

Graphical abstract
Keywords: Blood–brain barrier, Acoustic waves, Shockwave, Ultrasound, TRPV4, PKC-δ
Abstract
Introduction
Numerous studies have shown the ability of low-energy acoustic waves such as focused ultrasound or shockwave to transiently open blood-brain barrier (BBB) and facilitate drug delivery to the brain. Preclinical and clinical evidences have well demonstrated the efficacy and safety in treating various brain disorders. However, the molecular mechanisms of acoustic waves on the BBB are still not fully understood.
Objectives
The present study aimed at exploring the possible molecular mechanisms of acoustic wave stimulation on brains.
Methods: Briefly describe the experimental design
The left hemisphere of the rat‘s brain was treated with pulsed ultrasound from a commercial focused shockwave or a planar ultrasound device, and the right hemisphere served as a control. One hour after the mechanical wave stimulation or overnight, the rats were sacrificed and the brains were harvested for protein or histological analysis. Agonists and antagonists related to the signal transduction pathways of tight junction proteins were used to investigate the possible intracellular mechanisms.
Results
Intracellular signal transduction analysis shows calcium influx through transient receptor potential vanilloid 4 (TRPV4) channels, and the activation of PKC-δ pathway to mediate dissociation of ZO-1 and occludin after acoustic wave stimulation. The activation of TRPV4 or PKC-δ signaling further increased the expression level of TRPV4, suggesting a feedback loop to regulate BBB permeability. Moreover, the tight junction proteins dissociation can be reversed by administration of PKC-δ inhibitor and TRPV4 antagonist.
Conclusion
The present study shows the crucial role of TRPV4 in acoustic wave-mediated BBB permeability, specifically its effect on compromising tight junction proteins, ZO-1 and occludin. Our findings provide a new molecular perspective to explain acoustic wave-mediated BBB opening. Moreover, activation of TRPV4 by agonists may reduce the threshold intensity level of acoustic waves for BBB opening, which may prevent undesirable mechanical damages while maintaining efficient BBB opening.
Introduction
Previous studies have indicated that acoustic waves, such as ultrasound [1], [2], [3], [4], [5] or shockwave [6], can effectively induce BBB opening via transcranial application. The facilitated trans-BBB delivery of molecules are believed to go through paracellular and transcellular pathways after focused ultrasound (FUS) application in the presence of an ultrasound contrast agent (UCA) [7], [8]. Cavitation from UCA in response to ultrasound sonication has been shown to induce cell membrane deformation and permeability changes of the endothelium [9], [10]. The permeability change usually parallels the disintegration of tight junction proteins, ZO-1 and occludin [1], [7]. The Akt signaling pathway is activated in neuronal cells surrounding the disrupted BBB [1].
Calcium-mediated pathways, such as PKC pathway, have been reported to regulate epithelial tight junction integrity [11], [12], [13], [14]. For example, ischemia induces calcium influx into brain endothelial cells [13], which activates members of the PKC family and then induces various cellular mechanisms such as apoptosis and the loss of tight junction interactions, thus perturbing BBB integrity [13], [14], [15]. In contrast, prevention of BBB disruption can be achieved by calcium chelators [16]. However, the mechosensors responsible for ultrasound stimulation and the subsequent molecular pathways, leading to BBB disruption, are still not fully understood.
Transient receptor potential vanilloid 4 (TRPV4) is a calcium-permeable cation channel that has been shown to be widely expressed in the kidneys, lungs, hearts, vessels and nerve ganglia. In the brain region, TRPV4 is expressed in neurons [17], astrocytes [17], [18], choroid plexus [19] and vascular endothelial cells [20], [21]. The channel is characterized by multimodal activation properties such as osmolarity, temperature and acidity [22], [23], [24], [25]. Recent evidence demonstrates that the channel can also be activated by mechanical stress, and may have a crucial role in the modulation of BBB integrity [13], [21], [26]. Blockage of TRPV4 activity was found to potentially ameliorate brain injury in a variety of central nervous system disorders such as ischemic stroke [25], [27], [28], intracerebral hemorrhage (ICH) [20] and traumatic brain injury (TBI) [29], possibly through modulating BBB permeability and thus decreased edema.
Despite evidence showing the possible connection between TRPV4 and BBB integrity, the direct effect of acoustic wave stimulation on TRPV4 activation and its role on subsequent BBB opening remains unclear. We hypothesize that mechanical stimulation such as ultrasound or shockwave induces calcium influx through TRPV4 channels, subsequently activating the calcium-related intracellular signal pathways, leading to disruption of BBB integrity.
Materials & methods
Animals
All animal experimental procedures were conducted in accordance with the care and use guidelines of the Laboratory Animal Center at National Taiwan University College of Medicine and were approved by the Institutional Animal Care and Use Committee (IACUC, approval No. 20170091) of National Taiwan University College of Medicine. Male Sprague–Dawley rats (body weight 300–350 g) purchased from BioLASCO Taiwan Co., Ltd. were used in this study. Total of 65 rats were used for immunoprecipitation assay (50 rats) and immunofluorescence (15 rats).
Animal grouping and treatment protocol:
For shockwave treatment, fifty rats were equally categorized into group 1 (vehicle injected via I.V.), group 2 (rottlerin injected via I.V.), group 3 (HC067047 injected via I.V.) and group 4 (GSK1016790A injected via I.V.). For ultrasound treatment, fifteen rats were equally categorized into group 5 (vehicle injected via I.V.), group 6 (rottlerin injected via I.V.), and group 7 (HC067047 injected via I.V.)
Shockwave treatment
The shockwave device (PiezoWave, Richard Wolf GmbH, Knittlingen, Germany) setup is described in our previous report [6]. For in-vivo study, the condition of the shockwave-induced BBB opening was based on our previous reports with an energy dosage of 0.21 mJ/mm2 (intensity level 5), pulse repetition frequency (PRF) of 5 Hz, and 200 iterations without the presence of ultrasound contrast agents. These conditions provide a greater percentage of BBB opening without visible bleeding [6]. The left hemisphere of the brain was treated with shockwave as previously reported [6], and the right hemisphere served as a control. One hour after the shockwave treatment or overnight, the rats were sacrificed and the brains were harvested for protein or histological analysis.
For in-vitro study (Ca2+ influx assay and In-Cell ELISA assay), the test-wells were filled with medium and immediately sealed without any bubble by microplate sealing tape (NUNC) and then the shockwave probe was placed above the plate. The gap between the sealing tape and the probe was filled with ultrasound gel. Ten shockwave iterations were applied (intensity level 4, equivalent to 0.18 mJ/mm2, PRF= 5 Hz, N = 4). In some experiments, cells were pretreated with TRPV4 antagonist or agonist for 30 min using the following concentrations: 20 nM, 40 nM HC067047, and 0.15 μM, 1.5 μM and 3 μM GSK1016790A.
Ultrasound treatment
The ultrasound device US-700 was purchased from ITO Physiotherapy & Rehabilitation CO., LTD (Japan). The planar transducer was placed on the left side of the brain and ultrasound gel was applied at the interfaces between the bottom of the transducer and the rat scalp. The parameters of ultrasound are 1 MHz, 3 W/cm2 and 10% duty cycle, with 5 min sonication duration. One hour after the ultrasound treatment, the rats were sacrificed and the brains were harvested for protein or histological analysis. For in-vitro study (In-Cell ELISA assay), the test well settings were the same as the shockwave, then placed ultrasound probe above the plate, and the gap between the probe and the sealing tape is filled with ultrasound gel. The ultrasound parameters are 1 MHz, 2.1 W/cm2 and 5% duty cycle, with 3 min sonication duration. In some experiments, cells were pretreated with TRPV4 antagonist or agonist for 30 min using the following concentrations: 20 nM, 40 nM HC067047, and 0.15 μM, 1.5 μM and 3 μM GSK1016790A.
Drug administration
The TRPV4 antagonist HC067047, agonist GSK1016790A and rottlerin were purchased from Abcam (UK). The TRPV4 agonist GSK1016790A (3 μg/kg body weight), TRPV4 antagonist HC067047 (0.5 mg/kg body weight), PKC-δ inhibitor rottlerin (0.3 mg / kg body weight), CaCl2 (80 mg/kg body weight), EDTA (40 mg/kg body weight), EGTA (130 mg/kg body weight) were intravenously injected via the tail vein. The doses of the above-listed chemicals were selected based on previous reports [30], [31], [32], [33], [34], [35]. The GSK1016790A, HC-067047, and rottlerin were first dissolved in DMSO, and then in normal saline to a final volume of 1 ml with a DMSO concentration of 1%. The control groups were treated correspondingly with saline containing the same DMSO dose.
Immunoblot analysis
Brain tissues were homogenized in an ice-cold immunoprecipitation buffer (IP buffer) (containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 10% glycerol, 1% Nonidet p-40, phosphatase inhibitor cocktail 2 (Sigma) and protease inhibitor cocktail (Merck)). The homogenate was centrifuged at 15,000g for 30 min at 4 °C and the supernatant was determined using a coomassie protein assay reagent (Thermo). Equal amounts of extracted proteins were respectively loaded on 4–12% NuPAGE Bis-Tris Gels (Thermo) and subjected to immunoblotting. The following primary antibodies were used: rabbit anti-ZO1 (1:500, Thermo); mouse anti-occludin (1:2000, Thermo); rabbit PKC-δ (1:500, cell signaling); mouse anti-GAPDH (1:5000, Proteintech). Appropriate secondary antibodies (GE Healthcare) were incubated with the membranes for 1 h at room temperature. For Co-Immunoprecipitation assay, immunoprecipitated proteins were detected using VeriBlot for IP Detection Reagents (Abcam) without being contaminated by IgG heavy and light chains from the precipitated-antibodies. The signals were visualized using Amersham™ ECL Select Western Blotting Detection Reagent (GE Healthcare) according to the manufacturer’s instructions and recorded via UVP BioSpectrum Image system (UVP). Data were analyzed using Vision Works LS software (UVP).
Co-Immunoprecipitation (Co-IP) assay
Proteins were extracted from brain tissue as described above. The extractions containing 0.5 mg total protein in 500 μl IP buffer were first preabsorbed with protein G Mag Sepharose Xtra (GE Health care) for 1 h and then incubated with 5 μg rabbit anti-ZO-1 antibody (Thermo) overnight at 4 °C with constant shaking on a rotator. The samples were then incubated with protein G Mag Sepharose Xtra (GE Healthcare) for 4 h at 4 °C, and were collected using MagRack 6 (GE Healthcare). The samples were washed three times with IP buffer, eluted by elution buffer (100 mM glycine-HCl, pH 2.8) and neutralized by 1 M Tris-HCl, pH 9.0 buffer. Then the samples added sample reducing agent (Thermo) and mixed with 4X SDS sample buffer, boiled for 5 min and then analyzed with immune-blotting as explained previously.
Immunofluorescence staining
Two or eighteen hours after the shockwave application, the rats were sacrificed and the brains were harvested and fixed with 10% formalin at room temperature overnight. Samples were then embedded in paraffin and serial 7-μm transverse sections around the shockwave treatment site were mounted on slides. The sections were deparaffinized, rehydrated, antigen retrieved (120℃, 10 min) and washed in TBS, followed by washing with TBS containing 0.025% Triton X-100 for 10 min. The sections were blocked with 10% newborn calf serum (NCS) and 1% BSA in TBS for 2 h. The sections were incubated with primary antibody overnight at 4℃. After washing with TBS, the samples were incubated with the secondary antibody for 2 h at RT, washed with TBS and mounted with EverBrite™ Hardset Mounting Medium containing DAPI to label the nuclei (Biotium). Slides were viewed, and images were captured with LSM780 confocal microscope (Zeiss, Jena, Germany). The primary antibodies used for immunostaining and their dilutions were as follows: rabbit anti-ZO1 (1:50, Thermo), mouse anti-occludin (1:50, Thermo), rabbit TRPV4 (1:50, Thermo). The secondary antibodies used were Alexa Fluor 488-conjugated goat anti-mouse IgG (1:100, Thermo) and Alexa Fluor 555-conjugated goat anti-rabbit IgG (1:100, Thermo).
In vitro Ca2+ influx assay
Murine brain-derived endothelial bEnd.3 cells were cultured with Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1X Antibiotic-Antimycotic (Invitrogen). The bEnd.3 cells were seeded in clear flat-bottom black 96-well culture plate at a density of 2.4x104 cells/well (80–90% confluenct). After 18 h of incubation, cells were washed twice with HBSS and incubated with Fura2-AM dye solution (5 μM Fura2-AM (Invitrogen), 0.1%BSA, 0.05% Pluronic F-127(Sigma) in HBSS buffer) for 1 h at RT in the dark. The test-wells were washed twice with HBSS buffer, after which the entire well was filled with medium (without phenol red) and immediately sealed without any bubbles by microplate sealing tape (NUNC). Subsequently, the plates were inserted into a microplate reader (Infinite M200, Tecan), and the fluorescence (excitation: 340 nm or 380 nm, emission: 510 nm) was measured before and after the addition of various concentrations of test compounds or shockwave treatment. The data are showed as the ratio of Fura2 fluorescence due to excitation at 340 nm to that due to excitation at 380 nm.
In-cell ELISA assay
The expression of TRPV4 was measured using the In-cell ELISA Kit (Thermo), as described in the manufacturer’s protocol. The bEnd.3 cells were seeded in a 96-well culture plate at a density of 2.4 × 104 cells/well. After 18 h of incubation, cells were washed with PBS, then the wells were filled with medium and sealed without bubbles by microplate sealing tape. Shockwave treatment was conducted as previously described. At 18 h post-shockwave treatment, the cells were washed twice with PBS and fixed with 4% formaldehyde for 15 min at room temperature. The cells were incubated with permeabilization buffer (0.1% triton X-100 in Tris-buffered saline (TBS)) for 15 min at room temperature and then the plate was washed twice with TBS. Peroxidase suppressor (Thermo) was then added to each well and incubated for 20 min. The washing steps were repeated and the plate was incubated with blocking buffer at room temperature for 30 min. After blocking, plates were incubated with TRPV4 antibody (Thermo) overnight at 4 °C followed by HRP conjugated secondary antibody for 1 h at room temperature. The plates were washed and incubated with TMB substrate. After sufficient blue color development, stop solution was added to each well and absorbance was measured at 450 nm. Results across wells were then normalized to cell number based on whole-cell staining with Janus Green (Thermo).
Statistics
All numerical data are expressed as the mean ± standard deviation. Tests of significance were performed by Student’s t test or ANOVA followed by LSD post hoc test. P-values < 0.05 are considered statistically significant and are denoted with an asterisk.
Results
BBB tight junction integrity is perturbed by shockwave
The BBB opening induced by shockwave was demonstrated by Evans blue extravasation. Visual inspection of the 3 mm thick sections revealed a penetration of blue dye throughout left hemispheres (shockwave-treated), whereas contralateral hemispheres showed no blue stain (Fig. 1A). Reduction of protein–protein interaction between tight junction proteins means a loss of junctional integrity and increased paracellular permeability in BBB. Previous studies showed that application of FUS resulted in a decrease in the interaction of ZO-1 and occludin [1]. To evaluate whether shockwave could also compromise the interaction of tight junction proteins, the interaction of ZO-1 and occludin was analyzed by co-immunoprecipitation assay (Co-IP). Co-IP analysis with brain tissue homogenates showed that the interaction of the tight junction proteins ZO-1 and occludin in the shockwave treated region was significantly reduced (Fig. 1B and 1C), indicating a loss of junctional integrity and possibly an increase in paracellular permeability in the BBB. However, the total amounts of ZO-1 and occludin proteins were unaffected by shockwave treatment (Fig. 1B).
Fig. 1.

Tight junction integrity is disrupted by shockwave treatment. (A) Evans blue extravasation in the coronal sections of brain. (B) Co-immunoprecipitation of occludin and ZO-1. The interaction of ZO-1 and occludin is significantly reduced by shockwave treatment. Brain tissue lysates shows that total ZO-1 and occludin protein levels are not significantly changed in response to shockwave treatment. (C) The bar graph summarizes the Co-IP results, and the association between occludin and ZO-1 is decreased by shockwave treatment. n = 6 rats per group. Data are presented as the mean standard errors of the means. Significant differences (Student’s t test) are depicted with asterisks. p < 0.05 compared with the control group. (D) Interrupted patterns of tight junction proteins ZO-1 and occludin in brain after shockwave treatment is shown on confocal images. (Scale bars: 10 μm).
We also analyzed the distribution of ZO-1 and occludin proteins on brain sections by immunofluorescence stain (IF). The results showed that shockwave treatment group demonstrated a discontinuous, irregular distribution of ZO-1 and occludin and loss of co-localization. In contrast, the control group demonstrated a continuous and linear labeling of ZO-1 and occludin along the brain vessel (Fig. 1D). The result indicates that shockwave treatment disrupted the interaction between ZO-1 and occludin, which in turn increases in paracellular permeability in the BBB.
Shockwave-induced BBB opening is blocked by EGTA
Previous studies have found calcium signaling to be involved in BBB disruption. Calcium influx can trigger a number of calcium-sensitive signaling cascades that alter the integrity of tight junctions [13], [36]. Furthermore, Calcium homeostasis has been shown to mediate tight junction permeability [37], [38]. To evaluate whether shockwave induced BBB opening also occurs through the calcium-mediated signaling pathway, we perturbed calcium homeostasis by tail vein injection of CaCl2 or chelators (EDTA or EGTA) and observed the interaction of ZO-1 and occludin as previously shown. Unlike EDTA (Ethylenediaminetetraacetic acid), a non-selective cation chelator, EGTA (Ethylene glycol tetraacetic acid) has a high affinity for calcium ions. Shockwave-induced reduction in the interaction between ZO-1 and occludin was observed in groups pretreated with EDTA and CaCl2, similar to that of the control group, but the effect of shockwave was significantly reversed by EGTA treatment (Fig. 2A and B). The results indicate that calcium is crucially important for shockwave-induced tight junction disruption.
Fig. 2.

Calcium chelator- EGTA blocks shockwave-induced tight junction dissociation. (A) Co-immunoprecipitation of occludin and ZO-1. After shockwave treatment, the interaction of ZO-1 and occludin is significantly rescued by EGTA but EDTA and CaCl2 are not. (B) Bar graphs summarizing the ratio of occludin associated with ZO-1 in various pretreatment experiments after shockwave treatment. Data are presented as the mean standard errors of the means. Significant differences (Student’s t test) are depicted with asterisks. p < 0.05 compared with the control group.
ZO-1 and occludin are hyper-tyrosine phosphorylated and PKC-δ signaling is activated by shockwave treatment.
To determine whether shockwave exerts its effect via the PKC-δ signaling pathway, we measured the tyrosine phosphorylation of ZO-1 and occludin in the brain tissue by immunoprecipitation with anti-phosphotyrosine antibodies followed by immunoblotting with anti-ZO1 and anti-occludin antibodies. The tyrosine phosphorylation of ZO-1 and occludin in the brain tissue of the shockwave-treated group was significantly elevated with increased tyrosine phosphorylation of PKC-δ when compared with the group without shockwave treatment (Fig. 3A). The elevated tyrosine phosphorylation of ZO-1, occludin, and PKC-δ was blocked by EGTA administration (Fig. 3B), implying the involvement of calcium dependent PKC-δ signaling process.
Fig. 3.

Shockwave treatment induces tyrosine phosphorylation of tight junction proteins and PKC-δ proteins and are blocked by EGTA. (A) Anti-phosphotyrosine immunoprecipitates (IP:α-pTyr) form brain extracts of rat treated with shockwave are immunoblotted for ZO-1, occludin and PKC-δ proteins. Tyrosine phosphorylation of ZO-1, occludin and PKC-δ proteins are significantly increased after shockwave treatment. (B) Shockwave-induced tyrosine phosphorylation of tight junction and PKC-δ proteins are significantly decreased by pretreatment of EGTA. Western Blot analysis on brain tissue lysates shows that ZO-1 and occludin protein levels are not significantly changed in response to EGTA and shockwave treatment.
Previous studies demonstrated that PKC-δ mediated BBB disruption and tight junction integrity was preserved by the attenuation of PKC-δ activity (such as by δV1-1 or Bryostatin-1) [12], [39], [40]. Inhibition of PKC-δ by rottlerin also enhanced barrier function in Caco-2 cells [41]. Furthermore, previous studies have shown that tyrosine phosphorylation of PKC-δ is a mechanism for regulating the catalytic activity of PKC-δ [42], [43]. Therefore, we investigated the role of tyrosine phosphorylation of PKC-δ in shockwave-induced tight junction disruption. Our data show that tyrosine phosphorylation of PKC-δ was significantly enhanced by shockwave treatment, but was reduced by EGTA administration (Fig. 3A and B). This result suggests that PKC-δ signaling was activated by shockwave treatment.
Inhibition of PKC-δ activity by rottlerin blocked shockwave-induced BBB disruption.
To further confirm whether the PKC-δ signaling mediates shockwave-triggered BBB disruption, we administrated a PKC-δ inhibitor, rottlerin, via tail vein injection before shockwave treatment. The administration of rottlerin preserved the interaction of ZO-1 and occludin and reduced tyrosine phosphorylation of ZO-1 and occludin after shockwave treatment (Fig. 4A, B and C). The tyrosine phosphorylation of PKC-δ was also significantly reduced (Fig. 4C). In addition, the distribution of ZO-1 and occludin proteins after shockwave treatment was not significantly changed in the group treated with rottlerin compared with those without (Fig. 4E, data not shown). Taken together, the results confirm that the blockage of PKC-δ signaling by rottlerin arrests shockwave-induced BBB disruption.
Fig. 4.

PKC-δ signaling mediates shockwave-induced BBB opening. (A) Co-immunoprecipitation of occludin and ZO-1. Inhibition of PKC-δ activity by rottlerin preserves ZO1-occludin interaction after shockwave treatment. (B) Bar graphs summarizing the ratio of occludin associated with ZO-1 in rottlerin pretreatment experiments after shockwave treatment. n = 5 rats per group. Data are presented as the mean standard errors of the means. Statistical results show no significant differences (Student’s t test) (C) Anti-phosphotyrosine immunoprecipitates (IP:α-pTyr) experiments indicate that tyrosine phosphorylation of ZO-1, occludin and PKC-δ proteins are significantly reduced in rottlerin pretreatment group after shockwave treatment. (D) ZO-1, occludin and PKC-δ proteins are not significantly changed in rottlerin pretreatment experiments after shockwave application. (E) Immunostaining for ZO-1 and occludin proteins in brain tissue in rottlerin pretreatment experiments. (Scale bars: 10 μm).
TRPV4 inhibition prevents shockwave-induced BBB disruption.
TRPV4 is a mechanosensor of the endothelial cells which can be activated by shear stress [26], [44], [45]. It mediates calcium influx and increases endothelial permeability [20], [44], [46]. To determine whether the TRPV4 mediated shockwave-triggerred BBB disruption, we administrated a TRPV4 antagonist, HC067047, via tail vein injection before shockwave treatment. The interaction of ZO-1 and occludin after shockwave treatment was retained and tyrosine phosphorylation of ZO-1 and occludin was mainly reduced by administration of HC067047 (Fig. 5A, B and C). The distribution of ZO-1 and occludin proteins after shockwave treatment was not significantly changed in groups treated with HC067047 compared with those without shockwave treatment (Fig. 5E, data not shown). In addition, the tyrosine phosphorylation of PKC-δ after shockwave treatment was significantly reduced (Fig. 5C). These results suggest that blockade of TRPV4 reduces PKC-δ activation by affecting calcium influx.
Fig. 5.

TRPV4 activation regulates shockwave-induced BBB opening. (A) Co-immunoprecipitation of occludin and ZO-1. Blockage of TRPV4 activity by HC067047 retains ZO1-occludin interaction after shockwave treatment. (B) Bar graphs summarizing the ratio of occludin associated with ZO-1 in HC067047 pretreatment experiments after shockwave treatment. n = 5 rats per group. Data are presented as the mean standard errors of the means. Statistical results show no significant differences (Student’s t test) (C) Anti-phosphotyrosine immunoprecipitates (IP: α-pTyr) experiments indicate that tyrosine phosphorylation of ZO-1, occludin and PKC-δ proteins are significantly reduced in HC067047 pretreatment group after shockwave treatment. (D) ZO-1, occludin and PKC-δ proteins are not significantly changed in HC067047 pretreatment experiments after shockwave application. (E) Immunostaining for ZO-1 and occludin proteins in brain tissue in HC067047 pretreatment experiments. (Scale bars: 10 μm).
TRPV4-dependent Ca2+ influx is predominately induced by shockwave treatment.
To determine whether shockwave induces TRPV4-dependent calcium influx in the endothelium, we first measured calcium influx in response to the known TRPV4 activator GSK1016790A in bEnd.3 cells, a brain endothelium-like cell line that expresses TRPV4 to regulate calcium influx [47]. The bEND.3 cells were pre-loaded with calcium indicator fura2. Stimulation of bEnd.3 cells with 3 μM GSK1016790A was shown to induce significant calcium influx (Fig. 6A). Shockwave treatment also induces calcium influx, and is blocked by pretreatment with TRPV4 antagonist in a concentration-dependent manner (20 nM and 40 nM HC067047) (Fig. 6B), strongly suggesting that shockwave-induced calcium signaling was mediated by TRPV4. Taken together, the role of the shockwave is to stimulate Ca2+ influx into the cell through activating TRPV4, thereby activating the PKCδ signaling pathway and eventually causing the disruption of tight junctions.
Fig. 6.

TRPV4 mediates shockwave-induced calcium influx. Representative graphs showing the changes in [Ca2++]i levels in individual bEnd.3 cells in response to the TRPV4 agonist (GSK1016790A), antagonist (HC067047) or shockwave treatment. (A) Calcium influx is induced by bEnd.3 cells treated with 3 μM GSK1016790A. (B) Calcium influx is induced by shockwave treatment and this phenomenon is blocked by treatment with TRPV4 antagonist.
TRPV4 is significantly overexpressed by shockwave via the TRPV4/PKC-δ pathway.
A number of studies have indicated that TRPV4 is significantly overexpressed after brain injury, such as intracerebral hemorrhage [20], traumatic brain injury [29], [48] and middle cerebral artery occlusion [27], leading to BBB disruption. Interestingly, 18 h after shockwave treatment, the expression of TRPV4 increased significantly along brain vessels (Fig. 7A). In vitro study using In-Cell ELISA assay also shows the increased expression of TRPV4 by shockwave treatment. This enhancement could be reversed by the administration of inhibitors such as rottlerin (Fig. 7B) or HC067047 (Fig. 7C). These results again show that shockwave treatment could activate intracellular PKC-δ and TRPV4-mediated signaling.
Fig. 7.

The expression of TRPV4 is remarkably increased by TRPV4/ PKC-δ pathway after shockwave treatment. (A) Eighteen hours after shockwave application, the rats were sacrificed and the brains were harvested for immunostain analysis. TRPV4 is overexpressed by shockwave treatment. (Scale bars: 10 μm). (B, C) The expression of TRPV4 is analyzed through In-Cell ELISA assay in vitro. TRPV4 protein is overexpressed by shockwave treatment. Pretreatment of rottlerin or HC067047 inhibit shockwave-induced TRPV4 overexpression. (Student t-test, *P < 0.05, n = 4).
Combining TRPV4 agonist and shockwave reduces the intensity threshold of shockwave to open the BBB.
The possibility of brain injury induced by shockwave treatment is always a concern. Our previous study showed that shockwave could induce minor inflammation and apoptosis in rat brains [6]. By reducing the intensity level of shockwave required to open the BBB, the possibility of brain injury can be reduced. Our results indicate that the activation of TRPV4 by shockwave promote the disruption of tight junction, implied TRPV4 is a candidate for shockwave-mediated BBB opening to ameliorate the intensity level of shockwave. Thus, we used a selective agonist (GSK1016790A) to determine whether low-intensity shockwave can also induce BBB opening by activating TRPV4. Fig. 8A shown that GSK1016790A also promoted BBB opening at lower SW treatment level (level 1 = 0.1 mJ/mm2, 50 times), but was not in the control group. It is worth noting that all mice induced the opening of BBB by using lower SW level in combination with GSK1016790A, but only lower SW treatment could not (Fig. 8B). Statistical analysis using the Mann-Whitney U test showed that when used in combination with GSK1016790A, the BBB opening was significantly improved at lower SW treatment levels (p = 0.008). We also found that the discontinuity, irregular distribution, and loss of co-localization of ZO-1 and occludin patterns are still present in the group treated with shockwaves at a lower treatment level, below the threshold intensity of BBB opening using shockwave [6] (Fig. 8C). Therefore, the TRPV4 agonist can reduce the shockwave intensity threshold required for BBB opening.
Fig. 8.

TRPV4 activation by GSK1016790A promote low-intensity level of shockwave to achieve BBB opening. (A) Evans blue extravasation in the coronal sections of brain. (B) The successful BBB opening rate under low-intensity level of shockwave. (C) Representative images of immunofluorescence staining for ZO-1 and occludin in GSK1016790A administration groups with or without shockwave treatment. (Scale bars: 10 μm). The reduced colocalization of ZO-1 and occludin is still found in groups treated with low-intensity shockwave.
Ultrasound also induces the dissociation of ZO-1-occludin and mediated TRPV4 overexpression by TRPV4/PKC-δ pathway.
Finally, planar ultrasound is tested in similar way as shockwave with respect to BBB opening effect and the TRPV4/PKC-δ pathway. The interaction of ZO-1 and occludin was decreased by ultrasound treatment (Fig. 9 A), and inhibition of TRPV4 or PKC-δ activity rescued ultrasound-induced ZO-1-occludin dissociation (Fig. 9 B). The total amounts of ZO-1 and occludin proteins were unchanged by ultrasound treatment (data not shown). In-Cell ELISA assay in vitro revealed increased TRPV4 by ultrasound treatment, which is significantly reversed by treatment with TRPV4 antagonist (Fig. 9C).
Fig. 9.

Ultrasound also induce BBB opening through TRPV4/ PKC-δ pathway. Co-immunoprecipitation of occludin and ZO-1 (A and C). (A) The interaction of ZO-1 and occludin is significantly decrease by ultrasound treatment. (B) Bar graphs summarizing the ratio of occludin associated with ZO-1. n = 6 rats per group. Significant differences (Student’s t test) are depicted with asterisks. p < 0.05 compared with the control group. (C) Ultrasound-induced disassociated ZO-1 and occludin is rescued by administration of rottlerin and HC067047. (D) The expression of TRPV4 is induced via ultrasound treatment, and blocked by HC067047 administration. Significant differences (ANOVA followed by LSD post hoc test) are depicted with asterisks. p < 0.05 compared with the control group, n = 4.
Discussion
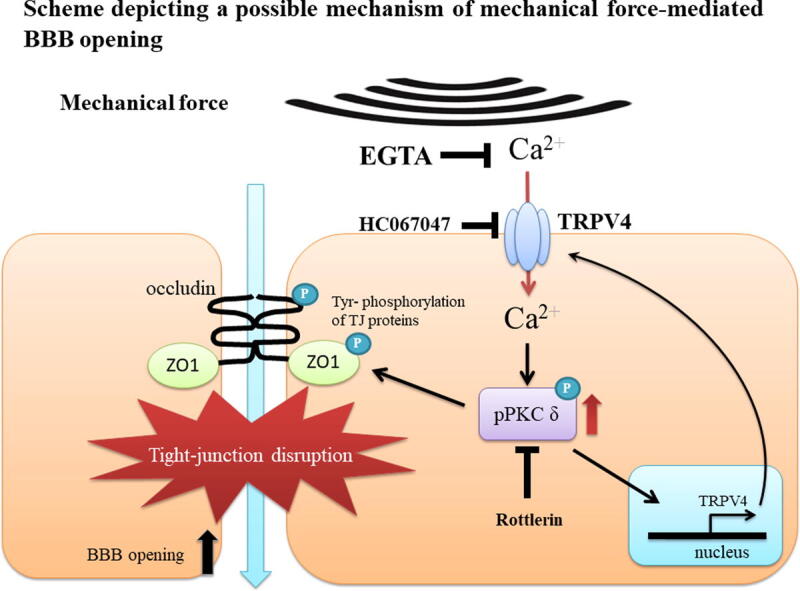
This study illustrates the role of TRPV4 in acoustic wave-mediated BBB opening and its possible mechanisms (Fig. 10). Acoustic waves stimulate TRPV4 and promote calcium influx to activate PKC-δ, which increases tyrosine phosphorylation of ZO-1 and occludin, resulting in dissociation of these tight junction proteins. Our study is the first to illustrate that both shockwave and ultrasound-induced BBB opening is modulated via TRPV4/PKC-δ mediated signaling pathway. We further show the overexpression of TRPV4 in vascular endothelial cells after acoustic waves stimulation, which was regulated by itself and PKC-δ activation, in a possible positive feedback loop. Notably, TRPV4 agonists decrease the intensity threshold of shockwave required for BBB opening, implying possible future clinical applications.
Fig. 10.

Scheme depicting a possible mechanism of mechanical wave mediated BBB opening. After mechanical wave treatment, TRPV4 is activated and triggers calcium influx into a vascular endothelial cell in the brain. Calcium induces PKC-δ activation, and then directly or indirectly phosphorylates ZO-1 and occludin proteins. The interaction between ZO-1 and occludin is disassociated by its phosphorylation, which then promotes paracellular permeability and BBB opening. Mechanical wave-mediated BBB opening is inhibited by administration of EGTA, rottlerin or HC067047. On the other hand, the expression of TRPV4 is enhanced by itself or PKC-δ activation.
It is worth noting that UCA or microbubbles were not introduced in our BBB-opening study using shockwave or ultrasound. In-vivo application of ultrasound mediated BBB opening usually requires UCA, where ultrasound-induced UCA oscillations promote cell membrane deformation and increase permeability of the endothelium [2], [9], [10], [49]. The addition of UCA increases the efficiency of BBB opening, but also increases the possibility of irreversible damage [6], [50]. The combination of ultrasound and UCA has been shown to induce apoptosis through a caspase-mediated pathway [51], [52]. UCA could produce organ hemorrhage by inducing capillary rupture through the cavitation effect, even at the intensity level of diagnostic ultrasound [53], [54]. In clinical practice, the advantages of applying shockwave or ultrasound without the addition of UCA are: (1) avoiding possible systemic side effects such as allergy, (2) lowing total cost, and (3) reducing vascular injury by cavitation. Shockwave has been shown to open the BBB without UCA [6]. However, due to its strong and long negative pressure, the cavitation effect produced by shockwaves may also result in tissue damage. Here, we propose a novel concept: activation of TRPV4 reduces the required intensity threshold of shockwaves for BBB opening, and thus may substantially reduce the possibility of tissue damage by shockwave.
There have been some interesting research in the computational study of shockwave induced BBB opening recently [55], [56]. Adhikari at al reported that when the passage of the shockwaves occurred but no bubble was present, it has almost no effect on the 2D structure of the occludin protein and will not damage the tight junction. However, when the same shockwave intensity is combined with nanobubbles, the bubble collapse will change the 2D structure of occludin protein and damaged the pairs of occludins [56]. Although microbubbles were not used in our study to increase the efficiency of shockwaves, in reality, there are gases dissolved in blood. During the period of shockwave treatment (200 times in total), the gas in the blood may participate to destroy the tight junction structure. Since the concentration and quality of the gas in the blood are not homogenized and stabilized, the shockwave cannot effectively open the BBB [6]. On the other hand, when combined with microbubbles, only 1 pulse of shockwave is enough to effectively open BBB or even cause brain injury by strong cavitation effect [57].
Our results show that the activation of TRPV4 by acoustic waves or agonist (GSK1016790A) does not affect the level of tight junction proteins in western blot (Fig. 1A, 3B, 4D and 5D) and immunostaining assays (Fig. 1C, 4E, 5E and 8). However, a previous study indicated that 4α-PDD activates TRPV4 to induce calcium influx and finally degrades or down-regulates tight junction proteins [58]. The possible explanation is that the short-term stimulus of acoustic waves is weak enough that it only affects tight junction interaction, without affecting the degradation or down-regulation. According to our previous reports, the shockwave parameters we employed in the current study did not significantly induce inflammation or apoptosis [6]. Severe brain injury which promotes calcium influx via TRPV4 activation leads to irreversible brain edema and tight junction protein degradation [20]. On the other hand, TRPV4 inhibition ameliorated BBB leakage after intracerebral hemorrhage [20], ischemia–reperfusion injury [59], and an in vitro ischemia model [25].
Following ultrasound and shockwave treatment, the expression of TRPV4 is significantly increased in a possible positive feedback loop in vascular endothelial cells (Fig. 7, Fig. 9C). Both the blockage of TRPV4 and PKC-δ activity attenuates acoustic wave-mediated TRPV4 overexpression (Fig. 7B, 7C and 9C). Our finding suggests that TRPV4 overexpression after acoustic wave treatment was regulated by itself and PKC-δ activation. Thus, this positive feedback loop will amplify signaling through the TRPV4/ PKC-δ signal transduction cascade. A previous study also shows that GSK1016790A, a TRPV4 agonist, activates TRPV4 to enhance self-expression [60]. Why the overexpression of TRPV4 is induced by acoustic waves warrants further investigation.
Molecular transport through the BBB may be realized either paracellularly or transcellularly (endocytosis/exocytosis). Previous reports have indicated that TRPV4 regulates receptor-mediated vesicular transport. The inactivation of TRPV4 decreased alpha2 macroglobin (A2M) transport [19]. In choroid plexus epithelial cells, TRPV4 activation promotes the recruitment of NA+/k+ ATPase to apical surface in an α-Klotho-dependent manner and induces the secretion of α-Klotho into cerebrospinal fluid [61]. Exosome-associated proteins expressions are up-regulated by 4α-PDD in a TRPV4-specific manner, suggesting TRPV4 mediated exocytosis [62]. In addition, the activation of TRPV4 in vivo promotes an increase in bile flow as well as ATP release and bicarbonate secretion [63]. Altogether, as a sensor, TRPV4 mediates intracellular molecular transport and extracellular secretion. This phenomenon suggests that, in this study, TRPV4 overexpression in BBB vascular endothelium in response to acoustic stimulation may also facilitate molecule transport through endocytosis and exocytosis, another possible mechanism for BBB opening. Further studies are needed. Our previous experiments indicated that after 24hr of shockwave treatment, tail vein injection Evans blue (EB) could not stain the brain, implying the BBB had closed [6]. However, this method does not explain the effect of TRPV4 on receptor-mediated vesicle transport. Although EB dye also binds to albumin as a tracer for analyzing receptor-mediated intracellular transport pathway [64], previous studies have indicated that albumin cannot transport via TRPV4-mediated receptor-mediated intracellular transport pathway [19]. Therefore, different method is needed for acoustic wave-mediated TRPV4 overexpression to analyze receptor-mediated vesicle transport infiltration in the future.
Neuron apoptosis is always a concern for brain stimulation by FUS. Early studies have shown that PKC-δ is activated by oxidative stress and induces caspase activity to cleave PKC-δ and form a constitutively active catalytic fragment (δCF). Then, the δCF is transported into the nucleus to induce apoptosis in various cell types [43], [65], [66]. In addition, ultrasound generates reactive oxygen species (ROS) and induces apoptosis via a caspase-mediated cell death pathway [67], [68]. The expression of PKC-δ is increased by ultrasound-induced calcium influx to regulate endocytosis [69]. Altogether, it is reasonable to assume that ultrasound generates ROS to activate caspase activity via PKC-δ signaling, and then caspases cleave PKC-δ to induce irreversible apoptosis and tissue disruption. Our experiments show that inhibition of PKC-δ by rottlerin prevents ultrasound-induced disassociation of ZO-1 and occludin (Fig. 9B), suggesting that PKC-δ is activated to regulate BBB opening, as previously reported [12], [40]. It is notable that activation of PKC-δ signaling can induce brain damage [12], [40], although FUS has previously been reported to promote BBB opening without apoptosis or brain damage under appropriate conditions [70], [71]. However, all reports of ultrasound-induced apoptosis in brain were analyzed by TUNEL assay, which was designed to detect apoptotic cells that undergo DNA fragmentation at late stages of apoptosis, but cannot detect early apoptosis and pro-apoptosis stage. More safety assessments are thus needed to explore the effects of FUS on apoptosis. On the other hand, shockwave did not significantly generate ROS in our preliminary experiments (data not shown) when compared with FUS, probably due to its short pulses and scarce iterations. This observation is beneficial for acoustically induced BBB opening in clinical applications. A previous study also shows that shockwave treatment did not induce the expression of oxidized proteins, NOX-1 and NOX-2 (oxidative stress indicators) in the brain [67].
One of the limitation of this study is that we only use inhibitors or agonist to analyze the related signal pathways. It will be more convincing if TRPV4 knockout mice were used. Moreover, TRPV4 channel activity is measured in bEnd.3 cell line, and not in native brain endothelium or in vivo. It would be more convincing if we use two photon laser scanning microscopy for in vivo analysis [72] or primary cerebral endothelial cells for in vitro study.
Acknowledgments
Acknowledgments
This work was supported by grant 105-2923-B-002 -001 -MY3 and 106-2314-B-002 -168 -MY3 from the Ministry of Science and Technology (MOST), NTUH.107-004075 from the National Taiwan University Hospital and BN-107-PP-03 from the National Health Research Institutes (NHRI). We thank Ai-Ling Hour for statistical analysis and the staff of the Biomedical Resource Core and imaging core at the First Core Laboratories, National Taiwan University College of Medicine, for technical assistance.
Declaration of Competing Interest
The authors have declared no conflict of interest.
Footnotes
Peer review under responsibility of Cairo University.
References
- 1.Jalali S., Huang Y., Dumont D.J., Hynynen K. Focused ultrasound-mediated bbb disruption is associated with an increase in activation of AKT: experimental study in rats. BMC Neurol. 2010;10:114. doi: 10.1186/1471-2377-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burgess A., Shah K., Hough O., Hynynen K. Focused ultrasound-mediated drug delivery through the blood-brain barrier. Expert Rev Neurother. 2015;15(5):477–491. doi: 10.1586/14737175.2015.1028369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan C.H., Ting C.Y., Lin C.Y., Chan H.L., Chang Y.C., Chen Y.Y. Noninvasive, targeted, and non-viral ultrasound-mediated GDNF-plasmid delivery for treatment of Parkinson's disease. Sci Rep. 2016;6:19579. doi: 10.1038/srep19579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tung Y.S., Vlachos F., Feshitan J.A., Borden M.A., Konofagou E.E. The mechanism of interaction between focused ultrasound and microbubbles in blood-brain barrier opening in mice. J Acoust Soc Am. 2011;130(5):3059–3067. doi: 10.1121/1.3646905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Treat L.H., McDannold N., Vykhodtseva N., Zhang Y., Tam K., Hynynen K. Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI-guided focused ultrasound. Int J Can. 2007;121(4):901–907. doi: 10.1002/ijc.22732. [DOI] [PubMed] [Google Scholar]
- 6.Kung Y., Lan C., Hsiao M.Y., Sun M.K., Hsu Y.H., Huang A.P. Focused shockwave induced blood-brain barrier opening and transfection. Sci Rep. 2018;8(1):2218. doi: 10.1038/s41598-018-20672-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheikov N., McDannold N., Sharma S., Hynynen K. Effect of focused ultrasound applied with an ultrasound contrast agent on the tight junctional integrity of the brain microvascular endothelium. Ultrasound Med Biol. 2008;34(7):1093–1104. doi: 10.1016/j.ultrasmedbio.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheikov N., McDannold N., Vykhodtseva N., Jolesz F., Hynynen K. Cellular mechanisms of the blood-brain barrier opening induced by ultrasound in presence of microbubbles. Ultrasound Med Biol. 2004;30(7):979–989. doi: 10.1016/j.ultrasmedbio.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 9.van Wamel A., Kooiman K., Emmer M., ten Cate F.J., Versluis M., de Jong N. Ultrasound microbubble induced endothelial cell permeability. J Control Release. 2006;116(2):e100–e102. doi: 10.1016/j.jconrel.2006.09.071. [DOI] [PubMed] [Google Scholar]
- 10.Lentacker I., De Cock I., Deckers R., De Smedt S.C., Moonen C.T. Understanding ultrasound induced sonoporation: definitions and underlying mechanisms. Adv Drug Deliv Rev. 2014;72:49–64. doi: 10.1016/j.addr.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Qiu L.B., Ding G.R., Li K.C., Wang X.W., Zhou Y., Zhou Y.C. The role of protein kinase C in the opening of blood-brain barrier induced by electromagnetic pulse. Toxicology. 2010;273(1–3):29–34. doi: 10.1016/j.tox.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 12.Qi X., Inagaki K., Sobel R.A., Mochly-Rosen D. Sustained pharmacological inhibition of deltaPKC protects against hypertensive encephalopathy through prevention of blood-brain barrier breakdown in rats. J Clin Invest. 2008;118(1):173–182. doi: 10.1172/JCI32636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown R.C. Calcium Signaling at the Blood-Brain Barrier in Stroke. In: Li Y.V., Zhang J.H., editors. Metal Ion in Stroke. New York, NY; Springer, New York: 2012. pp. 129–163. [Google Scholar]
- 14.Bright R., Mochly-Rosen D. The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke. 2005;36(12):2781–2790. doi: 10.1161/01.STR.0000189996.71237.f7. [DOI] [PubMed] [Google Scholar]
- 15.Rakkar K., Bayraktutan U. Increases in intracellular calcium perturb blood-brain barrier via protein kinase C-alpha and apoptosis. Biochim Biophys Acta. 2016;1862(1):56–71. doi: 10.1016/j.bbadis.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 16.Brown R.C., Mark K.S., Egleton R.D., Davis T.P. Protection against hypoxia-induced blood-brain barrier disruption: changes in intracellular calcium. Am J Physiol Cell Physiol. 2004;286(5):C1045–C1052. doi: 10.1152/ajpcell.00360.2003. [DOI] [PubMed] [Google Scholar]
- 17.Rakers C., Schmid M., Petzold G.C. TRPV4 channels contribute to calcium transients in astrocytes and neurons during peri-infarct depolarizations in a stroke model. Glia. 2017;65(9):1550–1561. doi: 10.1002/glia.23183. [DOI] [PubMed] [Google Scholar]
- 18.Shibasaki K., Ikenaka K., Tamalu F., Tominaga M., Ishizaki Y. A novel subtype of astrocytes expressing TRPV4 (transient receptor potential vanilloid 4) regulates neuronal excitability via release of gliotransmitters. J Biol Chem. 2014;289(21):14470–14480. doi: 10.1074/jbc.M114.557132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narita K., Sasamoto S., Koizumi S., Okazaki S., Nakamura H., Inoue T. TRPV4 regulates the integrity of the blood-cerebrospinal fluid barrier and modulates transepithelial protein transport. FASEB J. 2015;29(6):2247–2259. doi: 10.1096/fj.14-261396. [DOI] [PubMed] [Google Scholar]
- 20.Zhao H., Zhang K., Tang R., Meng H., Zou Y., Wu P. TRPV4 Blockade Preserves the Blood-Brain Barrier by Inhibiting Stress Fiber Formation in a Rat Model of Intracerebral Hemorrhage. Front Mol Neurosci. 2018;11:97. doi: 10.3389/fnmol.2018.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Neil R.G., Heller S. The mechanosensitive nature of TRPV channels. Pflugers Arch. 2005;451(1):193–203. doi: 10.1007/s00424-005-1424-4. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki M., Mizuno A., Kodaira K., Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278(25):22664–22668. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe H., Vriens J., Suh S.H., Benham C.D., Droogmans G., Nilius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002;277(49):47044–47051. doi: 10.1074/jbc.M208277200. [DOI] [PubMed] [Google Scholar]
- 24.Liedtke W., Choe Y., Marti-Renom M.A., Bell A.M., Denis C.S., Sali A. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103(3):525–535. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoshi Y., Okabe K., Shibasaki K., Funatsu T., Matsuki N., Ikegaya Y. Ischemic Brain Injury Leads to Brain Edema via Hyperthermia-Induced TRPV4 Activation. J Neurosci. 2018;38(25):5700–5709. doi: 10.1523/JNEUROSCI.2888-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibasaki K. TRPV4 ion channel as important cell sensors. J Anesth. 2016;30(6):1014–1019. doi: 10.1007/s00540-016-2225-y. [DOI] [PubMed] [Google Scholar]
- 27.Jie P., Lu Z., Hong Z., Li L., Zhou L., Li Y. Activation of transient receptor potential vanilloid 4 is involved in neuronal injury in middle cerebral artery occlusion in mice. Mol Neurobiol. 2016;53(1):8–17. doi: 10.1007/s12035-014-8992-2. [DOI] [PubMed] [Google Scholar]
- 28.Jie P., Tian Y., Hong Z., Li L., Zhou L., Chen L. Blockage of transient receptor potential vanilloid 4 inhibits brain edema in middle cerebral artery occlusion mice. Front Cell Neurosci. 2015;9:141. doi: 10.3389/fncel.2015.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu K.T., Huang T.C., Tsai Y.H., Yang Y.L. Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J Neurochem. 2017;140(5):718–727. doi: 10.1111/jnc.13920. [DOI] [PubMed] [Google Scholar]
- 30.Vizin R.C., Scarpellini Cda S., Ishikawa D.T., Correa G.M., de Souza C.O., Gargaglioni L.H. TRPV4 activates autonomic and behavioural warmth-defence responses in Wistar rats. Acta Physiol (Oxf). 2015;214(2):275–289. doi: 10.1111/apha.12477. [DOI] [PubMed] [Google Scholar]
- 31.Pankey E.A., Zsombok A., Lasker G.F., Kadowitz P.J. Analysis of responses to the TRPV4 agonist GSK1016790A in the pulmonary vascular bed of the intact-chest rat. Am J Physiol Heart Circ Physiol. 2014;306(1):H33–H40. doi: 10.1152/ajpheart.00303.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neckar J., Markova I., Novak F., Novakova O., Szarszoi O., Ost'adal B. Increased expression and altered subcellular distribution of PKC-delta in chronically hypoxic rat myocardium: involvement in cardioprotection. Am J Physiol Heart Circ Physiol. 2005;288(4):H1566–H1572. doi: 10.1152/ajpheart.00586.2004. [DOI] [PubMed] [Google Scholar]
- 33.Foglieni C., Fulgenzi A., Ticozzi P., Pellegatta F., Sciorati C., Belloni D. Protective effect of EDTA preadministration on renal ischemia. BMC Nephrol. 2006;7:5. doi: 10.1186/1471-2369-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewin E., Wang W., Olgaard K. Rapid recovery of plasma ionized calcium after acute induction of hypocalcaemia in parathyroidectomized and nephrectomized rats. Nephrol Dial Transplant. 1999;14(3):604–609. doi: 10.1093/ndt/14.3.604. [DOI] [PubMed] [Google Scholar]
- 35.Baraka Y.M., Bekemeier H. The influence of arrhythmogenic substances on the E.C.G. of rats calcified by high doses of vitamin D2. Arch Toxicol Suppl. 1978;1:365–368. doi: 10.1007/978-3-642-66896-8_80. [DOI] [PubMed] [Google Scholar]
- 36.Brown R.C., Davis T.P. Calcium modulation of adherens and tight junction function: a potential mechanism for blood-brain barrier disruption after stroke. Stroke. 2002;33(6):1706–1711. doi: 10.1161/01.str.0000016405.06729.83. [DOI] [PubMed] [Google Scholar]
- 37.Bleich M., Shan Q., Himmerkus N. Calcium regulation of tight junction permeability. Ann N Y Acad Sci. 2012;1258:93–99. doi: 10.1111/j.1749-6632.2012.06539.x. [DOI] [PubMed] [Google Scholar]
- 38.Keep R.F., Ulanski L.J., 2nd, Xiang J., Ennis S.R., Lorris Betz A. Blood-brain barrier mechanisms involved in brain calcium and potassium homeostasis. Brain Res. 1999;815(2):200–205. doi: 10.1016/s0006-8993(98)01155-x. [DOI] [PubMed] [Google Scholar]
- 39.Lucke-Wold B.P., Logsdon A.F., Smith K.E., Turner R.C., Alkon D.L., Tan Z. Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol. 2015;52(3):1119–1134. doi: 10.1007/s12035-014-8902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou W.H., Messing R.O. Hypertensive encephalopathy and the blood-brain barrier: is deltaPKC a gatekeeper? J Clin Invest. 2008;118(1):17–20. doi: 10.1172/JCI34516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki T., Hara H. Quercetin enhances intestinal barrier function through the assembly of zonula [corrected] occludens-2, occludin, and claudin-1 and the expression of claudin-4 in Caco-2 cells. J Nutr. 2009;139(5):965–974. doi: 10.3945/jn.108.100867. [DOI] [PubMed] [Google Scholar]
- 42.Konishi H., Yamauchi E., Taniguchi H., Yamamoto T., Matsuzaki H., Takemura Y. Phosphorylation sites of protein kinase C delta in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci USA. 2001;98(12):6587–6592. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinberg S.F. Mechanisms for redox-regulation of protein kinase C. Front Pharmacol. 2015;6:128. doi: 10.3389/fphar.2015.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michalick L., Erfinanda L., Weichelt U., van der Giet M., Liedtke W., Kuebler W.M. Transient receptor potential Vanilloid 4 and serum glucocorticoid-regulated kinase 1 are critical mediators of lung injury in overventilated mice in vivo. Anesthesiology. 2017;126(2):300–311. doi: 10.1097/ALN.0000000000001443. [DOI] [PubMed] [Google Scholar]
- 45.Mochizuki T., Sokabe T., Araki I., Fujishita K., Shibasaki K., Uchida K. The TRPV4 cation channel mediates stretch-evoked Ca2+ influx and ATP release in primary urothelial cell cultures. J Biol Chem. 2009;284(32):21257–21264. doi: 10.1074/jbc.M109.020206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suresh K., Servinsky L., Jiang H., Bigham Z., Yun X., Kliment C. Reactive oxygen species induced Ca(2+) influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2018;314(5):L893–L907. doi: 10.1152/ajplung.00430.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown R.C., Wu L., Hicks K., O'Neil R.G. Regulation of blood-brain barrier permeability by transient receptor potential type C and type v calcium-permeable channels. Microcirculation. 2008;15(4):359–371. doi: 10.1080/10739680701762656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szarka N., Pabbidi M.R., Amrein K., Czeiter E., Berta G., Pohoczky K. Traumatic brain injury impairs myogenic constriction of cerebral arteries: role of mitochondria-derived H2O2 and TRPV4-dependent activation of bkca channels. J Neurotrauma. 2018 doi: 10.1089/neu.2017.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meairs S. Facilitation of drug transport across the blood-brain barrier with ultrasound and microbubbles. Pharmaceutics. 2015;7(3):275–293. doi: 10.3390/pharmaceutics7030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kovacs Z.I., Kim S., Jikaria N., Qureshi F., Milo B., Lewis B.K. Disrupting the blood-brain barrier by focused ultrasound induces sterile inflammation. Proc Natl Acad Sci USA. 2017;114(1):E75–E84. doi: 10.1073/pnas.1614777114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feril L.B., Jr., Kondo T., Zhao Q.L., Ogawa R., Tachibana K., Kudo N. Enhancement of ultrasound-induced apoptosis and cell lysis by echo-contrast agents. Ultrasound Med Biol. 2003;29(2):331–337. doi: 10.1016/s0301-5629(02)00700-7. [DOI] [PubMed] [Google Scholar]
- 52.Honda H., Zhao Q.L., Kondo T. Effects of dissolved gases and an echo contrast agent on apoptosis induced by ultrasound and its mechanism via the mitochondria-caspase pathway. Ultrasound Med Biol. 2002;28(5):673–682. doi: 10.1016/s0301-5629(02)00509-4. [DOI] [PubMed] [Google Scholar]
- 53.Wible J.H., Jr., Galen K.P., Wojdyla J.K., Hughes M.S., Klibanov A.L., Brandenburger G.H. Microbubbles induce renal hemorrhage when exposed to diagnostic ultrasound in anesthetized rats. Ultrasound Med Biol. 2002;28(11):1535–1546. doi: 10.1016/s0301-5629(02)00651-8. [DOI] [PubMed] [Google Scholar]
- 54.Miller D.L., Dou C., Wiggins R.C. Glomerular capillary hemorrhage induced in rats by diagnostic ultrasound with gas-body contrast agent produces intratubular obstruction. Ultrasound Med Biol. 2009;35(5):869–877. doi: 10.1016/j.ultrasmedbio.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goliaei A., Adhikari U., Berkowitz M.L. Opening of the blood-brain barrier tight junction due to shock wave induced bubble collapse: a molecular dynamics simulation study. ACS Chem Neurosci. 2015;6(8):1296–1301. doi: 10.1021/acschemneuro.5b00116. [DOI] [PubMed] [Google Scholar]
- 56.Adhikari U., Goliaei A., Berkowitz M.L. Nanobubbles, cavitation, shock waves and traumatic brain injury. Phys Chem Chem Phys. 2016;18(48):32638–32652. doi: 10.1039/c6cp06704b. [DOI] [PubMed] [Google Scholar]
- 57.Huang A.P., Lai D.M., Hsu Y.H., Kung Y., Lan C., Yeh C.S. Cavitation-induced traumatic cerebral contusion and intracerebral hemorrhage in the rat brain by using an off-the-shelf clinical shockwave device. Sci Rep. 2019;9(1):15614. doi: 10.1038/s41598-019-52117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reiter B., Kraft R., Gunzel D., Zeissig S., Schulzke J.D., Fromm M. TRPV4-mediated regulation of epithelial permeability. FASEB J. 2006;20(11):1802–1812. doi: 10.1096/fj.06-5772com. [DOI] [PubMed] [Google Scholar]
- 59.Xie H., Lu W.C. Inhibition of transient receptor potential vanilloid 4 decreases the expressions of caveolin-1 and caveolin-2 after focal cerebral ischemia and reperfusion in rats. Neuropathology. 2018 doi: 10.1111/neup.12469. [DOI] [PubMed] [Google Scholar]
- 60.Adapala R.K., Thoppil R.J., Ghosh K., Cappelli H.C., Dudley A.C., Paruchuri S. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene. 2016;35(3):314–322. doi: 10.1038/onc.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imura A., Tsuji Y., Murata M., Maeda R., Kubota K., Iwano A. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316(5831):1615–1618. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- 62.Lee W.H., Choong L.Y., Jin T.H., Mon N.N., Chong S., Liew C.S. TRPV4 plays a role in breast cancer cell migration via Ca(2+)-dependent activation of AKT and downregulation of E-cadherin cell cortex protein. Oncogenesis. 2017;6(5) doi: 10.1038/oncsis.2017.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gradilone S.A., Masyuk A.I., Splinter P.L., Banales J.M., Huang B.Q., Tietz P.S. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci USA. 2007;104(48):19138–19143. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kinugasa S., Tojo A., Sakai T., Tsumura H., Takahashi M., Hirata Y. Selective albuminuria via podocyte albumin transport in puromycin nephrotic rats is attenuated by an inhibitor of NADPH oxidase. Kidney Int. 2011;80(12):1328–1338. doi: 10.1038/ki.2011.282. [DOI] [PubMed] [Google Scholar]
- 65.Reyland M.E. Protein kinase Cdelta and apoptosis. Biochem Soc Trans. 2007;35(Pt 5):1001–1004. doi: 10.1042/BST0351001. [DOI] [PubMed] [Google Scholar]
- 66.Kato K., Yamanouchi D., Esbona K., Kamiya K., Zhang F., Kent K.C. Caspase-mediated protein kinase C-delta cleavage is necessary for apoptosis of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2009;297(6):H2253–H2261. doi: 10.1152/ajpheart.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhong W., Chen X., Jiang P., Wan J.M., Qin P., Yu A.C. Induction of endoplasmic reticulum stress by sonoporation: linkage to mitochondria-mediated apoptosis initiation. Ultrasound Med Biol. 2013;39(12):2382–2392. doi: 10.1016/j.ultrasmedbio.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 68.Honda H., Kondo T., Zhao Q.L., Feril L.B., Jr., Kitagawa H. Role of intracellular calcium ions and reactive oxygen species in apoptosis induced by ultrasound. Ultrasound Med Biol. 2004;30(5):683–692. doi: 10.1016/j.ultrasmedbio.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 69.Lee J.L., Lo C.W., Inserra C., Bera J.C., Chen W.S. Ultrasound enhanced PEI-mediated gene delivery through increasing the intracellular calcium level and PKC-delta protein expression. Pharm Res. 2014;31(9):2354–2366. doi: 10.1007/s11095-014-1332-4. [DOI] [PubMed] [Google Scholar]
- 70.Fan C.H., Liu H.L., Ting C.Y., Lee Y.H., Huang C.Y., Ma Y.J. Submicron-bubble-enhanced focused ultrasound for blood-brain barrier disruption and improved CNS drug delivery. PLoS ONE. 2014;9(5) doi: 10.1371/journal.pone.0096327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDannold N., Arvanitis C.D., Vykhodtseva N., Livingstone M.S. Temporary disruption of the blood-brain barrier by use of ultrasound and microbubbles: safety and efficacy evaluation in rhesus macaques. Can Res. 2012;72(14):3652–3663. doi: 10.1158/0008-5472.CAN-12-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Russell J.T. Imaging calcium signals in vivo: a powerful tool in physiology and pharmacology. Br J Pharmacol. 2011;163(8):1605–1625. doi: 10.1111/j.1476-5381.2010.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]