091–11094 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McNally JP et al. Manipulating DNA damage-response signaling for the treatment of immune-mediated diseases. Proc Natl Acad Sci U S A 114, E4782–E4791 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shao L et al. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med 206, 1435–1449 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qi Q et al. Defective T Memory Cell Differentiation after Varicella Zoster Vaccination in Older Individuals. PLoS Pathog 12, e1005892 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu Y et al. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 8, 439–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chandra T et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 47, 203–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang R & Adams PD Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle 6, 784–789 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Adams PD Remodeling chromatin for senescence. Aging Cell 6, 425–427 (2007). [DOI] [PubMed] [Google Scholar]
- 113.Schonland SO et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci U S A 100, 13471–13476 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li Y et al. Deficient Activity of the Nuclease MRE11A Induces T Cell Aging and Promotes Arthritogenic Effector Functions in Patients with Rheumatoid Arthritis. Immunity 45, 903–916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang Z et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med 8, 331ra338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Coppe JP et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kuilman T et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008). [DOI] [PubMed] [Google Scholar]
- 118.Coppe JP, Desprez PY, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuilman T & Peeper DS Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 9, 81–94 (2009). [DOI] [PubMed] [Google Scholar]
- 120.Rodier F et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11, 973–979 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Akbar AN, Henson SM & Lanna A Senescence of T lymphocytes: implications for enhancing human immunity. Trends Immunol 37, 866–876 (2016). [DOI] [PubMed] [Google Scholar]
- 122.Martens PB, Goronzy JJ, Schaid D & Weyand CM Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum 40, 1106–1114 (1997). [DOI] [PubMed] [Google Scholar]
- 123.Liuzzo G et al. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation 101, 2883–2888 (2000). [DOI] [PubMed] [Google Scholar]
- 124.Goronzy JJ et al. Value of immunological markers in predicting responsiveness to influenza vaccination in elderly individuals. J Virol 75, 12182–12187 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pawelec G Age and immunity: What is “immunosenescence”? Exp Gerontol 105, 4–9 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Di Mitri D et al. Reversible senescence in human CD4+CD45RA+CD27- memory T cells. J Immunol 187, 2093–2100 (2011). [DOI] [PubMed] [Google Scholar]
- 127.Weng NP, Akbar AN & Goronzy J CD28(−) T cells: their role in the age-associated decline of immune function. Trends Immunol 30, 306–312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pereira BI & Akbar AN Convergence of Innate and Adaptive Immunity during Human Aging. Front Immunol 7, 445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gustafson CE et al. Immune Checkpoint Function of CD85j in CD8 T Cell Differentiation and Aging. Front Immunol 8, 692 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jeng MY et al. Metabolic reprogramming of human CD8(+) memory T cells through loss of SIRT1. J Exp Med 215, 51–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lanna A, Henson SM, Escors D & Akbar AN The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol 15, 965–972 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Henson SM et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8(+) T cells. J Clin Invest 124, 4004–4016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lanna A et al. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat Immunol 18, 354–363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study describes signaling pathways in terminally differentiated T cells that account for their senescence features, but are different from classical senescent cells.
- 134.Childs BG, Durik M, Baker DJ & van Deursen JM Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21, 1424–1435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kirkland JL & Tchkonia T Cellular Senescence: A Translational Perspective. EBioMedicine 21, 21–28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jeon OH et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xu M et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 24, 1246–1256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study shows the benefits of depleting senescent cells in model systems.
- 138.Weinberg A & Levin MJ VZV T cell-mediated immunity. Curr Top Microbiol Immunol 342, 341–357 (2010). [DOI] [PubMed] [Google Scholar]
- 139.Tchkonia T, Zhu Y, van Deursen J, Campisi J & Kirkland JL Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123, 966–972 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Winthrop KL et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol 66, 2675–2684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Winthrop KL et al. Herpes Zoster and Tofacitinib: Clinical Outcomes and the Risk of Concomitant Therapy. Arthritis Rheumatol 69, 1960–1968 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fleischmann R et al. Safety and efficacy of baricitinib in elderly patients with rheumatoid arthritis. RMD Open 3, e000546 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walters HE, Deneka-Hannemann S & Cox LS Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY) 8, 231–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mannick JB et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med 10 (2018). [DOI] [PubMed] [Google Scholar]
- 145.Araki K, Youngblood B & Ahmed R The role of mTOR in memory CD8 T-cell differentiation. Immunol Rev 235, 234–243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Henson SM, Macaulay R, Riddell NE, Nunn CJ & Akbar AN Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur J Immunol 45, 1441–1451 (2015). [DOI] [PubMed] [Google Scholar]