

Abstract
The COVID-19 pandemic caused by SARS-CoV-2 requires rapid development of specific therapeutics and vaccines. The main protease of SARS-CoV-2, 3CL Mpro, is an established drug target for the design of inhibitors to stop the virus replication. Repurposing existing clinical drugs can offer a faster route to treatments. Here, we report on the binding mode and inhibition properties of several inhibitors using room temperature X-ray crystallography and in vitro enzyme kinetics. The enzyme active-site cavity reveals a high degree of malleability, allowing aldehyde leupeptin and hepatitis C clinical protease inhibitors (telaprevir, narlaprevir, and boceprevir) to bind and inhibit SARS-CoV-2 3CL Mpro. Narlaprevir, boceprevir, and telaprevir are low-micromolar inhibitors, whereas the binding affinity of leupeptin is substantially weaker. Repurposing hepatitis C clinical drugs as COVID-19 treatments may be a useful option to pursue. The observed malleability of the enzyme active-site cavity should be considered for the successful design of specific protease inhibitors.
Keywords: room temperature X-ray crystallography, SARS-CoV-2, 3CL main protease, 3CL Mpro, protease inhibitor, hepatitis C clinical drugs, enzyme kinetics, drug design, repurposing clinical drugs
Graphical Abstract
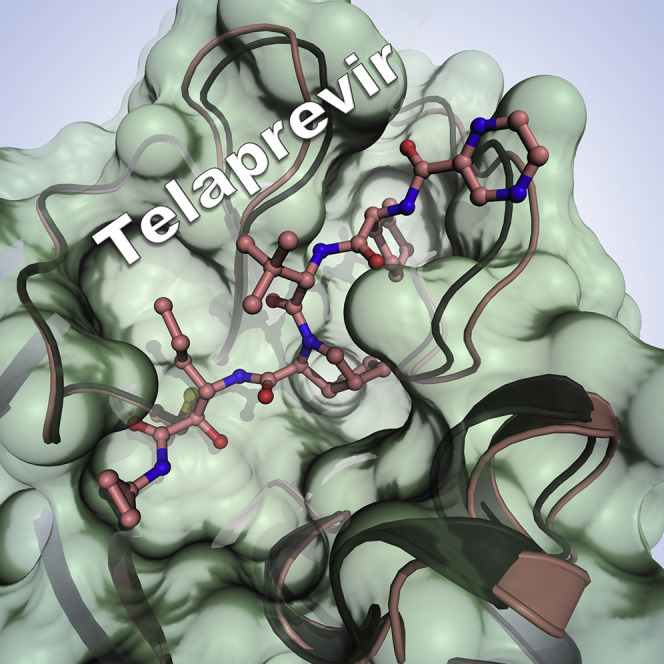
Highlights
-
•
X-ray structures of SARS-CoV-2 3CL Mpro-inhibitor complexes at room temperature
-
•
Telaprevir, narlaprevir, and boceprevir bind and efficiently inhibit the enzyme
-
•
3CL Mpro active-site cavity is malleable, accommodating large inhibitors
-
•
Hepatitis C clinical protease inhibitors can be repurposed to treat COVID-19
Kneller et al. used room temperature X-ray crystallography and in vitro enzyme kinetics to probe the binding of hepatitis C clinical protease inhibitors and the natural aldehyde leupeptin to the SARS-CoV-2 main protease (3CL Mpro). The study visualized significant malleability of the enzyme active-site cavity, providing insights for drug design.
Introduction
The etiological agent of coronavirus disease-19 (COVID-19) is the novel human coronavirus SARS-CoV-2, the origin of which is still being debated. Nonetheless, COVID-19 has become a pandemic of extraordinary proportions, causing worldwide disruptions in travel, economic activity, and social life (Belongia and Osterholm, 2020; Lotfi et al., 2020; Helmy et al., 2020; Wu et al., 2020; Rastogi et al., 2020; Coronaviridae Study Group of the International Committee on Taxonomy of Virus, 2020; Liu et al., 2020). SARS-CoV-2 is an enveloped positive-sense single-stranded RNA virus having one of the largest genomes of ~30 kb. SARS-CoV-2 is very similar to the earlier SARS-CoV that was responsible for a 2003 outbreak; the viruses share ~80% genomic identity, and certain proteins encoded in the viral genomes are over 90% homologous (Xu et al., 2020).
SARS-CoV-2 replication involves the synthesis of two large polyproteins, pp1a and pp1ab (~450 and ~790 kDa, respectively), which are inactive until the viral chymotrypsin-like cysteine protease enzyme (3CL Mpro or main protease) cleaves them into smaller functional proteins (Gorbalenya and Snijder, 1996; Muramatsu et al., 2016). 3CL Mpro is thus indispensable for the viral life cycle, and its inhibition can prevent the production of infectious virions. The lack of homologous human proteins and the vital role of the enzyme for SARS-CoV-2 replication make it one of the most promising targets for the design of specific protease inhibitors (Dai et al., 2020; Zhang et al., 2020; Jin et al., 2020a; Pillaiyar et al., 2016; Wang et al., 2017; Yang et al., 2005). The design of new antivirals and their development for clinical use may take years, emphasized by the fact that no SARS-CoV protease inhibitors have been US FDA approved after more than a decade of research efforts. Conversely, the repurposing of known clinical drugs developed for the treatment of other diseases may provide rapid therapeutic interventions to battle COVID-19 in emergency situations and, possibly, preexposure prophylaxis against SARS-CoV-2 infection (Huang et al., 2020; Jean and Hsueh, 2020).
Here, we present crystallographic and inhibition kinetics evidence for SARS-CoV-2 3CL Mpro inhibition by three hepatitis C clinical NS3/4A serine protease inhibitors (Scheme 1 ), telaprevir (Incivek, Vertex Pharmaceuticals; Kwong et al., 2019), boceprevir (Victrelis, Merck; Venkatraman, 2019), and narlaprevir (Arlansa, R-Pharm, Russia; de Bruijne et al., 2010; Isakov et al., 2016), and a naturally occurring microbial peptide aldehyde, leupeptin (Appleyard and Tisdale, 1985). We chose these compounds because of their peptidomimetic chemical nature and capability to act as reversible covalent inhibitors. We, therefore, hypothesized that they should bind to and inhibit SARS-CoV-2 3CL Mpro. Our near-physiological room temperature X-ray crystallographic structures establish the mechanism of action for these reversible covalent peptidomimetic inhibitors and demonstrate a unique ability of the enzyme active-site cavity to accommodate a variety of chemical groups by substantially distorting its shape and size compared with the ligand-free state (Kneller et al., 2020) through conformational changes in the P2 helix (residues 46–50), the P4 β-hairpin flap (residues 165–170), and the P5 loop (residues 189–194) (Figure 1 ). All four inhibitors bind in a stereospecific fashion, with only the S enantiomers observed for their covalent hemithioacetal and hemithioketal linkages, utilizing the available enzyme's substrate binding subsites S1′ through S5. Solution measurements demonstrate that hepatitis C clinical drugs are low-micromolar inhibitors, whereas leupeptin has the lowest affinity to SARS-CoV-2 3CL Mpro. Our results, therefore, lay a foundation for possible repurposing of the hepatitis C clinical drugs that were originally developed for the inhibition of hepatitis C NS3/4A protease. Our structural data also emphasize the requirement to consider the observed significant malleability of the enzyme's active-site cavity in the structure-assisted and computational design of improved specific 3CL Mpro inhibitors.
Scheme 1.

Chemical Diagrams of the Inhibitors Used in This Study
The reactive warheads to which the catalytic Cys145 covalently binds are colored red.
Figure 1.

The Structure of the 3CL Mpro Homodimer from SARS-CoV-2
Protomers are drawn in cartoon representation showing leupeptin (in ball-and-stick) covalently bound in both active-site cavities (PDB: 6XCH). One of the protomers is shown with a transparent surface. The regions of the active-site cavity that demonstrate significant conformational changes after inhibitor binding are colored green and labeled as P2 helix, P4 β-hairpin flap, and P5 loop.
Results
Leupeptin Binds to SARS-CoV-2 3CL Mpro in a Stereospecific Fashion
SARS-CoV-2 3CL Mpro shares 96% sequence homology with and has similar catalytic efficiency to that of the SARS-CoV enzyme. Its tertiary structure consists of the catalytic domains I and II and the helical domain III (Figure 1) (Xu et al., 2020; Muramatsu et al., 2016; Dai et al., 2020; Xue et al., 2007; Huang et al., 2004). The non-canonical catalytic dyad with residues Cys145 and His41 is buried in the active-site cavity that is located on the surface of the protein flanked by residues from both domains I and II. The active dimer has a two-fold axis that goes from top to bottom between the two monomers (Figure 1) and frequently occurs as a crystallographic two-fold axis. The C-terminal aldehyde group of leupeptin, N-acetyl-L-leucyl-L-leucyl-L-argininal (Ac-Leu-Leu-Arg-CHO, Scheme 1), readily reacts with Cys145, forming a hemithioacetal, with the aldehyde carbon atom becoming sp3 hybridized. P1 arginine and the P2 and P3 leucine side chains bind in the substrate-binding subsites S1, S2, and S3, respectively. Although the aldehyde can freely rotate in leupeptin, it reacts with Cys145 in SARS-CoV-2 3CL Mpro in a stereospecific fashion. Only the S enantiomer is observed in the electron density maps, with the hemithioacetal oxygen facing the enzyme's oxyanion hole (Menard and Storer, 1992) and making hydrogen bonds with the main-chain amide nitrogens of Gly143 and Cys145 (Figure 2 A). The geometry of the hemithioacetal suggests that its oxygen atom is deprotonated and thus negatively charged. The charge is partially stabilized by the hydrogen bonds with the oxyanion hole residues. The hemithioacetal moiety of leupeptin bound to Cys145 has the same geometry as the tetrahedral intermediate that forms along the peptide bond hydrolysis reaction pathway catalyzed by this enzyme. This tetrahedral intermediate is necessarily metastable as it spontaneously collapses into the acyl intermediate. Therefore, the hemiacetal linkage of leupeptin to Cys145 is probably also metastable, reducing the stability of the complex. Leupeptin makes three more direct hydrogen bonds with the main-chain atoms of His164 and Glu166 and a water-mediated interaction with the side chain of Glu189 (Figure 3 A). Interestingly, the ligand makes no direct hydrogen bonds with side chains of SARS-CoV-2 3CL Mpro. The P1 arginine side chain of leupeptin fits in the hydrophilic S1 substrate-binding subsite but makes no hydrogen bonds with the enzyme residues. In contrast, the P2 leucine fits well in the hydrophobic S2 subsite, and P3 leucine faces the bulk solvent.
Figure 2.

Binding Modes of Studied Inhibitors
Leupeptin (A, PDB: 6XCH), telaprevir (B, PDB: 6XQS), narlaprevir (C, PDB: 6XQT), and boceprevir (D, PDB: 6XQU) are shown in ball-and-stick representation. The 2FO-FC electron density maps for the inhibitors are shown as violet meshes and are all contoured at 1.4 σ.
Figure 3.

Hydrogen Bonding Interactions of the Studied Inhibitors (Blue Dashed Lines) with SARS-CoV-2 3CL Mpro Observed in the Room Temperature X-Ray Structures
(A) Complex with leupeptin, (B) complex with telaprevir, (C) complex with narlaprevir, and (D) complex with boceprevir. The insets show the stereochemistry of the covalent conjugates with Cys145. Distances are in Å.
We observe that binding of leupeptin to SARS-CoV-2 3CL Mpro results in significant conformational changes in several structural elements relative to their geometry in the ligand-free enzyme (Figure S1A) (Kneller et al., 2020). The P2 helix and the tip of the P4 β-hairpin flap shift their positions in the leupeptin-bound structure to expand the active-site cavity. The distance between Ser46 and Pro168 Cα atoms increases from 15.7 Å in the ligand-free structure to 17.1 Å when leupeptin is present. The Met49 and Met165 side chains move away from leupeptin to accommodate the chemical groups in positions P2 and P4 of the inhibitor. Conversely, the P5 loop does not move due to the lack of a P5 substituent in leupeptin.
Binding of Hepatitis C Protease Inhibitors to SARS-CoV-2 3CL Mpro
Telaprevir, narlaprevir, and boceprevir are β-ketoamide inhibitors and have a ketone warhead that undergoes a nucleophilic attack by the Cys145 thiolate to form a hemithioketal. Telaprevir and narlaprevir possess substituents in six positions, including chemical groups at P1′ and P1 through P5, whereas boceprevir lacks a chemical group at P5 (Scheme 1). The P1′ amides of these inhibitors occupy the S1′ substrate-binding subsite, and the amide carbonyl oxygens make hydrogen bonds with the enzyme's oxyanion hole. Similar to leupeptin, the drugs bind stereospecifically, with only the S enantiomer visible in the electron density maps (Figures 2B–2D). But unlike leupeptin, the hemithioketal oxygens point away from the oxyanion hole, allowing the formation of strong hydrogen bonds with His41 imidazole side chains with distances of 2.4–2.8 Å (Figures 3B–3D), with boceprevir forming the shortest and narlaprevir the longest hydrogen bonds. Such orientation of the hemithioketal groups suggests that their oxygens may be protonated through the abstraction of a hydrogen from His41. The 2.4 and 2.5 Å hydrogen bonds made by boceprevir and telaprevir also indicate the possible formation of low-barrier hydrogen bonds between their hemithioketal oxygens and the histidine Nε2 imidazole nitrogens. It is evident that the presence of P1′ amide groups in these inhibitors determines the stereospecificity of the hemithioketal formation, directing the oxygen atom toward His41 so that the P1′ amide carbonyl can form hydrogen bonds with the oxyanion hole. Moreover, the neutrally charged hemithioketal groups would be more stable than the negatively charged hemithioacetal of bound leupeptin, if indeed His41 protonates the hemithioketal oxygen.
Because telaprevir, narlaprevir, and boceprevir are peptidomimetic inhibitors with similar structures, they make very similar hydrogen bonds with the main-chain atoms of His164 and Glu166, as does leupeptin (Figures 3B–3D). The urea moiety in narlaprevir and boceprevir linking their P3 and P4 groups makes an extra hydrogen bond with the main-chain carbonyl of Glu166, not seen in the other two inhibitor complexes. Instead of the water-mediated interaction with Gln189 observed in the leupeptin complex, telaprevir forms a direct hydrogen bond to the Gln189 side-chain amide located at the start of the P5 loop. Telaprevir also makes a water-mediated interaction with the main chain of Gln192. The P5 pyrazine of telaprevir overshoots the active-site cavity and is positioned above the P5 loop, making only π-π stacking interactions with the main-chain amide moiety connecting Thr190 and Ala191. Conversely, narlaprevir and boceprevir do not interact with Gln189, whose side chain rotates away from the inhibitors.
It is instructive to analyze the P1-P5 chemical groups present in these hepatitis C inhibitors in relation to the S1-S5 substrate-binding subsites in SARS-CoV-2 3CL Mpro. These groups in all three inhibitors are hydrophobic and are not able to form hydrogen bonds with the enzyme, although several substrate-binding subsites provide opportunities for hydrogen bonding. The S1 subsite is lined by several hydrophilic side chains, including those of Ser144, His163, Glu166, and His172. Therefore, P1 groups bearing hydrophilic functionalities should be more preferred, as previously reported (Dai et al., 2020; Zhang et al., 2020; Jin et al., 2020a). The S2 subsite is more hydrophobic, but the presence of a Tyr54 side chain hydroxyl can be exploited as a potential hydrogen bond donor to an inhibitor's P2. Subsites S3 and S5 are located on the protein surface, but hydrogen bonding with the side chain of Gln189 is possible, as observed in the telaprevir complex. Thus, the side chain of Gln189 can be considered a hydrogen bond partner for an inhibitor's P3 group. The S4 subsite has both hydrophilic and hydrophobic groups. Met165, Leu167, and Phe185 make up the hydrophobic wall toward the interior of the protein, whereas the main-chain carbonyls of Val186, Arg188, and Thr190 and the side chain of Gln192 provide the hydrophilic wall at the enzyme's surface. The S4 subsite may therefore prefer inhibitors with amphiphilic P4 groups, e.g., saturated heterocycles.
Hepatitis C Protease Inhibitors Reshape the 3CL Mpro Active-Site Cavity
The structures of the four inhibitor complexes were superimposed on our previously published room temperature X-ray structure (Kneller et al., 2020) of the ligand-free SARS-CoV-2 3CL Mpro to visualize how the inhibitors change the conformation of the active-site cavity (Figures 4 and S1B–S1D). The inhibitors have a significant effect on the enzyme's active-site cavity geometry. There are dramatic shifts in the positions of the P2 helix, the tip of the P4 β-hairpin flap, and the P5 loop when inhibitors are bound relative to the ligand-free enzyme. Leupeptin is the smallest of the four inhibitors studied, causing the least conformational distortions. Telaprevir, narlaprevir, and boceprevir have a larger effect on the active-site cavity, with each resulting in unique active-site conformations due to differing inhibitor side chains. Telaprevir pushes the P2 helix the farthest by ~1.5 Å, whereas the tip of the P4 β-hairpin flap shifts the most under the steric pressure from boceprevir by ~2.4–2.6 Å. Narlaprevir, on the other hand, causes the end of the P5 loop (residue Ala193) to move away by ~2 Å due to the presence of the bulky P5 tert-butylsulfoxide group, but telaprevir makes the start of the P5 loop (residue Gln189) shift the most by ~1.1 Å. Therefore, the SARS-CoV-2 3CL Mpro active-site cavity shows significant malleability, as it can change its shape and expand by as much as 3.4 Å between the P2 helix and the tip of the P4 β-hairpin flap, and by as much as 2.5 Å between the tip of the P4 β-hairpin flap and the P5 loop (Figure 4).
Figure 4.

Superposition of the Four Inhibitor Complexes onto the Ligand-Free SARS-CoV-2 3CL Mpro (Black, PDB: 6WQF) in Cartoon Representation
The complex with leupeptin is shown in yellow, the complex with telaprevir in dark pink, the complex with narlaprevir in blue, and the complex with boceprevir in gray. Narlaprevir and telaprevir are represented in ball-and-stick, whereas the other two inhibitors are omitted for clarity. The conformational changes for the P2 helix, the tip of the P4 β-hairpin flap, and the P5 loop due to inhibitor binding are shown as red arrows and maximal shifts are indicated with values colored red. The expansion of the active-site cavity is represented by black arrows and the maximal shifts are indicated with values colored black.
Comparison of the SARS-CoV-2 3CL Mpro complexes studied here with those containing the covalent ketoamide inhibitor 13b (PDB: 6Y2F, Zhang et al., 2020) and aldehyde inhibitor GC-376 (PDB: 6WTT, Ma et al., 2020) demonstrates similar conformational changes observed in the active-site cavity (Figure S4), indicating that the SARS-CoV-2 3CL Mpro active-site cavity has an intrinsic ability to adapt its conformation to different chemical groups of inhibitors. Also, we note that telaprevir, narlaprevir, and boceprevir bind with the same stereochemistry to the catalytic Ser of the hepatitis C NS3/4A serine protease as they do in SARS-CoV-2 3CL Mpro, having their hemiketal oxygen atoms directed toward and hydrogen bonded with the catalytic His and their P1′ amide carbonyl groups hydrogen bonded with the oxyanion hole residues (Prongay et al., 2007; Romano et al., 2012). The aliphatic P1 groups of the three inhibitors are positioned in the hydrophobic substrate-binding subsite S1 in the hepatitis C protease, whereas subsite S1 is more hydrophilic in SARS-CoV-2 3CL Mpro. Unlike their binding mode in SARS-CoV-2 3CL Mpro, the other chemical groups of these inhibitors are located on the surface of the hepatitis C protease.
3CL Mpro Inhibition by Hepatitis C Protease Inhibitors and Leupeptin
Enzyme kinetics measurements of the SARS-CoV-2 3CL Mpro activity in vitro using a Förster resonance energy transfer substrate gave the k cat/K M value of 1,600 s−1·M−1, which is in agreement with the previously reported values (Figure S2) (Zhang et al., 2020; Ma et al., 2020). As expected, the inhibition kinetics measurements demonstrated that leupeptin is the lowest-affinity inhibitor of the four assayed, with an IC50 of 92 μM and 95% confidence interval (CI) of 80–106 μM (Figure S3). The three hepatitis C protease inhibitors efficiently inhibit SARS-CoV-2 3CL Mpro activity with micromolar affinity. Narlaprevir and boceprevir have similar inhibition potencies, with IC50 values of 5.1 (CI [4.3, 6.1]) and 3.1 μM (CI [2.8, 3.5]), respectively, and these two are better inhibitors than telaprevir, with an IC50 of 18 μM (CI [16, 21]). The measured IC50 values following 30 min preincubation with inhibitor for boceprevir or narlaprevir are similar to their recently reported IC50 of 4.1 and 5.7 μM, respectively (Ma et al., 2020). Initial binding of a covalent inhibitor is followed by covalent bond formation with the rate constant k 2. Similar to the previous report (Ma et al., 2020), we were not able to obtain accurate values of k 2 due to substrate depletion prior to the equilibrium between the non-covalent and the covalent enzyme-inhibitor complexes. Although the three studied hepatitis C inhibitors are not specific SARS-CoV-2 3CL Mpro inhibitors, their affinities, nonetheless, appear to be similar to those of many protease inhibitors that have been designed to specifically target SARS-CoV 3CL Mpro (Anand et al., 2003; Pillaiyar et al., 2016; Wang et al., 2017; Thanigaimalai et al., 2013; Jacobs et al., 2013; Jo et al., 2020).
Discussion
Two therapeutic intervention strategies for the treatment of COVID-19 are being considered by the scientific community. One such strategy involves the repurposing of the existing clinical drugs, which may provide a quicker route to available treatments to battle COVID-19. Although such drugs have not been specifically developed for the treatment of SARS-CoV-2 infection, they may prove useful in alleviating the disease symptoms by inactivating SARS-CoV-2 3CL Mpro, which is considered a challenging target for inhibitor design (Bzowka et al., 2020). Knowledge of the mechanism of action of such repurposed drugs is of paramount importance. We have obtained and analyzed room temperature X-ray structures of several peptidomimetic inhibitors in complex with SARS-CoV-2 3CL Mpro and measured their effectiveness in inhibiting the enzyme. X-ray crystallography at room temperature, which is close to the physiological temperature of ~37°C, has gained appreciation in recent years because low-temperature structures may not provide representative conformational states of the protein or a bound ligand and may contain artifacts due to the required use of cryoprotectant chemicals and temperature effects (Fraser et al., 2011; Keedy et al., 2014; Kovalevsky et al., 2018; Otten et al., 2018; Gerlits et al., 2019; Kneller et al., 2020). The structures reported here provide evidence that the studied hepatitis C clinical drugs can bind to SARS-CoV-2 3CL Mpro, and enzyme inhibition kinetics measurements demonstrate that they are micromolar inhibitors of the protease. Boceprevir and narlaprevir have the highest affinities to SARS-CoV-2 3CL Mpro and, perhaps, can be looked at as possible treatment options. For example, boceprevir and an antineoplastic drug, carmofur, were recently reported to inhibit SARS-CoV-2 viral replication in cell culture (Ma et al., 2020; Jin et al., 2020b). We also observed significant malleability of the SARS-CoV-2 3CL Mpro active site, including the P2 helix, the tip of the P4 β-hairpin flap, and the P5 loop, that permits the inhibitor molecules to reshape the binding cavity. The flexibility of these secondary structure elements allows the inhibitors to efficiently bind to SARS-CoV-2 3CL Mpro, but it should also be carefully considered in the design of specific SARS-CoV-2 3CL Mpro inhibitors. The P2 helix, the tip of the P4 β-hairpin flap, and the P5 loop can change their conformations independent of one another, as they are seen to shift differently when the studied complexes are compared. Perhaps, the SARS-CoV-2 3CL Mpro active-site cavity may use the ability to alter its shape to avoid interactions with the inhibitors.
In summary, we have succeeded in using room temperature crystallography to probe the conformational flexibility of the SARS-CoV-2 3CL Mpro active-site cavity at near-physiological temperature, which showed significant malleability in accommodating inhibitors not specifically designed to target this enzyme. We were able to analyze how the active-site cavity reacts to the binding of three hepatitis C clinical protease inhibitors (telaprevir, narlaprevir, and boceprevir) and a natural peptide aldehyde, leupeptin. Namely, we directly observed how ligand binding induces conformational changes in the P2 helix, the tip of the P4 β-hairpin flap, and the P5 loop in the active-site cavity. The conformational flexibility and dynamics of this enzyme active-site cavity should be considered in drug design efforts. It also appears that the docking of small-molecule fragments to the ligand-free protease structure may be a futile endeavor. Finally, our structures also point to possible improvements in the specific SARS-CoV-2 3CL Mpro inhibitor design.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli | New England Biolabs. Inc. | Cat#504294 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Leupeptin | VWR | Cat #J580 |
| Boceprevir | Sigma-Aldrich | Cat #ADV465749229 |
| Telaprevir | Apexbio Technology | Cat # A4031 |
| Narlaprevir | MedChemExpress | Cat # HY-10300 |
| DABCYL-KTSAVLQSGFRKM-E(EDANS) | Bachem | Cat #4045664 |
| Free EDANS acid | Sigma-Aldrich | Cat #A6517 |
| Deposited Data | ||
| SARS-CoV-2 3CL Mpro-Leupeptin @ 293K | This study | PDB: 6XCH |
| SARS-CoV-2 3CL Mpro-Telaprevir | This study | PDB: 6XQS |
| SARS-CoV-2 3CL Mpro-Narlaprevir | This study | PDB: 6XQT |
| SARS-CoV-2 3CL Mpro-Boceprevir | This study | PDB: 6XQU |
| SARS-CoV-2 3CL Mpro-13b | Zhang et al., 2020 | PDB: 6Y2F |
| SARS-CoV-2 3CL Mpro-GC-376 | Ma et al., 2020 | PDB: 6WTT |
| SARS-CoV-2 3CL Mpro-apo | Kneller et al., 2020 | PDB: 6WQF |
| Recombinant DNA | ||
| Plasmid pD451-SR | ATUM | N/A |
| Software and Algorithms | ||
| CCP4 | Winn et al., 2011 | http://www.ccp4.ac.uk/download/#os=windows |
| COOT | Emsley et al., 2010 | https://bernhardcl.github.io/coot/wincoot-download.html |
| CrysAlis Pro | Rigaku, Inc. | https://www.rigaku.com/products/smc/crysalis |
| PHENIX | https://www.phenix-online.org/ | |
| Molprobity | Chen et al., 2010 | http://molprobity.biochem.duke.edu/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrey Kovalevsky (kovalevskyay@ornl.gov).
Materials Availability
The study did not generate new unique reagents.
Data and Code Availability
The four protein structures have been deposited into the Protein Data Bank with accessions codes of 6XCH for the SARS-CoV-2 3CL Mpro complex with leupeptin, 6XQS for the complex with telaprevir, 6XQT for the complex with narlaprevir, and 6XQU for the complex with boceprevir. See Key Resources Table for more details.
Experimental Model and Subject Details
Escherichia coli BL21 DE3 competent cells were used in this study, and the cells were grown in LB media in shaking incubators. Additional details are provided in the Method Details section.
Method Details
General Information
Protein purification supplies were purchased from GE Healthcare (Piscataway, New Jersey, USA). Crystallization reagents were purchased from Hampton Research (Aliso Viejo, California, USA).
Cloning, Expression, and Purification of SARS-CoV-2 3CL Mpro
The 3CL Mpro (Nsp5 Mpro) from SARS CoV-2 was cloned into pD451-SR plasmid (Atum, Newark, CA), expressed and purified according to the published procedure (Kneller et al., 2020). To make the authentic N-terminus, the protease sequence is flanked by the maltose binding protein followed by the 3CL Mpro autoprocessing site SAVLQ↓SGFRK (arrow indicates the cleavage site) corresponding to the cleavage between NSP4 and NSP5 in the viral polyprotein. To make the authentic C-terminus, the enzyme construct codes for the human rhinovirus 3C PreScission protease cleavage site (SGVTFQ↓GP) connected to a His6 tag. The N-terminal flanking sequence is autoprocessed during expression in E. coli (BL21 DE3), whereas the C-terminal flanking sequence is removed by the treatment with PreScission protease (Millipore Sigma, St. Louis, MO). For crystallization, the authentic 3CL Mpro is concentrated to 4-6 mg/mL.
Crystallization
Room-temperature X-ray crystallography quality protein crystals were grown using the sitting-drop vapor diffusion method. Ligand-free 3CL Mpro at 4 mg/mL formed crystal cluster aggregates in 22% PEG3350, 0.1 M Bis-Tris pH 6.5, which were used to generate microseeds utilizing the Hampton Research Seed Bead™ kit. These ligand-free protease microseeds were used to nucleate 3CL Mpro in the presence of leupeptin, boceprevir, telaprevir, or narlaprevir. 1 mg of lyophilized leupeptin dissolved directly into 250 μL of 3CL Mpro at 5.4 mg/mL. Boceprevir, telaprevir, or narlaprevir dissolved in 100% dimethyl sulfoxide were mixed at 1:10 molar ratio with 3CL Mpro at 5.4-5.6 mg/mL and allowed to incubate at room temperature for a minimum of 1 hour. Precipitation, if present, was removed by centrifugation prior to setting up crystal trays. All crystals obtained for this study grew in drops containing 10 μL protein-ligand complex mixed with 10 μL 18-22% PEG3350, 0.1 M Bis-Tris pH 7.0 with 0.4 μL of 1:200 dilution microseeds. Single plate-shaped crystals appeared after 3 days at 14°C and were allowed to grow for an additional 4 days before harvesting them for crystallographic data collection.
X-ray Data Collection and Structure Refinement
To collect room-temperature diffraction datasets, crystals of 3CL Mpro in complexes with leupeptin, telaprevir, narlaprevir or boceprevir were mounted using the MiTeGen (Ithaca, NY) room-temperature capillary setup. Room temperature X-ray crystallographic data from these crystals were collected using a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator and Osmic VariMax optics. The diffraction images were obtained using an Eiger R 4M hybrid photon counting detector. Diffraction data were integrated using the CrysAlis Pro software suite (Rigaku Inc., The Woodlands, TX). Diffraction data were then reduced and scaled using the Aimless (Evans and Murshudov, 2013) program from the CCP4 suite (Winn et al., 2011); molecular replacement using PDB code 6WQF (Kneller et al., 2020) was then performed with Molrep (Winn et al., 2011) from the CCP4 program suite. The refinement of each protein structure was conducted using Phenix.refine from the Phenix (Adams et al., 2010) suite of programs and the COOT (Emsley et al., 2010) molecular graphics program. The geometry of each final structure was then carefully checked with Molprobity (Chen et al., 2010); the data collection and refinement statics are shown in Table S1.
Enzyme Activity Kinetics Assay
The enzymatic activity of SARS-CoV-2 Mpro was characterized by measuring initial rate with a previously established Förster resonance energy transfer (FRET) peptide substrate assay (Kuo et al., 2004) over a concentration range of 7.4 – 640 μM to determine K m and V max with 130 nM enzyme. The FRET substrate DABCYL-KTSAVLQSGFRKM-E(EDANS) trifluoroacetate salt was purchased from Bachem (PN 4045664) and dissolved at 10-14 mM concentration in DMSO. The assay buffer contained 20 mM Tris-HCl pH 7.3, 100 mM NaCl, 1 mM EDTA, and 2 mM reduced glutathione (6.15 mg added per 10 mL buffer fresh for each experiment) with 5% v/v final DMSO concentration. The assays were performed in 40 μL total volume in black half area 96-well plates (Greiner PN 675076) at 25°C. Fluorescence was detected every 24 s by a Biotek Synergy H1 plate reader with an excitation wavelength of 336 nm and an emission wavelength of 490 nm, 6.25 mm read height, low lamp energy, and 3 measurements per data point. After the background subtraction of the average of no enzyme negative controls, product formation was quantified using a 0.05 – 22 μM calibration curve of the free EDANS acid (Sigma PN A6517). Product concentrations were adjusted for inner filter absorbance effects with correction factors generated by comparing the fluorescence of 2 μM EDANS in solution with each concentration of substrate used to that with no substrate. To determine kinetic constants, initial velocity was measured at 20 to 640 μM substrate with 130 nM enzyme (8.8 μg/mL) in triplicate in two independent experiments to determine Vmax and KM. Reactions were initiated by adding 20 μL substrate solution to 20 μL enzyme solution in plates. GraphPad Prism 8.4.2 was used to perform nonlinear regression with the Henri-Michaelis-Menten equation (Equation 1) to give K M of 170 μM, 95% CI [130, 230] and V max of 36 nM/s, 95% CI [32, 41], yielding a second-order rate constant of k cat/K M = 1600 s-1 M-1 (Figure S2). This value is similar to those recently obtained by others (Zhang et al., 2020; Ma et al., 2020).
| (Equation 1) |
Enzyme Inhibition Assay
We examined the inhibition kinetics of four reversible covalent protease inhibitors, including leupeptin, a well-established cysteine and serine protease inhibitor, and three α-ketoamide HCV NS3/4A serine protease inhibitors, namely telaprevir, narlaprevir and boceprevir. Inhibition kinetics were performed by incubating 300 nM enzyme with 300 nM – 300 μM inhibitor in assay buffer at room temperature with gentle shaking for 30 minutes, briefly centrifuging, and then adding 20 μL of 80 μM substrate to initiate reactions, for final concentrations of 150 nM enzyme, 0.15-150 μM inhibitor, and 40 μM substrate. Two independent experiments were performed in triplicate. Initial velocities were normalized to the average initial rate of reactions without inhibitor. Nonlinear regression was performed with the IC50 equation (Equation 2) for each inhibitor across the concentration range, where is the normalized initial velocity at each inhibitor concentration. The inhibitors boceprevir (Sigma ADV465749229), narlaprevir (MedChemExpress HY-10300), and telaprevir (Apex Bio A4031) were stored as 60 mM stocks in DMSO at -20°C, and leupeptin (VWR J580) was stored at 20 mM in DMSO at -20°C.
| (Equation 2) |
The IC50 values obtained were: leupeptin, 92 μM, 95% CI [80, 106]; telaprevir, 18 μM, 95% CI [16, 21]; narlaprevir, 5.1 μM, 95% CI [4.3, 6.1]; and boceprevir, 3.1 μM, 95% CI [2.8, 3.5] (Figure S3). These values are similar to the recently measured IC50 values for narlaprevir (5.7 μM) and boceprevir (4.1 μM) (Ma et al., 2020).
Quantification and Statistical Analysis
For kinetics experiments, initial rates were determined by linear regression of the linear portion of enzyme reaction progress curves with <10% substrate conversion using the linest() formula in Excel. The relationships between initial rates and substrate concentration and between initial rates and inhibitor concentration were determined by nonlinear regression using GraphPad Prism 8.4.3. Statistical details including best-fit values for KM, Vmax, and IC50 and their 95% confidence intervals using the asymmetrical (profile-likelihood) method are given in Figures S2 and S3 and the associated figure legends.
Acknowledgments
This research was supported by the DOE Office of Science through the National Virtual Biotechnology Laboratory, a consortium of DOE national laboratories focused on the response to COVID-19, with funding provided by the Coronavirus CARES Act. This research used resources at the Spallation Neutron Source and the High Flux Isotope Reactor, which are DOE Office of Science User Facilities operated by the Oak Ridge National Laboratory. The Office of Biological and Environmental Research supported research at ORNL's Center for Structural Molecular Biology, a DOE Office of Science User Facility. This research used resources at the Second Target Station, which is a DOE Office of Science User Facilities Construction Project at Oak Ridge National Laboratory.
Author Contributions
Conceptualization, D.W.K., L.C., and A.K.; Methodology, D.W.K., S.G., G.P., and H.M.O’N.; Software, S.G. and L.C.; Validation, D.W.K., S.G., L.C., and A.K.; Formal Analysis, D.W.K., S.G., L.C., and A.K.; Investigation, D.W.K. and A.K.; Resources, G.P. and H.M.O’N.; Data Curation, S.G. and L.C.; Writing – Original Draft, D.W.K., L.C., and A.K.; Writing – Review & Editing, D.W.K., S.G., H.M.O’N., L.C., and A.K.; Visualization, D.W.K. and A.K.; Supervision, L.C. and A.K.; Project Administration, A.K.; Funding Acquisition, H.M.O’N., L.C., and A.K.
Declaration of Interests
The authors declare no competing interests.
Published: December 1, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.str.2020.10.007.
Supplemental Information
References
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr, Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main protease (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Appleyard G., Tisdale M. Inhibition of the growth of human coronavirus 229E by leupeptin. J. Gen. Virol. 1985;66:363–366. doi: 10.1099/0022-1317-66-2-363. [DOI] [PubMed] [Google Scholar]
- Belongia E.A., Osterholm M.T. COVID-19 and flu, a perfect storm. Science. 2020;368:1163. doi: 10.1126/science.abd2220. [DOI] [PubMed] [Google Scholar]
- de Bruijne J., Bergmann J.F., Reesink H.W., Weegink C.J., Molenkamp R., Schinkel J., Tong X., Li J., Treitel M.A., Hughes E.A. Antiviral activity of narlaprevir combined with ritonavir and pegylated interferon in chronic hepatitis C patients. Hepatology. 2010;52:1590–1599. doi: 10.1002/hep.23899. [DOI] [PubMed] [Google Scholar]
- Bzowka M., Mitusinska K., Raczynska A., Samol A., Tuszynski J.A., Gora A. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. Int. J. Mol. Sci. 2020;21:3099. doi: 10.3390/ijms21093099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of Virus The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Zhang B., Jiang X.-M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P.R., Murshudov G.N. How good are my data and what is the resolution? Acta Crystallogr. Section D. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J.S., van den Bedem H., Samelson A.J., Lang P.T., Holton J.M., Echols N., Alber T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl. Acad. Sci. USA. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlits O., Kong X., Cheng X., Wymore T., Blumenthal D.K., Taylor P., Radic Z., Kovalevsky A. Productive reorientation of a bound oxime reactivator revealed in room temperature X-ray structures of native and VX-inhibited human acetylcholinesterase. J. Biol. Chem. 2019;294:10607–10618. doi: 10.1074/jbc.RA119.008725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Snijder E.J. Viral cysteine proteases. Perspect. Drug Discov. Des. 1996;6:64–86. doi: 10.1007/BF02174046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmy Y., Fawzy M., Elaswad A., Sobieh A., Kenney S.P., Shehata A.A. The COVID-19 pandemic: a ccomprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J. Clin. Med. 2020;9:1225. doi: 10.3390/jcm9041225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F., Li Y., Leung E.L.-H., Liu X., Liu K., Wang Q., Lan Y., Li X., Yu H., Cui L., Luo H., Luo L. A review of therapeutic agents and Chinese herbal medicines against SARS-COV-2 (COVID-19) Pharmacol. Res. 2020;158:104929. doi: 10.1016/j.phrs.2020.104929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wei P., Fan K., Liu Y., Lai L. 3C-like proteinase from SARS coronavirus Catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- Isakov V., Koloda D., Tikhonova N., Kikalishvili T., Krasavina E., Lekishvili K., Malaya I., Ryska M., Samsonov M., Tolkacheva V. Pharmacokinetics of the new hapatitis C virus NS3 protease inhibitor narlaprevir following single-dose use with or without ritonavir in patients with liver cirrhosis. Antimicrob. Agents Chemother. 2016;60:7098–7104. doi: 10.1128/AAC.01044-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean S.-S., Hsueh P.-R. Old and re-purposed drugs for the treatment of COVID-19. Exp. Rev. Anti-infect. Ther. 2020:1–5. doi: 10.1080/14787210.2020.1771181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y., Zhu Y., Zhu C., Hu T., Du X. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- Jo S., Kim S., Shin D.H., Kim M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enz. Inhibit. Med. Chem. 2020;35:145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy D.A., van den Bedem H., Sivak D.A., Petsko G.A., Ringe D., Wilson M.A., Fraser J.S. Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR. Structure. 2014;22:899–910. doi: 10.1016/j.str.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D.W., Phillips G., O’Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalevsky A., Aggarwal M., Velazquez H., Cuneo M.J., Blakeley M.P., Weiss K.L., Smith J.C., Fisher S.Z., McKenna R. ‘To be or not to be’ protonated: atomic details of human carbonic anhydrase-clinical drug complexes by neutron crystallography and simulation. Structure. 2018;26:383–390. doi: 10.1016/j.str.2018.01.006. e3. [DOI] [PubMed] [Google Scholar]
- Kuo C.-J., Chi Y.-H., Hsu J.T.-A., Liang P.-H. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A.D., Perni R.B., Graham C.S. Development and marketing of INCIVEK (telaprevir; VX-950): a first-generation HCV protease inhibitor, in Combination with PEGylated interferon and ribavirin. In: Sofia M., editor. HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry. vol. 31. Springer; 2019. pp. 261–291. [Google Scholar]
- Liu C., Zhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6:315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfi M., Hamblin M.R., Rezaei N. COVID-19:transmission, prevention, and potential therapeutic opportunities. Clin. Chim. Acta. 2020;508:254–266. doi: 10.1016/j.cca.2020.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir. GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard R., Storer A.C. Oxyanion hole interactions in serine and cysteine protease. Biol. Chem. Hoppe-sayler. 1992;373:393–400. doi: 10.1515/bchm3.1992.373.2.393. [DOI] [PubMed] [Google Scholar]
- Muramatsu T., Takemoto C., Kim Y.-T., Wang H., Nishli W., Terada T., Shirouzu M., Yokoyama S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. U.S.A. 2016;113:12997–13002. doi: 10.1073/pnas.1601327113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten R., Liu L., Kenner L.R., Clarkson M.W., Mavor D., Tawfik D.S., Kern D., Fraser J.S. Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun. 2018;9:1–11. doi: 10.1038/s41467-018-03562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prongay A.J., Guo Z., Yao N., Pichardo J., Fischmann T., Strickland C., Myers J., Weber P.C., Beyer B.M., Ingram R. Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-([3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (SCH 503034) II. Kye steps in structure-based optimization. J. Med. Chem. 2007;50:2310. doi: 10.1021/jm060173k. [DOI] [PubMed] [Google Scholar]
- Rastogi Y.R., Sharma A., Nagraik R., Aygun A., Sen F. The novel coronavirus 2019-nCoV: its evolution and transmission into humans causing global COVID-19 pandemic. Int. J. Environ. Sci. Technol. 2020 doi: 10.1007/s13762-020-02781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano K.P., Ali A., Aydin C., Soumana D., Ozen A., Deveau L.M., Silver C., Cao H., Newton A., Petropoulos C.J. The molecular basis of drug resistance against hepatisis C virus NS3/4A protease inhibitors. PLOS Pathol. 2012;8:02832. doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Chen S.-E., Naser-Tavakolian A. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: desing, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013;68:372–384. doi: 10.1016/j.ejmech.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman S. Discovery of boceprevir, a ketoamide-derived HCV NS3 protease inhibitor, for treatment of genotype 1 infections. In: Sofia M., editor. HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry. vol 31. Springer; 2019. pp. 293–315. [Google Scholar]
- Wang L., Bao B.-B., Song G.-Q., Chen C., Zhang X.-M., Lu W., Wang Z., Cai Y., Li S., Fu S. Discovery of unsymmetrical aromtic disulfides as novel inhibitors of SARS-CoV main protease: chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem. 2017;137:450–461. doi: 10.1016/j.ejmech.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Zhao S., Teng T., Abdalla A.E., Zhu W., Xie L., Wang Y., Guo X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses. 2020;12:244. doi: 10.3390/v12020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Jin Y., Bartlam M., Rao Z. Production of authentic SARS-CoV Mpro with enzhanced activity: application as a novel tag-cleavage endopeptidase for protein overexpression. J. Mol. Biol. 2007;366:965–975. doi: 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J. Desing of wide-spectrum inhibitors targeting coronavirus main proteases. PLOS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The four protein structures have been deposited into the Protein Data Bank with accessions codes of 6XCH for the SARS-CoV-2 3CL Mpro complex with leupeptin, 6XQS for the complex with telaprevir, 6XQT for the complex with narlaprevir, and 6XQU for the complex with boceprevir. See Key Resources Table for more details.