

Abstract
The formation and maintenance of synapses require long-distance delivery of newly synthesized synaptic proteins from the soma to distal synapses, raising the fundamental question of whether impaired transport is associated with neurodevelopmental disorders such as autism. We previously revealed that syntabulin acts as a motor adaptor linking kinesin-1 motor and presynaptic cargos. Here, we report that defects in syntabulin-mediated transport and thus reduced formation and maturation of synapses are one of core synaptic mechanisms underlying autism-like synaptic dysfunction and social behavioral abnormalities. Syntabulin expression in the mouse brain peaks during the first two weeks of postnatal development and progressively declines during brain maturation. Neurons from conditional syntabulin−/− mice (stb cKO) display impaired transport of presynaptic cargos, reduced synapse density and active zones, and altered synaptic transmission and long-term plasticity. Intriguingly, stb cKO mice exhibit core autism-like traits, including defective social recognition and communication, increased stereotypic behavior, and impaired spatial learning and memory. These phenotypes establish a new mechanistic link between reduced transport of synaptic cargos and impaired maintenance of synaptic transmission and plasticity, contributing to autism-associated behavioral abnormalities. This notion is further confirmed by the human missense variant STB-R178Q, which is found in an autism patient and loses its adaptor capacity for binding kinesin-1 motors. Expressing STB-R178Q fails to rescue reduced synapse formation and impaired synaptic transmission and plasticity in stb cKO neurons. Altogether, our study suggests that defects in syntabulin-mediated transport mechanisms underlie the synaptic dysfunction and behavioral abnormalities that bear similarities to autism.
Keywords: autism, axonal transport, presynaptic cargos, social novelty, synapse formation, synaptic dysfunction, syntabulin
One Sentence Summary:
Xiong et al report that altered axonal transport of presynaptic cargos is one of the core mechanisms underlying autism-like synaptic dysfunction and social behavioral abnormalities.
Introduction
The formation of new synapses and maintenance and remodeling of mature synapses requires seamless integration of axonal transport of presynaptic cargos [1, 2]. Since synapses are distant from the neuronal cell body, newly synthesized presynaptic components must be transported from the soma to the presynaptic terminals driven by kinesin motors moving along lengthy axonal processes [3, 4]. Among these presynaptic components is the scaffolding protein Bassoon, which functions as an organizer of the active zone (AZ) [5, 6] and appears first at newly formed presynaptic terminals [7, 8]. Bassoon undergoes axonal transport in 80-nm dense-core organelles containing multiple components of the AZ, thus ensuring formation and maintenance of presynaptic terminals [9, 10]. However, the fundamental question of whether impaired axonal transport of these presynaptic cargos contributes to synaptic dysfunction and pathology in neurodevelopmental disorders remains largely unaddressed.
Autism spectrum disorders (ASDs) are a group of childhood-onset neurodevelopmental disorders characterized by impaired social interactions and communication along with restricted interests and stereotyped repetitive behaviors [11]. Although ASDs have a strong genetic basis [12, 13], the underlying cellular and neurological mechanisms are largely unknown in most ASD cases. De novo missense mutations have been hypothesized to underlie a substantial fraction of risk for developing sporadic ASDs [14, 15]. Several ASD-associated genes have been linked to impaired synaptic transmission and characterized in the formation of postsynaptic adhesion structures, dendritic spines, and synaptic contacts during neuronal development [16–20]. While postsynaptic mechanisms play an important role in the susceptibility to ASDs [21, 22], it remains unknown whether altered axonal transport of presynaptic cargos, and thus reduced formation, maturation, and maintenance of presynaptic terminals, contributes to ASD-linked pathogenesis. Investigations into axonal transport mechanisms in an in vivo model system will provide an important tool to identify core presynaptic defects at the onset of ASDs.
Syntabulin (STB) is a kinesin-1 (KIF5) motor adaptor that selectively links the motor to transport cargos containing Bassoon and multiple presynaptic components, therefore contributing to presynaptic assembly and maintenance in cultured neurons [23]. However, the role of STB in an in vivo CNS system, particularly in regulating synaptic density in young adult mice, maintaining maturation of active zones, modulating synaptic transmission and long-term plasticity in hippocampal circuits, and mediating social behavioral phenotypes remains unknown. Interestingly, the stb gene maps to 8q23.2 within the autism susceptibility loci 8q22–24 [24–26]. A recent whole exome sequencing study identified a de novo STB missense variant in a human autism patient [27]. Thus, there is an urgent and critical need to establish a mechanistic link between impaired transport of synaptic cargos and ASD-like impairment of social interactions and communication. Generating a brain-specific conditional stb cKO mouse line provides an in vivo model system to investigate synaptic and social behavioral abnormalities, thereby advancing our understanding of how transport mechanisms contribute to altered social interactions and communication.
Results
Loss of STB impairs axonal transport and reduces synapse formation
To investigate whether impaired axonal transport of presynaptic cargos is associated with ASD-like pathogenesis, we generated a Nestin-Cre; stbLoxP/LoxP cKO mouse, in which brain-selective inactivation of the stb gene was achieved by deleting Exon 5, leading to a frameshifting that generates a premature stop codon in exon 6 after crossing stbLoxP/LoxP and Nestin-Cre mice (Fig. S1a). Immunoblots of hippocampal homogenates indicate a loss of STB expression in the cKO mice (Fig. S1b). The cKO animals were viable and fertile with a normal life span and Mendelian ratios at weaning. However, the cKO mice displayed a slightly reduced body weight starting at postnatal day 21 (P21) (Figs. S1c and d), possibly due to an inability to compete for food and water during early development. The cKO animals showed normal gross morphology in somatosensory cortex, caudate putamen, dentate gyrus, and CA1 region of the hippocampus (Figs. S1e–j). Loss of STB expression in cKO brains was further confirmed in the cerebral cortex, hippocampus, cerebellum, and spinal cords isolated from cKO mice at 2.5 months of age (Fig. S2a).
To examine whether deleting the stb gene impairs synapse formation and maturation, we cultured hippocampal neurons from E18 control and cKO embryos. While cKO neurons differentiated normally in culture, the relative density of Bassoon-labeled presynaptic cargos (p<0.001) and synaptophysin-labeled presynaptic terminals (p<0.001) was consistently reduced along axonal processes at 15 days in vitro (DIV15) (Figs. 1a, b and d). In addition, the density of PSD95-labeled postsynaptic sites along dendritic profiles was also reduced (p<0.001) in cKO neurons (Figs. 1c and d), indicating impaired synapse formation. To characterize axonal transport, we applied dorsal root ganglion (DRG) neurons, which provide an ideal model system for studying axonal transport because (1) almost all neurites are tau-positive axons [28] and (2) neurons are directly derived from young adult mice where STB expression is developmentally regulated. STB depletion was confirmed in cKO DRG neurons at DIV3 isolated from P50 cKO mice (Fig. S2b). Live imaging of DRG neurons isolated from adult P50 mice revealed a selective reduction of anterograde but not retrograde transport of Bassoon-labeled presynaptic cargos along axons of cKO neurons. Axonal delivery of Bassoon cargos, as measured by Bassoon influx events (100 μm in 3 min) (p=0.001) and anterograde transport towards the distal tip (p=0.001), were impaired in DRG neurons with STB depletion (Figs. 1e and f, Videos S1 and 2). Defective axonal transport was confirmed in developing hippocampal neurons at DIV7, which showed a selective reduction of anterograde but not retrograde transport of Bassoon-labeled presynaptic cargos along axons of cKO neurons when compared to control neurons (p<0.001) (Figs. 1g and S2c).
Fig 1. stb cKO neurons display impaired axonal transport of presynaptic cargos, reduced density of synapses and spines, and altered structures of AZs.
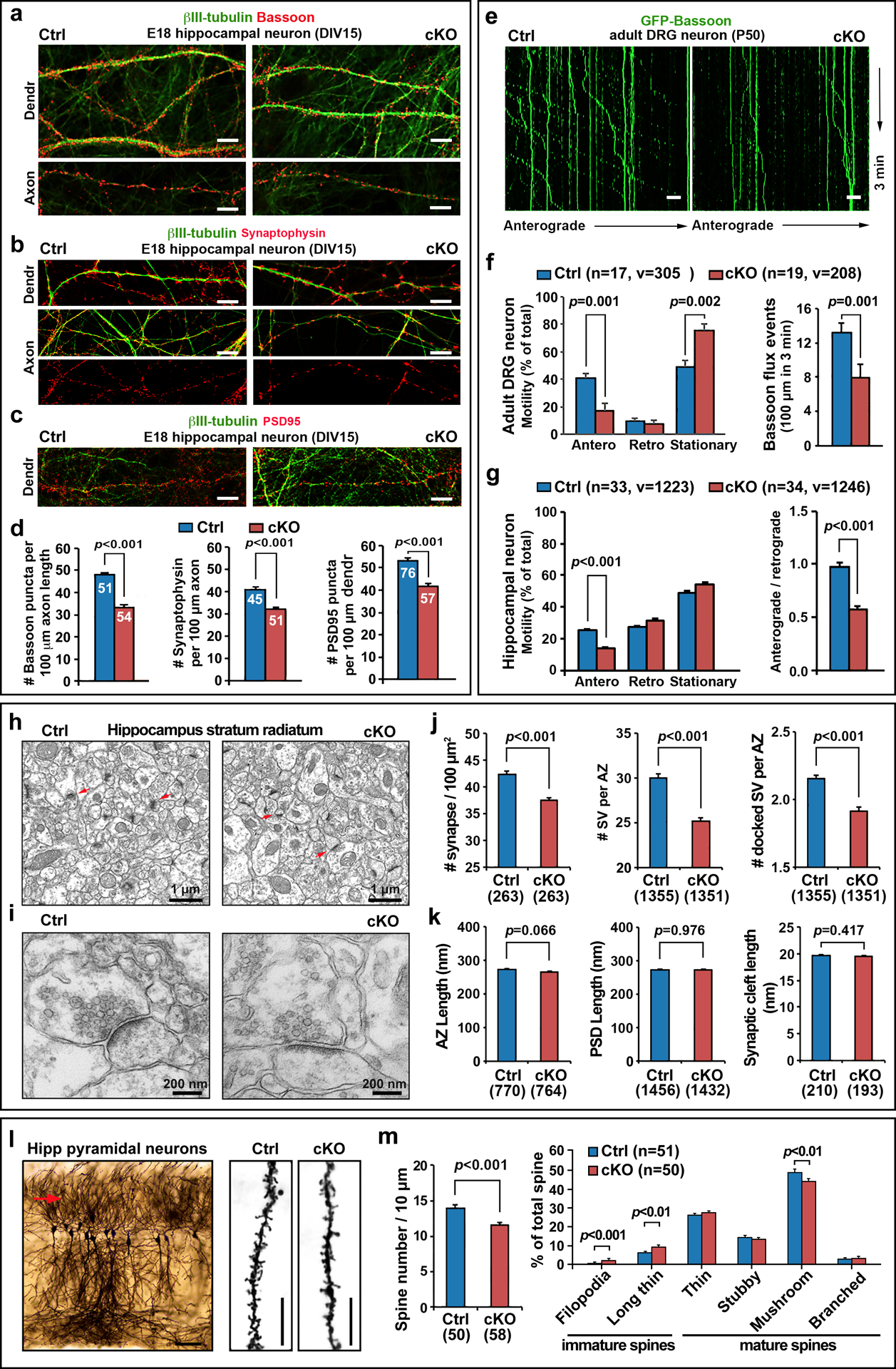
a-d, Reduced density of Bassoon (p<0.001) (a) and synaptophysin (p<0.001) (b) along axons and PSD95 (p<0.001) along dendritic profiles (c) in stb cKO hippocampal neurons. Neurons cultured from E18 control and cKO mouse embryos were co-immunostained at DIV15 with the antibodies indicated. e, f, Reduced axonal delivery of Bassoon-labeled presynaptic cargos in DRG neurons isolated from adult cKO mice at P50 (anterograde transport, p=0.001; flux rate, p=0.001). DRG neurons at DIV0 were transfected with GFP-Bassoon and time-lapse imaged at DIV3–4 to measure the motility and axonal flux rate of Bassoon organelles (f). In kymographs (e), vertical lines represent stationary organelles; oblique lines or curves to the right indicate anterograde transport from the soma towards distal terminals (also see Videos S1 and 2). g, Selective reduction of anterograde (p<0.001), but not retrograde, transport of Bassoon-labeled presynaptic cargos in cKO hippocampal neurons. Neurons were transfected with GFP-Bassoon at DIV3 and time-lapse imaged at DIV7. h-k, Representative electron micrographs (h, i) and quantitative analyses (j, k) showing reduced density of AZs (red arrows) in 100-μm2 micrograph images (h, j) and reduced total and docked synaptic vesicles per AZ (i, j) from the hippocampus stratum radiatum in cKO mice at 2.5-months of age. l, m, Golgi silver impregnation and quantitative analysis of spine density and maturation in CA1 pyramidal neurons of cKO mice. The red arrow (l) indicates distal basal dendrites (>60 μm from soma) where the spine density was counted. Spine length was defined as the distance from the tip of the spine head to the interface with the dendritic stalk. Filopodia (>2 μm) and long-thin spines (>1 μm in length and <0.2 μm in head diameter) were counted as immature spines, while thin, stubby, mushroom and branched spines were counted as mature spines. Note that both the spine density and percentage of mature spines (mushroom) were reduced (p<0.001, p<0.01, respectively) in stb cKO mice. Data was collected from the total number of axons or dendrites indicated within bars (d) and in parentheses (m), or the number of vesicles (v) in the total number of neurons (n) as indicated in parentheses (f, g) from 3 independent experiments, and presented as mean ± sem. Electron micrograph data were collected from the total number of electron micrographs (j, left panel) and total number of AZs indicated below the bars (j, k). The difference between genotypes was analyzed by an unpaired two-tailed Student’s t-test. Scale bars: 50 μm (l left), 10 μm (a-c, e, l right), 1 μm (h), and 200 nm (i).
Electron micrographs (EMs) revealed striking ultrastructural changes in AZs of hippocampal CA1 stratum radiatum of cKO mice at 2.5 months of age, which showed decreased AZ density (p<0.001) and reduced number of total (p<0.001) and docked (p<0.001) SVs per AZ (Figs. 1h–j) without detectable changes in the average lengths of the AZ, postsynaptic density (PSD), and synaptic cleft (Fig. 1k). During development, dendritic spines undergo activity-dependent maturation and remodeling [29, 30]. To determine whether reduced presynaptic terminals and impaired AZ structures affect the formation and maturation of dendritic spines, we performed Golgi staining of CA1 pyramidal neurons. Filopodia (>2μm) and long-thin spines (>1μm in length and <0.2μm in head diameter) were counted as immature spines, while thin, stubby, mushroom and branched spines were counted as mature spines. Both the total spine density and percentage of mature spines were significantly reduced (p<0.001, p<0.05, respectively) in the cKO mice (Figs. 1l and m). These observations are consistent with a recent in vivo live imaging study demonstrating that altered spine maintenance is a common phenotype in different autism mouse models [31].
In WT mouse brains, STB expression peaks at P0–14 and then undergoes a progressive decline during brain maturation from P14 to 8 months (Figs. S2d–g), an expression pattern consistently observed in various brain regions including the cerebral cortex, hippocampus, cerebellum, and spinal cord. Both STB and Bassoon in the cerebral cortex and hippocampus were readily detected between P7/14 and 2 months, a critical stage for synapse formation and maturation during brain development [32, 33]. We further confirmed reduced presynaptic distribution of Bassoon using synapse-enriched synaptosomal preparations from 2.5-month-old adult cKO mouse brains. Quantitative analyses of immunoblots demonstrated that Bassoon was depleted (p=0.039) in synapses from three pairs of the cKO mouse brains (Figs. S3a and b), further suggesting synaptic alternations after deleting the stb gene. However, in whole brain homogenates of adult stb cKO mice, Bassoon was not significantly reduced (Figs. S3c and d), confirming selective defects in the axonal delivery and synaptic targeting of Bassoon without affecting total Bassoon expression in stb cKO mouse brains. Given that Bassoon is one key presynaptic scaffolding protein [5, 6], its depletion in synaptosomal preparations is consistent with reduced density of presynapses and altered structures of AZs.
We further examined the relative enrichment of neurotransmitter receptors and transporters in synaptosomal preparations by blotting for GluR1/2, NR1, 2A, 2B, VGLUT1, and VGAD67. Among these receptors, only the AMPAR subunit GluR2 displayed a mild but significant reduction in synaptosomes (p=0.034), but not in total brain homogenates, indicating impaired synaptic delivery or retention (Figs. S3a–d). STB was also reported to mediate protein interacting with C-kinase 1 (PICK1)-containing vesicular transport [34]. PICK1 plays a critical role in the surface trafficking of AMPA receptors associated with synaptic plasticity including long-term potentiation (LTP) and long-term depression (LTD) [35–37]. We examined PICK1 expression and distribution in both control and stb cKO mouse brains and cortical neurons. Immunoblots from three pairs of age-matched Ctrl and stb cKO mice showed no significant change (p=0.7524) in PICK1 expression (Fig. S3e). By co-immunostaining PICK1 and the dendritic marker MAP2 in cortical neurons at DIV12, we further demonstrated that depleting the stb gene did not alter the mean intensity of PICK1 in axons or in dendrites (Figs. S3f and g). Interestingly, PICK1 vesicular structures were almost entirely stationary in dendrites throughout the duration of time-lapse recordings in developing control cortical neurons at DIV7 (data not shown), an observation supported by a previous study showing no dendritic transport of PICK1 vesicles [34]. Thus, we speculate that PICK1 is not primarily involved in dendrtic transport of postsynaptic cargos.
An autism-linked STB mutant impairs axonal transport and fails to rescue cKO phenotypes
Interestingly, in a recent collaborative autism research program with the US military, whole exome sequencing performed on 210 autism patients identified 7 clinically relevant genetic variants including a de novo missense mutation in the human stb gene that changes a conserved arginine into glutamine (R178Q) within its KIF5-binding domain (KBD) (Figs. 2a and b) [27]. To determine whether this de novo stb variant has an impact on axonal transport of presynaptic cargos, we examined whether STB-R178Q is a motor adaptor loss-of-function mutation by performing a GST pull-down assay. The STB-R178Q mutation abolished the binding capacity of STB to the cargo binding domain of KIF5 heavy chain (KHC) (p=0.004) (Fig. 2c). We further examined the physiological relevance of STB-R178Q in mediating transport of presynaptic cargos in live DRG neurons isolated from young adult mice at P40, where endogenous STB expression is reduced. Expressing STB-R178Q significantly reduced axonal Bassoon influx rate (p=0.006) and selectively impaired anterograde (p<0.001), but not retrograde (p=0.280), axonal transport of Bassoon cargos when compared to neurons expressing wild-type STB (Figs. 2d–f). Next, we performed four lines of rescue experiments in stb cKO neurons to characterize synapse formation or density following expression of wild-type STB or human autism-associated mutant STB-R178Q. First, we examined the rescue effect of STB-R178Q in stb cKO DRG neurons isolated from young adult mice. While expressing wild-type STB in cKO DRG neurons effectively rescued axonal Bassoon influx rate (p<0.001) and anterograde Bassoon transport (p<0.001), expressing STB-R178Q failed to reverse Bassoon influx rate (p=0.678) and anterograde transport (p=0.826) (Figs. 2g–i; Videos S3 and 4). Second, expressing wild-type STB in cKO cortical neurons rescued defective anterograde transport (p<0.001) (Figs. 2j–l) and reduced density (p<0.001) (Figs. 2m and n) of Bassoon-labeled presynaptic cargos along axons. However, expressing STB-R178Q failed to rescue these phenotypes. Altogether, these three rescue experiments indicate that the human autism-associated variant STB-R178Q is a loss-of-function mutation that fails to rescue cell biology phenotypes in stb cKO cortical and DRG neurons.
Fig 2. The human autism-linked STB missense variant is a loss-of-function mutant.
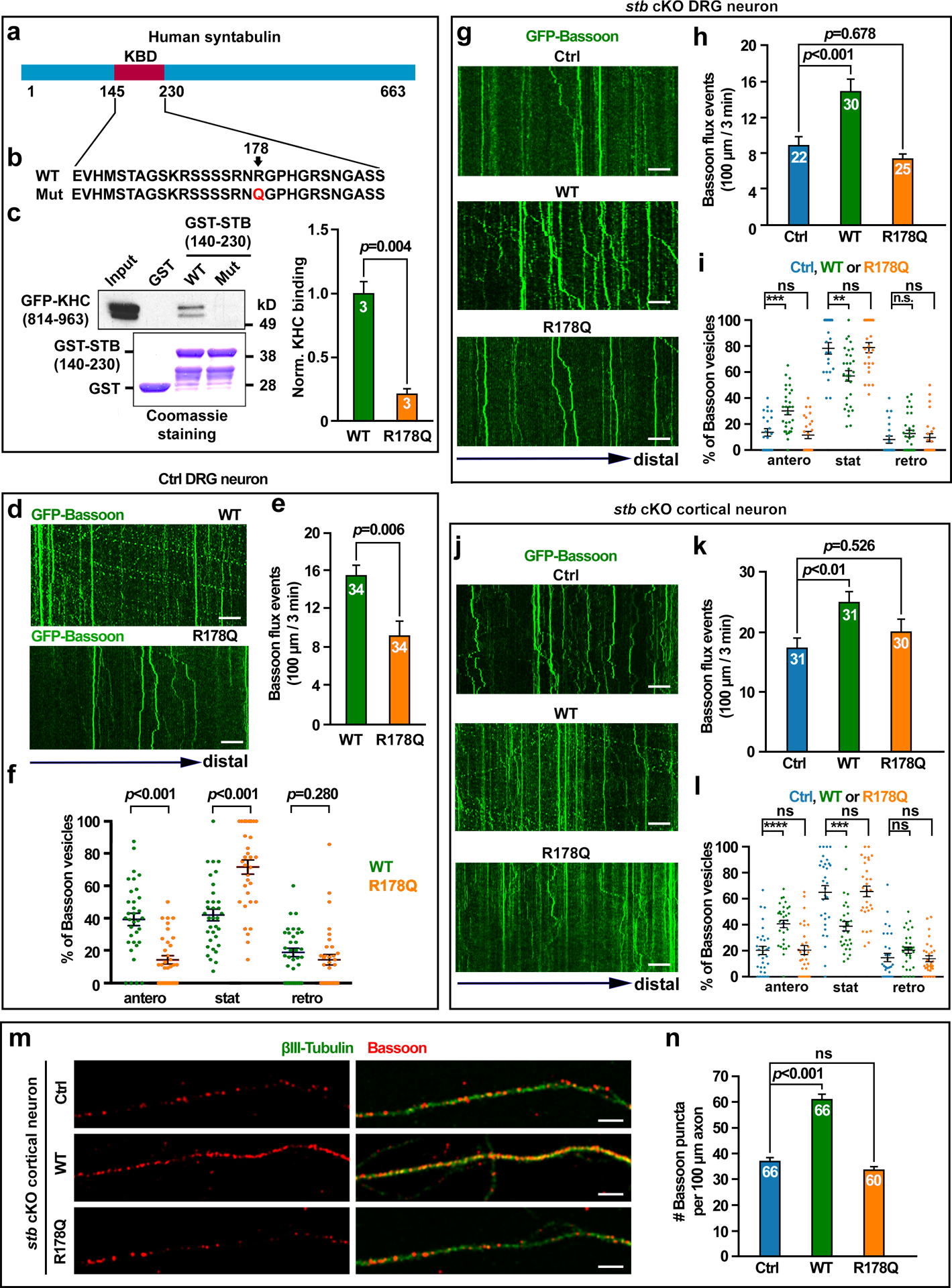
a, b, Schematic diagram of human STB (a) and the human autism-associated de novo variant R178Q (b) that changes arginine at 178 into glutamine within the KIF5-binding domain (KBD). c, GST pull-down assay showing impaired binding capacity of STB-R178Q to the KIF5 motor heavy chain (KHC) (p=0.004). GFP-tagged STB or mutant STB-R178Q (140–230), or GST alone was incubated with lysates of HEK cells expressing GFP-KHC (814–930), the cargo-binding domain of the KIF5 heavy chain. The bound protein complexes were blotted with an anti-GFP antibody. Coomassie blue staining shows loaded GST-STB or GST-STB-R178Q, or GST. Pull-down of GFP- KHC (814–930) was quantified from three repeats, calibrated and normalized to the band in the STB pull-down group. d-f, Representative kymographs (d) and quantitative analyses (e, f) showing impaired axonal delivery of Bassoon-labeled presynaptic cargos in live DRG neurons expressing mutant STB-R178Q. DRG neurons were isolated from adult mice at P40 and transfected with GFP-Bassoon together with STB or STB-R178Q at DIV0. Note that STB-R178Q selectively reduced anterograde (p<0.001) but not retrograde (p=0.280) transport of Bassoon organelles in axons (f), thus reducing flux rate into distal axons (p=0.006) (e). g-i, Representative kymographs (g) and quantitative analyses (h, i) showing failed rescue of impaired Bassoon transport in cKO DRG neurons by expressing STB-R178Q. DRG neurons were isolated from adult cKO mice at P40 and co-transfected with GFP-Bassoon and control vector, STB, or STB-R178Q at DIV0. Note that expressing STB-R178Q failed to rescue axonal flux rate (One-way ANOVA, Ctrl vector vs. STB, p<0.001; Ctrl vector vs. STB-R178Q, p=0.678) (h) and anterograde transport of presynaptic cargos (One-way ANOVA, Ctrl vector vs. STB, p<0.001; Ctrl vector vs. STB-R178Q, p=0.826) (i) (also see Videos S3 and 4). j-l, Representative kymographs (j) and quantitative analyses (k, l) showing failed rescue of defective Bassoon transport in cKO cortical neurons by expressing STB-R178Q. Embryonic cortical neurons were co-transfected with GFP-Bassoon and control vector, STB, or STB-R178Q at DIV4 and imaged at DIV6–7. Note that expressing STB-R178Q failed to rescue axonal flux rate (One-way ANOVA, Ctrl vector vs. STB, p=0.007; Ctrl vector vs. STB-R178Q, p=0.526) (k) and anterograde transport of presynaptic cargos (One-way ANOVA, Ctrl vector vs. STB, p<0.001; Ctrl vector vs. STB-R178Q, p=0.998) (l). Axonal Bassoon flux rate (100μm/3min) (e, h, k) and relative Bassoon motility (f, i, l) of anterograde (antero), retrograde (retro), or stationary (stat) were examined by time-lapse imaging at 2 sec intervals for a total of 3 min. m, n, Representative images (m) and quantitative analysis (n) showing failed rescue of reduced Bassoon density in axons of cKO cortical neurons by expressing STB-R178Q at DIV14. Note that expressing STB effectively reverses the phenotype (One-way ANOVA, Ctrl vector vs. STB, p<0.001; Ctrl vector vs. STB-R178Q, p=0.212). Data were collected from the total number of axons indicated within bars from 3 experiments and presented as mean ± sem and analyzed with an unpaired t-test with Welch’s correction (e, f), one-way ANOVA with Tukey’s multiple comparisons test (h, i, k, l) or Dunnett’s multiple comparisons test (n), or Student’s t test (c). **, p<0.01; ***, p<0.001, ns, not significant. Scale bars: 10 μm.
Given that GluR2 displayed a reduction in synaptosomes (Figs. S3a–d), we further examined whether expressing STB-R178Q in control cortical neurons induces synaptic GluR2 reduction by immunostaining for GluR2 or PSD95 at DIV15. While expressing STB-R178Q reduced the density of GluR2 (p<0.01) and PSD95 (p<0.001) compared to neurons expressing control vector, the mean intensity of GluR2 was not reduced (p>0.05) (Fig. S3h). These results suggest that STB primarily contributes to synapse formation or maintenance, although we could not exclude the possibility that the delivery of a specific set of postsynaptic cargos containing GluR2 was also affected by deleting the stb gene.
STB was also reported as a KIF5 motor adaptor driving mitochondrial transport in neurons [38]. We examined axonal mitochondrial density and presynaptic mitochondrial distribution in hippocampal neurons at DIV14. Although presynaptic density was reduced (p<0.001) in stb cKO neurons, there was no significant change in axonal mitochondrial density using two mitochondrial markers DsRed-Mito (p>0.05) and TOM20 (p>0.05), nor was presynaptic distribution of mitochondria changed (p>0.05) (Figs. S4a–d). We further examined the mitochondrial distribution in cortical neurons expressing STB-R178Q. Neurons were infected with a lentivirus encoding STB-R178Q at DIV4, followed by co-immunostaining for TOM20 and βIII-tubulin at DIV10. Consistently, no significant change was observed in mitochondrial density in axons expressing STB-R178Q (p=0.0774) compared to neurons expressing control vector (Figs. S4e and f). Mitochondrial receptors Miro1/2 and adaptors Trak1/2 likely compensate for genetic STB loss in driving mitochondrial transport into axons in stb cKO neurons [39]. These data further support our notion that STB-R178Q is a loss-of-function but not a dominant-negative mutant that is unable to compete with Miro1/2 and Trak1/2 in kinesin-driven mitochondrial transport.
Mice with loss of STB exhibit autism-like synaptic dysfunction and impaired plasticity
Next, we characterized synaptic transmission and plasticity by focusing on hippocampal Schaffer collateral–CA1-pyramidal (SC–CA1) synapses, a well-studied brain area implicated in social behavior [40–42]. CA1 pyramidal cells from age-matched male control and cKO mice (8–10 weeks) displayed similar mean amplitudes of both miniature excitatory postsynaptic currents (mEPSCs) and the miniature inhibitory postsynaptic currents (mIPSCs) (Figs. 3a, b, and S5a), suggesting normal quantal content or postsynaptic function in cKO mice. However, the frequency of both mEPSCs and mIPSCs were significantly decreased in cKO mice (p=0.002, p=0.032, respectively), suggesting a similar reduction in the density of excitatory and inhibitory synapses. This notion is supported by EM analyses showing reduced presynaptic density and docked synaptic vesicles per AZ (Figs. 1h–k). In addition, input–output (I/O) curves of field EPSPs at SC-CA1 synapses displayed a decreased amplitude across a series of stimulation intensities (Two-way ANOVA, genotype: F1,48=4.648, p=0.036) and a slightly downward shift (Fig. 3c), indicating a reduction in basal synaptic transmission in cKO mice. In contrast, the intrinsic excitability of pyramidal neurons was rarely affected (Figs. S5b, d and e) and the NMDAR/AMPAR ratio was unaltered in cKO mice (Fig. S5c), suggesting no detectable effect of stb deletion on the equilibrium between NMDAR and AMPAR components. Intriguingly, long-term potentiation (LTP) induced by high-frequency stimulation was severely impaired (p=0.006) (Fig. 3d) and long-term depression (LTD) induced by low-frequency stimulation was almost abolished in cKO mice (p=0.003) (Fig. 3e). Because LTD induced by low-frequency stimulation activates both NMDARs and mGluRs, we isolated mGluR-dependent LTD by bath-applying the mGluR5 agonist (RS)-3,5-dihydroxyphenylglycine (DHPG), which demonstrated that mGluR LTD was unaffected by deleting the stb gene (Fig. S5f).
Fig 3. Stb cKO mice exhibit deficits in synaptic transmission and long-term plasticity.
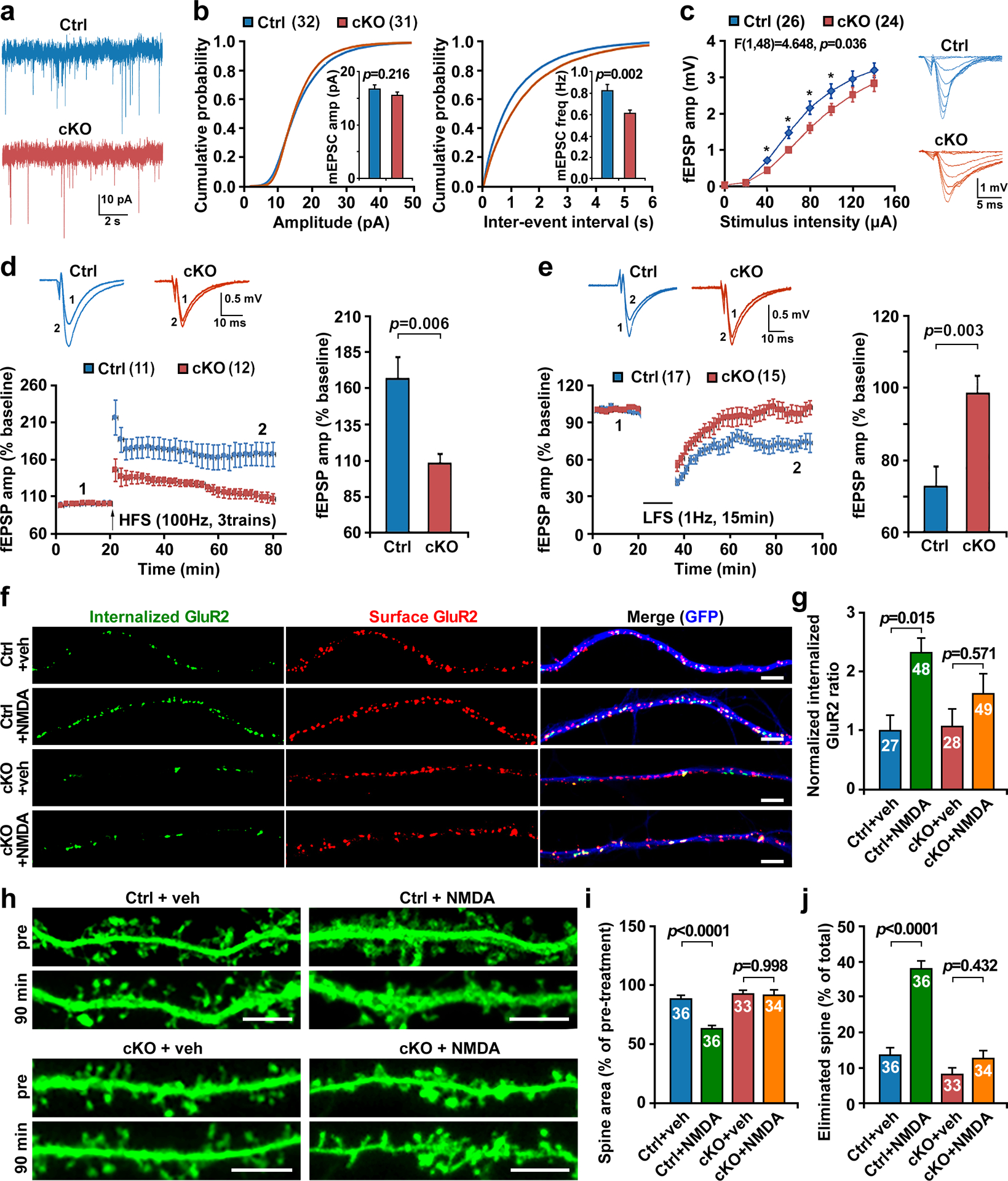
a, b, Representative mEPSC traces (a) and cumulative probability plots of mEPSC amplitudes and inter-event intervals (b) in CA1 pyramidal neurons from acute hippocampal slices of age-matched (8–10 weeks) male control and cKO mice. Note that mEPSC frequency (p=0.002) but not amplitude was decreased in cKO neurons (Ctrl, 32 neurons from 5 mice; cKO, 31 neurons from 4 mice). c, Representative traces (right) and summary plots (left) showing down-shifted input-output in the cKO hippocampal SC-CA1 pathway by extracellular field recording (Ctrl, 26 slices from 3 mice; cKO, 24 slices from 3 mice). fEPSPs were recorded by stimulating the SC-CA1 pathway with increasing intensities as indicated. Two-way ANOVA revealed a main effect of genotype (F1, 48=4.648, p=0.036). d, Impaired LTP of the cKO hippocampal SC-CA1 pathway by extracellular field recordings (Ctrl, 11 slices from 4 mice; cKO, 12 slices from 5 mice). Representative traces (upper left) were taken before (1) and 50 min after (2) HFS and normalized fEPSP amplitudes were plotted every 2 min (lower left). LTP was induced by high-frequency stimulation (HFS: 100Hz, 3 trains). The mean ± sem was plotted for the magnitude of LTP at the last 10 min post-induction (p=0.006, right). e, Impaired LTD of the cKO hippocampal SC-CA1 pathway (Ctrl, 17 slices from 6 mice; cKO, 15 slices from 5 mice). Representative traces (upper left) were taken before (1) and 50 min after (2) LFS and normalized fEPSP amplitudes were plotted every 2 min (lower left). LTD was induced by low-frequency stimulation (LFS: 1Hz, 15 min). The mean ± sem was plotted for the magnitude of LTD at the last 10 min post-induction (p=0.003, right). f, g, Representative images (f) and quantitative analysis (g) showing impaired NMDA (50 μM, 10 min)-induced internalization of AMPAR subunit GluR2 in cKO hippocampal neurons at DIV17–18 (One-way ANOVA, Ctrl: veh vs. NMDA, p=0.015; cKO: veh vs. NMDA, p=0.571). The intensity of internalized GluR2 were normalized to the total intensity of internalized and surface GluR2. h-j, Representative images (h) and quantitative analysis (i, j) showing impaired NMDA-induced dendritic spine remodeling in cKO hippocampal neurons at DIV17–20. Control or stb cKO neurons were transfected with GFP vector at DIV10, followed by live imaging at DIV17–20 at 37°C. Neurons were then stimulated with 50 μM NMDA for 5 min, followed by a 90-min washout period and fixed. The same neurons were re-imaged where the spines in the secondary dendrites were analyzed using ImageJ to evaluate the spine morphological alterations before and after NMDA treatment. At least two secondary dendrites starting 30–150 μm away from the soma were analyzed for each neuron. Data were collected from the total number of dendrites indicated within bars (g, i, j) from 3 experiments and presented as mean ± sem. The difference between genotypes was analyzed by an unpaired two-tailed Student’s t-test (b, d, e), two-way ANOVA with genotype and treatment as independent variables (c), or one-way ANOVA with Tukey’s multiple comparisons test (g, i, j). Scale bars: 5 μm (f, h).
Synaptic plasticity is predominantly expressed through changes in the number, location, and properties of postsynaptic receptors [43]. While LTP involves the insertion of more AMPARs into the synapse, LTD may involve the removal or endocytosis of synaptic AMPARs [44]. Since NMDAR-dependent LTD is impaired in hippocampal slices from stb cKO mice, we sought to examine AMPA receptor internalization using an antibody feeding assay to analyze the endocytosis of AMPA receptor subunit GluR2. The rationale for examining GluR2 is that (1) GluR2 displays a significant reduction in synaptosomes isolated from stb cKO mice (Figs S3a–d); and (2) expressing STB-R178Q in control neurons reduced the density of GluR2 (p<0.01) compared to neurons expressing GFP control (Fig. S3h). Cultured hippocampal neurons at DIV17–18 were stimulated with NMDA (50 μM for 10 min) to induce “chemical LTD,” which shares a similar molecular mechanism with electrically induced LTD [45]. NMDA-induced GluR2 internalization was significantly increased in control neurons, but not in stb cKO neurons (One-way ANOVA, Ctrl: veh vs. NMDA, p=0.015; cKO: veh vs. NMDA, p=0.571) (Figs. 3f and g). It was reported that PICK1 interacts with GluR2 and this interaction is required for NMDAR-dependent endocytosis of synaptic GluR2 [46–48]. Given that STB has been reported to interact with PICK1 [34], we speculate that STB may also regulate PICK1-mediated AMPAR endocytosis during LTD.
Activity-dependent modification of dendritic spines is an important cellular mechanism for brain development and cognition. The size and geometry of dendritic spines are coupled to synaptic strength [49, 50] and modulated during synapse development and synaptic plasticity [51]. NMDAR-LTD has been reported to be accompanied by prolonged remodeling of spines, including spine shrinkage and loss [52, 53]. Considering educed spine maturation (Fig. 1m) and impaired LTD in hippocampal slices from stb cKO mice (Fig. 3e), we sought to examine whether the long-lasting spine plasticity associated with LTD was also impaired in stb cKO mice. We monitored dendritic spine morphology under a “chemical LTD” condition stimulated by NMDA (50 μM for 5 min) in cultured hippocampal neurons at DIV17–20. By comparing dendritic spines before and 90 min after NMDA treatment, we found that NMDA treatment in control neurons induced robust spine shrinkage (p<0.0001) and spine retraction (p<0.0001). However, the same treatment in stb cKO neurons failed to induce such spine plasticity; NMDA treatment did not induce spine shrinkage (p=0.998) or spine retraction (p=0.423) (Figs. 3h–j), indicating impaired long-lasting spine remodeling associated with LTD (Figs, 3h–j). Altogether, these results suggest that while STB primarily contributes to synapse formation, its role in trafficking may also affect the maintenance of activity-dependent LTP and NMDAR-LTD, although the underlying cellular mechanisms remain unknown.
Human autism-linked stb variant fails to rescue synaptic dysfunction in cKO neurons
The human autism-associated STB-R178Q variant is a loss-of-function mutant of the axonal transport of presynaptic cargos. To test whether expressing this mutant contributes to autism-linked synaptic dysfunction, we conducted four electrophysiological studies in control hippocampal neurons. First, by performing paired whole cell recordings at DIV14–19, we found that expressing STB-R178Q resulted in a marked reduction in EPSC amplitude compared to neurons expressing GFP control (p=0.009) (Fig. 4a). Second, we examined the frequency of quantal events to evaluate the number of presynaptic release sites by measuring evoked asynchronous release through substitution of extracellular Ca2+ with strontium (Sr2+), a manipulation that increases delayed asynchronous release after a major peak current. Analysis of Sr2+-induced asynchronous events has been used to characterize presynaptic quantal events or density of presynaptic release sites [54, 55]. We observed a significant reduction in Sr2+-induced and evoked initial synaptic response recorded from paired neurons expressing STB-R178Q relative to the neurons expressing GFP control (p=0.004) (Fig. 4b left). The frequency of Sr2+-induced asynchronous events was also decreased in presynaptic neurons expressing STB-R178Q compared with those expressing GFP (p<0.0001) (Fig. 4b, inset). Reduced frequency of quantal events is further reflected by a rightward shift of the cumulative inter-event-interval distribution, a phenotype attributed to a reduced density of presynaptic release sites or impaired presynaptic function. In addition, both LTP (p=0.014) and LTD (p=0.003) were diminished in paired control neurons expressing STB-R178Q (Figs. 4c and d). Altogether, these electrophysiological data consistently support the notion that expressing the human autism-associated STB-R178Q variant in control neurons induces synaptic dysfunction.
Fig 4. The human autism-associated STB variant fails to rescue synaptic dysfunction in cKO neurons.
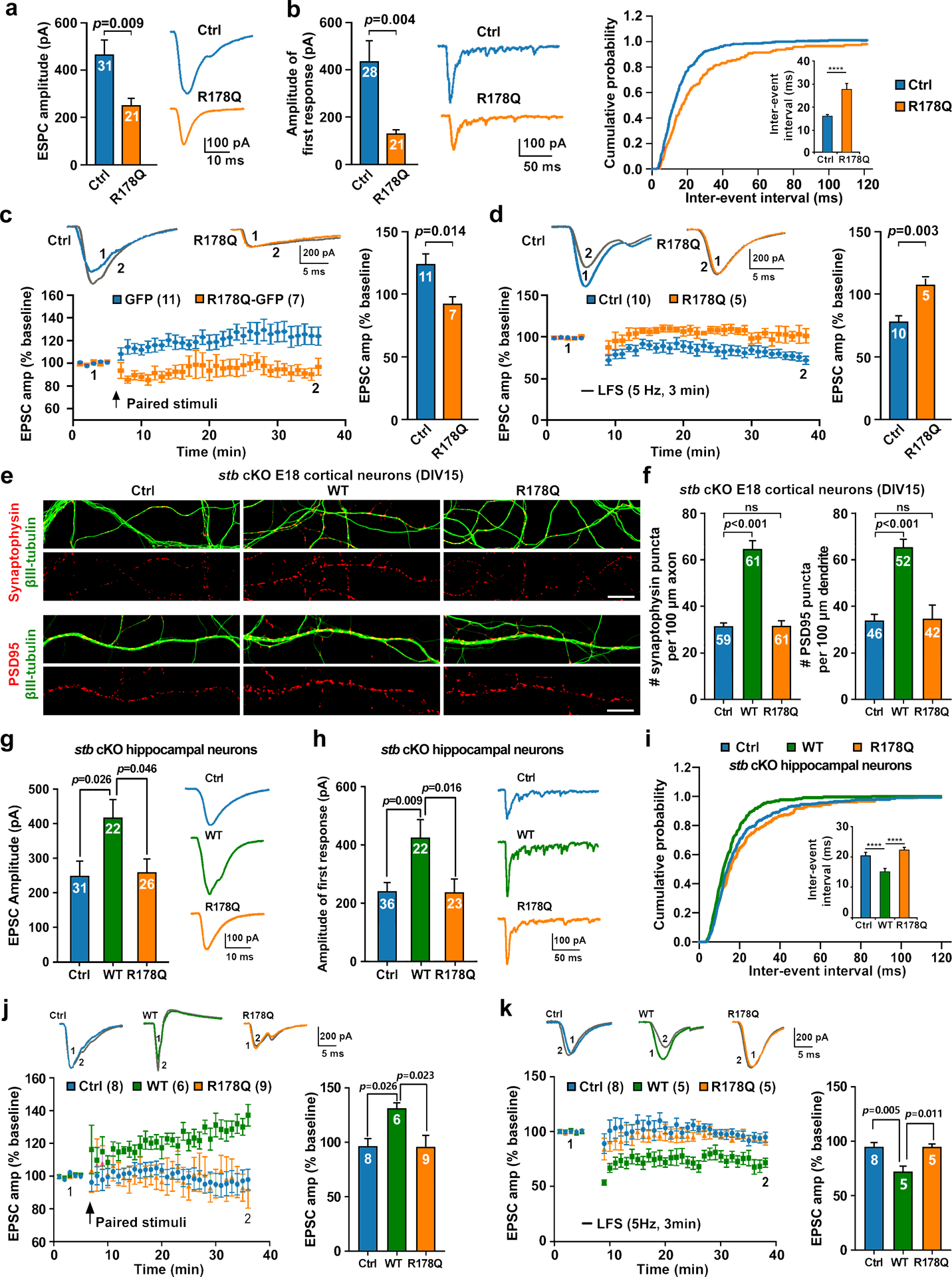
a-d, Expressing STB-R178Q in control hippocampal neurons impairs synaptic transmission and plasticity. Reduced EPSC amplitude was recorded from paired hippocampal neurons expressing STB-R178Q (Ctrl vector, n=31; R178Q, n=21, p=0.009) (a). Right: Representative EPSC traces. Decreased Sr2+-induced and evoked first EPSC amplitude (left, Ctrl vector, n=28; R178Q, n=21, p=0.004) and reduced frequency of the Sr2+-induced asynchronous events (right inset, inter-event-interval, p<0.001) were recorded from paired control hippocampal neurons expressing STB-R178Q (b). Middle: Representative EPSC traces. Impaired LTP was recorded from paired control hippocampal neurons expressing STB-R178Q (c) (Ctrl vector, n=11; R178Q, n=7). Representative traces (upper left) were taken before (1) and 25 min after (2) paired stimuli and normalized EPSC amplitudes were plotted every 1 min (lower left). The mean ± sem was plotted for the magnitude of LTP at the last 5 min post-induction (p=0.014, right). Impaired LTD was recorded from paired control hippocampal neurons expressing STB-R178Q (d) (Ctrl vector, n=10; R178Q, n=5). Representative traces (upper left) were taken before (1) and 25 min after (2) LFS and normalized EPSC amplitudes were plotted every 1 min (lower left). The mean ± sem was plotted for the magnitude of LTD at the last 5 min post-induction (p=0.003, right). e, f, Representative images (e) and quantitative analysis (f) showing failed rescue of reduced formation and density of synapses in stb cKO cortical neurons by expressing STB-R178Q. The cKO neurons were infected with a lentivirus expressing STB or STB-R178Q at DIV4, followed by co-immunostaining with βIII-tubulin and synaptophysin or PSD95 at DIV15. Note that expressing STB, but not STB-R178Q, in stb cKO cortical neurons effectively rescues the reduced density of synaptophysin along axons (One-way ANOVA, Ctrl vector vs. STB, p<0.001; Ctrl vector vs. STB-R178Q, p=0.997) and PSD95 along dendritic profiles (One-way ANOVA, Ctrl vector vs. STB, p<0.001; Ctrl vector vs. STB-R178Q, p=0.987). g-i, Expressing STB-R178Q in paired cKO hippocampal neurons failed to rescue decreased EPSC amplitude (One-way ANOVA, STB vs. Ctrl vector, p=0.026; STB vs. STB-R178Q, p=0.046) (g), reduced Sr2+-induced and evoked first EPSC amplitude (One-way ANOVA, STB vs. Ctrl vector, p=0.009; STB vs. STB-R178Q, p=0.016) (h), and reduced frequency of Sr2+-induced asynchronous events (inset, inter-event-interval, one-way ANOVA, STB vs. Ctrl vector, p<0.001; STB vs. STB-R178Q, p<0.001) (i). Representative EPSC traces are shown on the right (g, h). j, k, Expressing STB-R178Q in paired cKO hippocampal neurons failed to rescue impaired LTP (j) and LTD (k) (for LTP: Ctrl vector, n=8; STB, n=6; STB-R178Q, n=9; for LTD: Ctrl vector, n=8; STB, n=5; STB-R178Q, n=5). Representative traces (upper left) in LTP (j) were taken before (1) and 25 min after (2) paired stimuli and normalized EPSC amplitudes were plotted every 1 min (lower left). The mean ± sem was plotted for the magnitude of LTP at the last 5 min post-induction (right, one-way ANOVA, STB vs. Ctrl vector, p=0.026; STB vs. STB-R178Q, p=0.023). Representative traces (upper left) in LTD (k) were taken before (1) and 25 min after (2) LFS and normalized EPSC amplitudes were plotted every 1 min (lower left). The mean ± sem was plotted for the magnitude of LTD at the last 5 min post-induction (right, one-way ANOVA, STB vs. Ctrl vector, p=0.005; STB vs. STB-R178Q, p=0.011). The difference between genotypes was analyzed by an unpaired two-tailed Student’s t-test (a-d) and/or by One-way ANOVA with treatment as the independent variable (f-k). Scale bars: 10 μm.
We next performed both imaging and electrophysiological studies to determine whether expressing STB-R178Q in stb cKO neurons is able to rescue impaired synaptic formation and synaptic function. First, we characterized the density of presynaptic marker synaptophysin and postsynaptic marker PSD95 along neuronal processes. Expressing STB, but not STB-R178Q, in stb cKO cortical neurons effectively rescued reduced synapse formation and density (Figs. 4e and f). Thus, these studies combined with live imaging rescue experiments (Figs. 2g–l) consistently support that the human autism-associated variant STB-R178Q is a loss-of-function mutant that fails to rescue impaired axonal transport of presynaptic cargos and reduced synapse formation in stb cKO neurons. Second, we evaluated synaptic transmission and plasticity in stb cKO hippocampal neurons following expression of STB or STB-R178Q. By paired whole cell recordings through stimulating presynaptic neurons (from-70 to 30 mV; 1.5 ms) at DIV14–19, expressing STB, but not STB-R178Q or GFP control, significantly increased EPSC amplitude (Fig. 4g), suggesting that expressing STB-R178Q failed to rescue impaired basal synaptic transmission in stb cKO neurons. We next examined the frequency of evoked asynchronous release to evaluate the number of presynaptic release sites by substitution of extracellular Ca2+ with Sr2+. In parallel with the increase of EPSC amplitude (Fig. 4h), we observed a significant increase in Sr2+-induced and evoked initial synaptic response recorded from the paired neurons expressing STB relative to neurons expressing GFP control or STB-R178Q. The frequency of Sr2+-induced asynchronous events was increased in presynaptic neurons expressing STB compared with those expressing GFP or STB-R178Q (Fig. 4i). Increased frequency of quantal events was further reflected by a leftward shift of the cumulative inter-event-interval distribution. In addition, we characterized LTP and LTD via paired stimulation of hippocampal neurons at DIV14–19. Impaired LTP and LTD in paired stb cKO neurons was effectively rescued by expressing STB, but not GFP control or STB-R178Q (Figs. 4j and k). Together, these rescue experiments further support our notion that the human autism-associated STB-R178Q variant is a motor adaptor loss-of-function mutant, thus establishing a mechanistic link between axonal transport of presynaptic cargos and maintenance of synaptic transmission and plasticity.
STB cKO mice exhibit core autistic behavioral traits
As deficits in social interaction and communication, as well as restricted interests and repetitive behaviors, represent typical autistic features in humans [11], we conducted an extensive battery of analyses of these behavioral traits in age-matched male control and stb cKO mice (8–10 weeks). First, we tested mice with a three-chamber social interaction assay [56] to assess voluntary initiation of social interaction and the ability to discriminate social novelty (Fig. 5a). Mice were allowed to explore and initiate social contact with a partner (stranger 1) placed inside a wired cage or an identical empty wired cage (empty). The cKO mice preferred to explore the mouse-containing chamber (Ctrl: S1 vs. E, p=0.003; cKO: S1 vs. E, p=0.016) and interacted more extensively with peer mice than with the empty cage to an extent comparable to that of control mice (Ctrl: S1 vs. E, p<0.001; cKO: S1 vs. E, p<0.001), suggesting normal social ability (Fig. 5b). When a second stranger mouse (stranger 2) was introduced into the unoccupied chamber, control mice displayed a preference for the new stranger (Ctrl: S2 vs. S1, p<0.001). In contrast, cKO mice spent almost identical amounts of time in both chambers interacting with stranger 1 and stranger 2 (cKO: S2 vs. S1, p=0.334) (Fig. 5c, left panel). In particular, cKO mice spent significantly reduced time in close interaction with stranger 2 (p<0.001) compared to controls (Fig. 5c, right panel). Given that the control and cKO mice performed similarly in exploring the novel object (Fig. S6a), this social interaction assay suggests a defect in social, but not object, novelty recognition in cKO mice. Deficient social memory is a key factor contributing to abnormal social novelty recognition [57]. We next performed a five-trial social memory assay to examine whether stb cKO mice display altered social learning and memory (Fig. 5d). The control mice displayed normal social memory, as demonstrated by a marked habituation (decreased close exploration) during the first four trials to a stimulus mouse (stranger 1) and a striking dishabituation (increased close interaction) upon the presentation of a novel animal (stranger 2) in the fifth trial. Although cKO mice showed significant habituation to a stimulus mouse (stranger 1), there was no dishabituation to a novel animal as cKO mice spent significantly less time in close interaction with stranger 2 in the fifth trial compared to control mice (Two-way ANOVA, genotype: F1,20=7.1, p=0.01; genotype × trial interaction: F4,80=8.2, p<0.0001) (Fig. 5e). Altogether, these assays suggest that cKO mice have lost their interest in repeated social interactions and have deficits in social novelty recognition between two strangers.
Fig 5. stb cKO mice exhibit core autistic traits.
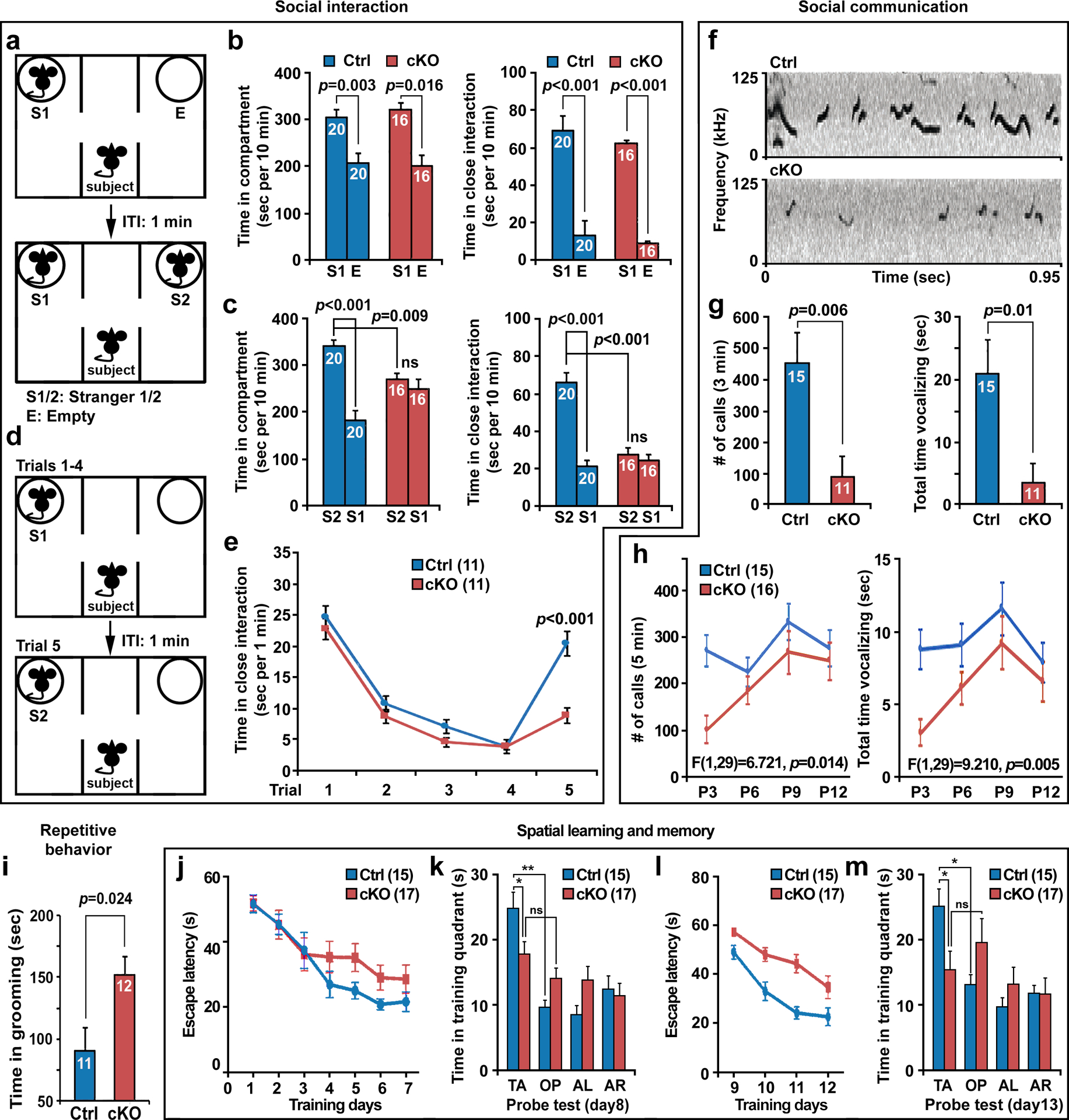
a-c, stb cKO mice display impaired social novelty preference. (a) Diagram of the three-chamber social interaction test for the sociability and social novelty preference. ITI, inter-trial interval. (b) Both control and cKO mice (8–10 weeks) showed a similar preference to an age- and gender-matched S1, measured as more time spent in the chamber containing S1 (Ctrl, S1 vs. E, p=0.003; cKO, S1 vs. E, p=0.016) and in close interaction with S1 (Ctrl, S1 vs. E, p<0.001; cKO, S1 vs. E, p<0.001). (c) cKO mice displayed impaired social novelty preference (S1 versus S2). Note that while control mice spent more time in the chamber containing a novel mouse (S2) than in the chamber with a familiar mouse (S1) (Ctrl, S2 vs. S1, p<0.001), cKO mice displayed no preference for S1 or S2 chambers (S2 vs. S1, p=0.627) and spent less time in close interaction with both S1 and S2 (S2 vs. S1, p=0.334). d, e, Diagram (d) and the five-trial social memory assays (e) showing that while both genotypes habituated to the same mouse (trials 1–4), cKO mice exhibited impairment in dishabituation to a novel mouse (trial 5). Two-way ANOVA revealed a main effect of genotype (F1, 20=7.1, p=0.01) and a main effect of genotype × trial interaction (F4, 80=8.2, p<0.0001). f-h, cKO mice displayed deficits in social communication. Representative images (f) and quantitative analysis (g) of USVs emitted by a Ctrl or cKO male mouse to female urine demonstrated that cKO mice emitted less USVs (p=0.006) and spent less total time vocalizing (p=0.01) during a 3-min period of exposure to fresh female urine. (h) cKO pups emitted less USVs and spent less total time vocalizing during a 5-min period of acute separation from their mother when tested from P3 to P12 with a 3-day interval. Two-way ANOVA revealed a main effect of genotype (F1, 29=6.721, p=0.014; F1, 29=9.210, p=0.005, respectively). (i) cKO mice displayed increased repetitive behavior. Note that cKO mice spent more time in the repetitive self-grooming behavior (p=0.024). j-m, cKO mice exhibited impaired initial spatial memory and reversal spatial memory in the hidden platform version of the water maze. (j) cKO mice spent a trend but not significant longer latency to reach the platform during spatial learning (Two-way ANOVA, genotype: F1, 30=1.187, p=0.2847). (k) While control mice showed clear preference for the target quadrant versus opposite quadrant (p=0.001), cKO mice showed no preference and spent less time in the target quadrant compared to controls during the probe test on day 8 (Ctrl vs. cKO, p=0.028). (l) cKO mice exhibited an impaired reversal spatial learning curve (Two-way ANOVA, genotype: F1, 30=14.62, p=0.0006). (m) cKO mice showed no preference and spent less time in the target quadrant than controls during the probe test on day 13 (Ctrl vs. cKO, p=0.017). AL, adjacent left quadrant; AR, adjacent right quadrant; OP, opposite quadrant; TA, target quadrant. The total number of age-matched male control and cKO mice tested is indicated within bars or in parentheses. Data are presented as mean ± sem and the difference between genotypes was revealed by an unpaired two-tailed Student’s t-test and/or two-way ANOVA with genotype and treatment as independent variables.
Social communication plays an essential role in successful social interaction. Deficient communication is common in autistic patients and has been reported in autism-linked mouse models [58, 59]. Rodents emit ultrasonic vocalizations (USVs) in specific social contexts. Adult male interaction-induced USVs are thought to attract females. Thus, adult male mice would emit high levels of USVs when exposed to female mice or even to female urine [60]. When exposed to fresh urine from a stranger female mouse, adult cKO male mice emitted fewer USVs (p=0.006) and spent significantly reduced time vocalizing (p=0.01) compared with age-matched control male mice (Figs. 5f and g), suggesting defects in social communication. We further tested social communication in young mouse pups that emit USVs when separated from their mother and littermates during social isolation. During an acute (5-min) separation from their mother, cKO pups emitted significantly fewer USVs (Two-way ANOVA, genotype: F1,29=6.721, p=0.014) and spent significantly less time vocalizing (Two-way ANOVA, genotype: F1,29=9.210, p=0.005) compared to control pups when tested from P3 to P12 (Fig. 5h). These data indicate communication deficits in cKO pups and adult mice. In addition, cKO mice spent significantly more time self-grooming compared to control mice (p=0.024) (Fig. 5i), suggesting a pattern of repetitive behavior common in ASDs. Proper social communication depends on olfactory cues, and thus olfactory function is crucial for normal social behaviors [61]. Control and cKO mice displayed a similar ability to detect buried food under a deep layer of cage bedding, a test of non-social odor detection (Fig. S6b), and discriminated non-social and social odors in an olfactory habituation and dishabituation test (Fig. S6c). Therefore, the deficit in social recognition in cKO mice is unlikely due to defective sensing of social or nonsocial odors.
Given that cognitive deficits are often associated with ASDs, we next conducted the Morris water maze (MWM) test to examine spatial learning and memory. Control and cKO mice exhibited similar swimming velocity (Fig. S6d). After 7 days of an initial spatial learning phase (Fig. 5j), cKO mice exhibited significant impairment in spatial memory, evidenced by spending equivalent amounts of time in the target and opposite quadrants at Day 8 during the probe test (Ctrl: TA vs. OP, p=0.001; cKO: TA vs. OP, p=0.637) (Fig. 5k). Disruption of LTD impairs reversal learning and behavioral flexibility [62, 63], which represents an ability to replace a previously acquired rule with a new one, in adaption to a new environmental context. We further assessed cognitive flexibility by performing spatial reversal in MWM test. On Day 9 of MWM testing, the hidden platform was moved to the opposite quadrant of the pool. We found that stb cKO mice showed impaired escape latency time in learning the new platform location at Day 9–12 (Two-way ANOVA, genotype: F1,30=14.62, p=0.0006) (Fig. 5l) and spent equivalent amounts of time in the new target and opposite quadrants at Day 13 during the probe test (Ctrl: TA vs. OP, p=0.037; cKO: TA vs. OP, p=0.907) (Fig. 5m). These data indicate that cKO mice exhibited an impairment in both initial spatial memory and spatial reversal memory in the MWM test.
We next examined the performance of stb cKO mice in contextual fear extinction, an alternative approach to test behavioral flexibility. The stb cKO mice showed impaired acquisition curves during the fear conditioning phase, indicated by less time spent freezing after each foot shock (Two-way ANOVA, genotype: F1,36=11.13, p=0.002) (Fig. S6e, left panel). In addition, the stb cKO mice displayed significantly less time spent freezing during the first 5 min of the extinction phase, indicating an impairment in contextual fear memory retrieval (p=0.041) (Fig. S6e, right panel). Although there was no significant difference following contextual fear extinction (Two-way ANOVA, genotype: F1,36=1.131, p=0.2947), the control mice showed a robust reduction in freezing time during the last 5 min compared to the first 5 min during extinction (the 8th 5 min vs. the 1st 5 min, p=0.001), In contrast, the stb cKO mice did not show a significant reduction in freezing time (the 8th 5min vs. the 1st 5min, p=0.362). Thus, impaired tasks in both initial spatial learning and memory and contextual fear learning and memory in stb cKO mice are consistent with their impaired hippocampal LTP. Altogether, these studies establish a mechanistic link between defective axonal transport of presynaptic cargos and autism-like behavioral and cognitive deficits.
Alterations in emotional, locomotor and sensory behaviors also contribute to impaired social interaction and cognitive function. To exclude these possibilities, we conducted five additional behavioral studies. The cKO mice displayed (1) no significant differences in aggressive behavior compared to controls in the cotton bud biting test (Fig. S6f); (2) normal motor coordination and balance in the rotarod test (Fig. S6g); (3) normal locomotor activity in an open field test (Fig. S6h), (4) no anxiety-like behaviors in elevated plus maze test and open field test (Fig. S6i); and (5) no sensory deficits during the prepulse inhibition test (Fig. S6j). These behavioral assays support that impaired social novelty recognition in stb cKO mice is unlikely due to defects in olfactory, emotional, locomotor or sensory functions.
Discussion
Targeted delivery of synaptic proteins is a prerequisite for the formation and maturation of synapses during brain development and synaptic maintenance and remodeling in mature brains. Our current findings provide new mechanistic insights into how defective axonal transport and the associated reductions in synapse formation and maintenance are a potential trigger for synaptic dysfunction contributing to ASD-like behavioral phenotypes. We revealed that stb cKO mice display (1) altered synaptic transmission in hippocampal SC–CA1 synapses, and (2) behavioral alterations in social interaction and communication. These phenotypes are attributed to defective axonal transport of presynaptic cargos, reduced synapse density, and altered AZ structures. We further demonstrated that the clinically relevant human missense variant STB-R178Q is a loss-of-function STB mutant that impairs axonal transport of presynaptic cargos. Expressing STB-R178Q in stb cKO neurons failed to rescue impaired axonal transport, reduced synapse formation, and altered synaptic transmission and plasticity, thus supporting the physiological relevance of autism-like phenotypes. Collectively, this disrupted synaptic transmission and abnormal behaviors establish the stb cKO mouse as a promising autism-like model for investigations into the core presynaptic defects at the onset of ASDs.
Several ASD-associated genes in the neurexin–neuroligin–PSD95–SAPAP–SHANK axis have been well characterized in the formation of postsynaptic scaffolding or adhesion, dendritic spines, and neuronal contacts during development. Copy number variants and de novo mutations of these genes have been linked to altered synaptic transmission accounting for autism pathogenesis [64–66]. Mice with neuroligin dysfunction exhibit imbalance between excitatory and inhibitory synaptic transmission in postsynaptic neurons [16, 67]. Knockdown of Shank impairs dendritic spine formation (Shank3) or reduces dendritic spine size (Shank1), which is accompanied by disruption of excitatory synaptic transmission [68, 69]. While these postsynaptic mechanisms contribute to ASD susceptibility [21, 22], a mechanistic link between altered axonal transport and autism etiology has not been established. Proper neuronal function and synaptic transmission require seamless integration of the transport of synaptic components and organelles, as well as the assembly and regulation of synaptic structures and function. Therefore, understanding the regulation of axon transport and the extent to which it contributes to the formation of new synapses in developing neurons as well as the maintenance and modulation of synapses in mature neurons is an emerging research frontier related to the presynaptic basis of neurodevelopmental disorders.
Synaptogenesis is a critical process in the first two weeks of postnatal development in rodent [32, 33]. Consistently, STB expression in the mouse brain peaks during the first two weeks of postnatal development and then progressively declines during maturation, remaining low throughout adulthood, suggesting a critical role of STB in synapse formation and maturation. STB deletion reduces synapse density and alters AZ structure in adult mouse brains. In mature hippocampal neurons at DIV15, deleting the stb gene reduced the density of presynaptic boutons. This reduction persisted into adulthood when TEM imaging was used to examine the hippocampal CA1 stratum radiatum region, indicating long-lasting presynaptic changes in the brain. Reduced postsynaptic density, as measured by PSD95 labeling, was also observed in cKO hippocampal neurons, a phenotype consistent with decreased spine density and maturation in hippocampal CA1 regions from stb cKO mice. Our findings consistently suggest that STB regulates transport of synaptic cargos in developing neurons critical for synaptic formation and maturation.
The density of presynaptic AZs, as well as their internal components, closely relate to the efficiency of synaptic transmission. Stb cKO neurons showed reduced total and docked SVs in AZs, which are dynamic structures undergoing remodeling to shape SV release [70]. For example, Bassoon promotes rapid SV recruitment at AZs of central excitatory synapses [71]. The rate of SV reloading was almost halved after deleting bassoon, causing a pronounced depression during high-frequency stimulation. Our findings demonstrated that Bassoon is depleted in synaptosomes, but not in whole brain homogenates of adult stb cKO mice, further confirming selective defects in axonal delivery of Bassoon cargos without a global effect on total Bassoon expression in stb cKO mice. This reduction may partially contribute to the failure of post-potentiation after high frequency stimulation in the hippocampal CA3-CA1 circuit. Remodeling of synaptic strength could involve modification of existing synapses or formation of new synapses, both of which require axonal transport of newly synthesized presynaptic proteins. Thus, impairing axonal transport results in reduced maintenance of long-term synaptic strength in response to activity-dependent stimuli.
Our live imaging studies in stb cKO neurons, combined with expression of the clinically relevant human missense variant STB-R178Q, consistently support the notion that defects in STB-mediated axonal transport of presynaptic cargos, and thus the reduced formation and maturation of synapses are core synaptic mechanisms underlying autism-like synaptic dysfunction. Interestingly, our biochemical analysis also shows a slight reduction of GluR2 level in synaptosomal preparations from adult stb cKO mouse brains. STB deletion impairs NMDA-induced internalization of GluR2 and dendritic spine remodeling in mature cKO hippocampal neurons. Consistently, overexpressing STB-R178Q mutant in WT neurons leads to reduced GluR2 puncta along dendrites. These phenotypes collectively suggest a potential role of STB in the dendritic trafficking and surface expression or recycling of AMPA receptors at mature synapses, thus contributing to the maintenance of long-term synaptic plasticity. Several kinesin motor proteins have been identified to mediate the dendritic transport of neurotransmitter receptors to synapses. For example, KIF17 is the motor that drives transport of cargos carrying NMDAR subunit 2B (NR2B) along dendrites to synapses [72], while NMDAR NR2A is transported by KIF3B motor to synapses [73]; both transport mechanisms are essential for synapse development and synaptic function. However, the identity of transport cargos and their motors for dendritic transport of AMPA receptors remain further investigations to determine (1) whether STB also acts as an adaptor for kinesin motor families that mediate long-distance dendritic transport, and (2) which postsynaptic cargos could carry GluR2/PSD95 for this dendritic trafficking. In addition, our results may suggest an alternative role of STB in GluR2 local membrane trafficking or internalization in postsynaptic spines during LTD induction. Kinesin-4 motor KIF21B was reported to facilitate AMPAR endocytosis and reduce postsynaptic strength, thereby forming a mechanistic link to LTD expression [74]. Interestingly, STB was also reported to mediate the trafficking of PICK1-containing vesicles through interaction with PICK1 [34], which plays a critical role in the surface expression and functioning of AMPA receptors during synaptic plasticity including LTP and LTD [35–37, 46–48]. We found that deletion of the stb gene did not alter the expression and distribution of PICK1 in either axons or dendrites and that PICK1 vesicular structures are almost entirely stationary in dendrites throughout the duration of time-lapse recordings in developing cortical neurons at DIV7, which is consistent with a previous study showing no dendritic transport of PICK1 vesicles [34]. Therefore, our study suggests that STB-mediated motor and PICK1 coupling may play a secondary role in surface expression/trafficking of AMPA receptors during synaptic plasticity. Experimentally addressing these mechanistic issues represents promising directions for future investigations.
The behavioral and synaptic phenotypes of stb cKO mice recapitulate the core features of human ASDs. Stb cKO mice exhibited impaired social novelty recognition, defective social communication, increased repetitive behaviors, and reduced spatial learning and memory. It is believed that different circuits may be differentially affected in ASDs, resulting in a mixture of disabilities in humans with ASDs. We demonstrated that these core behavioral phenotypes occurred in the absence of deficits in recognition memory, olfaction, locomotion and motor learning, and anxiety, indicating that neuronal circuits in stb cKO mice are not globally impaired. Loss of function of STB by the de novo missense R178Q mutation consistently supports the notion that axonal transport is a core presynaptic pathway in autism-like susceptibility. Therefore, our study reveals a transport mechanism that contributes to synaptic dysfunction and behavioral abnormalities with similarities to autism. Autism is a systemic mental disorder and autism-like behavioral abnormalities are unlikely to stem from dysfunction of a single brain region [75]. Given that social deficits are the most prominent impairments to emerge early in the development of autism, the prefrontal cortex, amygdala, and hippocampus are three major brain regions intensively studied in mediating social cognition [76–78]. Future research could aim to target these regions specifically with human STB-R178Q knock-in mice in order to provide a genetic tool to investigate neural circuitry dysfunction associated with autism-linked behavioral phenotypes.
Supplementary Material
Acknowledgments:
We thank members of the Sheng lab for technical assistance and constructive discussion, Zezhi Li for assistance in analyzing human sequence data, Eckart Gundelfinger for EGFP-Bassoon, Richard Youle for pCMV-DsRed-Mito, Jun Xia for YFP-PICK1, the NIH/NICHD rodent behavioral core facility and Daniel Tadese Abebe for assistance in animal behavioral tests, the NINDS Electron Microscopy Facility and Susan Cheng for assistance in TEM analysis, and Kelly Chamberlain and Joseph Roney for critical reading/editing.
Funding: This work was supported by the Intramural Research Program of NINDS, NIH ZIA NS003029, and ZIA NS002946 (Z-H. Sheng).
Abbreviations:
- ASD
autism spectrum disorder
- AZ
active zone
- cKO
conditional knockout
- DIV
days in vitro
- DRG
dorsal root ganglion
- EM
electron micrograph
- KIF5
kinesin-1
- LTD
long-term depression
- LTP
long-term potentiation
- mEPSC
miniature excitatory postsynaptic current
- mIPSC
miniature inhibitory postsynaptic current
- P90
postnatal day 90
- PSD
postsynaptic density
- SC–CA1
Schaffer collateral–CA1
- SV
synaptic vesicle
- STB
syntabulin
- USV
ultrasonic vocalization
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
Data and materials availability: All data are available in the manuscript or the supplementary materials.
Animal care and use were carried out in accordance with NIH guidelines and approved by the NIH, NINDS/NIDCD Animal Care and Use Committee.
Supplementary information is available at MP’s website.
REFERENCES AND NOTES
- 1.Goda Y, Davis GW. Mechanisms of synapse assembly and disassembly. Neuron. 2003; 40(2): 243–264. [DOI] [PubMed] [Google Scholar]
- 2.Chia PH, Li PP, Shen K. Cellular and molecular mechanisms underlying presynapse formation. J Cell Biol. 2013; 203(1): 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziv NE, Garner CC. Cellular and molecular mechanisms of presynaptic assembly. Nat Rev Neurosci. 2004; 5(5): 385–399. [DOI] [PubMed] [Google Scholar]
- 4.Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998; 140(3): 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin Y, Garner CC. Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Bi. 2008; 24: 237–262. [DOI] [PubMed] [Google Scholar]
- 6.Gundelfinger ED, Fejtova A. Molecular organization and plasticity of the cytomatrix at the active zone. Curr Opin Neurobiol. 2012; 22(3): 423–430. [DOI] [PubMed] [Google Scholar]
- 7.Fenster SD, Chung WJ, Zhai R, Cases-Langhoff C, Voss B, Garner AM et al. Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron. 2000; 25(1): 203–214. [DOI] [PubMed] [Google Scholar]
- 8.Gundelfinger ED, Reissner C, Garner CC. Role of Bassoon and Piccolo in Assembly and Molecular Organization of the Active Zone. Front Synaptic Neurosci. 2015; 7: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altrock WD, tom Dieck S, Sokolov M, Meyer AC, Sigler A, Brakebusch C et al. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron. 2003; 37(5): 787–800. [DOI] [PubMed] [Google Scholar]
- 10.Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE et al. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001; 29(1): 131–143. [DOI] [PubMed] [Google Scholar]
- 11.Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC et al. The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000; 30(3): 205–223. [PubMed] [Google Scholar]
- 12.Miles JH. Autism spectrum disorders--a genetics review. Genet Med. 2011; 13(4): 278–294. [DOI] [PubMed] [Google Scholar]
- 13.Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015; 21(2): 185–191. [DOI] [PubMed] [Google Scholar]
- 14.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012; 485(7397): 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014; 515(7526): 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007; 318(5847): 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peca J, Feliciano C, Ting JT, Wang WT, Wells MF, Venkatraman TN et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011; 472(7344): 437–U534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai NP, Wilkerson JR, Guo WR, Maksimova MA, DeMartino GN, Cowan CW et al. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012; 151(7): 1581–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013; 493(7432): 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davenport EC, Szulc BR, Drew J, Taylor J, Morgan T, Higgs NF et al. Autism and Schizophrenia-Associated CYFIP1 Regulates the Balance of Synaptic Excitation and Inhibition. Cell Rep. 2019; 26(8): 2037–2051 e2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zoghbi HY. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science. 2003; 302(5646): 826–830. [DOI] [PubMed] [Google Scholar]
- 22.Bourgeron T A synaptic trek to autism. Curr Opin Neurobiol. 2009; 19(2): 231–234. [DOI] [PubMed] [Google Scholar]
- 23.Su QN, Cai Q, Gerwin C, Smith CL, Sheng ZH. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat Cell Biol 2004; 6(10): 941–53. [DOI] [PubMed] [Google Scholar]
- 24.Ylisaukko-oja T, Alarcon M, Cantor RM, Auranen M, Vanhala R, Kempas E et al. Search for autism loci by combined analysis of Autism Genetic Resource Exchange and Finnish families. Ann Neurol. 2006; 59(1): 145–155. [DOI] [PubMed] [Google Scholar]
- 25.Delgado MS, Camprubi C, Tumer Z, Martinez F, Mila M, Monk D. Screening individuals with intellectual disability, autism and Tourette’s syndrome for KCNK9 mutations and aberrant DNA methylation within the 8q24 imprinted cluster. Am J Med Genet. B 2014; 165(6): 472–478. [DOI] [PubMed] [Google Scholar]
- 26.Chen CH, Chen HI, Chien WH, Li LH, Wu YY, Chiu YN et al. High resolution analysis of rare copy number variants in patients with autism spectrum disorder from Taiwan. Sci Rep. 2017; 7(1):11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herman GE, Hansen-Kiss E, Sadee W, Barrie E. A collaborative translational autism research program for the military. 2016. https://apps.dtic.mil/dtic/tr/fulltext/u2/a631878.pdf
- 28.Perlson E, Jeong GB, Ross JL, Dixit R, Wallace KE, Kalb RG et al. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J Neurosci. 2009; 29(31): 9903–9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen-Cory S The developing synapse: Construction and modulation of synaptic structures and circuits. Science. 2002; 298(5594): 770–776. [DOI] [PubMed] [Google Scholar]
- 30.Wong ROL, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002; 3(10): 803–812. [DOI] [PubMed] [Google Scholar]
- 31.Isshiki M, Tanaka S, Kuriu T, Tabuchi K, Takumi T, Okabe S. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat Commun. 2014; 5. [DOI] [PubMed] [Google Scholar]
- 32.Knaus P, Betz H, Rehm H. Expression of synaptophysin during postnatal-development of the mouse brain. J Neurochem. 1986; 47(4): 1302–1304. [DOI] [PubMed] [Google Scholar]
- 33.Watson RE, DeSesso JM, Hurtt ME, Cappon GD. Postnatal growth and morphological development of the brain: A species comparison. Birth Defects Res B Dev Reprod Toxicol. 2006; 77(5): 471–484. [DOI] [PubMed] [Google Scholar]
- 34.Xu J, Wang N, Luo JH, Xia J. Syntabulin regulates the trafficking of PICK1-containing vesicles in neurons. Sci Rep. 2016; 6: 20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia J, Zhang XQ, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999; 22(1): 179–187. [DOI] [PubMed] [Google Scholar]
- 36.Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL et al. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008; 57(6): 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volk L, Kim CH, Takamiya K, Yu YL, Huganir RL. Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc Natl Acad Sci U S A. 2010; 107(50): 21784–21789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai Q, Gerwin C, Sheng ZH. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol. 2005; 170(6): 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng ZH. The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 2017; 27(6): 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phelps SM, Campbell P, Zheng DJ, Ophir AG. Beating the boojum: Comparative approaches to the neurobiology of social behavior. Neuropharmacology. 2010; 58(1): 17–28. [DOI] [PubMed] [Google Scholar]
- 41.Etherton M, Foldy C, Sharma M, Tabuchi K, Liu XR, Shamloo M et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci U S A. 2011; 108(33): 13764–13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012; 486(7402): 261–265. [DOI] [PubMed] [Google Scholar]
- 43.Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008; 33(1): 18–41. [DOI] [PubMed] [Google Scholar]
- 44.Luscher C, Malenka RC. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol. 2012; 4(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M et al. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000; 3(12): 1291–1300. [DOI] [PubMed] [Google Scholar]
- 46.Hanley JG. Molecular mechanisms for regulation of AMPAR trafficking by PICK1. Biochem Soc T. 2006; 34: 931–935. [DOI] [PubMed] [Google Scholar]
- 47.Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008; 10(3): 259–U257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fiuza M, Rostosky CM, Parkinson GT, Bygrave AM, Halemani N, Baptista M et al. PICK1 regulates AMPA receptor endocytosis via direct interactions with AP2 alpha-appendage and dynamin. J Cell Biol. 2017; 216(10): 3323–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asrican B, Lisman J, Otmakhov N. Synaptic strength of individual spines correlates with bound Ca2+-calmodulin-dependent kinase II. J Neurosci. 2007; 27(51): 14007–14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001; 4(11): 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Segal M Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005; 6(4): 277–284. [DOI] [PubMed] [Google Scholar]
- 52.Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004; 44(5): 749–757. [DOI] [PubMed] [Google Scholar]
- 53.Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004; 44(5): 759–767. [DOI] [PubMed] [Google Scholar]
- 54.Xu-Friedman MA, Regehr WG. Probing fundamental aspects of synaptic transmission with strontium. J Neurosci. 2000; 20(12): 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hagler DJ, Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001; 85(6): 2324–2334. [DOI] [PubMed] [Google Scholar]
- 56.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004; 3(5): 287–302. [DOI] [PubMed] [Google Scholar]
- 57.Thor DH, Holloway WR. Social memory of the male laboratory rat. J Comp Physiol Psych. 1982; 96(6): 1000–1006. [Google Scholar]
- 58.Mody M, Belliveau JW. Speech and language impairments in autism: Insights from behavior and neuroimaging. N Am J Med Sci. (Boston) 2013; 5(3): 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung W, Choi SY, Lee E, Park H, Kang J, Park H et al. Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat Neurosci. 2015; 18(3): 435–443. [DOI] [PubMed] [Google Scholar]
- 60.Maggio JC, Maggio JH, Whitney G. Experience-based vocalization of male-mice to female chemosignals. Physiol Behav. 1983; 31(3): 269–272. [DOI] [PubMed] [Google Scholar]
- 61.Devor M, Murphy MR. The effect of peripheral olfactory blockade on the social behavior of the male golden hamster. Behav Biol. 1973; 9(1): 31–42. [DOI] [PubMed] [Google Scholar]
- 62.Dong Z, Bai Y, Wu X, Li H, Gong B, Howland JG et al. Hippocampal long-term depression mediates spatial reversal learning in the Morris water maze. Neuropharmacology. 2013; 64: 65–73. [DOI] [PubMed] [Google Scholar]
- 63.Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S et al. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008; 58(1): 104–117. [DOI] [PubMed] [Google Scholar]
- 64.Baudouin SJ, Gaudias J, Gerharz S, Hatstatt L, Zhou KK, Punnakkal P et al. Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science. 2012; 338(6103): 128–132. [DOI] [PubMed] [Google Scholar]
- 65.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012; 4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee E, Lee J, Kim E. Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol Psychiat. 2017; 81(10): 838–847. [DOI] [PubMed] [Google Scholar]
- 67.Hines RM, Wu LJ, Hines DJ, Steenland H, Mansour S, Dahlhaus R et al. Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J Neurosci. 2008; 28(24): 6055–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet. 2011; 20(15): 3093–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hung AY, Futai K, Sala C, Valtschanoff JG, Ryu J, Woodworth MA et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J Neurosci. 2008; 28(7): 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matz J, Gilyan A, Kolar A, McCarvill T, Krueger SR. Rapid structural alterations of the active zone lead to sustained changes in neurotransmitter release. Proc Natl Acad Sci U S A. 2010; 107(19): 8836–8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hallermann S, Fejtova A, Schmidt H, Weyhersmuller A, Silver RA, Gundelfinger ED et al. Bassoon speeds vesicle reloading at a central excitatory synapse. Neuron. 2010; 68(4): 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin X, Takei Y, Kido MA, Hirokawa N. Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron 2011; 70(2): 310–325. [DOI] [PubMed] [Google Scholar]
- 73.Alsabban AH, Morikawa M, Tanaka Y, Takei Y, Hirokawa N. Kinesin Kif3b mutation reduces NMDAR subunit NR2A trafficking and causes schizophrenia-like phenotypes in mice. EMBO J. 2020; 39(1):e101090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morikawa M, Tanaka Y, Cho HS, Yoshihara M, Hirokawa N. The Molecular Motor KIF21B Mediates Synaptic Plasticity and Fear Extinction by Terminating Rac1 Activation. Cell Rep. 2018; 23(13): 3864–3877. [DOI] [PubMed] [Google Scholar]
- 75.Belger A, Carpenter KL, Yucel GH, Cleary KM, Donkers FC. The neural circuitry of autism. Neurotox Res. 2011; 20(3): 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Amodio DM, Frith CD. Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci. 2006; 7(4): 268–277. [DOI] [PubMed] [Google Scholar]
- 77.Felix-Ortiz AC, Tye KM. Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci. 2014; 34(2): 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000; 10(1): 47–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.