

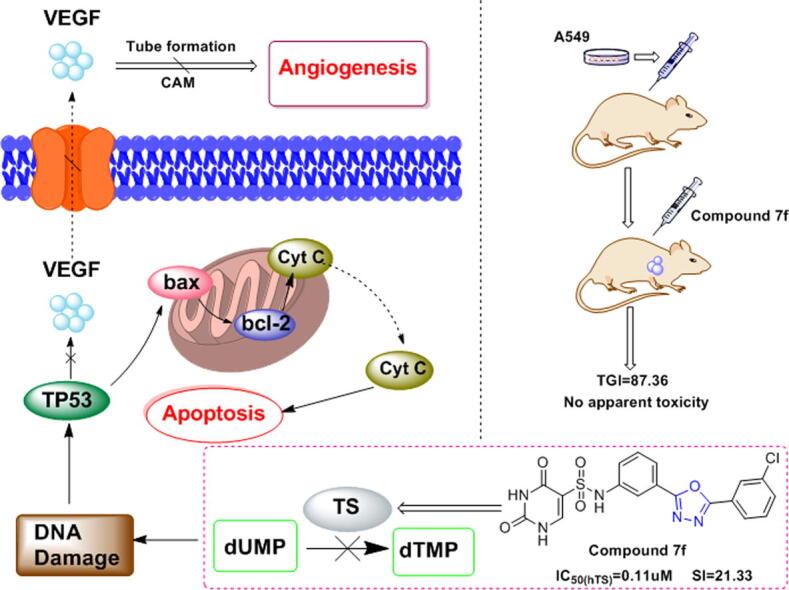
Graphical abstract
Abstract
Introduction
The development of a new type of Thymidylate synthase (TS) inhibitor that could inhibit cancer cells' proliferation and anti-angiogenesis is of great significance for cancer's clinical treatment.
Objectives
Our research hopes to develop a TS inhibitor that is more effective than the current first-line clinical treatment of pemetrexed (PTX) and provide a new reference for the clinical treatment of non-small cell lung cancer (NSCLC).
Methods
We obtained a series of novel TS inhibitors by chemical synthesis. Moreover, TS assay and molecular docking to verify the target compound's inhibitory mode. Use MTT assay, colony-forming assay, flow cytometry, and western blot to verify the compound's inhibitory effect on cancer cell proliferation and its mechanism; and explore the compound’s effect on angiogenesis in vitro and in vivo. Further, explore the hit compound's anti-cancer ability through the xenograft tumor model and the orthotopic cancer murine model.
Results
A series of N-(3-(5-phenyl-1,3,4-oxadiazole-2-yl) phenyl)-2,4-dihydroxypyrimidine-5-sulfamide derivatives were synthesized as TS inhibitors for the first time. All target compounds significantly inhibited hTS enzyme activity and demonstrated significant antitumor activity against five cancer cell lines. Notably, 7f had a high selectivity index (SI) and unique inhibitory effects on eight NSCLC cells. In-depth research indicated that 7f could induce apoptosis by the mitochondrial pathway in A549 and PC-9 cells through the upregulation of wild-type P53 protein expression. Additionally, 7f was shown to inhibit angiogenesis in vitro and in vivo. In vivo studies, compared to PTX, 7f significantly inhibited tumor growth in A549 cell xenografts and had a higher therapeutic index (TGI). Moreover, 7f could prolong the survival of the orthotopic lung cancer murine model more effectively than PTX.
Conclusion
The anti-angiogenic effect of 7f provides a new reference for the development of TS inhibitors and the clinical treatment of NSCLC.
Introduction
Thymidylate synthase (TS) plays a pivotal role in DNA synthesis [1]. Searching from The Cancer Genome Atlas (TCGA) database that the expression of TS is significantly higher in cancer tissues than in cancer-adjacent tissues (Fig. 1A) [2]. TS is the sole rate-limiting enzyme responsible for the de novo synthesis of thymine monophosphate (dTMP) in humans. It catalyzes the conversion of uracil deoxynucleoside monophosphate (dUMP) to methylene dTMP. Phosphorylated dTMP is converted to deoxythymidine triphosphate (dTTP), which is involved in DNA synthesis (Fig. 1B). Therefore, TS expression is significantly increased in sites where DNA replication and metabolism are active, such as the nucleus and mitochondria. Because cancer cells replicate a large amount of DNA, relatively healthy tissues have strong TS expression [1]. When TS activity is inhibited, cancer cell proliferation can be effectively controlled, making it is a classic and practical target of anticancer therapies. Currently, TS inhibitors are broad-spectrum anticancer drugs for clinical treatment.
Fig. 1.

The mechanism and design of TS inhibitors. (A) Search the difference of TS enzyme expression in cancer tissue and healthy tissue from TCGA database; blue was normal tissue; red was cancer tissue (B) Mechanism of action of TS inhibitors (C) Structure optimization of target compounds.
Angiogenesis plays a vital role in the development and progression of cancer. Without a blood supply, tumor cells will die due to ischemia and hypoxia [3]. Therefore, accelerating the re-proliferation of tumor cells during chemotherapy and antiangiogenic therapy can inhibit the repopulation of residual tumor cells, thereby improving the therapeutic effect of chemotherapy drugs and delaying the disease's progression. Tumor cells are capable of releasing angiogenic factors and inducing paravascular angiogenesis [3]. In our previous work, a series of derivatives based on uracil was reported [4], [5]. Among these, the structure of (N-phenyl- (2,4-dihydroxypyrimidine-5-sulfonamido) phenylephrine derivative) had the potential to inhibit VEGF secretion, which in turn could inhibit tumor angiogenesis, this finding that suggests that this bone structure had excellent value (Fig. 1C). Ultimately, our research hope to develop TS inhibitors that are more efficacious than the current clinical first-line treatment pemetrexed (PTX).
Therefore, this study continued to use the lead compound structure for structural optimization (Fig. 1C) to obtain more antitumor TS inhibitors for clinical drug research. Thus, this work mainly focused on modifying the connected moiety of the diphenyl fragment. In previous studies, we obtained a series of novel uracil derivatives with 1,2,3-triazole moieties, and have excellent anticancer activity and enzyme inhibition in vitro. In the subsequent animal experiments, we found that these compounds do not have the significance of continuing in vivo studies due to poor solubility and other reasons. However, from the structure of this type of compound, we found that when the derived fragment has a structure such as a triazole, the overall anticancer effect of the compound will be significantly increased, so we decided to continue to introduce this kind of heterocyclic fragment with similar structure in order to expect to find candidate compounds with excellent antitumor activity and good in vivo effects [6]. In the pharmaceutical field, 1,3,4-oxadiazole and its derivatives are widely used as an indispensable class of heterocyclic compounds [7], [8], [9], [10]. Besides, 1,3,4-oxadiazole heterocycles are strong bioisosteres of amides and esters, contributing substantially to increasing pharmacological activity by participating in hydrogen bonding interactions with the receptors [11], [12]. Therefore, in this study, introduced 1,3,4-oxadiazole groups in this structure.
Here, a series of N-(3-(5-phenyl-1,3,4-oxadiazole-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfamide derivatives as novel TS inhibitors (Fig. 1) was first reported. In vitro, the ability of the target compound to inhibit hTS and its effects on the viability of the five cancer cell lines and corresponding healthy cell lines were evaluated. A structure–activity relationship study screened the hit compound 7f. Moreover, 7f had an excellent inhibitory effect on eight non-small cell lung cancer (NSCLC) cell lines. The flow cytometry and western blot analysis was used to determine the impact of 7f on the cell cycle and the apoptotic pathway. Furthermore, the effect of 7f on tumor angiogenesis was explored in vitro and in vivo. Finally, in vivo pharmacological studies were used to assess the antitumor ability of the 7f on A549 cell xenografts. And study the treatment ability of 7f through the orthotopic lung cancer murine model.
Material and methods
Chemistry.
All raw materials required for chemical synthesis were purchased from Shipek Chemical Co., Ltd. (Zhengzhou, China). The 1H NMR spectra were recorded at 600 MHz and recorded the 13C NMR spectra at 150 MHz on a Varian NMR spectrometer. DMSO‑d6 was used as a solvent to measure all chemical shifts, recorded ESI-MS data on a Finnigan MAT LC-MS. Analytical TLC had performed on silica gel 60 F254 plates (Qingdao Haiyang Chemical Co., Ltd., China) and visualized by UV and potassium permanganate staining. Flash column chromatography was performed on Gel No. 60 (40–63 mm) (Qingdao Haiyang Chemical Co., Ltd., China). The melting point was determined by an electrothermal melting point instrument and was not corrected. High-resolution mass spectrometry (HRMS) was performed using an Agilent Accurate-Mass Q-TOF 6530 (Agilent, Santa Clara, California, USA) in ESI mode.
HPLC analysis of the purity of compounds
An ESSENTIA LC-15C (Shimadzu Corporation, Japan) was used for HPLC detection. A chromatogram (200 mm × 4.6 mm, 5 um) was obtained on a Waters BDS C18 column. The detection was performed at 37℃ and a wavelength of 254 nm. The mobile phase consisted of 0.1% phosphoric acid in water/methanol (50/50, v/v) with a flow rate of 1.0 mL/min. The injection volume was 10 µL. The generated spectrum was loaded into the supplementary file.
Synthesis of 3-nitrobenzohydrazide (4)
To a solution of methyl 3-nitrobenzoate (3) (10 g, 50.72 mmol) in methanol (200 mL), added hydrazine monohydrate (80%) (13.82 g, 220.82 mmol) in the mixture. The reaction mixture was heated and refluxed under an oil-bath in methanol for 6–8 h, and used TLC the monitored reaction. After the reaction, the solvent was removed under vacuum, and compound (4) was obtained as light yellow solid (8.24 g, yield: 82.40%). Intermediate compounds spectral data were included in the supporting information.
Synthesis of benzoyl chloride (2a)
A mixture of benzoic acid (1a) (5.00 g, 40.94 mmol) and thionyl chloride (SOCl2) (20.00 mL) was stirred at 50℃ in an oil-bath and catalyzed with a small amount of DMF. The reaction lasted 3–5 h. After the reaction was completed, the solvent was removed under vacuum, and compound (2a) was obtained as a light yellow liquid (5.19 g, yield: 89.93%). Prepared compound (2b-2 m) was in the same manner.
Synthesis of 2-(3-nitrophenyl)-5-phenyl-1,3,4-oxadiazole (5a)
Under the condition of the ice bath, the solution of CH2Cl2 (30.00 mL) containing the product (2a) (4.50 g, 32.01 mmol) was slowly added to the mixture of CH2Cl2 (150.00 mL) containing compound (4) (5.81 g, 32.01 mmol) and Et3N (9.72 g, 96.04 mmol) in an ice bath. The reaction was stirred at 35℃ in an oil-bath for 8 h, after which methanesulfonyl chloride (TsCl) (5.88 g, 51.32 mmol) was added, and the reaction continued for 8 h. After the reaction was completed, the solvent was removed under vacuum, and the residue was purified by a silica gel column to obtain the pure compound (5a) as a yellow solid (7.05 g, yield: 82.36%). The preparation of the compound (5b-5 m) was carried out in the same manner.
Synthesis of 3-(5-phenyl-1,3,4-oxadiazol-2-yl)aniline (6a)
Next, 80% hydrazine hydrate (2.33 g, 46.55 mmol) and sequentially added 10% Pd/C (20 mg) to a solution of the compound (5a) (6.22 g, 23.27 mmol) in methanol. The reaction mixture was refluxed in an oil-bath at 50℃ for 2–3 h, and TLC monitored the reaction. After the reaction was completed, the solution was filtered to remove Pd/C, and the solvent was removed under vacuum. The compound (6a) was obtained as a pale yellow solid (3.62 g, yield: 65.58%). Prepared the compound (6b-6 m) was in the same manner. Intermediate compounds spectral data were added to the supporting information.
Synthesis of 2,4-dioxo-N-(3-(5-phenyl-1, 3,4-oxadiazol-2-yl) phenyl)-1, 2,3,4-tetrahydropyrimidine-5-sulfuramide (7a)
Here, 2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-sulfonyl chloride (2.65 g, 12.58 mmol) was added to compound (6a) (3.00 g, 12.58 mmol) and pyridine (3.00 g, 37.75 mmol) in DMF (20.00 mL). Stirred the reaction mixture at room temperature for 12 h, and TLC monitored the reaction. After the reaction completed, the liquid was slowly poured into water (150 mL) and precipitated the solid. Adjusted the pH of the solution to 2–3 by adding 10% dilute hydrochloric acid. Filtered the solid and washed thoroughly with water. Added the solid into a 10% NaOH solution, and the filtrate was obtained by filtration. Then adjusted the pH to 2–3 with 10% dilute hydrochloric acid, the solid was filtered and dried, and compound (7a), an off-white solid, was obtained (2.52 g, yield: 48.46%). The preparation of the compound (7b-7 m) was performed in the same manner.
Synthesis of N-(3-(5-phenyl-1,3,4-oxadiazol-2-yl)phenyl)- 2,4-dihydroxypyrimidine-5-sulfuramide (7a)
An off-white solid, 2.52 g, yield: 48.46%. MP: 164.5–168.3℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.97 (s, 1H), 11.65 (s, 1H), 10.57 (s, 1H), 8.18 (s, 1H), 8.08 (d, J = 6.4 Hz, 2H), 7.88 (s, 1H), 7.78 (d, J = 7.5 Hz, 1H), 7.63 (d, J = 6.7 Hz, 3H), 7.53 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.52, 164.14, 159.09, 150.81, 149.10, 139.11, 132.56, 130.86, 129.94(2C), 127.14(2C), 124.57, 123.75, 122.87, 122.24, 117.33, 111.07. ESI-HRMS calcd for C18H12N5O5S [M−H]- 410.0637, found: 410.0614. Purity: 97.24%.
N-(3-(5-(o-tolyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine −5-sulfonamide (7b)
A white solid, 2.16 g, yield: 42.52%. MP: 149.8–154.4℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.92 (s, 1H), 11.65 (s, 1H), 10.58 (s, 1H), 8.17 (s, 1H), 8.01 (d, J = 7.3 Hz, 1H), 7.88 (s, 1H), 7.76 (d, J = 7.3 Hz, 1H), 7.52 (d, J = 4.4 Hz, 2H), 7.48–7.41 (m, 2H), 7.39 (d, J = 7.4 Hz, 1H), 2.67 (s, 3H). 13C NMR (150 MHz, DMSO‑d6) δ 164.79, 163.73, 159.08, 150.82, 149.10, 139.12, 138.15, 132.28, 132.03, 130.87, 129.32, 127.00, 124.61, 122.92, 122.68, 122.14, 117.26, 111.07, 21.97. ESI-HRMS calcd for C19H14N5O5S [M−H]- 424.0794, found: 424.0766. Purity: 97.62%.
N-(3-(5-(m-tolyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7c)
A white solid, 1.91 g, yield: 37.60%. MP: 167.6–171.1℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.91 (s, 1H), 11.65 (s, 1H), 10.56 (s, 1H), 8.18 (s, 1H), 7.91–7.84 (m, 3H), 7.77 (d, J = 7.4 Hz, 1H), 7.55–7.47 (m, 2H), 7.43 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 7.7 Hz, 1H), 2.40 (s, 3H). 13C NMR (150 MHz, DMSO‑d6) δ 164.57, 164.05, 159.08, 150.83, 149.11, 139.39, 139.10, 133.20, 130.83, 129.81, 127.39, 124.58, 124.31, 123.65, 122.79, 122.22, 117.37, 111.08, 21.32. ESI-HRMS calcd for C19H14N5O5S [M−H]- 424.0794, found: 424.0787. Purity: 99.52%.
N-(3-(5-(p-tolyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7d)
A white solid, 2.57 g, yield: 50.59%. MP: 168.1–173.9℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.91 (s, 1H), 11.65 (s, 1H), 10.55 (s, 1H), 8.18 (d, J = 3.9 Hz, 1H), 7.96 (d, J = 8.0 Hz, 2H), 7.87 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.42 (d, J = 7.9 Hz, 2H), 7.38 (d, J = 8.0 Hz, 1H), 2.38 (s, 3H). 13C NMR (150 MHz, DMSO‑d6) δ 164.59, 163.88, 159.08, 150.83, 149.12, 142.72, 139.09, 130.83, 130.46(2C), 127.07(2C), 124.61, 122.76, 122.17, 120.99, 117.28, 111.08, 21.61. ESI-HRMS calcd for C19H14N5O5S [M−H]- 424.0794, found: 424.0783. Purity: 96.56%.
N-(3-(5-(2-chlorophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7e)
A white solid, 2.45 g, yield: 49.80%. MP: 174.5–183.3℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.89 (s, 1H), 11.65 (s, 1H), 10.60 (s, 1H), 8.17 (s, 1H), 8.10 (d, J = 7.6 Hz, 1H), 7.88 (s, 1H), 7.74 (t, J = 8.5 Hz, 2H), 7.66 (t, J = 7.6 Hz, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.53 (t, J = 7.9 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 163.91, 162.13, 158.37, 150.13, 148.33, 138.58, 133.15, 131.72, 131.20, 131.06, 130.27, 127.75, 123.73, 122.41, 122.31, 121.60, 116.87, 110.60. ESI-HRMS calcd for C18H11ClN5O5S [M−H]- 444.0248, found: 444.0230. Purity: 97.04%.
N-(3-(5-(3-chlorophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7f)
A white solid, 2.19 g, yield: 44.51%. MP: 173.5–186.1℃. 1H NMR (600 MHz, DMSO‑d6) δ 12.06 (s, 1H), 11.64 (s, 1H), 10.56 (s, 1H), 8.15 (s, 1H), 8.09 (s, 1H), 8.05 (d, J = 7.6 Hz, 1H), 7.89 (s, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.73 (d, J = 7.4 Hz, 1H), 7.67 (s, 1H), 7.53 (t, J = 7.9 Hz, 1H), 7.40 (d, J = 7.9 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.45, 163.41, 159.09, 150.76, 149.07, 139.11, 134.54, 132.36, 132.02, 130.86, 126.65, 125.85, 125.72, 124.39, 122.97, 122.42, 117.50, 111.03. ESI-HRMS calcd for C18H11ClN5O5S [M−H]- 444.0248, found: 444.0228. Purity: 100.00%.
N-(3-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7 g)
A white solid, 2.25 g, yield: 45.73%. MP: 178.2–184.9℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.90 (s, 1H), 11.65 (s, 1H), 10.56 (s, 1H), 8.19 (s, 1H), 8.08 (s, 2H), 7.87 (s, 1H), 7.73 (d, J = 47.7 Hz, 3H), 7.52 (s, 1H), 7.39 (s, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.29, 163.77, 159.08, 150.84, 149.14, 139.11, 137.26, 130.85, 130.10(2C), 128.91(2C), 124.45, 122.92, 122.65, 122.26, 117.29, 111.09. ESI-HRMS calcd for C18H11ClN5O5S [M−H]- 444.0248, found: 444.0233. Purity: 99.41%.
N-(3-(5-(2-fluorophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7 h)
An off-white solid, 2.34 g, yield: 46.33%. MP: 149.7–159.5℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.92 (s, 1H), 11.65 (s, 1H), 10.60 (s, 1H), 8.17 (s, 1H), 8.11 (t, J = 6.9 Hz, 1H), 7.88 (s, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.70 (d, J = 5.1 Hz, 1H), 7.55–7.48 (m, 2H), 7.45 (t, J = 7.6 Hz, 1H), 7.40 (dd, J = 8.1, 1.1 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.26, 161.19 (d, J = 5.3 Hz), 160.59, 159.08, 150.83, 149.13, 139.15, 134.74 (d, J = 8.6 Hz), 130.91, 130.11, 125.82 (d, J = 3.3 Hz), 124.38, 122.83, 122.16, 117.64 (d, J = 20.4 Hz), 117.32, 112.09 (d, J = 11.5 Hz), 111.05. ESI-HRMS calcd for C18H11FN5O5S [M−H]- 428.0543, found: 428.0523. Purity: 98.18%.
N-(3-(5-(3-fluorophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7i)
An off-white solid, 1.99 g, yield: 39.41%. MP: 173.5–186.2℃. 1H NMR (600 MHz,DMSO‑d6) δ 1H NMR (600 MHz, DMSO) δ 11.89 (s, 1H), 11.65 (s, 1H), 10.56 (s, 1H), 8.19 (d, J = 4.1 Hz, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.90–7.85 (m, 2H), 7.81 (d, J = 7.7 Hz, 1H), 7.68 (dd, J = 13.8, 7.9 Hz, 1H), 7.55–7.48 (m, 2H), 7.39 (dd, J = 8.1, 1.1 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.41, 163.58 (d, J = 3.2 Hz), 161.92, 159.08, 150.83, 149.14, 139.10, 132.34 (d, J = 8.3 Hz), 130.85, 125.79 (d, J = 8.8 Hz), 124.40, 123.42, 122.97, 122.37, 119.51 (d, J = 21.1 Hz), 117.39, 113.93 (d, J = 24.3 Hz), 111.05. ESI-HRMS calcd for C18H11FN5O5S [M−H]- 428.0543, found: 428.0522. Purity: 98.57%.
N-(4-(5-(4-fluorophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7j)
An off-white solid, 2.31 g, yield: 45.74%. MP: 176.8–184.7℃. 1H NMR (600 MHz, DMSO‑d6) δ 12.13 (d, J = 5.1 Hz, 1H), 11.64 (s, 1H), 10.57 (s, 1H), 8.17–8.11 (m, 3H), 7.86 (s, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.50 (dt, J = 17.6, 8.4 Hz, 3H), 7.38 (dd, J = 8.1, 1.4 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 165.46, 164.16, 163.79, 159.10, 150.74, 149.03, 139.11, 130.85, 129.88 (d, J = 9.2 Hz) (2C), 124.50, 122.92, 122.25, 120.45 (d, J = 2.9 Hz), 117.30 (2C), 117.15, 111.06. ESI-HRMS calcd for C18H11FN5O5S [M−H]- 428.0543, found: 428.0522. Purity: 95.05%.
N-(3-(5-(2-bromophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7 k)
An off-white solid, 2.03 g, yield: 43.66%. MP: 130.4–136.5℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.91 (s, 1H), 11.65 (s, 1H), 10.60 (s, 1H), 8.17 (s, 1H), 8.04 (dd, J = 7.7, 1.6 Hz, 1H), 7.90 (dd, J = 9.5, 1.2 Hz, 2H), 7.75 (d, J = 7.8 Hz, 1H), 7.63 (td, J = 7.6, 0.9 Hz, 1H), 7.57 (td, J = 7.8, 1.7 Hz, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.40 (dd, J = 8.2, 1.3 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.54, 163.36, 159.07, 150.83, 149.13, 139.19, 134.90, 133.89, 132.29, 130.96, 128.80, 124.99, 124.34, 122.92, 122.18, 121.29, 117.31, 111.04. ESI-HRMS calcd for C18H11BrN5O5S [M−H]- 489.9649, found: 489.9632. Purity:100.00%.
N-(3-(5-(3-bromophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7 l)
An off-white solid, 1.79 g, yield: 38.49%. MP: 171.2–174.6℃. 1H NMR (600 MHz, DMSO‑d6) δ 12.10 (s, 1H), 11.64 (s, 1H), 10.56 (s, 1H), 8.21 (s, 1H), 8.15 (s, 1H), 8.08 (d, J = 7.8 Hz, 1H), 7.89 (s, 1H), 7.83 (dd, J = 15.5, 7.8 Hz, 2H), 7.59 (t, J = 7.9 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.40 (dd, J = 8.1, 1.2 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.43, 163.28, 159.10, 150.74, 149.03, 139.10, 135.22, 132.18, 130.85, 129.42, 126.18, 125.90, 124.38, 122.97, 122.89, 122.43, 117.54, 111.04. ESI-HRMS calcd for C18H11BrN5O5S [M−H]- 489.9722, found: 489.9637. Purity: 95.09%.
N-(3-(5-(4-bromophenyl)-1,3,4-oxadiazol-2-yl)phenyl)-2,4-dihydroxypyrimidine-5-sulfonamide (7 m)
An off-white solid, 1.64 g, yield: 35.27%. MP: 163.5–167.3℃. 1H NMR (600 MHz, DMSO‑d6) δ 11.85 (s, 1H), 11.64 (s, 1H), 10.56 (s, 1H), 8.19 (s, 1H), 8.01 (d, J = 6.8 Hz, 2H), 7.88–7.82 (m, 3H), 7.78 (d, J = 6.3 Hz, 1H), 7.52 (s, 1H), 7.38 (d, J = 6.5 Hz, 1H). 13C NMR (150 MHz, DMSO‑d6) δ 164.30, 163.90, 159.07, 150.84, 149.15, 139.11, 133.03(2C), 130.86, 129.04(2C), 126.17, 124.45, 123.00, 122.92, 122.26, 117.28, 111.08. ESI-HRMS calcd for C18H11BrN5O5S [M−H]- 489.9722, found: 489.9653. Purity: 100.00%.
Cell culture and MTT assay
All cancer cell lines were obtained from the American Type Culture Collection (ATCC). All healthy cell lines were obtained from the Shanghai Cell Resource Bank. Cultured cells using the culture guidelines provided by the supplier and performed relevant mycoplasma tests once a month.
The cultured cells were collected in the logarithmic growth phase, digested with 0.25% trypsin, and diluted into a single cell suspension with phosphate-buffered saline (PBS). Seeded cells into a 96-well plate at a concentration of 4 × 103 cells per well (100 µL per well), and placed the culture plate in an incubator containing 5% CO2 and cultured at 37℃. After the cells were attached entirely (12 h), discarded the nutrient solution in each well, and 100 μL of different concentrations of the compound (0.625 μM, 1.25 μM, 2.5 μM, 5 μM, and 10 μM) were added to the cells, except for the 0.1% condition. Then added DMSO alone to the cells of the control group, and each concentration to 5 wells. In addition, 5 wells in each 96-well plate received only 100 µL of nutrient solution as a blank control. After incubating the cells with the compound for 24 h, removed the plate and added 20 µL of MTT solution (5 mg/mL) (Cat. No. AR1156, Wuhan Bost Biotechnology Co., Ltd., China) to each well. After 4 h of incubation at 37℃, slowly discarded the nutrient solution from each well, and added 150 µL of DMSO to each well. Furthermore, we then shook the plate on a shaker for 20 min. When the crystals were completely dissolved, a blank well was used to zero the plate reader. The optical density (OD) of each well was detected at 490 nm. The IC50 value was determined as the inhibitor's concentration, which produced 50% inhibition—calculated IC50 values for the compounds using probability units and weighted regression methods. Determined the CC50 value as the concentration of the compound at which 50% of the healthy cells died. The MTT detection method described previously was used.
TS assay
Enzyme activity was measured by ultraviolet spectrophotometry at a wavelength of 340 nm. Recombinant human TS (hTS) was purchased from ProSpec-Tany Company. Temporary buffer containing a mixture of 5 mM DTT, 20 mM MgCl2, 40 mM Tris-HCl, and 75 μM NaEDTA [13] was freshly prepared. Then, 100 μL of dUMP, 200 µL of human TS (hTS) and 100 μL of different concentrations of target compound were added sequentially to 500 μL of buffer. The final reaction system contained 100 μM dUMP, 40 nM human TS, and different concentrations of target compounds (0.625 μM, 1.25 μM, 2.5 μM, 5 μM, and 10 μM). After incubation at 37℃ for 5 min, the reaction was terminated, and UV spectroscopic analysis was carried out at 340 nm by a full-wavelength microplate reader (Multiskan Spectrum, Thermo, USA).
Molecular docking
MOE (Molecular Operating Environment, Version 2016.08; Canadian Chemical Computing Group) software was used to perform molecular modeling simulation on 7f in a TS binding bag. The docking calculation used the crystal structure (PDB code: 1JUJ) in conjunction with TS and PTX [14]. Specific operation procedures and data indicators are included in the supplementary materials.
Colony-forming assay
A549 or PC-9 cells of each group were taken in the logarithmic growth phase to prepare a cell suspension and seeded 10,000 cells in a petri dish. Different concentrations (0 μM, 0.5 μM, 1.0 μM, and 1.5 μM) of 7f were applied to A549 or PC-9 cells at 37℃ for 24 h. Cells were then collected, and 100 cells were inoculated into each petri dish containing culture medium and then cultured generally for 2 weeks. The supernatant was discarded, and fixed cells with 4% paraformaldehyde and stained with Giemsa staining solution for 10 min.
Cell cycle analysis and Annexin V-FITC/propidium iodide (PI) staining
Treated A549 cells and PC-9 cells with 0 μM, 0.5 μM, 1.0 μM, or 1.5 μM 7f for 24 h; then, according to the operating instructions of the kit (CA1020, Solarbio, China), the cells were stained by PI (Solarbio, China) or Annexin V-FITC/PI (Solarbio, China) and analyzed by flow cytometry (Becton Dickinson and Company, USA).
Mitochondrial membrane potential detection (JC-10)
After treated A549 or PC-9 cells with 0 μM, 0.5 μM, 1.0 μM, or 1.5 µM 7f, followed procedures of the mitochondrial membrane potential detection kit (JC-10 assay, CA1310-100, Solarbio, China). The stained cells were then analyzed by flow cytometry to test the cells' mitochondrial membrane potential changes.
Western blot analysis
After 0 μM, 0.5 μM, 1.0 μM, or 1.5 µM 7f treatment of A549 or PC-9 cells, performed western blot analysis according to relevant general operating procedures. Primary antibodies (100 μL/cm2) included those against c-myc (ab185656, Abcam Company, USA), cyclin D (ab16663, Abcam Company, USA), cyclin E (ab33911, Abcam Company, USA), Ki67 (ab16667, Abcam Company), P53 (SC-126, Santa Cruz Company, USA), MDM2 (ab38618, Abcam Company, USA), cleaved caspase-3 (YT6161, Immunoway Company, USA), caspase-3 (YT6113, Immunoway Company, USA), bax (50599–2-lg, Proteintech Company, USA), VEGFR-2 (#9698, Cell Signaling Technology, USA), β-actin (60012–1-lg, Proteintech Company, USA), bcl-2 (12789–1-AP, Proteintech Company, USA), VDCA-1 (ab154856, Abcam Company, USA), p-VEGFR-2 (#3770, Cell Signaling Technology, USA), and Cytochrome C (ab133504, Abcam Company, USA).
VEGF secreted content detection
Collected supernatants of A549 or PC-9 cells after treatment with different concentrations (0 μM, 0.5 μM, 1.0 μM or 1.5 µM) of 7f and concentrated with an ultrafiltration concentrator. Then, 1 mL of cell supernatant at each concentration was collected and concentrated to 200 μL. Experimental procedures were then followed according to the ELISA kit protocol (ab222510, Abcam Company, USA).
Chick embryo chorioallantoic membrane (CAM) assay
After treated A549 cells with different concentrations (0 μM, 0.5 μM, 1.0 μM, or 1.5 µM) of 7f, 1 mL of cell supernatant at each concentration was collected and concentrated to 200 μL. A window was carefully opened on the broad surface of a 3-day-old eggshell (SPF eggs) and sealed with a transparent film. The sealed eggs were cultured in an incubator for 5 days. On day 6, the concentrated cell supernatant was added dropwise from the exposed window to the embryo. CAM measurements were then analyzed over 4 days [15], determined the angiogenesis index by calculating the average number of microvessels visible in the defined area.
Tube formation
After A549 cells were treated with different concentrations (0 μM, 0.5 μM, 1.0 μM, or 1.5 μM) of 7f, 100 μL of cell supernatant of each concentration was collected. Seeded HUVECs (1 × 104 cells) on Matrigel (#354234, Corning, USA) surfaces and cultured for 12 h with or without A549 cell supernatant [16]. The formation of tubes was then observed and photographed with a microscope.
Tumor xenograft model
Five-week-old immunodeficient BALB/c female nude mice weighing 20 g to 22 g were purchased and maintained under specific pathogen-free (SPF) conditions. Then, 2 × 107 A549 cells were implanted subcutaneously in nude mice (n = 5), with 200 μL of PBS as the vehicle. Tumor volume was then measured every 3 days. For a total of 3 weeks, mice in each group received a weekly intravenous injection of either 200 μL of PBS, 7f (80 mg/kg) with PBS as a vehicle, or 200 μL of PBS as a vehicle for PTX (80 mg/kg) [17]. After treatment, mice were euthanized, and the xenograft tumors were dissected.
The tumor growth inhibition value (TGI) = (1-tumor weight in treatment group/tumor weight in control group) * 100%.
Immunohistochemistry
Sections from formalin-fixed and paraffin-embedded rat lung tissue were dewaxed by incubating at 65℃ overnight and then rehydrating in continuous xylene and ethanol rinses. After incubation with hydrogen peroxide, the slides were washed with PBS and then incubated with 0.4% Triton X-100. After washing with PBS, the sections were incubated with a blocking solution (Dako Protein Block, Dako, Glostrup, Denmark) for 30 min at 37℃. The sections were incubated with primary antibodies (anti-Ki67 and anti-cleaved caspase3, both from CST, diluted 1:200) at 4℃ overnight, and washed several times with PBS and then with the corresponding biotinylated secondary antibody (diluted at 1:500). Sections were then rinsed with PBS multiple times. After adding the avidin–biotin complex (Vector Laboratories, USA), diaminobenzidine (DAB) detection reagent (Enzo Life Sciences, USA) was used to visualize the slices fixed using fixative (Vector Laboratories, Burlingame, USA).
Toxicity test
Five-week-old ICR mice received vehicle (200 μL of PBS) daily, 7f (80 mg/kg) or PTX (80 mg/kg) weekly for 3 weeks (n = 5). Blood was collected from euthanized mice by cardiac puncture under deep isoflurane-induced anesthesia. After performed blood coagulation at 4℃, serum was collected by centrifugation at 12000 rpm for 20 min at 4℃. An automated biochemical analyzer was used (Chemray 240, Rayto, China) to analyze ALT, AST, Glu, and CRE levels (ALT: serum alanine aminotransferase; AST: aspartate aminotransferase; Glu: blood glucose; CRE: urine creatinine).
Orthotopic lung cancer murine model and prognosis experiment
Five-week-old ICR mice (n = 10, per group) were given a tail vein injection of 1 × 106 Lewis lung cancer (LLC) cells [18], [19]. After three weeks of regular feeding under SPF conditions, 200 μL of PBS, 80 mg/kg PTX, or 80 mg/kg 7f was injected intraperitoneally weekly for three consecutive weeks. Survival days of the mice from the first day after injection was recorded and used to draw the survival curve.
Statistical analysis
Data were analyzed using GraphPad Prism 5 from GraphPad Software (La Jolla, CA). All grayscale analysis was performed using ImageJ from ImageJ software (National Institutes of Health, USA). A t-test was used to measure the normal distribution of the two groups, and one-way analysis of variance (ANOVA) was used to analyze comparisons between the groups. Numerical data are expressed as a ratio or percentage. P < 0.05 was considered statistically significant.
Results and discussion
Synthesis and verification of target compounds
Carried out the chemical synthesis of 2,4-dioxo-N-(3- (5-phenyl-1,3,4-oxadiazol-2-yl) phenyl)-1,2,3,4-tetrahydropyrimidine-5- sulfonamide derivative (7a-7 m), as shown in Scheme 1.
Scheme 1.

Reagents and conditions: (I) SOCl2, DMF, 50℃, 3–5 h; (II) N2H4·H2O, CH3OH, 75℃, 8 h; (III) Et3N, CH2Cl2, 35℃,8h; MsCl, 8 h. (IV) Pd/C, N2H4·H2O, C2H5OH, 50℃, 2–4 h; (V) C5H5N, DMF, 37℃, 12 h.
Briefly, various substituted benzoic acids (1a-1 m) were reacted with thionyl chloride to obtain intermediates (2a-2 m) [20]. The commercially available methyl 3-nitrobenzoate (3) and hydrazine monohydrate were refluxed in methanol to obtain the corresponding compound (4) [4], [21]. Performed One-pot synthesis of critical intermediates (5a-5 m) from compounds (2a-2 m) and 4, and there are many ways to synthesis the oxadiazoles in the synthesis reaction [22], [23]. The methane sulfonyl chloride (MsCl) was selected to carry out the ring closure [24], [25], which ensures high yield and is critical to one-pot synthesis. In methanol, the nitro group of compounds (5a-5 m) was reduced to an amino group using hydrazine hydrate and a 10% Pd/C catalyst. The method has high yield and convenient operation and is suitable for large-scale production [26].
The 2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-sulfonyl chloride and compound (6a-6 m) underwent the Hinsberg reaction under alkaline conditions, and the target compounds (7a-7 m) were obtained [27], [28]. Preparation of the critical intermediate 2,4-dihydroxypyrimidine-5-sulfonyl chloride is described in detail in our previous research [4], [5].
Characterized Compounds using 1H NMR, 13C NMR, and HRMS, and reverse-phase HPLC was used to determine their purity.
Preliminary assessment of compound biological activity and drug selectivity
This study used a TS assay to screen the inhibitory effects of all target compounds (7a-7 m) on TS enzymes. The results of the inhibition assay, shown as IC50 values, are summarized in Table 1. Pemetrexed (PTX) was used as a positive control to examine the inhibitory effect of the compound on the cell viability of five cancer cell lines (A549, OVCAR-3, SGC7901, MCF-7, and HepG2) at the cellular level (evaluated by MTT assay), and used IC50 values to indicate the inhibitory ability of the compound on cancer cells. The same method was used to determine the compounds' inhibitory effects on five healthy cell lines (HPAEpiC, IOSE80, GES-1, MCF10A, and LO2). Expressed results as CC50 values for determining the toxicity of the compound in healthy cells. All results are shown as IC50 or CC50 values and recorded in Table 1 and Table 2, respectively.
Table 1.
The IC50 value of compounds (7a-7 m) and PTX against hTS enzyme and five cancer cell lines.
| Compounds | IC50a(µM) |
|||||
|---|---|---|---|---|---|---|
| hTS | A549b | OVCAR-3c | SGC7901d | MCF-7e | HepG2f | |
| 7a | 3.15 ± 0.61 | 7.80 ± 0.22 | 6.03 ± 0.47 | >10 | 7.38 ± 0.49 | >10 |
| 7b | 2.75 ± 0.29 | 5.33 ± 0.14 | 5.91 ± 0.94 | 7.00 ± 0.28 | 5.24 ± 0.73 | 5.78 ± 0.55 |
| 7c | 1.46 ± 0.37 | 3.23 ± 0.11 | 5.06 ± 0.22 | 5.63 ± 0.07 | 5.11 ± 1.27 | 3.43 ± 0.21 |
| 7d | 1.79 ± 0.12 | 5.73 ± 0.10 | 5.63 ± 0.07 | 6.71 ± 0.10 | 5.74 ± 0.81 | 4.71 ± 0.59 |
| 7e | 0.65 ± 0.43 | 1.37 ± 0.12 | 2.47 ± 0.26 | 2.61 ± 0.17 | 2.49 ± 0.04 | 3.61 ± 0.22 |
| 7f | 0.11 ± 0.03 | 0.78 ± 0.07 | 1.09 ± 0.18 | 1.26 ± 0.17 | 1.55 ± 0.06 | 2.06 ± 0.31 |
| 7 g | 0.44 ± 0.10 | 1.08 ± 0.12 | 1.66 ± 0.21 | 1.65 ± 0.11 | 1.93 ± 0.17 | 2.75 ± 0.15 |
| 7 h | 5.18 ± 0.73 | 7.35 ± 0.19 | 9.36 ± 0.14 | 9.77 ± 0.19 | 8.24 ± 1.27 | >10 |
| 7i | 4.61 ± 1.10 | 6.12 ± 0.61 | 8.03 ± 0.39 | 8.17 ± 0.96 | 8.11 ± 1.03 | 8.24 ± 1.27 |
| 7j | 5.61 ± 0.60 | 9.66 ± 1.02 | 8.75 ± 0.49 | 9.84 ± 0.63 | 9.84 ± 0.63 | >10 |
| 7 k | 1.16 ± 0.37 | 1.91 ± 0.08 | 4.02 ± 0.85 | 4.28 ± 0.62 | 4.15 ± 0.32 | 5.15 ± 0.20 |
| 7 l | 0.93 ± 0.28 | 1.15 ± 0.08 | 2.86 ± 0.87 | 2.47 ± 0.09 | 2.29 ± 0.55 | 4.38 ± 0.43 |
| 7 m | 1.29 ± 0.12 | 1.87 ± 0.12 | 3.74 ± 0.16 | 4.60 ± 0.16 | 5.57 ± 0.48 | 4.96 ± 0.59 |
| PTX | 2.44 ± 0.88 | 3.29 ± 0.15 | 5.35 ± 0.60 | 6.96 ± 0.43 | 4.18 ± 0.14 | 3.73 ± 0.31 |
a: IC50 = Mean ± SD, after 24 h of treatment. The values were average from at least 3 independent experiments.
b: Human lung cancer cell lines.
c: Human ovarian cancer cell lines.
d: Human gastric cancer cell lines.
e: Human breast cancer cell lines.
f: Human liver cancer cell lines.
Table 2.
The CC50 value of compounds (7a-7 m) and PTX against HPAEpiC, IOSE80, GES-1, MCF10A and LO2 cell lines.
| Compounds | CC50 (µM)g |
||||
|---|---|---|---|---|---|
| HPAEpiCh | IOSE80i | GES-1j | MCF10Ak | LO2l | |
| 7a | 35.74 ± 0.32 | >100 | >100 | 57.13 ± 1.32 | >100 |
| 7b | 40.44 ± 0.47 | 37.14 ± 0.75 | 33.83 ± 0.85 | 23.97 ± 0.36 | >100 |
| 7c | 42.06 ± 0.58 | 25.16 ± 0.65 | 17.52 ± 0.54 | 25.03 ± 0.28 | 16.94 ± 0.18 |
| 7d | 24.44 ± 1.20 | 20.36 ± 0.53 | 23.64 ± 0.70 | 35.90 ± 0.09 | 20.61 ± 0.72 |
| 7e | 20.87 ± 0.95 | 18.94 ± 0.42 | 16.29 ± 0.47 | 14.52 ± 0.95 | 18.22 ± 0.57 |
| 7f | 16.62 ± 0.65 | 15.42 ± 0.53 | 22.60 ± 0.48 | 20.31 ± 0.71 | 20.11 ± 0.44 |
| 7 g | 19.28 ± 0.95 | 18.72 ± 0.31 | 24.11 ± 0.45 | 26.97 ± 1.20 | 19.27 ± 0.51 |
| 7 h | 56.01 ± 2.33 | 71.52 ± 2.19 | >100 | >100 | – |
| 7i | – | – | 97.82 ± 2.93 | 73.56 ± 1.75 | – |
| 7j | 50.01 ± 2.12 | >100 | – | – | – |
| 7 k | 13.64 ± 0.32 | 11.91 ± 0.23 | 12.21 ± 0.33 | 10.18 ± 0.34 | 15.60 ± 0.39 |
| 7 l | 14.22 ± 0.24 | 9.29 ± 0.12 | 6.71 ± 0.09 | 5.71 ± 0.21 | 7.22 ± 0.49 |
| 7 m | 14.50 ± 0.25 | 12.30 ± 0.19 | 12.91 ± 0.54 | 5.24 ± 0.36 | 10.36 ± 0.38 |
| PTX | 26.44 ± 1.10 | 22.69 ± 0.95 | 21.32 ± 0.88 | 30.10 ± 0.56 | 31.75 ± 1.23 |
g: CC50 = Mean ± SD (after 24 h of treatment). The values were average from at least 3 independent experiments.
h: Human alveolar epithelial cell lines.
i: Human normal ovarian epithelial cell lines.
j: Human gastric mucosal cell lines.
k: Human normal breast epithelial cell lines.
l: Human normal liver cell lines.
The drug selection index (SI) represents a safe range for evaluating and judging drug effects. The SI value is CC50/IC50. The larger the index is, the more extensive the safety range. SI values of all compounds are summarized in Table 3. Overall, all compounds had the highest drug selectivity for lung cancer. The SI value of compounds 7f and 7 g were higher than that of 10 other compounds for the five cancer cell lines. Among them, compound 7f had the highest SI value for these five cancer cell lines.
Table 3.
Selectivity index (SI) of compound 7f, and PTX.
| Compounds | SIm |
||||
|---|---|---|---|---|---|
| HPAEpiC | IOSE80 | GES-1 | MCF10A | LO2 | |
| 7a | 4.582051 | – | – | 7.741192 | – |
| 7b | 7.587242 | 6.284264 | 4.832857 | 4.574427 | – |
| 7c | 13.02167 | 4.972332 | 3.111901 | 4.898239 | 4.938776 |
| 7d | 4.265271 | 3.616341 | 3.5231 | 6.254355 | 4.375796 |
| 7e | 15.23358 | 7.668016 | 6.241379 | 5.831325 | 5.047091 |
| 7f | 21.30769 | 14.14679 | 17.93651 | 13.10323 | 9.762136 |
| 7 g | 17.85185 | 11.27711 | 14.61212 | 13.97409 | 7.007273 |
| 7 h | 7.620408 | 7.641026 | – | – | – |
| 7i | – | – | 11.97307 | 9.070284 | – |
| 7j | 5.177019 | – | – | – | – |
| 7 k | 7.141361 | 2.962687 | 2.852804 | 2.453012 | 3.029126 |
| 7 l | 12.36522 | 3.248252 | 2.716599 | 2.49345 | 1.648402 |
| 7 m | 7.754011 | 3.28877 | 2.806522 | 0.940754 | 2.08871 |
| PTX | 8.036474 | 4.241121 | 3.089855 | 7.200957 | 8.512064 |
m: SI = CC50/IC50.
To sum up, the synthesized compounds demonstrated robust inhibitory ability against hTS activity as well as the activity of five cancer cell types in this study. Overall, the inhibitory effect of compounds on healthy cells was lower than that on cancer cells. This result clarified that the optimized compounds could contribute to an in-depth discussion of the structure–activity relationship.
Structure-activity relationship (SAR) study and molecular docking of target compounds
The next research is to use SAR studies to discuss the relationship between the structure and activity of the compounds to obtain hit compounds. This analysis found that when an F atom replaced R, the compound's anti-proliferative ability was reduced, and the F atom may be more hydrophobic than other groups. Besides, a Br group also reduced inhibitory activity, potentially indicating that the hydrophobic pocket may not be sufficient to accommodate the Br group, while the Cl group was well tolerated. As summarized in Table 2, when the R substituent was a Br atom, these compounds demonstrated relatively high cytotoxicity to healthy cells compared to that with other substituents.
Using compound 7f, which was the most potent TS enzyme inhibitor, the possible interaction between optimized 7f and hTS was predicted as an example. 1JUJ is a human thymidylate synthase that combines dUMP and PTX. Because PTX is an antifolate based on pyrrolo (2,3-d) pyrimidine, this study chose 1JUJ as the docking molecule and used MOE to perform molecular simulation of 7f and PTX in hTS binding pockets. According to the docking results, when the phenyl-sulfonamide bonded group is in the Meta position, it fits well in the active site. A meta-linked uracil fragment forms three powerful H bonds with Ala312 and Asp218 residues, and a phenyl-sulfonamide-bonded group is conjugated with Ile108 to form an H-aromatic hydrocarbon. Concurrently, the 1,3,4-oxadiazole group and Leu221 also form H-aromatic conjugates. Compound 7f has an alternating π-π stacking interaction between Phe225 and the terminal benzyl ring to maintain binding affinity (Fig. 2A). This mode of interaction explains the difference in TS inhibitory activity between 7f and PTX.
Fig. 2.

Compound 7f had a more inhibitory effect on NSCLC than PTX. (A) The docking posture of PTX and compound 7f with TS (PDB ID: 1JUJ): The green structure was PTX, while the yellow was compound 7f. (B): a. and b. The effect of compound 7f and PTX on hTS enzyme activity or the proliferation of A549 cells was compared at each concentration point. c. The inhibitory effects of compounds 7f and PTX on the cell viability of five cancer cells at each concentration point. d. Comparison of drug selectivity index of compound 7f and PTX against five healthy cells (where *p < 0.05, **p < 0.01 and ***p < 0.005 compared to control). (C) Inhibition of cell viability of eight NSCLC cells by compound 7f at different concentrations. (D) Colony-forming assay shows compound 7f could inhibit A549 or PC-9 cell proliferation.
Furthermore, 7f had a more exceptional ability to inhibit hTS enzyme activity and five cancer cell lines activities than PTX at each concentration (Fig. 2B), especially in A549 cells. Also, the inhibitory effect of 7f on eight types of NSCLC cells was investigated. These results showed that 7f inhibited the viability of these eight NSCLC cells, with the most significant effect observed in A549 and PC-9 cells (Fig. 2C). Further colony-forming assay results showed that 7f effectively inhibited A549 or PC-9 cells' proliferation in a concentration-dependent manner (Fig. 2D). In general, compound 7f had excellent research potential as a hit compound.
Effect of compound 7f on the cell cycle and induction of A549 cells and PC-9 cells analyzed by flow cytometry
The cell cycle is divided into three phases: the pre-DNA synthesis phase (G0/G1 phase), the DNA synthesis phase (S phase), and the late DNA synthesis phase (G2/M phase). When a drug inhibits TS enzyme activity, it affects the transition from the G1 phase to the S phase. Inhibition of the cell cycle process eventually leads to blockage of DNA synthesis and induces cells to move toward apoptosis. Investigate whether 7f could induce cell proliferation inhibition, used different concentrations of 7f to treat A549 cells or PC-9 cells for 24 h. Different staining methods were then used to analyze the cell cycle and apoptosis by flow cytometry.
The results showed that the percentage of cells in the G0/G1 phase increased in a concentration-dependent manner. The opposite was exact for cells in S and G2/M phase (Fig. 3A and B). In general, compound 7f could directly inhibit the proliferation of A549 and PC-9 cells by causing cell cycle arrest. PI staining results show that with the increase of 7f concentration, the percentage of cells in the G0/G1 phase increases, while S and G2/M are opposite (Fig. 3A.a and B.a). Annexin V-FITC/PI staining results showed that as the 7f concentration increased, the apoptotic cell ratio increased significantly (Fig. 3A.b and B.b).
Fig. 3.

Flow cytometry analysis of the inhibitory effect of compound 7f on A549 and PC-9 cells. (A) and (B): a. PI staining showed that 7f blocks the cycle of A549 or PC-9 cells. (A) and (B): b. Annexin V-FITC/PI dual staining showed that compound 7f induced apoptosis in A549 or PC-9 cells. (A) and (B): c. JC-10 staining demonstrated the effect of compound 7f on A549 or PC-9 cell mitochondrial membrane potential. Three independent experiments were performed, each column represented the mean ± SD of triplicate determinations (where *p < 0.05, **p < 0.01 and ***p < 0.005 compared to control).
Effect of compound 7f on the mitochondrial membrane potential of A549 cells and PC-9 cells was identified by JC-10 staining
Previous studies have shown that TS is also highly expressed in tumor cell mitochondria (1). In addition to energizing cells, mitochondria are involved in cell differentiation, cell signaling, and apoptosis and regulate cell growth and the cell cycle.
To elucidate whether apoptosis induced by compound 7f was associated with intrinsic mitochondrial apoptosis, JC-10 staining was used to detect the intracellular mitochondrial membrane potential. JC-10 is a fluorescent probe that can rapidly and sensitively detect changes in cell mitochondrial membrane potential (△Ψm) and can be used for apoptosis early. Flow cytometry results showed that △Ψm changes in A549 and PC-9 cells were changed in a concentration-dependent manner (Fig. 3A.c and B.c). This result indicated that 7f could cause a significant change in mitochondrial membrane potential, and concentration-dependent change was concentration-dependent.
The effect of compound 7f on cell cycle-related proteins of A549 cells and PC-9 cells was analyzed by western blot
The c-myc protein, cyclin E protein, cyclin D1 protein, and P53 protein are critical proteins in the G1/S checkpoint. The biological function of wild-type P53 (TP53) is similar to that of a “genomic protector.” If DNA is damaged, TP53 prevents DNA replication and arrests the cell cycle in the G1 phase, providing sufficient time to repair the damaged DNA. If the repair fails, TP53 then induces apoptosis. Fig. 4A showed that the TP53 protein was overexpressed in both A549 cells and PC-9 cells, while the expression of the mutant P53 (MP53) protein was not detectable in PC-9 cells. Additionally, 7f was able to downregulate the c-myc protein, cyclin E protein, and cyclin D1 protein expression [29]. However, although intracellular MDM2 protein content was significantly downregulated at high concentrations, there was no significant change compared to that of TP53.
Fig. 4.

Compound 7f could cause cycle arrest and induces apoptosis in the mitochondrial pathway in A549 or PC-9 cells. (A) a. Effect of compound 7f on cell cycle checkpoint associated proteins expression in A549 or PC-9 cells. b. Effects of compound 7f on MDM2 protein expression in A549 or PC-9 cells. (B) Effect of compound 7f on the apoptotic protein expression in A549 or PC-9 cells. (C) Compound 7f could enhance mitochondrial release of Cyto C and induce apoptosis (where *p < 0.05, **p < 0.01 and ***p < 0.005 compared to control).
In summary, 7f could upregulate the P53 protein in A549 and PC-9 cells in a concentration-dependent manner, preventing the transition of the cell cycle from G1 to S phase. The results were significantly different (P < 0.001).
The effect of compound 7f on mitochondrial apoptotic pathway-related proteins in A549 cells and PC-9 cells was analyzed by western blot
Western blot analysis was used to verify, at the protein level, that 7f activated the mitochondrial apoptotic pathway. As shown in Fig. 4B, as the concentration of the 7f increased, apoptosis-related protein expression was activated. Moreover, based on the separation of the cytoplasm and mitochondria, the relative content of cytochrome c (Cyto C) protein in the cytoplasm or mitochondria was detected. We isolated and purified mitochondria from cancer cells while retaining the cytoplasm. Protein extraction was performed separately, with β-actin used as the internal reference for cytoplasmic proteins and voltage-dependent anion-selective channel 1(VDAC-1) used as an internal reference for mitochondrial proteins for analysis by western blot. VDCA-1 forms ion channels in the outer mitochondrial membrane (OMM) and cell membrane and is a scaffold for mitochondrial proteins. The results showed that Cyto C expression was significantly increased in the cytoplasm and decreased significantly in the mitochondria, which was in a concentration-dependent manner (Fig. 4C).
Effects of compound 7f on angiogenesis in vitro and in vivo
Angiogenesis plays an essential role in the development and progression of non-small cell lung cancer [2], [3]. Vascular endothelial growth factor (VEGF) can stimulate angiogenesis through human umbilical vein endothelial cell (HUVEC) proliferation or vascular endothelial growth factor receptor 2 (VEGFR2) phosphorylation. Prior studies have shown that TP53 is negatively correlated with VEGF expression [30]. Simultaneously, inhibiting tumor growth and limiting blood supply can enhance the effectiveness of drugs in treating tumors.
As described in the above studies, 7f could activate the expression of TP53. Therefore, this study used the ELISA to analyze VEGF levels secreted by A549 and PC-9 cells in vitro, and co-cultured HUVECs with cancer cell supernatants. We studied the effect of cancer cell supernatant on the chick chorioallantoic membrane (CAM) model in vivo. As shown in Fig. 5A, 7f could inhibit the secretion of VEGF by cancer cells and inhibited tube formation in vitro by inhibiting the phosphorylation of VEGFR2 in HUVECs (Fig. 5B). This effect was concentration-dependent. Similarly, the in vivo model results also demonstrated that the level of angiogenesis decreased with increasing concentrations of 7f (Fig. 5C). These results demonstrated that 7f was capable of inhibiting tumor angiogenesis.
Fig. 5.

Compound 7f could inhibit angiogenesis in vitro and in vivo. (A) ELISA determined the content of VEGF in the tumor supernatant. (B) The effect of tumor supernatant on the formation of the tube of HUVECs in vitro at different drug concentrations. (C) Effect of 7f on tumor supernatant-induced angiogenesis assessed on the CAM model. The bar graphs represent the angiogenic index by counting vessel branch points using Image-Pro Plus software in a double-blinded manner. Data represent the mean of their replicates (where *p < 0.05, **p < 0.01 and ***p < 0.005 compared to control).
Effect of compound 7f on tumor growth in an A549 tumor xenograft model
The combined results of the above studies in vitro demonstrated that the 7f had a significant effect on inhibiting cell proliferation and that the SI was exceptionally high. However, this study ultimately hopes to gain a better TS inhibitor than PTX. Therefore, this study further studied the antitumor effects of 7f and PTX in the A549 tumor xenograft model. Ki67 is a nuclear protein encoded by the MKI-67 gene and is often used in pathological immunohistochemistry to indicate the degree of cell proliferation activity. The tumor growth inhibition value (TGI) is used to evaluate the inhibitory effect of test drugs on tumor growth in vivo. The results showed that 7f could conspicuously inhibit the proliferation of A549 xenografts and had little impact on body weight (Fig. 6A). The TGI of the PTX was 60.47 at a dose of 80 mg/kg, while the TGI of the 7f was as high as 87.36 at the same dose. IHC analysis results showed that the expression of Ki67 was decreased by treatment with 7f (Fig. 6B). Conversely, the expression of cleaved-caspase-3 in tumors was increased considerably. In addition, the same quality of the tumor tissue was analyzed by western blot. As a result, the expression of Ki67 was significantly decreased, and the expression of cleaved-caspase-3 was increased, as shown in Fig. 6D.
Fig. 6.

Compound 7f could inhibit the growth of xenograft tumors and LLC transplanted lung cancers. (A) Effects of the 7f on body weight changes in nude mice. (B) The xenograft volume changes and TGI value in PTX- or 7f-treated nude mice (n = 5). (C) IHC analyses evaluate the expression of cleaved-caspase-3 and Ki67 in the tumors from PTX- or 7f-treated nude mice. (D) Western blot was used to analyze the protein expression of cleaved caspase-3 and Ki67 in tumors. (E) Treatment with 7f prolongs survival in mice with implanted LLC cells. Treatment with 7f prolonged survival relative to treatment with PBS or PTX (P < 0.005). (F) Mechanism of the 7f in inducing apoptosis and inhibiting angiogenesis in the NSCLC. Where *p < 0.05, **p < 0.01 and ***p < 0.005 compared to control.
Effect of compound 7f on orthotopic lung cancer model in mice.
This study performed biochemical blood tests on mice injected with PBS, PTX, or 7f for three consecutive weeks to evaluate the compound's toxicity. Concurrently, the research established an orthotopic lung cancer murine model in situ by injecting Lewis lung cancer (LLC) cells through the tail vein. After three weeks of administration, observed the survival of the LLC model mice and the mice survival analyses were performed. As summarized in Table 4, the 7f could prolong LLC-implanted orthotopic lung cancer mice's survival more effectively than PTX (p = 0.0037) (Fig. 6E). Besides, there was no significant hepatotoxicity or renal toxicity.
Table 4.
Results of biochemical tests and mice survival days’ analysis of compound 7f and PTX.
| Items | Control | PTX | 7f | Normal Range |
|---|---|---|---|---|
| ALT(U/L) | 65.53 ± 25.3 | 49.11 ± 17.2 | 62.14 ± 9.5 | 33.70 ~ 98.70 |
| AST(U/L) | 139.65 ± 24.20 | 151.16 ± 21.10 | 103.10 ± 18.60 | 69.70 ~ 322.90 |
| Glu (mmol/L) | 5.19 ± 1.27 | 6.99 ± 0.98 | 7.01 ± 1.02 | 2.65 ~ 7.62 |
| CRE(μmol/L) | 22.47 ± 2.36 | 17.02 ± 5.36 | 15.35 ± 2.10 | 19.43 ~ 64.97 |
| Median survival (Days) | 15.5 | 39 | 45.5 | – |
| P value | – | 0.0037** | ||
Conclusion
In summation, this study designed and synthesized a novel TS inhibitor. The hit compound 7f was more effective than PTX and had a higher drug selectivity index. The compound 7f could promote the expression of wild-type P53 protein in NSCLC cells with P53 mutations, thereby activating apoptosis in the mitochondrial pathway. Simultaneously, 7f could effectively inhibit tumor proliferation and angiogenesis in vivo and in vitro. Moreover, the 7f could prolong the survival rate in the orthotopic lung murine cancer model and had lower toxicity than PTX. The effectiveness of the hit compound 7f and its antiangiogenesis effect provide a new reference for the development of TS inhibitors and the clinical treatment of NSCLC.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No. 81573687), the project of Liaoning distinguished professor, and Key Research and Development Project of Liaoning.
Compliance with Ethics Requirements
All animal studies were conducted with the approval of the Laboratory Animal Welfare and Ethical Committee of China Medical University (No. CMU-2018127).
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2020.07.008.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Chen D., Jansson A., Sim D., Larsson A., Nordlund P. Structural analyses of human thymidylate synthase reveal a site that may control conformational switching between active and inactive states. J Biol Chem. 2017;292(32):13449–13458. doi: 10.1074/jbc.M117.787267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chandrashekar D.S., Bashel B., Balasubramanya S.A.H., Creighton C.J., Rodriguez I.P., Chakravarthi B.V.S.K. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huss W.J., Hanrahan C.F., Barrios R.J., Simons J.W., Greenberg N.M. Angiogenesis and prostate cancer: Identification of a molecular progression switch. Cancer Res. 2001;61(6):2736–2743. [PubMed] [Google Scholar]
- 4.Li X.Y., Liang J.W., Mohamed O.K., Zhang T.J., Lu G.Q., Meng F.H. Design, synthesis and biological evaluation of N-phenyl-(2,4-dihydroxypyrimidine-5-sulfonamido)benzoyl hydrazide derivatives as thymidylate synthase (TS) inhibitors and as potential antitumor drugs. Eur J Med Chem. 2018;154:267–279. doi: 10.1016/j.ejmech.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 5.Li XY, Zhang TJ, Mohamed O K, Lu GQ, Xu HL Wang DP, Meng FH. Discovery of N-phenyl-(2,4-dihydroxypyrimidine-5-sulfonamido) phenylurea-based thymidylate synthase (TS) inhibitor as a novel multi-effects antitumor drugs with minimal toxicity. Cell Death & Disease. 2019;10(7):532. [DOI] [PMC free article] [PubMed]
- 6.Lu G.Q., Li X.Y., Mohamed O.K., Wang D.P., Meng F.H. Design, synthesis and biological evaluation of novel uracil derivatives bearing 1, 2, 3-triazole moiety as thymidylate synthase (TS) inhibitors and as potential antitumor drugs. Eur J Med Chem. 2019;171:282–296. doi: 10.1016/j.ejmech.2019.03.047. [DOI] [PubMed] [Google Scholar]
- 7.Gurupadaswamy H.D., Girish V., Kavitha C.V., Raghavan S.C., Khanum S.A. Synthesis and evaluation of 2,5-di(4-aryloylaryloxymethyl)-1,3,4-oxadiazoles as anti-cancer agents. Eur J Med Chem. 2013;63(Complete):536–543. doi: 10.1016/j.ejmech.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 8.Dawood K.M., Gomha S.M. Synthesis and Anti-cancer Activity of 1,3,4-Thiadiazole and 1,3-Thiazole Derivatives Having 1,3,4-Oxadiazole Moiety. J Heterocycl Chem. 2014;52(5):1400–1405. [Google Scholar]
- 9.Pidugu V.R., Yarla N.S., Pedada S.R., Kalle A.M., Krishna Satya A. Design and Synthesis of Novel HDAC 8 Inhibitory 2,5-disubstituted-1,3,4-Oxadiazolescontaining Glycine and Alanine hybrids with Anti Cancer Activity. Bioorg Med Chem. 2016;24(21):5611–5617. doi: 10.1016/j.bmc.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 10.Shah T.A., Ahmad Z., Mir N.A., Muneer M., Rath N.P. One step synthesis of highly functionalized thiazolo[3,2- b][1,2,4]triazole, triazolo[1,5-a]pyrimidine and triazolo[3,4- b][1,3,4]thiadiazine†[J] RSC Adv. 2015;5(130):107931–107937. [Google Scholar]
- 11.Bajaj Shalini, Asati Vivek, Singh Jagadish, Roy Partha Pratim. 1,3,4-Oxadiazoles: An emerging scaffold to target growth factors, enzymes and kinases as anticancer agents. Eur J Med Chem. 2015;97:124–141. doi: 10.1016/j.ejmech.2015.04.051. [DOI] [PubMed] [Google Scholar]
- 12.Warmus Joseph S., Flamme Cathlin, Zhang Lu Yan, Barrett Stephen, Bridges Alexander, Chen Huifen, Gowan Richard, Kaufman Michael, Sebolt-Leopold Judy, Leopold Wilbur, Merriman Ronald, Ohren Jeffrey, Pavlovsky Alexander, Przybranowski Sally, Tecle Haile, Valik Heather, Whitehead Christopher, Zhang Erli. 2-Alkylamino- and alkoxy-substituted 2-amino-1,3,4-oxadiazoles—O-Alkyl benzohydroxamate esters replacements retain the desired inhibition and selectivity against MEK (MAP ERK kinase) Bioorg Med Chem Lett. 2008;18(23):6171–6174. doi: 10.1016/j.bmcl.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Gangjee A., Yu J., Mcguire J.J. Design, synthesis, and X-ray crystal structure of a potent dual inhibitor of thymidylate synthase and dihydrofolate reductase as an antitumor agent. J Med Chem. 2000;43(21):3837–3851. doi: 10.1021/jm000200l. [DOI] [PubMed] [Google Scholar]
- 14.Sayre P.H., Finer-Moore J.S., Fritz T.A., Biermann D., Gates S.B., MacKellar W.C. Multi-targeted antifolates aimed at avoiding drug resistance form covalent closed inhibitory complexes with human and Escherichia coli thymidylate synthases. J. Mol. Biol. 2001;313:813–829. doi: 10.1006/jmbi.2001.5074. [DOI] [PubMed] [Google Scholar]
- 15.Tufan A., Satiroglu-Tufan N. The Chick Embryo Chorioallantoic Membrane as a Model System for the Study of Tumor Angiogenesis, Invasion and Development of Anti-Angiogenic Agents. Curr Cancer Drug Targets. 2005;5(4):249–266. doi: 10.2174/1568009054064624. [DOI] [PubMed] [Google Scholar]
- 16.Swanson N., Hogrefe K., Javed Q. In vitro evaluation of vascular endothelial growth factor (VEGF)-eluting stents[J] Int J Cardiol. 2003;92(2–3):247–251. doi: 10.1016/s0167-5273(03)00102-5. [DOI] [PubMed] [Google Scholar]
- 17.Teicher B.A., Chen V., Shih C., Menon K., Amsrud T. Treatment regimens including the multitargeted antifolate ly231514 in human tumor xenografts. Clin Cancer Res. 2000;6(3):1016–1023. [PubMed] [Google Scholar]
- 18.Swoap O.F. A collaborative study on the use of mice in acute toxicity testing [J] J Am Pharm Assoc Am Pharm Assoc. 1955;44(1):11–16. doi: 10.1002/jps.3030440106. [DOI] [PubMed] [Google Scholar]
- 19.Justilien Verline, Fields Alan P. Utility and Applications of Orthotopic Models of Human Non-Small Cell Lung Cancer (NSCLC) for the Evaluation of Novel and Emerging Cancer Therapeutics[J]. Current Protocols. Pharmacology. 2013;62(1) doi: 10.1002/0471141755.ph1427s62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L., Xie Y.-B., Huang N.-Y., Yan J.-Y., Hu W.-M., Liu M.-G. Catalytic aza-Wittig Reaction of Acid Anhydride for the Synthesis of 4H-benzo[d][1,3]oxazin-4-ones and 4-benzylidene-2-aryloxazol-5(4H)-ones. ACS Catal. 2016;6(6):4010–4016. [Google Scholar]
- 21.El-Din M.M.G., El-Gamal M.I., Abdel-Maksoud M.S., Yoo K.H., Oh C.H. Synthesis and in vitro antiproliferative activity of new 1,3,4-oxadiazole derivatives possessing sulfonamide moiety. Eur J Med Chem. 2015;90:45–52. doi: 10.1016/j.ejmech.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Du Q.R., Li D.-D., Pi Y.-Z., Li J.-R., Sun J., Fang F. Novel 1,3,4-oxadiazole thioether derivatives targeting thymidylate synthase as dual anticancer/antimicrobial agents. Bioorg Med Chem. 2013;21(8):2286–2297. doi: 10.1016/j.bmc.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Liao B.-R., He H.-B., Yang L.-L., Gao L.-X., Chang L., Tang J. Synthesis and structure–activity relationship of non-phosphorus-based fructose-1,6-bisphosphatase inhibitors: 2,5-Diphenyl-1,3,4-oxadiazoles. Eur J Med Chem. 2014;83:15–25. doi: 10.1016/j.ejmech.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 24.He X., Li X.Y., Liang J.W., Cao C., Li S., Zhang T.J. Design, synthesis and anticancer activities evaluation of novel 5H-dibenzo[b, e]azepine-6,11-dione derivatives containing 1,3,4-oxadiazole units. Bioorg Med Chem Lett. 2018;28(5):847–852. doi: 10.1016/j.bmcl.2018.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Stabile P., Lamonica A., Ribecai A., Castoldi D., Guercio G., Curcuruto O. Mild and convenient one-pot synthesis of 1,3,4-oxadiazoles. Tetrahedron Lett. 2010;51(37):4801–4805. [Google Scholar]
- 26.Tois J., Vahermo M., Koskinen A. Novel and Convenient Synthesis of 4(1H)Quinolones. Tetrahedron Lett. 2005;46(5):735–737. [Google Scholar]
- 27.Hayashi Shigeo, Ueno Naomi, Murase Akio, Nakagawa Yoko, Takada Junji. Novel acid-type cyclooxygenase-2 inhibitors: Design, synthesis, and structure–activity relationship for anti-inflammatory drug. Eur J Med Chem. 2012;50:179–195. doi: 10.1016/j.ejmech.2012.01.053. [DOI] [PubMed] [Google Scholar]
- 28.Chwastek M., Pieczykolan M., Stecko S. The synthesis of 5-amino-dihydrobenzo[b]oxepines and 5-amino-dihydrobenzo[b]azepines via Ichikawa rearrangement and ring-closing metathesis. J Org Chem. 2016;81(19):9046–9074. doi: 10.1021/acs.joc.6b01691. [DOI] [PubMed] [Google Scholar]
- 29.Linke S.P., Clarkin K.C., Di Leonardo A., Tsou A., Wahl G.M. A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev. 1996;10(8):934–947. doi: 10.1101/gad.10.8.934. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi Y., Tsujii M., Kodama T., Akasaka T., Takehara T. p53 functional deficiency in human colon cancer cells promotes fibroblast-mediated angiogenesis and tumor growth. Carcinogenesis. 2016;10(10) doi: 10.1093/carcin/bgw085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.