

Graphical abstract
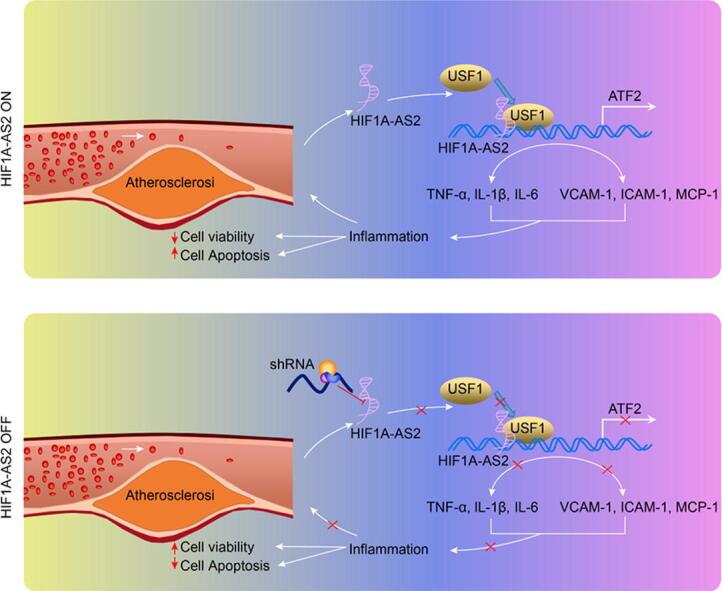
The map illustrating mechanisms associated with lncRNA HIF1A-AS2-mediated inflammation in the atherosclerosis. LncRNA HIF1A-AS2 forms a complex with USF1, which is recruited into the ATF2 promoter to elevate ATF2 expression, thus promoting the development of atherosclerotic inflammation. Meanwhile, downregulation of lncRNA HIF1A-AS2 decreases the expression of ATF2 by reducing the binding of USF1 to the ATF2 promoter regions, thereby inhibiting atherosclerotic inflammation, as reflected by decreased inflammatory factors TNF-α, IL-1β and IL-6 in serum and protein levels of adhesion molecules VCAM-1, ICAM-1, and MCP-1, as well as increased cell viability and reduced apoptosis in ox-LDL-induced inflammation in ECs, SMCs, and HCAECs.
Keywords: Atherosclerosis, Long noncoding RNA, Hypoxia-inducible factor 1 alpha-antisense RNA 2, Inflammation, Transcription factor, Upstream transcription factor 1, Activating transcription factor
Abbreviations: lncRNAs, long noncoding RNAs; ECs, endothelial cells; SMCs, smooth muscle cells; HCAECs, human coronary artery endothelial cells; LDL, low-density lipoprotein; HIF1A-AS2, hypoxia-inducible factor 1 alpha-antisense RNA 2; CAD, coronary artery disease; ATF2, activating transcription factor 2; USF1, upstream stimulatory factor 1; sh, short hairpin RNA; HFD, high fat diet; ND, normal diet; PBS, phosphate buffered saline; DMEM, Dulbecco’s modified Eagle’s medium; VCAM-1, vascular cell adhesion molecule 1; ICAM-1, intercellular adhesion molecule-1; MCP-1, monocyte chemoattractant protein-1; IgG, immunoglobulin G; HE, Hematoxylin-eosin; ELISA, enzyme linked immunosorbent assay; ATCC, American Type Culture Collection; ox-LDL, oxidized-low-density lipoprotein; si-NC, small interfering RNA-negative control; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; IL-6, interleukin-6; RT-qPCR, reverse transcription quantitative polymerase chain reaction; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; ChIP, Chromatin immunoprecipitation; RIP, RNA binding protein immunoprecipitation; CCK-8, cell counting kit-8
Abstract
Introduction
In atherosclerotic lesions, extensive inflammation of the vessel wall contributes to plaque instability. Long noncoding RNAs (lncRNAs) play important roles in diverse biological processes in atherosclerosis.
Objectives
Here, we aim to identify the functional role and regulatory mechanisms of lncRNA hypoxia-inducible factor 1 alpha-antisense RNA 2 (HIF1A-AS2) in atherosclerotic inflammation.
Methods
An atherosclerotic mouse model was induced in ApoE-/- mice by high fat diet (HFD). Endothelial cells (ECs), human aortic smooth muscle cells (SMCs) or human coronary artery endothelial cells (HCAECs) were exposed to ox-LDL to develop the in vitro model. The effects of lncRNA HIF1A-AS2 on inflammation were evaluated by determining levels of inflammatory factors tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) and levels of adhesion molecules vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and macrophage cationic peptide 1 (MCP-1).
Results
It was established that lncRNA HIF1A-AS2 and ATF2 were highly expressed in atherosclerotic ApoE-/- mice. Downregulating lncRNA HIF1A-AS2 in ox-LDL-exposed ECs, SMCs and HCAECs inhibited inflammation by reducing levels of pro-inflammatory factors and adhesion molecules. LncRNA HIF1A-AS2 bound to the transcription factor USF1 to elevate ATF2 expression. USF1 overexpression counteracted the suppressive effect of lncRNA HIF1A-AS2 silencing on ox-LDL-induced inflammation. Knockdown of lncRNA HIF1A-AS2 or ATF2 could also attenuate inflammation in atherosclerotic mice. Collectively, the present study demonstrates that downregulation of lncRNA HIF1A-AS2 represses the binding of USF1 to the ATF2 promoter region and then inhibits ATF2 expression, thereby suppressing atherosclerotic inflammation.
Conclusion
This study suggests lncRNA HIF1A-AS2 as an promising therapeutic target for atherosclerosis.
Introduction
Atherosclerosis, a chronic inflammatory disease, manifests with inflammation and accumulation of plaques in walls of arterial vessels [1], which is a main pathology underlying many cerebrovascular and cardiovascular diseases [2]. Risk factors for atherosclerosis include elevated circulating low-density lipoprotein (LDL) and triglyceride levels, smoking, obesity, and aging. Inflammation is involved in the pathogenesis of atherosclerosis as well as its complications, and therefore anti-inflammatory therapies have recently been proposed to improve the prognosis of individuals with atherosclerosis [3]. Accumulating evidence reveals the role of long noncoding RNAs (lncRNAs) in controlling atherosclerosis and inflammation-related diseases, thus highlighting them as important therapeutic targets [4], [5].
Over the past decade, lncRNAs have been extensively reported to be essential factors participating in a variety of pathophysiological processes. Dysregulation of lncRNAs is involved in many human diseases, including cardiovascular diseases [6], [7]. Besides, lncRNAs exert critical influence on the initiation of atherosclerosis through the inflammatory pathways [8]. For example, the expression of lncRNA LINC00599 is upregulated in aortic atherosclerotic lesions, but its downregulation accelerates atherosclerosis and aggravates inflammation [9]. Furthermore, the level of lncRNA myocardial infarction associated transcript was obviously increased in the serum from patients with atherosclerotic plaque, while knockdown of this lncRNA mitigated atherosclerosis progression [10]. A cardiac-related lncRNA hypoxia-inducible factor 1 alpha-antisense RNA 2 (HIF1A-AS2) has emerged as a potential modulator of inflammation [11]. More importantly, lncRNA HIF1A-AS2 has been proposed to serve as a critical biomarker for coronary artery disease (CAD) [12]. Hence, this current study aimed to find out whether lncRNA HIF1A-AS2 participates in regulating the inflammation of atherosclerosis. Bioinformatics analysis was adopted and predicted that lncRNA HIF1A-AS2 can regulate activating transcription factor 2 (ATF2). ATF2 is a member of the alkaline-leucine zipper class of proteins and is broadly expressed in a variety of tissues and involved in inflammatory responses [13]. Recent research has implicated ATF2 in the modulation of fatty diet-induced atherogenesis [14]. Another study has suggested that ATF2 regulates anti-inflammatory activities [15]. Transcription factors are the leading factors for adjusting gene expression in cells, with effects on transcription through their binding to precise genomic target sites [16]. Interestingly, bioinformatics analysis, i.e. the LncMAP website, also predicted that ATF2 could regulate the transcription factor upstream stimulatory factor 1 (USF1). USF1 is a type of transcription factor, which is related to human CAD [17]. USF1 has also been reported as a mediator involved in an anti-inflammatory strategy for the treatment of acute lung injury [18]. Therefore, we aimed to determine whether the lncRNA HIF1A-AS2/USF1/ATF2 regulatory network was involved in the control of inflammation in atherosclerosis, hoping to offer an enhanced understanding of the mechanisms underlying atherosclerosis, and in a search for new treatment.
Materials and methods
Ethics statement
All animal experiments were approved by the Animal Ethics Committee of the First Affiliated Hospital of Zhengzhou University and strictly followed the principle of minimizing number of animals used and minimizing the pain and suffering.
Short hairpin RNA (shRNA) construction
The shRNA targeting HIF1A-AS2 (sh-HIF1A-AS2) (sequence: 5′-AAGCTGATCAAAGGGGCCTGGTC-3′), shRNA targeting ATF2 (sh-ATF2) (sequence: 5′-CTTCTGTTGTAGAAACAAC-3′) as well as shRNA negative control (sh-NC) were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China).
Establishment of atherosclerosis mouse models
Eighty specific pathogen free ApoE−/− C57BL/6J male mice (aged 4–6 weeks, weighing 16–21 g) purchased from Peking University Medical Department were raised in the specific pathogen free animal laboratory with a humidity of 40–60% at 18–23 °C. The atherosclerosis mouse model was developed in 60 ApoE−/− male mice by feeding them with high fat diet (HFD) containing 0.25% cholesterol and 15% fat for 16 weeks [10], [19], [20]. The remaining 20 mice were fed with normal diet (ND) and used as controls. At the 4th week after HFD, 10 modeled mice were randomly selected and injected with lentivirus expressing sh-NC (2 × 107 TU/mouse, tail vein injection), and another 10 modeled mice were injected with lentivirus expressing sh-HIF1A-AS2 (2 × 107 TU/mouse, tail vein injection). No mice died during the experiment. After 16 weeks, the mice were anesthetized and euthanized, and 0.7 mL blood was collected by renal artery puncture. The thoracic aorta was immediately excised and cryopreserved with liquid nitrogen. A flow chart of animal experiments is shown in Supplementary Fig. 1.
Immunohistochemistry
After the euthanasia of mice, the aorta root was immediately excised and fixed by immersion in 4% paraformaldehyde. The paraffin-embedded sections were subjected to immunohistochemical staining. After dewaxing in xylene and dehydration in gradient ethanol, the sections were washed with tap water for 2 min. The sections were successively immersed in 3% methanol H2O2 for 20 min, distilled water for 2 min, and 0.1 M phosphate buffered saline (PBS) for 3 min. Next, the sections were water-bathed in antigen repair solution and cooled down with tap water, followed by blocking with normal goat serum (C-0005, Shanghai Haoran Biotechnology Co., Ltd., Shanghai, China) at ambient temperature for 20 min. The tissue sections were incubated overnight at 4 °C with rabbit polyclonal antibody to ATF2 (1:200, ab47476), rabbit monoclonal antibody to vascular cell adhesion molecule 1 (VCAM-1) (1:500, ab134047), rabbit monoclonal antibody to intercellular adhesion molecule-1 (ICAM-1) (1:2000, ab179707), and rabbit polyclonal antibody to monocyte chemoattractant protein-1 (MCP-1) (1:100, ab7202), all of which were purchased from Abcam Inc. (Cambridge, UK). The sections were washed 3 times in 0.1 M PBS, 5 min each time, and subsequently incubated with secondary antibody goat anti-rabbit immunoglobulin G (IgG) (1:2000, ab205718) at 37 °C for 20 min, followed by 3 PBS washes. The sections were then incubated with horseradish peroxidase-labeled streptavidin protein working solution (0343-10000U, Imun Biotechnology Co., Ltd., Beijing, China) at 37 °C for 20 min, followed by 3 PBS washes. Diaminobenzidine (ST033, Guangzhou Whiga Technology Co., Ltd., Guangzhou, China) was used for color development, followed by washing with water. The sections were counter-stained for 1 min with hematoxylin (PT001, Shanghai Bogoo Biotechnology Co., Ltd., Shanghai, China) and then blued in 1% ammonia water and washed with water. After being dehydrated with an ethanol series, the sections were cleared in xylene and sealed with neutral gum. Finally, the sections were observed under a microscope. The positive rate was calculated with the percentage of immunopositive area (0%, 0; 1–25%, 1; 26–50%, 2; 51–75%, 3; 76–100%, 4), multiplied by staining intensity (0, negative; 1, weak positive; 2, medium positive; 3, strong positive). The sections were scored independently by two pathologists.
Oil red O staining
The area of the facial plaque was quantified by oil red O staining. The aorta root was immediately excised and fixed in 4% paraformaldehyde after the mice were euthanized. Briefly, after careful removal of the adventitial tissue, the aorta was opened longitudinally to expose the entire lumen surface for Oil Red O staining (Sigma-Aldrich St. Louis, MO, USA). The area of atherosclerotic plaque was measured in pixels using Image J.
The frozen slices at 6-μm thick were dried by a cold air blower, and fixed in 10% formalin-1% CaCl2 solution for 15 min. After that, the slices were rinsed with 60% isopropanol for 2 min, and stained with oil red O working solution for 10 min, followed by rinses in 60% isopropanol again. The slices were sealed with glycerogelatin and visualized under an optical microscope (Olympus, Tokyo, Japan).
Hematoxylin-eosin (HE) staining
Thoracic aorta specimens fixed with formaldehyde were conventionally dehydrated with gradient ethanol (70%, 80%, 90%, 95%, and 100%, 5 min each time), cleared with xylene twice (10 min each time), and then dipped in wax. After paraffin embedding, the sections were sliced to slices at a thickness of 4 μm and the slices were baked at 60 °C for 1 h. The baked slices were dewaxed using xylene, hydrated with the gradient ethanol, followed by hematoxylin staining for ten 10 min. After rinsing with distilled water for 1 min, the slices were treated with 1% hydrochloric acid alcohol for 20 s, and rinsed with distilled water for 1 min. After being blued in 1% ammonia for 30 s and washed for 1 min, the slices were dyed with eosin for 3 min, washed with distilled water for 1 min, and dehydrated with gradient ethanol, 2 min each time. The slices were cleared twice with xylene (5 min/time), and sealed with the neutral gum. The aortic sinus lesions were observed under an optical microscope (Olympus, Tokyo, Japan).
Cell culture and transfection
Endothelial cells (ECs; CRL-1730), smooth muscle cells (SMCs; CRL-1999) and human coronary artery endothelial cells (HCAECs; PCS-100020) were obtained from the American Type Culture Collection (Manassas, VA, USA). ECs, SMCs and HCAECs were cultured with Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum at 37 °C in a 5% CO2 incubator.
The cells were seeded in a six-well plate for 24 h before transfection. When the cell confluence had reached about 50%, ECs, SMCs and HCAECs were transfected following the instructions of the lipofectamine 2000 reagents (Invitrogen Inc., Carlsbad, CA, USA). At 24 h after transfection, the cells were exposed to 50 μg/mL oxidized-low-density lipoprotein (ox-LDL) (Beijing Xiesheng Bio-Technology Limited, Beijing, China) for 24 h, or continued normal incubation [21].
LncRNA HIF1A-AS2-specific small interfering RNA (siRNAs) including si-HIF1A-AS2-1 (sense AAGAGAUCUGUGGCUCAGUUCCUUU and antisense AAAGGAACUGAGCCACAGAUCUCUU) and si-HIF1A-AS2-2 (sense GAGUUGGAGGUGUUGAAGCAAAUAU and antisense AUAUUUGCUUCAACACCUCCAACUC), si-NC, overexpression plasmids including oe-USF1 (pcDNA3.1-USF1) and oe-ATF2 (pcDNA3.1-ATF2) as well as oe-NC (pcDNA3.1-NC) were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China).
Enzyme linked immunosorbent assay (ELISA)
Whole blood was collected and centrifuged at 3000 to 5000 rpm for 30 min at room temperature to collect serum samples. Measurement of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) levels in mouse serum was performed according to the instructions of the kits, including the mouse TNF-α ELISA Kit (ab208348), mouse IL-1β ELISA kit (ab100704), and mouse IL-6 ELISA kit (ab100712). The optical density values were recorded at 450 nm microplate reader and analyzed using Origin 9.5 software. The levels of TNF-α, IL-1β, IL-6 and other inflammatory factors in ECs, SMCs and HCAECs were also detected by ELISA using the corresponding human ELISA kits for TNF-α (ab181421), IL-1β (ab46052) and IL-6 (ab178013).
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
The total RNAs were isolated from tissues and ECs, SMCs and HCAECs using an ultra-pure RNA extraction kit (QIAGEN, Duesseldorf, Germany), as instructed by the manufacturer. The reverse transcription was carried out according to the instructions of TaqMan MicroRNA Assays Reverse Transcription Primer (4366596, Thermo Fisher Scientific, Waltham, MA, USA). The produced cDNA was subjected to real-time PCR, following the instruction manual of SYBR® Premix Ex TaqTM II Kit (RR820A, Xingzhi Biotechnology Co., Ltd., Huaian, China). RT-qPCR was performed on an ABI PRISM® 7300 PCR instrument (Shanghai Kunke Instrument Equipment Co., Ltd., Shanghai, China). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was regarded as the endogenous reference. The relative expression of target genes was calculated by 2−ΔΔCt method using the formula as follows: ΔΔCT = ΔCt experimental group − ΔCt control group, wherein ΔCt = Ct target gene − Ct internal reference. Primers were synthesized by Aoke Biotechnology Co., Ltd (Zhenjiang, Jiangsu Province, China) (Table 1).
Table 1.
Primer sequences for reverse transcription quantitative polymerase chain reaction.
| Gene | Sequence |
|---|---|
| HIF1A-AS2 (human) | F: 5′-TCTGTGGCTCAGTTCCTTTTGT-3′ |
| R: 5′-ATGTAGGAAGTGCCAGAGCC-3′ | |
| HIF1A-AS2 (mouse) | F: 5′-AGGACCTAAGGCTCTGGCAC-3′ |
| R: 5′-GGGATGAGTGAAGCAGTTCTCA-3′ | |
| ATF2 (human) | F: 5′-AGATTTATTAATTTTTCTGTGCTCAA-3′ |
| R: 5′-ACACCCCCATTTATTAAAACACC-3′ | |
| ATF2 (mouse) | F: 5′-ATGGCAGTGGATTGGTTAGG-3′ |
| R: 5′-AGTTGTGTGAGCTGGAGAAG-3′ | |
| GAPDH (human) | F: 5′-ACAGTCAGCCGCATCTTCTT-3′ |
| R: 5′-GTTAAAAGCAGCCCTGGTGA-3′ | |
| GAPDH (mouse) | F: 5′-GAGCCAAAAGGGTCATCATC-3′ |
| R: 5′-TAAGCAGTTGGTGGTGCAGG-3′ |
Note: HIF1A-AS2, HIF1A antisense RNA 2; ATF2, activating transcription factor 2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; F: forward; R: reverse.
Cell counting kit-8 (CCK-8) assay
Cell viability was measured using the CCK-8 kit (Dojindo, Tokyo, Japan) according to the manufacturer's instructions. Briefly, ECs, SMCs and HCAECs were seeded in 96-well plates for 48 h, and the living cells were detected by incubation with CCK-8 reagent for 2 h under the specified conditions. Finally, a microplate reader was used to measure the absorbance at 450 nm to evaluate cell viability.
Flow cytometry
Apoptosis was determined by Annexin V-fluorescein isothiocyanate/propidium iodide apoptosis detection kit (Beyotime, Shanghai, china). Trypsin-digested cells in the absence of ethylenediaminetetraacetic acid were washed with PBS, resuspended in 100 µL of 1 × binding buffer, and then added with 5 μL of Annexin V-FITC and 10 μL of PI. After 15 min incubation in the dark, apoptosis was detected with a flow cytometer (BD Biosciences, Franklin Lake, Franklin Lake, New Jersey, USA), and data analysis was performed using FlowJo software (Stanford University, San Francisco, California, USA).
Western blot analysis
Tissues as well as ECs, SMCs and HCAECs were lysed by 1 mL cell lysis buffer containing 50 mmol/L Tris, 150 mmol/L NaCl, 5 mmol/L elhylenediamine tetraacetic acid, 0.1% sodium dodecyl sulfate, 1% NP-40, 5 Gg/mL aprotinin, and 2 mmol/L phenylmethylsulfonyl fluoride for 4 min at 4 °C. The protein concentration in the supernatant was measured using a bicinchoninic acid kit (20201ES76, Yeason Biotechnology Co., Ltd., Shanghai, China). The protein sample was mixed with the 10% SDS gel loading buffer and boiled at 100 °C for 10 min, after which the proteins were electrophoretically separated, and transferred to the nitrocellulose membrane. After being blocked with 5% skim milk overnight at 4 °C, the membrane was incubated with rabbit anti-VCAM-1 (1:2000, ab134047), ICAM-1 (1:1000, ab25375), and MCP-1 (1:2000, ab25124), ATF2 (1:1000, ab47476), USF (1:10,000, ab125020) and GAPDH (1:10,000, ab181602) overnight at 4 °C. The antibodies used above were purchased from Abcam Inc. (Cambridge, UK). The next day, the membrane was washed with PBS for 3 times (5 min/time) at room temperature whereupon it was incubated with horseradish peroxidase-labeled goat anti-rabbit IgG (1:10,000, ab205718, Abcam Inc., Cambridge, UK) at 37 °C for 1 h. After 3 washes in PBS buffer at ambient temperature, 10 min each time, the membrane was immersed in enhanced chemiluminescence reaction solution (Pierce, Waltham, MA, USA) at room temperature for 5 min and exposed by a luminometer (Shanghai Jingke Chemical Technology Co., Ltd., Shanghai China). With GAPDH as an endogenous reference, the ratio of the gray value of the target band to that of the inner reference band was regarded as the relative expression level of the protein.
RNA binding protein immunoprecipitation (RIP) assay
The binding of lncRNA to protein was detected by RIP kit (EMD Millipore, Billerica, MA, USA). The cells were washed with pre-cooled PBS and then the supernatant was discarded. The cells were lysed with RIP lysis buffer for 5 min, and centrifuged at 14,000 rpm for 10 min at 4 °C. A part of the cell lysate was taken out as an input, and the other part was co-precipitated by incubation with the specific antibody mouse anti-USF-1 (sc-101197, Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) or rabbit anti-IgG (ab172730, Abcam, Inc., Cambridge, UK). In brief, 50 μL of magnetic beads were resuspended in 100 μL RIP wash buffer and incubated with 2 μg antibody. The magnetic bead-antibody complex was resuspended in 900 μL RIP Wash Buffer and incubated with 100 μL cell lysate overnight at 4 °C. The sample was washed 3 times, and placed on a magnetic stand to collect the magnetic bead-protein complex. The sample and Input were separately detached by proteinase K, and the RNA was extracted for subsequent RT-qPCR determination of lncRNA.
RNA pull-down assay
The sense chain and the antisense chain of biotinylated lncRNA HIF1A-AS2 were mixed with cell lysates from ECs, SMCs and HCAECs. The biotinylated lncRNA HIF1A-AS2-protein complex was then pulled down using streptavidin agarose beads (Thermo Fisher Scientific, Waltham, MA, USA). The protein was then eluted from the RNA-protein complex and immunoblotted using antibody to USF1.
Dual-luciferase reporter assay
The USF1 binding sites on the ATF2 gene promoter were predicted by UCSC (http://genome.ucsc.edu/) and JASPAR (http://jaspar.genereg.net/) websites. The oe-NC and oe-USF1 were respectively co-transfected with ATF2-2 Kb luciferase reporter plasmid or the recombinant luciferase reporter plasmid with the truncated binding site. 48 h after transfection, cells were harvested and lysed. The luciferase activity was detected using a luciferase assay kit (K801-200, BioVision, Milpitas, CA, USA) and analyzed in a dual luciferase reporter gene system (Promega, Madison, WI, USA). Renilla luciferase was used as an internal reference, and the degree of activation of the target reporter gene was compared based on the ratio of the relative luciferase units of firefly luciferase divided by the relative luciferase units of Renilla luciferase.
Chromatin immunoprecipitation (ChIP)
When ECs reached 70–80% confluence, they were cross-linked with 1% formaldehyde at room temperature for 10 min. Next, the chromatin was randomly broken by ultrasonication for 10 s, at intervals of 10 s, for 15 times in total. The chromatin fragments were centrifuged at 13,000 rpm at 4 °C, and the supernatant was collected and incubated overnight at 4 °C with rabbit anti-IgG (ab109489, 1:100, Abcam Inc., Cambridge UK) or mouse anti-USF-1 (sc-101197, Santa Cruz Biotechnology). The endogenous DNA-protein complex was precipitated by Protein Agarose/Sepharose. After centrifugation, the supernatant was discarded, and the non-specific complex was removed. The cross-linking was reversed at 65 °C overnight. The DNA fragment was purified by phenol/chloroform extraction and analyzed by PCR. The primers used in this assay included ATF2 primer 1 (site 1) (forward ACCCGCCTTCTTCTCCTTA; reverse CCCTTCTTGCCTTCCTTCTG) and ATF2 primer 2 (site 1): (forward GAGGACTGAGTTGGCTTATACAC; reverse TGAGCCTGAAGTTCAAGAATGA).
Statistical analysis
All data were analyzed by SPSS 21.0 software (IBM Corporation, Chicago, IL, USA). Measurement data were expressed as mean ± standard deviation. Comparison of unpaired data obeying the normal distribution between two groups was performed using unpaired t test. Data comparisons among multiple groups were performed using one-way analysis of variance followed by Tukey's post hoc test. Correlation between indicators was analyzed using Pearson correlation coefficient. A value of p < 0.05 was considered to be statistically significant.
Results
LncRNA HIF1A-AS2 and ATF2 are highly expressed in atherosclerotic mice
First, we developed an ApoE−/− mouse model of atherosclerosis. After modeling, the body weight and composition of the mice were detected, the results of which illustrated a gradual upward inclination in the body weight of mice fed with HFD with the increase of the weight of white adipose tissue (WAT), brown adipose tissue (BAT) and liver, as compared with the fed with ND, while other organs such as muscle were not affected (Fig. 1A). Meanwhile, measurement on the food intake of HFD-fed mice suggested that the weight gain was not correlated with the food intake (Fig. 1B). Then Oil red O staining and HE staining suggested that the area of arterial tissue damage in the ApoE−/− mice fed with HFD was increased compared with that in the ApoE−/− mice fed with ND (Fig. 1C–E). Besides, ELISA showed the levels of TNF-α, IL-1β and IL-6 in the serum of ApoE−/− mice fed with HFD were higher than those in the ApoE−/− mice fed with ND (Fig. 1F). Simultaneously, immunohistochemistry detection showed that the expression of adhesion molecules VCAM-1, ICAM-1, and MCP-1 in arterial tissues of ApoE−/− mice fed with HFD was higher than that in ApoE−/− mice fed with ND (Fig. 1G). Therefore, the mouse model of atherosclerosis was successfully established. The expression of lncRNA HIF1A-AS2 and ATF2 in arterial tissues was determined by RT-qPCR. As shown in Fig. 1H, lncRNA HIF1A-AS2 and ATF2 were both highly expressed in the arterial tissues of ApoE−/− mice fed with HFD compared with ApoE−/− mice fed with ND. The expression of lncRNA HIF1A-AS2 was positively correlated with the expression of ATF2 (Fig. 1I). Besides, immunohistochemistry showed higher expression of ATF2 in arterial tissues from the ApoE−/− mice fed with HFD compared with the ApoE−/− mice fed with ND (Fig. 1J). The above results indicated that lncRNA HIF1A-AS2 and ATF2 were highly expressed in the mouse model of atherosclerosis.
Fig. 1.

High expression of lncRNA HIF1A-AS2 and ATF2 are identified in atherosclerotic mice. (A) Body weight and composition of ApoE−/− mice fed with ND or HFD. (B) Measurement of the food intake of ApoE−/− mice fed with ND or HFD. (C) Lipid deposition in the aorta observed after Oil red O staining. (D) The thoracic aortic injury area detected by HE staining (100×). (E) Lipid deposition in the thoracic aorta observed after Oil red staining (100×). (F) ELISA detection of TNF-α, IL-1β, and IL-6 levels in the serum of the ApoE−/− mice fed with ND or HFD. (G) Immunohistochemical detection of VCAM-1, ICAM-1, and MCP-1 expression in arterial tissues of the ApoE−/− mice fed with ND or HFD (400×). (H) The expressions of lncRNA HIF1A-AS2 and ATF2 in arterial tissues of the ApoE−/− mice fed with ND or HFD determined by RT-qPCR. (I) Correlation analysis of lncRNA HIF1A-AS2 expression and ATF2 expression. (J) The expression of ATF2 in arterial tissues of the ApoE−/− mice fed with ND or HFD detected by immunohistochemistry (400×). *p < 0.05 vs. the ApoE−/− mice fed with ND. Data (mean ± standard deviation) between two groups were analyzed using unpaired t test. n = 20 for mice fed with ND or HFD.
LncRNA HIF1A-AS2 repression inhibits ox-LDL-induced inflammation in ECs, SMCs and HCAECs
To investigate the effect of lncRNA HIF1A-AS2 on atherosclerosis, we downregulated the expression of lncRNA HIF1A-AS2 in ECs, SMCs and HCAECs using siRNA. RT-qPCR displayed that the expression of lncRNA HIF1A-AS2 was decreased in cells transfected with either si-HIF1A-AS2-1 or si-HIF1A-AS2-2 compared with cells transfected with si-NC (Fig. 2A). si-HIF1A-AS2-1 was selected for the subsequent study due to its higher silencing efficiency. Then ECs, SMCs and HCAECs were exposed to ox-LDL, and the expression of lncRNA HIF1A-AS2 was measured by RT-qPCR, which showed that lncRNA HIF1A-AS2 expression was upregulated in these cells that were exposed to ox-LDL, but it was downregulated in response to siRNA-mediated silencing of lncRNA HIF1A-AS2 (Fig. 2B). The results of ELISA revealed that the levels of inflammatory cytokines TNF-α, IL-1β and IL-6 were increased in ECs, SMCs and HCAECs after ox-LDL treatment, which were blocked after silencing of lncRNA HIF1A-AS2 (Fig. 2C). At the same time, the data obtained from Western blot analysis exhibited that the protein expression of adhesion molecules VCAM-1, ICAM-1 and MCP-1 was increased in ECs, SMCs and HCAECs after ox-LDL treatment, which was inhibited after lncRNA HIF1A-AS2 knockdown (Fig. 2D). Furthermore, Oil red O staining analysis showed that the lipid level was increased in cells treated with ox-LDL while it was reduced in ox-LDL-treated cells following HIF1A-AS2 knockdown (Fig. 2E). Collectively, lncRNA HIF1A-AS2 knockdown inhibited inflammation in ox-LDL-treated ECs, SMCs and HCAECs.
Fig. 2.

Inhibition of lncRNA HIF1A-AS2 curbs ox-LDL-induced inflammation in ECs, SMCs and HCAECs. (A) RT-qPCR determination of the expression of lncRNA HIF1A-AS2 in cells transfected with si-HIF1A-AS2-1 or si-HIF1A-AS2-2. (B) The expression of lncRNA HIF1A-AS2 in ECs, SMCs and HCAECs transfected with si-HIF1A-AS2 measured by RT-qPCR. (C) ELISA detection of the expression of TNF-α, IL-1β and IL-6 in cell culture supernatants after lncRNA HIF1A-AS2 downregulation in ECs, SMCs and HCAECs. (D), The protein expression of VCAM-1, ICAM-1, and MCP-1 in ECs, SMCs and HCAECs after inhibiting lncRNA HIF1A-AS2 measured by Western blot analysis. (E) Lipid level in ox-LDL-treated cells following HIF1A-AS2 knockdown analyzed by Oil red O staining (400×). *p < 0.05 vs. cells transfected with si-NC or cells without treatment. #p < 0.05 vs. ox-LDL-treated cells transfected with si-NC. Data (mean ± standard deviation) were obtained from three independent experiments and data between two groups were analyzed using unpaired t test.
Downregulating lncRNA HIF1A-AS2 mitigates ox-LDL-induced inflammation in ECs, SMCs and HCAECs by inhibiting ATF2
After silencing lncRNA HIF1A-AS2 in ox-LDL-exposed ECs, SMCs and HCAECs, we detected the expression of ATF2 using Western blot analysis. The results showed that ox-LDL treatment elevated ATF2 protein expression, while inhibition of lncRNA HIF1A-AS2 reversed the promotion caused by ox-LDL treatment (Fig. 3A). Furthermore, downregulation of lncRNA HIF1A-AS2 in ox-LDL-exposed ECs, SMCs and HCAECs reduced the protein expression of ATF2 and reduced the expression of inflammation-related factors TNF-α, IL-1β and IL-6 as well as that of the adhesion molecules VCAM-1, ICAM-1 and MCP-1. Overexpression of ATF2 increased the expression of TNF-α, IL-1β, IL-6, VCAM-1, ICAM-1 and MCP-1. However, silencing of lncRNA HIF1A-AS2 reversed the upregulating effect of ATF2 on the expression of the aforementioned inflammatory cytokines and adhesion molecules. The reductions in the aforementioned inflammatory cytokines and adhesion molecules induced by downregulation of lncRNA HIF1A-AS2 were rescued by overexpression of ATF2 (Fig. 3B–D). The above results together demonstrated that suppression of lncRNA HIF1A-AS2 inhibited inflammatory responses in ox-LDL-exposed ECs, SMCs and HCAECs by reducing the expression of ATF2.
Fig. 3.

Knockdown of lncRNA HIF1A-AS2 alleviates ox-LDL-induced inflammation in ECs, SMCs and HCAECs by inhibiting ATF2. (A) The protein expression of ATF2 determined by Western blot analysis after lncRNA HIF1A-AS2 was silenced in ox-LDL-exposed ECs, SMCs and HCAECs. (B) Measurement of ATF2 protein expression by Western blot analysis in response to downregulation of lncRNA HIF1A-AS2 in ox-LDL-exposed ECs, SMCs and HCAECs. (C) ELISA detection of the expression of TNF-α, IL-1β and IL-6 in EC and SMC culture supernatants exposed to ox-LDL. (D) The protein expression of VCAM-1, ICAM-1, and MCP-1 in ox-LDL-exposed ECs, SMCs and HCAECs measured by Western blot analysis. *p < 0.05 vs. the untreated cells or ox-LDL-exposed cells co-transfected with si-NC and oe-NC. #p < 0.05 vs. the cells ox-LDL-exposed transfected with si-NC or ox-LDL-exposed cells co-transfected with si-HIF1A-AS2 and oe-NC. & p < 0.05 vs. the ox-LDL-exposed cells co-transfected with si-NC and oe-ATF2. Data (mean ± standard deviation) were obtained from three independent cell experiments and comparison among multiple groups was analyzed with one-way analysis of variance.
LncRNA HIF1A-AS2 knockdown contributes to enhanced cell viability, but reduced apoptosis by inhibiting ATF2 expression
To better elucidate the role of lncRNA HIF1A-AS2 in cell viability and apoptosis, we adopted CCK-8 and flow cytometry. Results showed that silencing of lncRNA HIF1A-AS2 potently increased cell viability and reduced apoptosis in ox-LDL-induced inflammation in ECs, SMCs, and HCAECs, and that overexpression of ATF2 remarkably inhibited cell viability and promoted apoptosis. Treatment with oe-ATF2 could reverse the effect of si-HIF1A-AS2 on cell viability and apoptosis (Fig. 4A–F). Coherently, lncRNA HIF1A-AS2 knockdown enhanced cell viability, but reduced apoptosis by inhibiting ATF2 expression.
Fig. 4.

Silencing of lncRNA HIF1A-AS2 enhances cell viability, but reduces apoptosis by inhibiting ATF2 expression. (A) Viability of ECs after ox-LDL-induced inflammation determined using CCK-8 assay. (B) Viability of SMCs after ox-LDL-induced inflammation determined using CCK-8 assay. (C) Viability of HCAECs after ox-LDL-induced inflammation determined using CCK-8 assay. (D) Cell apoptosis of ECs after ox-LDL-induced inflammation determined using flow cytometry. (E) Cell apoptosis of SMCs after ox-LDL-induced inflammation determined using flow cytometry. (F) Cell apoptosis of HCAECs after ox-LDL-induced inflammation determined using flow cytometry. *p < 0.05 vs. ox-LDL-exposed cells co-transfected with si-NC + oe-NC. #p < 0.05 vs. ox-LDL-exposed cells co-transfected with si-HIF1A-AS2 + oe-NC. &p < 0.05 vs. the ox-LDL-exposed cells co-transfected with si-NC + oe-ATF2. Data (mean ± standard deviation) were obtained from three independent cell experiments and comparison among multiple groups was analyzed with one-way analysis of variance.
LncRNA HIF1A-AS2 regulates the expression of ATF2 via binding to the transcription factor USF1
The database LncMAP predicted that lncRNA HIF1A-AS2 might regulate ATF2 expression via the transcription factor USF1 (Fig. 5A). To test this prediction, the protein expression of USF1 and ATF2 in ECs, SMCs and HCAECs exposed to ox-LDL was measured by Western blot analysis, which showed that ox-LDL treatment increased the protein expression of USF1 and ATF2 (Fig. 5B). Furthermore, lncRNA HIF1A-AS2 was found to bind to the transcription factor USF1 according to RIP and RNA pull-down experiments (Fig. 5C, D). Prediction based on the JASPAR website showed 4 potential putative USF1 binding sites on the ATF2 promoter (Fig. 5E). The dual-luciferase reporter assay found that when the site 2 was deleted, the luciferase activity of the ATF2 promoter region was unaffected by USF1 overexpression (Fig. 5F). Subsequently, the ChIP assay showed that USF1 could bind to sites 1 and 2 on the promoter region of ATF2. The binding of USF1 to site 2 was weakened, but the binding of USF1 to site 1 was unaffected after lncRNA HIF1A-AS2 silencing (Fig. 5G). These results indicated that lncRNA HIF1A-AS2 regulated the expression of ATF2 via the transcription factor USF1.
Fig. 5.

LncRNA HIF1A-AS2 modulates the expression of ATF2 via binding to USF1. (A) Prediction of the lncRNA-TF-mRNA interaction based on the LncMAP database. (B) The protein expression of USF1 and ATF2 in ECs, SMCs and HCAECs exposed to ox-LDL measured by Western blot analysis. (C) The binding of lncRNA HIF1A-AS2 to USF1 in ECs, SMCs and HCAECs detected by RNA pull-down. (D) The binding of lncRNA HIF1A-AS2 to USF1 in ECs detected by RIP assay. (E) The binding site of USF1 on the ATF2 promoter predicted from JASPAR website. (F) The binding of USF1 to the ATF2 promoter region detected by the dual-luciferase reporter assay. (G) The binding of USF1 to the ATF2 promoter in ECs with or without silencing lncRNA HIF1A-AS2 detected by ChIP assay. *p < 0.05 vs. the untreated cells or ox-LDL-exposed cells treated with oe-NC or IgG. Data (mean ± standard deviation) were obtained from three independent experiments and those between groups were analyzed by unpaired t test.
LncRNA HIF1A-AS2 knockdown downregulates ATF2 expression to inhibit ox-LDL-induced inflammation in ECs, SMCs and HCAECs via USF1
To further investigate whether lncRNA HIF1A-AS2 affects inflammation in ox-LDL-exposed ECs, SMCs and HCAECs via binding to USF1, lncRNA HIF1A-AS2 was silenced and/or USF1 was overexpressed in the ox-LDL-exposed ECs, SMCs and HCAECs. Then the expression of lncRNA HIF1A-AS2 was determined by RT-qPCR and the expression of USF1 and ATF2 was determined by RT-qPCR and Western blot analysis in these cells. The results showed that the expression of lncRNA HIF1A-AS2 was decreased in cells after transfection with si-HIF1A-AS2, as a result of which, the expression of ATF2 was reduced, whereas the expression of USF1 was not affected. The expression of USF1 and ATF2 was increased in cells after overexpression of USF1, while overexpression of USF1 counteracted the inhibitory effect of si-HIF1A-AS2 on ATF2 expression (Fig. 6A, B). Further experiments demonstrated that silencing of lncRNA HIF1A-AS2 decreased the levels of proinflammatory proteins TNF-α, IL-1β, IL-6 and adhesion molecules VCAM-1, ICAM-1, and MCP-1 in the cell supernatants, but those reductions were neutralized by overexpression of USF1. Overexpression of USF1 elevated the expression of above-mentioned proinflammatory proteins and adhesion molecules, but their upregulation caused by USF1 overexpression was diminished by downregulation of lncRNA HIF1A-AS2 in the cell supernatants (Fig. 6C, D). Collectively, downregulation of lncRNA HIF1A-AS2 reduced the expression of ATF2 via mediating USF1, resulting in inhibition of ox-LDL-induced inflammation in ECs, SMCs and HCAECs.
Fig. 6.

Knockdown of lncRNA HIF1A-AS2 downregulates ATF2 expression via USF1 to inhibit inflammation in ox-LDL-exposed ECs, SMCs and HCAECs. (A) The expression of lncRNA HIF1A-AS2 in ox-LDL-exposed ECs, SMCs and HCAECs determined by RT-qPCR. (B) Measurement of USF1 and ATF2 protein expression in ox-LDL-exposed ECs, SMCs and HCAECs by Western blot analysis. (C) The expression of TNF-α, IL-1β and IL-6 in the supernatant of ox-LDL-exposed ECs, SMCs and HCAECs detected by ELISA. (D) The protein expression of VCAM-1, ICAM-1, and MCP-1 in ox-LDL-exposed ECs, SMCs and HCAECs measured by Western blot analysis. *p < 0.05 vs. the ox-LDL-exposed cells co-transfected with si-NC and oe-NC. #p < 0.05 vs. the ox-LDL-exposed cells co-transfected with si-HIF1A-AS2 and oe-NC. & p < 0.05 vs. the ox-LDL-exposed cells co-transfected with si-NC and oe-USF1. Data (mean ± standard deviation) were obtained from three independent experiments and those among multiple groups analyzed by one-way analysis of variance.
LncRNA HIF1A-AS2 silencing or ATF2 silencing inhibits inflammation in atherosclerotic mice in vivo
To further validate the effects of lncRNA HIF1A-AS2 and ATF2 on atherosclerotic inflammation, in vivo experiments were performed in an ApoE−/− mouse model of atherosclerosis. ApoE−/− mice fed with HFD were injected with lentiviruses expressing sh-NC, sh-HIF1A-AS2 or sh-ATF2 at 4th week after HFD. The expression of lncRNA HIF1A-AS2 and ATF2 in the arterial tissues of mice was then determined by RT-qPCR, which showed that the expression of lncRNA HIF1A-AS2 was successfully decreased by injection of lentivirus expressing sh-HIF1A-AS2, and ATF2 expression was reduced by infection with lentivirus expressing sh-ATF2 (Fig. 7A). Oil red O staining and HE staining showed that compared with atherosclerotic mice infected with lentivirus expressing sh-NC, the area of arterial tissue damage was reduced in the atherosclerotic mice infected with lentivirus expressing either sh-HIF1A-AS2 or sh-ATF2 (Fig. 7B–D). Meanwhile, ELISA analysis of atherosclerotic mouse plasma samples showed that the levels of TNF-α, IL-1β and IL-6 were lowered by silencing either lncRNA HIF1A-AS2 or ATF2 (Fig. 7E). As shown by Western blot analysis in Fig. 7F, the protein expression of VCAM-1, ICAM-1, and MCP-1 was decreased in mouse arterial tissues after silencing either lncRNA HIF1A-AS2 or ATF2. These results collectively indicated that knocking down either lncRNA HIF1A-AS2 or ATF2 inhibited inflammatory responses in atherosclerotic mice.
Fig. 7.

Knockdown of lncRNA HIF1A-AS2 or ATF2 inhibits inflammation in atherosclerotic ApoE−/− mice in vivo. (A) Determination of lncRNA HIF1A-AS2 and ATF2 expression in mouse arterial tissues by RT-qPCR. (B) Lipid deposition in injured aorta areas observed after Oil red O staining. (C) HE staining for detection of thoracic aortic injury area (100×). (D) Lipid deposition in the thoracic aorta observed after Oil red O staining (100×). (E) ELISA detection of TNF-α, IL-1β, IL-6 expression in the serum of ApoE−/− mice fed with HFD. (F) VCAM-1, ICAM-1, and MCP-1 protein expression in mouse arterial tissues measured by Western blot analysis. *p < 0.05 vs. the ApoE−/− mice fed with HFD and injected with lentivirus vector expressing sh-NC; #p < 0.05 vs. the ApoE−/− mice fed with HFD and injected with lentivirus vector expressing sh-HIF1A-AS2. Data (mean ± standard deviation) among multiple groups were analyzed by one-way analysis of variance. n = 20 for mice in each group.
Discussion
Atherosclerosis is thought to be a leading cause of cardiovascular diseases. Even though constant progresses have been made in the treatment of cardiovascular disease, there remains a great need to develop alternative strategies to limit inflammation and other proatherogenic changes in atherosclerosis [22]. LncRNAs have been identified to be regulatory transcripts in various cardiovascular conditions and some related risk factors such as atherosclerosis, CAD, and heart failure [23]. In this study, we revealed a modulatory role of lncRNA HIF1A-AS2 in the inflammation in atherosclerosis. We uncovered that downregulation of lncRNA HIF1A-AS2 inhibited ATF2 by restricting the binding of the USF1 to the ATF2 promoter region, thus suppressing atherosclerotic inflammation.
This study presented evidence indicating that lncRNA HIF1A-AS2 was up-regulated in the ApoE−/− mouse model of atherosclerosis. The dysregulation of lncRNA HIF1A-AS2 in several cancers or pathological conditions has been reported previously. For instance, lncRNA HIF1A-AS2 is upregulated in gastric carcinomas [24], and bladder cancer [25], and furthermore, lncRNA HIF1A-AS2 exerts oncogenic roles in those malignancies. Besides, a recent research has demonstrated that lncRNA HIF1A-AS2 is up-regulated in human umbilical vascular endothelial cells exposed to hypoxia, in which lncRNA HIF1A-AS2 acts as a promoter of angiogenesis [26]. Also, a previous study has suggested that lncRNA HIF1A-AS2 is upregulated in CAD patients [12]. In this study, we found out that lncRNA HIF1A-AS2 was a pathogenetic noncoding RNA in atherosclerosis by promoting inflammation.
Additionally, we provided evidence that downregulation of lncRNA HIF1A-AS2 inhibited ox-LDL-induced inflammation in ECs, SMCs and HCAECs, as shown by the decreased expression of inflammatory factors TNF-α, IL-1β and IL-6 as well as adhesion molecules VCAM-1, ICAM-1, and MCP-1. LncRNAs have been identified to play critical roles in inflammation. For example, downregulation of lncRNA TINCR inhibits the proinflammatory response through restraining the secretion of IL-6, TNF-α and CXCL1 [27]. Consistent with our study, lncRNA HIF1A-AS2 has been recently demonstrated to elevate the expression of the proinflammatory protein IL-6 [28]. VCAM-1 and ICAM-1 are critical cell adhesion molecules, which exert pro-atherosclerotic effects by triggering inflammation [29]. Therapeutically targeting the pro-inflammatory molecules TNF-α and IL-6 as well as adhesion molecules could achieve an anti-atherosclerotic action [30].
Additionally, we demonstrated in the current study that lncRNA HIF1A-AS2 elevated the expression of ATF2 to promote inflammation via binding to the transcription factor USF1. ATF2 was identified to be highly expressed in atherosclerosis in this study. ATF2 has previously been extensively investigated in a variety of developmental and pathological conditions [31]. A recent investigation found that ATF2 was upregulated in a mouse neuroinflammation model [13]. The promotive function of ATF2 in inflammation has also been identified in another study [13]. ATF2 decreases the expression of ATF3, leading to an inflammatory state [32]. Previous research has uncovered that arterial inflammation can be promoted in endothelial cells via c-Jun NH-terminal kinase-ATF2 signaling [33]. The present study also proved that decreasing ATF2 could inhibit the release of inflammatory proteins in atherosclerotic mice. Although the binding of ATF2 and USF1 to β-catenin has been identified in a previous study, their direct interaction had not previously been verified [34]. In our study, the ChIP assay provided evidence that USF1 was enriched in the ATF2 promoter region. Moreover, through RIP and RNA pull-down assays, lncRNA HIF1A-AS2 was proven to bind to USF1. Knockdown of lncRNA HIF1A-AS2 diminished the binding of USF1 to ATF2 promoter, thereby inhibiting ATF2, which was involved in the anti-atherosclerotic action mediated by lncRNA HIF1A-AS2 silencing.
To sum up, we unveiled the proinflammatory role of lncRNA HIF1A-AS2 in atherosclerosis. Downregulating lncRNA HIF1A-AS2 decreased the expression of ATF2 by reducing the binding of the USF1 to the ATF2 promoter region, thereby inhibiting the atherosclerotic inflammation (Fig. 8). Therefore, lncRNA HIF1A-AS2 and ATF2 should be considered as promising therapeutic targets for atherosclerosis. The epigenetic modulation using lncRNAs not only serves as novel targets for the treatment of atherosclerosis, but has also been proven effective in the clinical setting [35]. Nonetheless, the current study only presents a theoretical basis for understanding the lncRNA HIF1A-AS2-mediated mechanism in inflammation of atherosclerosis. Many other biomolecules as well as signaling pathways involved in angiogenesis that is associated with the growth of atherosclerotic plaques remain to be investigated. Furthermore, the role of lncRNA HIF1A-AS2 should be better elucidated in a clinical setting.
Fig. 8.

The map illustrating mechanisms associated with lncRNA HIF1A-AS2-mediated inflammation in the atherosclerosis. The upper panel shows that lncRNA HIF1A-AS2 forms a complex with USF1, which is then recruited into the ATF2 promoter to elevate ATF2 expression, thus promoting the development of atherosclerotic inflammation. The lower panel shows that downregulation of lncRNA HIF1A-AS2 reduces the expression of ATF2 by reducing the binding of USF1 to the ATF2 promoter regions, thereby inhibiting atherosclerotic inflammation, corresponding to decreased inflammatory factors TNF-α, IL-1β and IL-6 in serum and protein levels of adhesion molecules VCAM-1, ICAM-1, and MCP-1, as well as increased cell viability and reduced apoptosis in ox-LDL-induced inflammation in ECs, SMCs, and HCAECs.
Author contributions
Pengcheng Li and Junhui Xing conceived and designed research. Jielei Zhang and Jianwu Jiang performed experiments. Xuemeng Liu analyzed data. Di Zhao interpreted results of experiments. Yanzhou Zhang prepared figures. Pengcheng Li and Junhui Xing drafted manuscript. Jielei Zhang and Jianwu Jiang edited and revised manuscript. All authors approved final version of manuscript.
Funding
None.
Compliance with ethics requirements
All animal experiments were approved by the Animal Ethics Committee of the First Affiliated Hospital of Zhengzhou University and strictly followed the principle of minimizing number of animals used and minimizing the pain and suffering.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2020.07.015.
Contributor Information
Di Zhao, Email: zzz_zhaodi@yeah.net.
Yanzhou Zhang, Email: zhangyanzhoudr@126.com.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
Supplementary Fig. 1.

Flow chart of animal experiments.
References
- 1.Khyzha N., Alizada A., Wilson M.D., Fish J.E. Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med. 2017;23(4):332–347. doi: 10.1016/j.molmed.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Pothineni N.V.K., Subramany S., Kuriakose K., Shirazi L.F., Romeo F., Shah P.K. Infections, atherosclerosis, and coronary heart disease. Eur Heart J. 2017;38(43):3195–3201. doi: 10.1093/eurheartj/ehx362. [DOI] [PubMed] [Google Scholar]
- 3.Dou Y., Chen Y., Zhang X., Xu X., Chen Y., Guo J. Non-proinflammatory and responsive nanoplatforms for targeted treatment of atherosclerosis. Biomaterials. 2017;143:93–108. doi: 10.1016/j.biomaterials.2017.07.035. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z., Salisbury D., Sallam T. Long noncoding RNAs in atherosclerosis: JACC review topic of the week. J Am Coll Cardiol. 2018;72(19):2380–2390. doi: 10.1016/j.jacc.2018.08.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marques-Rocha J.L., Samblas M., Milagro F.I., Bressan J., Martinez J.A., Marti A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015;29(9):3595–3611. doi: 10.1096/fj.14-260323. [DOI] [PubMed] [Google Scholar]
- 6.Uchida S., Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. 2015;116(4):737–750. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 7.Haemmig S., Feinberg M.W. Targeting LncRNAs in cardiovascular disease: options and expeditions. Circ Res. 2017;120(4):620–623. doi: 10.1161/CIRCRESAHA.116.310152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H., Han S., Sun Q., Yao Y., Li S., Yuan C. Long non-coding RNA CDKN2B-AS1 reduces inflammatory response and promotes cholesterol efflux in atherosclerosis by inhibiting ADAM10 expression. Aging (Albany NY) 2019;11(6):1695–1715. doi: 10.18632/aging.101863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shan K., Jiang Q., Wang X.Q., Wang Y.N., Yang H., Yao M.D. Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell Death Dis. 2016;7(6) doi: 10.1038/cddis.2016.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye Z.M., Yang S., Xia Y.P., Hu R.T., Chen S., Li B.W. LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 2019;10(2):138. doi: 10.1038/s41419-019-1409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quan Y., Song K., Zhang Y., Zhu C., Shen Z., Wu S. Roseburia intestinalis-derived flagellin is a negative regulator of intestinal inflammation. Biochem Biophys Res Commun. 2018;501(3):791–799. doi: 10.1016/j.bbrc.2018.05.075. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y., Zhang L., Wang Y., Ding H., Xue S., Yu H. KCNQ1OT1, HIF1A-AS2 and APOA1-AS are promising novel biomarkers for diagnosis of coronary artery disease. Clin Exp Pharmacol Physiol. 2019;46(7):635–642. doi: 10.1111/1440-1681.13094. [DOI] [PubMed] [Google Scholar]
- 13.Li M., Zhang D., Ge X., Zhu X., Zhou Y., Zhang Y. TRAF6-p38/JNK-ATF2 axis promotes microglial inflammatory activation. Exp Cell Res. 2019;376(2):133–148. doi: 10.1016/j.yexcr.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Raghavan S., Singh N.K., Gali S., Mani A.M., Rao G.N. Protein kinase Ctheta via activating transcription factor 2-mediated CD36 expression and foam cell formation of Ly6C(hi) cells contributes to atherosclerosis. Circulation. 2018;138(21):2395–2412. doi: 10.1161/CIRCULATIONAHA.118.034083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y., Lee J., Rhee M.H., Yu T., Baek K.S., Sung N.Y. Molecular mechanism of protopanaxadiol saponin fraction-mediated anti-inflammatory actions. J Ginseng Res. 2015;39(1):61–68. doi: 10.1016/j.jgr.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le D.D., Shimko T.C., Aditham A.K., Keys A.M., Longwell S.A., Orenstein Y. Comprehensive, high-resolution binding energy landscapes reveal context dependencies of transcription factor binding. Proc Natl Acad Sci USA. 2018;115(16):E3702–E3711. doi: 10.1073/pnas.1715888115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laurila P.P., Soronen J., Kooijman S., Forsstrom S., Boon M.R., Surakka I. USF1 deficiency activates brown adipose tissue and improves cardiometabolic health. Sci Transl Med. 2016;8(323):323ra13. doi: 10.1126/scitranslmed.aad0015. [DOI] [PubMed] [Google Scholar]
- 18.Tiruppathi C., Soni D., Wang D.M., Xue J., Singh V., Thippegowda P.B. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol. 2014;15(3):239–247. doi: 10.1038/ni.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gui Y., Yao S., Yan H., Hu L., Yu C., Gao F. A novel small molecule liver X receptor transcriptional regulator, nagilactone B, suppresses atherosclerosis in apoE-deficient mice. Cardiovasc Res. 2016;112(1):502–514. doi: 10.1093/cvr/cvw183. [DOI] [PubMed] [Google Scholar]
- 20.Getz G.S., Reardon C.A. Use of mouse models in atherosclerosis research. Methods Mol Biol. 2015;1339:1–16. doi: 10.1007/978-1-4939-2929-0_1. [DOI] [PubMed] [Google Scholar]
- 21.Li S., Sun W., Zheng H., Tian F. Microrna-145 accelerates the inflammatory reaction through activation of NF-kappaB signaling in atherosclerosis cells and mice. Biomed Pharmacother. 2018;103:851–857. doi: 10.1016/j.biopha.2018.03.173. [DOI] [PubMed] [Google Scholar]
- 22.Moss J.W., Ramji D.P. Nutraceutical therapies for atherosclerosis. Nat Rev Cardiol. 2016;13(9):513–532. doi: 10.1038/nrcardio.2016.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Du W., Yang B. Long non-coding RNAs as new regulators of cardiac electrophysiology and arrhythmias: Molecular mechanisms, therapeutic implications and challenges. Pharmacol Ther. 2019;203:107389. doi: 10.1016/j.pharmthera.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 24.Chen M., Zhuang C., Liu Y., Li J., Dai F., Xia M. Tetracycline-inducible shRNA targeting antisense long non-coding RNA HIF1A-AS2 represses the malignant phenotypes of bladder cancer. Cancer Lett. 2016;376(1):155–164. doi: 10.1016/j.canlet.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 25.Chen X., Liu M., Meng F., Sun B., Jin X., Jia C. The long noncoding RNA HIF1A-AS2 facilitates cisplatin resistance in bladder cancer. J Cell Biochem. 2019;120(1):243–252. doi: 10.1002/jcb.27327. [DOI] [PubMed] [Google Scholar]
- 26.Li L., Wang M., Mei Z., Cao W., Yang Y., Wang Y. lncRNAs HIF1A-AS2 facilitates the up-regulation of HIF-1alpha by sponging to miR-153-3p, whereby promoting angiogenesis in HUVECs in hypoxia. Biomed Pharmacother. 2017;96:165–172. doi: 10.1016/j.biopha.2017.09.113. [DOI] [PubMed] [Google Scholar]
- 27.Fang Y.X., Zou Y., Wang G.T., Huang S.H., Zhou Y.J., Zhou Y.J. lnc TINCR induced by NOD1 mediates inflammatory response in 3T3-L1 adipocytes. Gene. 2019;698:150–156. doi: 10.1016/j.gene.2019.02.047. [DOI] [PubMed] [Google Scholar]
- 28.Wu R., Ruan J., Sun Y., Liu M., Sha Z., Fan C. Long non-coding RNA HIF1A-AS2 facilitates adipose-derived stem cells (ASCs) osteogenic differentiation through miR-665/IL6 axis via PI3K/Akt signaling pathway. Stem Cell Res Ther. 2018;9(1):348. doi: 10.1186/s13287-018-1082-z. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Ling S., Nheu L., Komesaroff P.A. Cell adhesion molecules as pharmaceutical target in atherosclerosis. Mini Rev Med Chem. 2012;12(2):175–183. doi: 10.2174/138955712798995057. [DOI] [PubMed] [Google Scholar]
- 30.Chistiakov D.A., Melnichenko A.A., Grechko A.V., Myasoedova V.A., Orekhov A.N. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp Mol Pathol. 2018;104(2):114–124. doi: 10.1016/j.yexmp.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Watson G., Ronai Z.A., Lau E. ATF2, a paradigm of the multifaceted regulation of transcription factors in biology and disease. Pharmacol Res. 2017;119:347–357. doi: 10.1016/j.phrs.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyata Y., Fukuhara A., Otsuki M., Shimomura I. Expression of activating transcription factor 2 in inflammatory macrophages in obese adipose tissue. Obesity (Silver Spring) 2013;21(4):731–736. doi: 10.1002/oby.20274. [DOI] [PubMed] [Google Scholar]
- 33.Cuhlmann S., Van der Heiden K., Saliba D., Tremoleda J.L., Khalil M., Zakkar M. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: a novel mode of NF-kappaB regulation that promotes arterial inflammation. Circ Res. 2011;108(8):950–959. doi: 10.1161/CIRCRESAHA.110.233841. [DOI] [PubMed] [Google Scholar]
- 34.Hwang J.R., Chou C.L., Medvar B., Knepper M.A., Jung H.J. Identification of beta-catenin-interacting proteins in nuclear fractions of native rat collecting duct cells. Am J Physiol Renal Physiol. 2017;313(1):F30–F46. doi: 10.1152/ajprenal.00054.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skuratovskaia D., Vulf M., Komar A., Kirienkova E., Litvinova L. Promising directions in atherosclerosis treatment based on epigenetic regulation using MicroRNAs and long noncoding RNAs. Biomolecules. 2019;9(6) doi: 10.3390/biom9060226. [DOI] [PMC free article] [PubMed] [Google Scholar]