

Abstract
Primary thyroid neoplasms with actionable NTRK rearrangements are rare, and their clinical behavior, histologic characteristics, and molecular landscape are not well understood. We report an institutional series of eleven NTRK-rearranged thyroid carcinomas (NRTC) by performing clinicopathologic review and next-generation sequencing for targeted mutations and gene rearrangements. The NRTC encompass a histomorphologic spectrum of ten papillary thyroid carcinomas (PTC), including one with high-grade features, and one secretory carcinoma (SC), in ten adults and one adolescent. All NRTC were characterized by an unusual multinodular growth pattern, extensive lymphovascular invasion, and cervical lymph node metastases at initial presentation. Immunophenotypically, while most cases were positive for TTF1 and PAX8, the SC case was negative/weak for these markers and instead diffusely expressed GATA3, mammaglobin and S100. Observed gene rearrangements included ETV6-NTRK3 (n = 4, including the SC), TPR-NTRK1 (n = 2), RBPMS-NTRK3 (n = 2), SQSTM1-NTRK1 (n = 1), SQSTM1-NTRK3 (n = 1), and EML4-NTRK3 (n = 1). Mutation profiling revealed a concurrent TERT promotor mutation C228T in two (22%) patients and a novel frameshift MEN1 deletion in one. All patients received total thyroidectomy and radioactive iodine. Despite frequent development of persistent/recurrent disease (9 cases, 82%) and distant metastases (6 cases; 55%), no tumor-related death occurred over a median (range) follow-up of 44 (11 to 471) months. Three patients received NTRK inhibitor therapy, with the SC case showing complete resolution and two other patients experiencing 33% and 69.7% decrease of disease burden. Although the range of features is variable, NRTC appear to be clinically aggressive tumors with high metastatic rate but relatively low mortality with NTRK inhibitor therapy. The histologic findings of multinodular growth and extensive lymphovascular spread, seen in all NRTC, including PTC and SC, may serve as useful histomorphologic clues to prompt NTRK status testing. We also present the first report of concurrent TERT promotor activating mutation which did not appear to confer entrectinib resistance to NRTC.
Introduction
Rearrangements involving the neurotrophic-tropomyosin receptor kinase (NTRK) gene family (NTRK1, NTRK2, and NTRK3) are well-known drivers in a wide diversity of human cancers [1]. NTRK fusions with various partner genes induce oncogenesis by producing chimeric oncoproteins with a constitutively activated kinase function, and lead to downstream stimulation of cellular proliferation via the RAS/RAF/MAPK pathway [2]. In the thyroid, NTRK-driven malignancies are rare [3–10]. In one recent genomic analysis of 33,997 solid tumors, NTRK1-3 rearrangements were found in 13 of 571 (2.28%) thyroid cases [1]. A similar frequency was reported by another study of 496 adult thyroid tumors in The Cancer Genome Atlas (TCGA), which noted NTRK1–3 rearrangements in 2.34% [11]. In the special populations of pediatric papillary thyroid carcinomas (PTC) and post-Chernobyl reactor accident PTC, NTRK fusions seemed slightly more common, reported in 2–26% and 3–15%, respectively [5–7]. Rearrangements discovered in thyroid tumors have thus far included EML4-NTRK3 [12], ETV6-NTRK3 [5, 6, 8, 13], IRF2BP2-NTRK1 [12], TPR-NTRK1 [5, 8, 10], TPM3-NTRK1 [7, 10], TFG-NTRK1 [10, 13], TRIM33-NTRK1 [13] and SQSTM1-NTRK3 [4, 12].
Determination of NTRK activation status has recently gained high clinical utility since the emergence of targeted inhibitor therapy. Among many investigated agents, larotrectinib and entrectinib have successfully received approval from the United States Food and Drug Administration approval for treating NTRK-rearranged solid tumors [14–17]. To identify targetable patients, several laboratory approaches, such as pan-TRK immunohistochemistry and nucleic acid sequencing methods, have attracted significant research interest in validation for clinical use with varied sensitivity observations [1, 18, 19]. Considering the substantial cost of laboratory diagnostics and the very low pre-test incidence of NTRK-rearranged thyroid carcinoma (NRTC), it remains an ongoing pursuit to develop a practical approach for recognizing NRTC based on clinicopathologic findings so that ancillary testing might be performed in a selective and algorithmic manner. Nevertheless, few studies have systemically examined the histologic spectrum of NRTC. While most published NRTC were allegedly PTC, there have been 11 case reports of primary thyroid secretory carcinomas (SC) with ETV6-NTRK3 fusions and displaying the same immunophenotype as SC of the salivary gland [20]. Both NTRK-rearranged PTC and primary thyroid SC have been associated with aggressive clinical behavior in small cohorts and isolated reports, but the literature is still scarce and in need of additional evidence [5, 9, 10, 20, 21]. Studies of NTRK-rearranged tumors have typically shown great responsiveness to larotrectinib and entrectinib with very little primary resistance seen in thyroid tumors [15–18, 22] apart from one study showing ~20% resistance to larotrectinib in NRTC [16]. Unlike secondary resistance which has been linked to acquired, on-target kinase domain mutations that interfere with drug binding [23] and bypassing mutations such as BRAF V600E and KRAS G12A/D [24], the mechanism of primary resistance remains poorly understood. Possible causes of primary progressive disease may include lack of expression of the NTRK fusion at the protein level, which may be assessed by pan-TRK immunohistochemical (IHC) analysis [22].
In this study, we conducted a thorough analysis of a series of NRTC from a single institution. The objective was twofold: (1) to establish an integrative clinical, histologic, immunophenotypic and genetic profile as an aid for future diagnostic evaluation, and (2) to describe long-term patient outcome in NRTC including response to NTRK inhibitor therapy.
Material and methods
Case identification
The study was approved by the Massachusetts General Hospital (MGH) Institutional Review Board (protocol No. 2012P001024). We retrospectively queried the MGH molecular pathology database for primary thyroid tumors that were consented by patients to undergo the targeted gene rearrangement next-generation sequencing analysis (described below) during the years 2013–2019 and reviewed the test results. For cases that showed NTRK rearrangements, diagnostic slides were retrieved from the MGH surgical pathology archive and evaluated by three pathologists (VN, PMS, and YHC). At MGH, cases were selected for molecular testing by the primary pathologists based on unusual morphology, advanced presentation, or an absence of BRAF V600E mutation by mutant-specific immunohistochemistry (described below). For this study, we have limited our case search to cases with an established thyroid carcinoma diagnosis at the time of genetic profiling, whose aim was mainly to find therapeutic targets. Patient demographic information, clinical notes, surgical, and molecular pathology reports were obtained from the electronic medical record system at MGH.
Immunohistochemistry
IHC stains were performed using 4-μm-thick formalin-fixed paraffin-embedded (FFPE) sections of selected representative tissue blocks from the 11 NTRK-rearranged thyroid tumors. A Bond 3 automated stainer (Leica Microsystems, Bannockburn, IL) and compatible primary antibodies for S100 protein (polyclonal and prediluted; Ventana, Oro Valley, AZ), mammaglobin (clone 304-1A5; dilution 1:250; Dako, Carpinteria, CA), GATA3 (clone L50-823, dilution 1:200; Cell Marque, Rocklin, CA), PAX8(polyclonal, dilution 1:1000; Proteintech, Rosemont, IL), TTF1 (clone 8G7G3/1; dilution 1:300; Dako, Carpinteria, CA), BRAF V600E (clone VE1; dilution 1:200; Abcam, Cambridge, United Kingdom), thyroglobulin (dilution 1:800; Cell Marque, Rocklin, CA) and HBME1 (prediluted; Ventana, Oro Valley, AZ) were used. Appropriate positive and negative controls were included. Stained slides were reviewed by three pathologists (VN, PMS, and YHC) and graded as negative (0% tumor cells stained), focal (<50%) and diffuse (>50%).
Targeted gene rearrangement next-generation sequencing (NGS) analysis
As previously described [25], an Anchored Multiplex PCR (AMP) assay was performed for targeted fusion transcript detection using the NGS technology. Total nucleic acid was extracted from FFPE tumor tissue, reverse transcribed with random hexamers, and converted to double-stranded complementary DNA (cDNA). The cDNA was end-repaired, adenylated, and ligated with a half-functional adapter. Two hemi-nested PCR reactions were carried out using the ArcherDx FusionPlex Solid Tumor Kit primers to create a sequencing library that targeted the following specific genes (exons): ALK (19–22, intron 19), BRAF (7–12, 15), EGFR (2–7 exon skipping/vIII variant, 7–9, 16, 20, 24, 25), EWSR1 (4–14), FGFR2 (2, 8–10, 17), MAML2 (2,3), MET (exon 14 skipping), NRG1 (1–3, 6), NUTM1 (3), RET (8–13), ROS1 (31–37), AKT3 (1–3), ARHGAP26 (2, 10–12), AXL (19,20), BRAF (7–12, 15), BRD3 (9–12), BRD4 (10, 11), ERG (2–11), ESR1 (3–6), ETV1 (3–13), ETV4 (2, 4–10), ETV5 (2, 3, 7–9), ETV6 (1–7), FGFR1 (2, 8–10, 17), FGFR3 (8–10, 17, intron 17), FGR (2), INSR (12–22), JAZF1 (2–4), MAML2 (2,3), MAST1 (7–9, 18–21), MAST2 (2, 3, 5, 6), MET (13, 15), MSMB (2–4), MUSK (7–9, 11–14), MYB (7–9, 11–16), NOTCH1 (2, 4, 26–31, internal exon 3–27 deletion), NOTCH2 (5–7, 26–28), NRG1 (1–3, 6), NTRK1 (8,10–13), NTRK2 (11–17), NTRK3 (13–16), NUMBL (3), PDGFRA (7, exon 8 deletion, 10–14), PDGFRB (8–14), PIK3CA (2), PKN1 (10–13), PPARG (1–3), PRKCA (4–6), PRKCB (3), RAF1 (4–7, 9–12), RELA (3, 4), RSPO2 (1, 2), RSPO3 (2), TERT (2), TFE3 (2–8), TFEB (1,2), THADA (28), and TMPRSS2 (1–6). Illumina MiSeq 2 × 147 base paired-end sequencing results were aligned to the hg19 human genome reference using BWA-MEM [26]. Fusion detection results were reviewed by a molecular pathologist (DDS).
SNaPshot mutation profiling
An AMP-based, clinically validated SNaPshot assay for targeted single nucleotide variants (SNVs), insertion/deletions (indels), and copy number variations (CNVs) detection in tumor-derived genomic DNA were performed for all the 11 NRTCs using the ArcherDx and Illumina NextSeq platforms as previously described [27]. The assay covered SNV and indel gene targets within the following genes (exons): ABL1 (4–7), AKT1 (3,6), ALK (21–23,25), APC (16), ARID1A (1–20), ATM (1–63), ATRX (1–35), AURKA (2,5–8), BRAF (11,15), BRCA1 (2–23), BRCA2 (2–27), CCNB1 (2,[3-partial],5,[6-partial],7), CCND2 ([2-partial],3–4,[5-partial]), CCND3 (2–5-partial), CCNE1 (3–8,10,12), CDH1 (1–16), CDK4 (2–7), CDK6 (6), CDKN2A (1–3), CIC (1–20), CSF1R (7,22), CTNNB1 (3), DAXX (1–8), DDR2 (12–18), DDX3×(1–17), EGFR (3,7,15,18–21), ERBB2 (8,10,19–21,24), ERBB3 (2–3,7–8), ERBB4 (3–4,6–9,15,23), ESR1 (8), EZH2 (16), FBXW7 (1–11), FGFR1 (4,7–8,13,15,17), FGFR2 (7,9,12,14), FGFR3 (7–9,14–16,18), FLT3 (11,14,16,20), FOXL2 (1), GNA11 (5), GNAQ (4–5), GNAS (6–9), H3F3A (2), HNF1A (3–4), HRAS (2–3), IDH1 (3–4), IDH2 (4), JAK2 (11,13–14,16,19), JAK3 (4,13,16), KDR (6–7,11,19,21,26–27,30), KEAP1 (2–6), KIT (2,8–11,13–15,17–18), KRAS (2–5), MAP2K1 (2,3,6–7), MAP3K1 (1–20), MDM2 (2–4,6,8,10), MDM4 ([4-partial], 5–6, [7,9–11-partial]), MEN1 (2–10), MET (2,11,14,16,19,21), MITF (1-partial), MLH1 (12), MPL (10), MSH6 (1–10), MSI, MYC (1–3), MYCN (3), NF1 (1–58), NF2 (1–15), NKX2–1 (1-partial), NOTCH1 (25–27,34), NPM1 (11), NRAS (2–5), PDGFRA (12,14–15,18,23), PIK3CA (2,5,7–8,10,14,19,21), PIK3R1 (1–10), POLE (9–14), PTCH1 (1–23), PTEN (1–9), PTPN11 (3,13), RB1 (1–27), RET (10–11,13–16), RHOA (2–3), RNF43 (2–10), ROS1 (36–38), SDHB (1–8), SMAD2 (7), SMAD4 (2–12), SMARCA4 (3–36), SMARCB1 (2,4,5,9), SMO (3,5–6,9,11), SRC (14), STAG2 (3–34), STK11 (1–9), SUFU (1–12), TERT (1), TP53 (1–11), TP63 (1–14), TSC1 (3–23), TSC2 (2–42), TSHR (10), and VHL (1–3). The CNV gene targets covered by this test were as follows: ABL1, AKT1, ALK, APC, ARID1A, ATM, ATRX, AURKA, BRAF, BRCA1, BRCA2, CAMTA1, CCNB1, CCND1, CCND2, CCND3, CCNE1, CDK4, CDKN2A, CDK6, CIC, CDH1, CSF1R, DAXX, DDR2, DDX3x, EGFR, ERBB2 (HER-2), ERBB3, ERBB4, FBXW7, FGF19, FGFR1, FGFR2, FGFR3, FLT3, FOXL2, GLI2, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, JAK2, JAK3, KDR, KEAP1, KIT, KRAS, MAP2K1, MAP3K1, MDM2, MDM4, MEN1, MET, MITF, MLH1, MSH6, MYC, MYCN, NF1, NF2, NKX2–1, NOTCH1, NRAS, PDGFRA, PIK3CA, PIK3R1, PLAUR, POLE, PTCH1, PTEN, PTPN11, RB1, RET, RHOA, RNF43, SDHB, SMAD2, SMAD4, SMARCA4, SMARCB1, SMO, SRC, STAG2, STK11, SUFU, TERT, TP53, TP63, TSC1, TSC2, and VHL. Sequencing results were reviewed by a molecular pathologist (DDS).
Results
Clinical presentation
A total of 351 primary thyroid carcinomas were subjected to the targeted gene rearrangement next-generation sequencing assay at MGH during the years 2013 through 2019. The originally rendered pathologic diagnoses include 186 PTC, 54 medullary carcinomas, 38 anaplastic carcinomas, 24 oncocytic (Hurthle cell) carcinomas, 23 follicular carcinomas, 21 poorly differentiated carcinomas, and 5 unclassifiable carcinomas. NTRK rearrangements were detected in 11 (3.1%) cases, all of which had been originally diagnosed as PTC (Table 1).
Table 1.
Clinical characteristics of NTRK-rearranged thyroid carcinomas.
| Case # | Age (years) | Sex | Radiation exposure | Size (cm) | Stage | Distant metastasis | Treatment | Follow-up (months) | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 42 | M | − | 7.0 | T3N1b | Lung, brain® | RAI; entrectinib | 471 | Recurrence ×2; AWD |
| 2 | 14 | F | − | 4.2 | T3N1b | Lung (D) | RAI | 80 | Recurrence; AWD |
| 3 | 32 | F | − | 3.5 | T3N1ba | − | RAI | 39 | NED |
| 4 | 74 | F | − | 10.5 | T3N1b | − | RAI; XRT | 13 | Recurrence; AWD |
| 5 | 60 | F | − | 7.0 | T3N1a | Lung® | RAI | 117 | Recurrence ×2; AWD |
| 6 | 22 | F | − | 2.1 | T3N1aa | − | RAI | 44 | NED |
| 7 | 37 | F | − | 3.9 | T2N1a | − | RAI | 44 | AWD |
| 8 | 43 | F | − | 6.6 | T3N1b | Lung (D) | RAI | 11 | AWD |
| 9 | 54 | M | +b | 4.0 | T3N1ba | Lung® | RAI; larotrectinib | 142 | Recurrence ×2; AWD |
| 10 | 24 | M | − | 5.4 | T3N1b | Lung (D) | RAI | 34 | AWD |
| 11 | 71 | F | − | 3.2 | T3N1a | − | RAI; XRT; larotrectinib | 68 | Recurrence ×2; NED |
R at recurrence, D at diagnosis, RAI radioactive iodine, XRT external radiation therapy, AWD alive with disease, NED alive with no evidence of disease.
The case was staged as T3 per the American Joint Committee on Cancer (AJCC) 7th edition guidelines at the time of diagnosis and managed accordingly, although they would have been deemed as T2 based on the AJCC 8th edition criteria had they occurred more recently.
History of radiotherapy in adolescence for acne.
The 11 NRTC included a 14-year-old adolescent and 10 adults ranging from 22 to 74 years in age (median 42 years). A 2.7-fold female predominance was observed (male-to-female ratio was 3:8). The cases were radiation-naïve except for one patient (case 9), who had low-dose radiotherapy for acne during adolescence (>30 years before developing thyroid cancer). All patients presented with a large and infiltrative mass pathologically staged as T2 (9%) or T3 (91%). At the initial presentation, all cases had cervical lymph node involvement, which was limited to the central compartment (N1a) in 4 (36%) patients and spreading to the lateral compartments (N1b) in 7 (64%) patients. Pulmonary metastases were present in 3 (27%) patients at the initial presentation and occurred later in the disease course to three more patients. The clinical characteristics of all cases are summarized in Table 1.
Histologic, immunophenotypic and molecular features
The observed NTRK rearrangements included ETV6-NTRK3 (n = 4, including the SC), TPR-NTRK1 (n = 2), RBPMS-NTRK3 (n = 2), SQSTM1-NTRK1 (n = 1), SQSTM1-NTRK3 (n = 1), and EML4-NTRK3 (n = 1). Although breakpoint positions were varied, the kinase domain was preserved in all cases (Fig. 1). No other NTRK alteration or other gene arrangement was found in the 11 NRTC. Regardless of fusion type, all cases showed a remarkably similar appearance at low magnification, characterized by multiple infiltrative tumor nodules and extensive lymphovascular spread within intrathyroidal and extrathyroidal vessels of variable caliber (Figs. 2–5). Direct extrathyroidal extension, at least microscopic, was identified in 9 (82%) cases. At high magnification, cases with the same gene fusion tended to present similar cellular architectural properties, as described below. The histologic and IHC findings of all cases are summarized in Tables 2 and 3, respectively.
Fig. 1. Observed NTRK rearrangements in the cases.
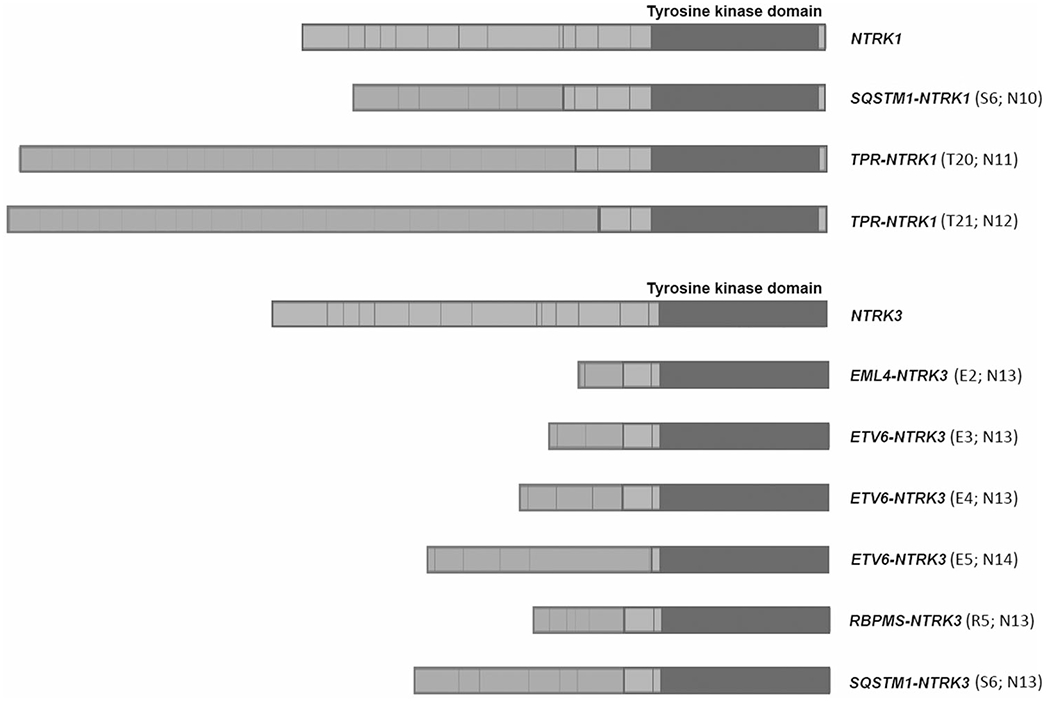
Numbers in parentheses indicate the exons involved at fusion breakpoints. The tyrosine kinase domain of NTRK1/3 remained intact in all the cases.
Fig. 2. NRTC with predominantly follicular pattern associated with ETV6-NTRK3 and RBPMS-NTRK3 fusions.
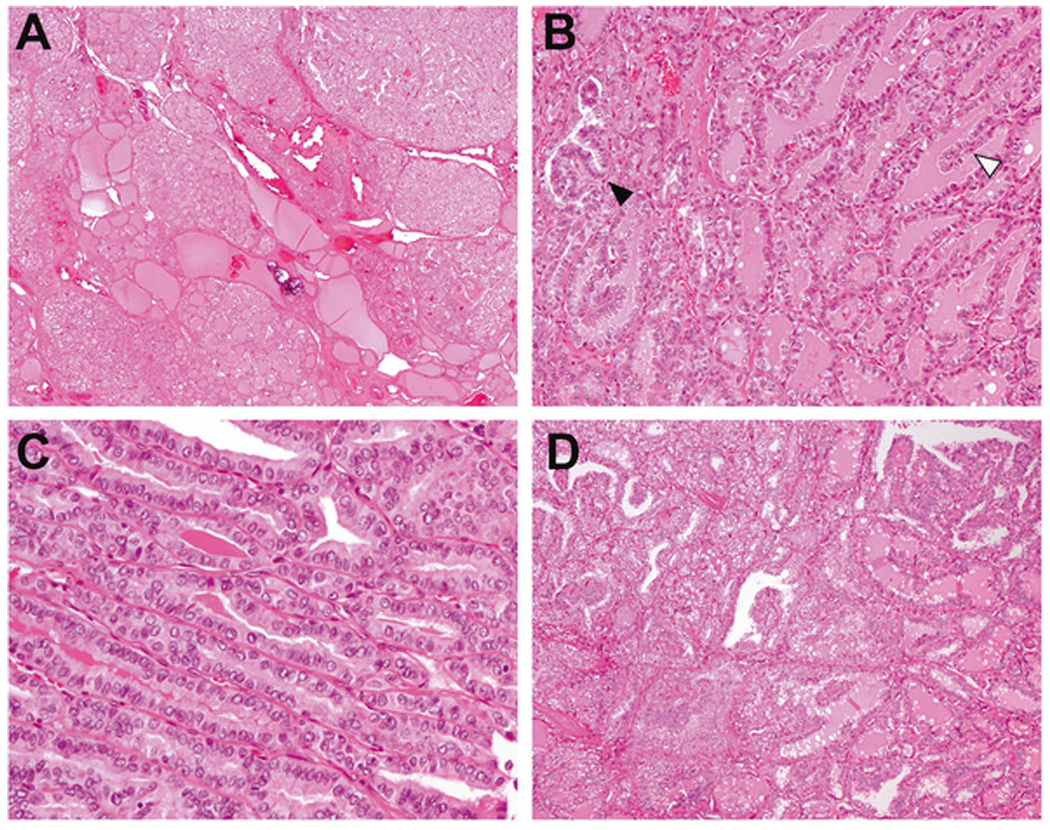
a–c Papillary thyroid carcinomas with ETV6-NTRK3 fusion showed multiple irregularly shaped tumor nodules infiltrating the thyroid (a). Growth pattern was mainly follicular with isolated foci of true papillae (black arrowhead) and hyperplasia-like epithelial folding (white arrowhead) (b). Tall cell features were focally seen in case 4 (c). d Papillary thyroid carcinomas with RBPMS-NTRK3 fusion had similar histologic appearance composed of well-formed neoplastic follicles and scattered papillae.
Fig. 5. NRTC with predominantly insular pattern associated with SQSTM1-NTRK3 fusion (case 8).
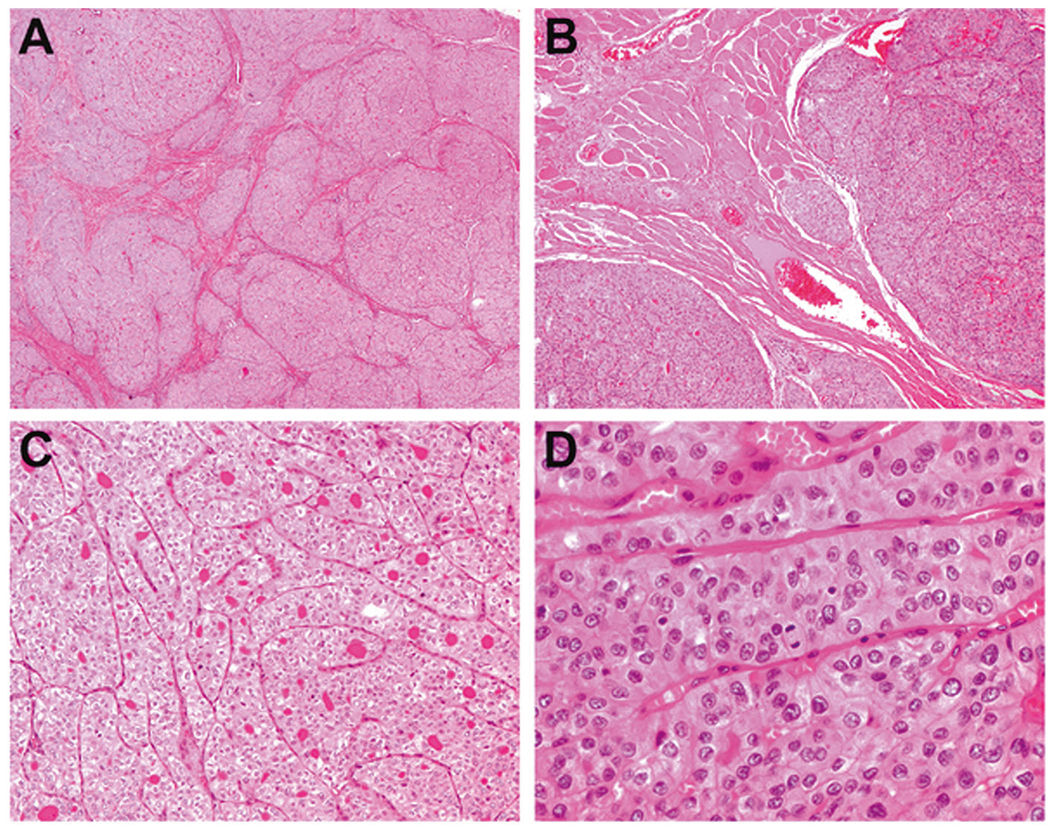
The tumor is multinodular and divided by fibrous septa (a). There is extensive extrathyroidal extension into the strap muscle (b). Higher magnification revealed an insular growth pattern with scattered microfollicles (c). The tumor cells are oncocytic, with convoluted nuclei, finely granular chromatin, and increased mitotic activity (up to 4 mitoses per 10 high-power fields) (d).
Table 2.
Histologic findings and observed fusions in NTRK-rearranged thyroid carcinomas.
| Case # | Original diagnosis | Revised diagnosis | Multinodularity | Fusion | Growth Pattern(s) | LVI | PNI | ETE | Margin |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PTC | PTC | na | EML4-NTRK3 | Primary tumor not availablea; brain metastasis was oncocytic forming microfollicles and papillae | + | − | − | − |
| 2 | PTC | PTC | + | ETV6-NTRK3 | Predominantly (90%) follicular with scatted papillae (10%); psammoma calcifications | + | − | +st | + |
| 3 | PTC | PTC | + | ETV6-NTRK3 | Predominantly (90%) follicular with scatted papillae (10%); psammoma calcifications | + | − | +st | − |
| 4 | PTC | PTC | + | ETV6-NTRK3 | Predominantly (70%) follicular with scatted papillae (20%); 10% tall cell; no psammoma calcification | + | − | +sm | + |
| 5 | PTC | PTC | + | RBPMS-NTRK3 | Primary tumor not availablea; lymph node metastasis was predominantly (90%) follicular with scatted papillae (10%) | + | − | +sm | + |
| 6 | PTC | PTC | + | RBPMS-NTRK3 | Predominantly (90%) follicular with scatted papillae (10%); psammoma calcifications | + | − | +st | − |
| 7 | PTC | PTC | + | SQSTM1-NTRK1 | Labyrinthine pattern formed by coalescent true and abortive papillae; no psammoma calcification | + | − | − | + |
| 8 | PTC | PTC | + | SQSTM1-NTRK3 | Insular and oncocytic with scattered microfollicles; increased mitotic activity; no psammoma calcification | + | − | +sm | + |
| 9 | PTC | PTC | + | TPR-NTRK1 | Packeted papillae with psammoma calcifications | + | − | +st | − |
| 10 | PTC | PTC | + | TPR-NTRK1 | Packeted papillae with psammoma calcifications | + | + | +st | + |
| 11 | PTC | SC | + | ETV6-NTRK3 | Microcystic; intracellular and extracellular eosinophilic material; no psammoma calcifications | + | − | +sm | + |
PTC papillary thyroid carcinoma, SC secretory carcinoma, na not available, LVI lymphovascular invasion, PNI perineural invasion, ETE extrathyroidal extension, microscopic or macroscopic, +st present in perithyroidal soft tissue, +sm present in strap muscle.
For cases #1 and #5, the histologic slides for the primary tumor were not available for the study, and written reports were reviewed.
Table 3.
Immunohistochemical analysis of NTRK-rearranged thyroid carcinomas.
| Case # | TTF1 | PAX8 | Thyroglobulin | HBME1 | BRAF V600E | Mammaglobin | S100 | GATA3 |
|---|---|---|---|---|---|---|---|---|
| 1 | ++ | ++ | + | ++ | − | − | − | − |
| 2 | ++ | ++ | ++ | ++ | − | − | − | − |
| 3 | ++ | ++ | ++ | ++ | − | − | − | − |
| 4 | ++ | ++ | ++ | ++ | − | − | − | − |
| 5 | ++ | ++ | ++ | ++ | − | − | − | − |
| 6 | ++ | ++ | ++ | ++ | − | − | − | − |
| 7 | ++ | ++ | + | + | Naa | − | − | − |
| 8 | ++ | ++ | ++ | ++ | − | − | − | − |
| 9 | ++ | ++ | ++ | ++ | − | − | − | − |
| 10 | ++ | ++ | ++ | ++ | − | − | − | − |
| 11 | + weak | − | − | na | − | ++ | ++ | ++ |
na not available, ++ diffuse, + focal, − negative.
Although BRAF V600E IHC is not available for case 7, SNaPshot testing was negative for BRAF V600E mutation.
ETV6-NTRK3 fusion thyroid carcinomas
ETV6-NTRK3 was the most common gene rearrangement, detected in 4 cases (case 2, 3, 4, 11). Histologically, cases 2–4 were PTC that showed multinodular growth with predominantly follicular architecture (Fig. 2a). There were scattered isolated foci of papillary formation, including true papillae, abortive/micropapillae, and hyperplasia-like epithelial folding (Fig. 2b). Case 4 showed focal tall cell change, accounting for about 10% of tumor volume (Fig. 2c). Psammoma bodies were present in cases 2 and 3 but absent in case 4. By immunohistochemistry, cases 2–4 showed diffuse expression of thyroid markers (TTF1, PAX8, thyroglobulin) and membranous labeling for HBME1.
Case 11, a primary thyroid SC, also showed multinodular growth but was histologically unique for prominent intratumoral fibrosis and admixed microcystic and tubulopapillary patterns (Fig. 3). The tumor cells showed elongated, frequently grooved nuclei (Fig. 3b), which had led to an original diagnosis of PTC. However, other PTC features, such as nuclear crowding and clearing, were lacking. Instead, the nuclei had finely stippled chromatin and prominent, central nucleoli. In addition, there was abundant vacuolar cytoplasm with eosinophilic intracytoplasmic and extracellular secretion that was thyroglobulin-negative (Fig. 3b). Unlike the other ETV6-NTRK3 fusion cases, case 11 expressed only focal weak TTF1 and was negative for PAX8 and thyroglobulin. In contrast, there was diffuse immunopositivity for S100, mammaglobin and GATA3 (Fig. 3). No primary salivary gland tumor was found after thorough clinical and radiologic survey of the patient, indicating a primary thyroid SC of the salivary type.
Fig. 3. Primary thyroid secretory carcinoma with ETV6-NTRK3 fusion (case 11).
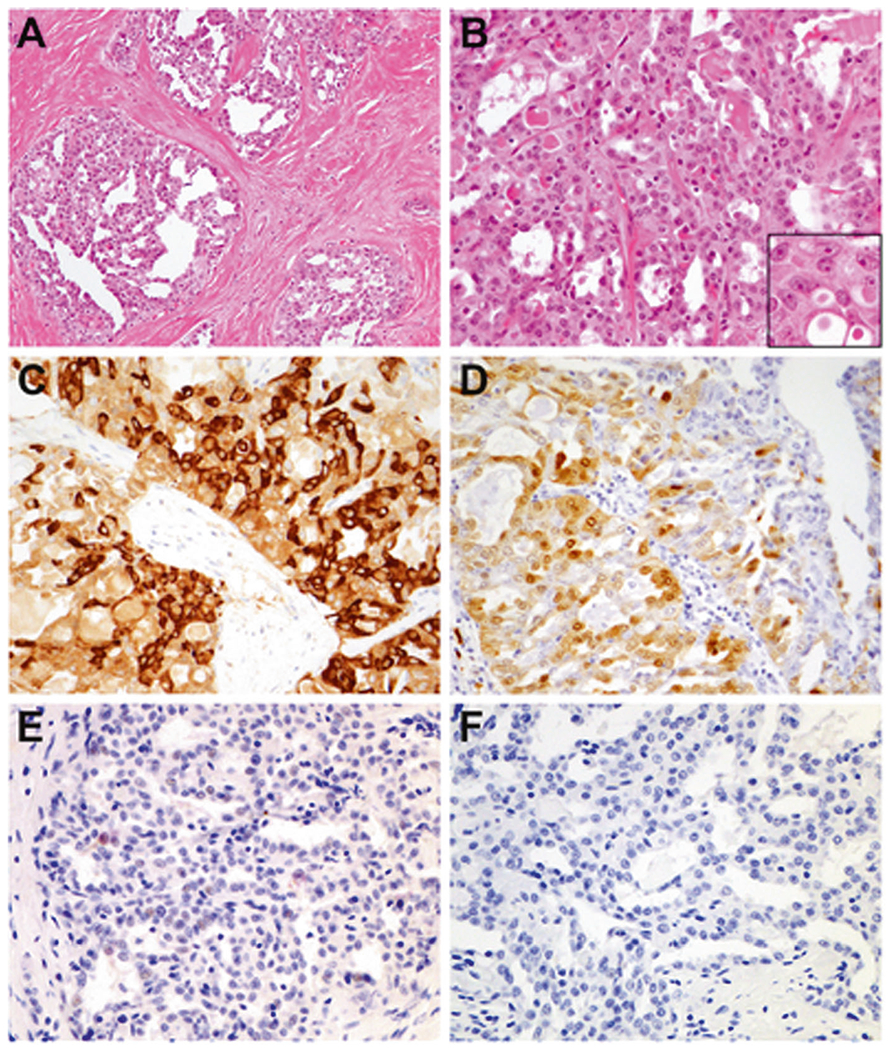
The tumor demonstrated a lobulated appearance with prominent intratumoral fibrosis (a). Admixed microcystic and tubulopapillary patterns were seen, and the cells showed elongated, often grooved nuclei with finely speckled chromatin and central nucleoli (b, inset). The cytoplasm was abundant and vacuolar, frequently containing intracytoplasmic eosinophilic globules in a signet ring-like appearance (b, inset). By immunohistochemistry, there was diffuse expression of mammaglobin (c) and S100 (d), and focal weak TTF1 (e). PAX8 was negative (f).
RBPMS-NTRK3 fusion thyroid carcinomas
The RBPMS-NTRK3 fusion was detected in two PTC (5 and 6). At the time of the study, glass slides for the case 5 primary tumor were unavailable. Therefore, for case 5, we utilized the primary thyroidectomy pathology report and studied the histology from the two cervical recurrences at 4 and 8 years after the initial diagnosis; all IHC and molecular analyses were performed on the second recurrence. Histologically, both cases displayed a multinodular growth with predominantly follicular architecture and scattered papillae, bearing close similarity to the ETV6-NTRK3 fusion PTC (Fig. 2d). Immunophenotypically, both cases were positive for TTF1, PAX8, thyroglobulin and HBME1, and were negative for S100, mammaglobin and GATA3.
TPR-NTRK1 fusion thyroid carcinomas
The TPR-NTRK1 fusion was detected in two histologically similar cases (9 and 10). At low magnification, both cases showed multiple nodules of packeted papillae divided by fibrotic septa (Fig. 4a). Psammomatous calcifications were numerous. One interesting finding was scattered glomeruloid structures formed by dilated neoplastic follicles each containing an intraluminal cluster of partially fused papillae often forming a pseudocribriform pattern (Fig. 4b). Both cases expressed TTF1, PAX8, thyroglobulin, and HBME1, and were negative for S100, mammaglobin, and GATA3.
Fig. 4. NRTC with predominantly papillary pattern associated with TPR-NTRK1 and SQSTM1-NTRK1 fusions.
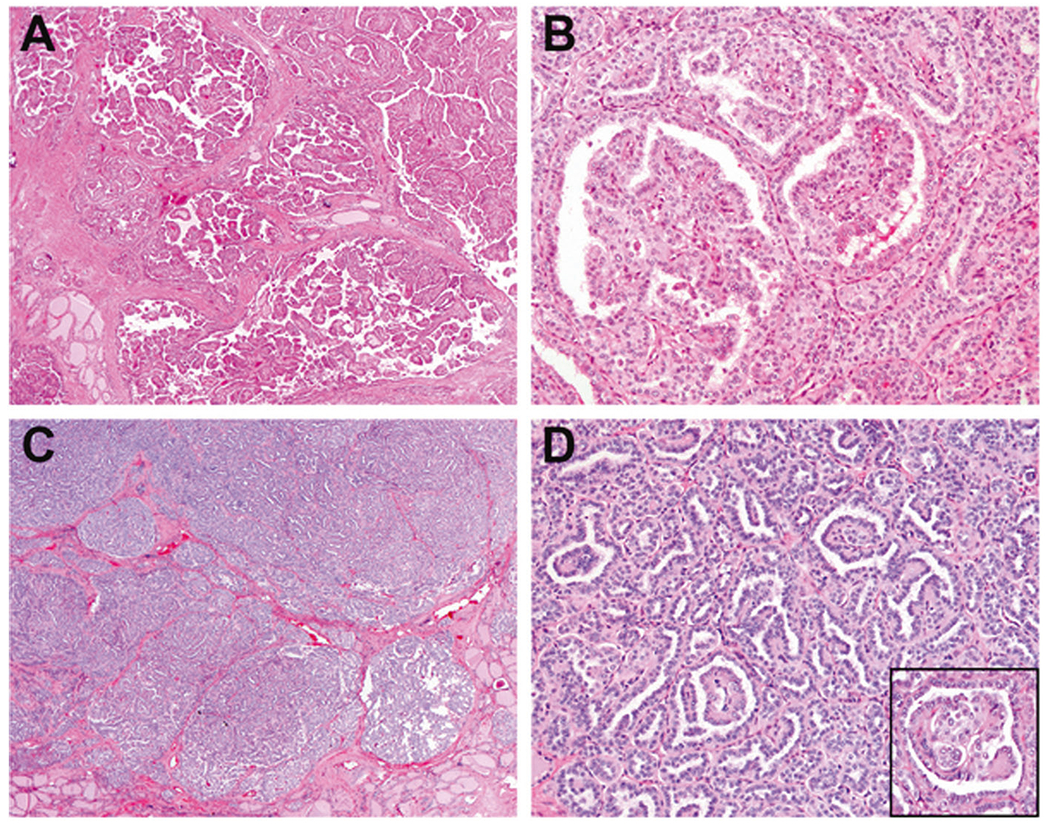
TPR-NTRK1 fusion thyroid carcinoma, showing multiple infiltrative nodules of well-formed papillae (a) and scattered glomeruloid structures (b). SQSTM1-NTRK1 fusion thyroid carcinoma exhibited multinodular growth (c) and a labyrinthine pattern formed by coalescent true and abortive papillae with glomeruloid structures (d, inset).
SQSTM1-NTRK1 fusion thyroid carcinomas
Case 7 exhibited overlapping histology with the TPR-NTRK1 fusion cases. The tumor was formed by multiple ill-defined nodules (Fig. 4c), which at high magnification showed a labyrinthine appearance formed by coalescent true and abortive papillae (Fig. 4d). Scattered glomeruloid structures were present (Fig. 4d inset). Colloid production and psammoma calcifications were absent. Immunostains revealed very focal labeling for thyroglobulin and HBME1 and diffuse positivity for TTF1 and PAX8. S100, mammaglobin and GATA3 were negative.
SQSTM1-NTRK3 fusion thyroid carcinomas
Case 8 was histologically unique in the series, consisting of numerous islands of oncocytic tumor cells in solid, trabecular and insular patterns with scattered microfollicles and increased mitotic activity (4 mitoses per 10 high power fields) (Fig. 5). Papillary nuclear features, such as nuclear irregularity and clearing, were identified (Fig. 5d). Given the prominent insular growth pattern and invasiveness, including increased mitotic activity, a diagnosis of poorly differentiated thyroid carcinoma (PDTC) was considered. However, lacking necrosis and with multifocal features of papillary thyroid carcinoma, the final diagnosis was PTC with high-grade features. Immunohistochemistry revealed diffuse positivity for TTF1, PAX8, thyroglobulin and HBME1, and negativity for S100, mammaglobin and GATA3.
EML4-NTRK3 fusion thyroid carcinomas
EML4-NTRK3 fusion was detected in an intracranial metastatic thyroid carcinoma to the right lateral ventricle in case 1, occurring 36 years after his initial diagnosis in 1981. The histologic slides of the primary tumor and for the first recurrence in 2000 were no longer archived at the time of the study, and therefore tumor characteristics were collected from the electronic medical records and summarized in Table 2. The brain metastasis showed oncocytic tumor cells forming microfollicles and papillae (Fig. 6). Immunophenotypically, there was diffuse positivity for TTF1, PAX8, and HBME1, and focal expression of thyroglobulin. S100, mammaglobin and GATA3 were negative.
Fig. 6. Brain metastasis from an EML4-NTRK3 fusion thyroid carcinoma (case 1).
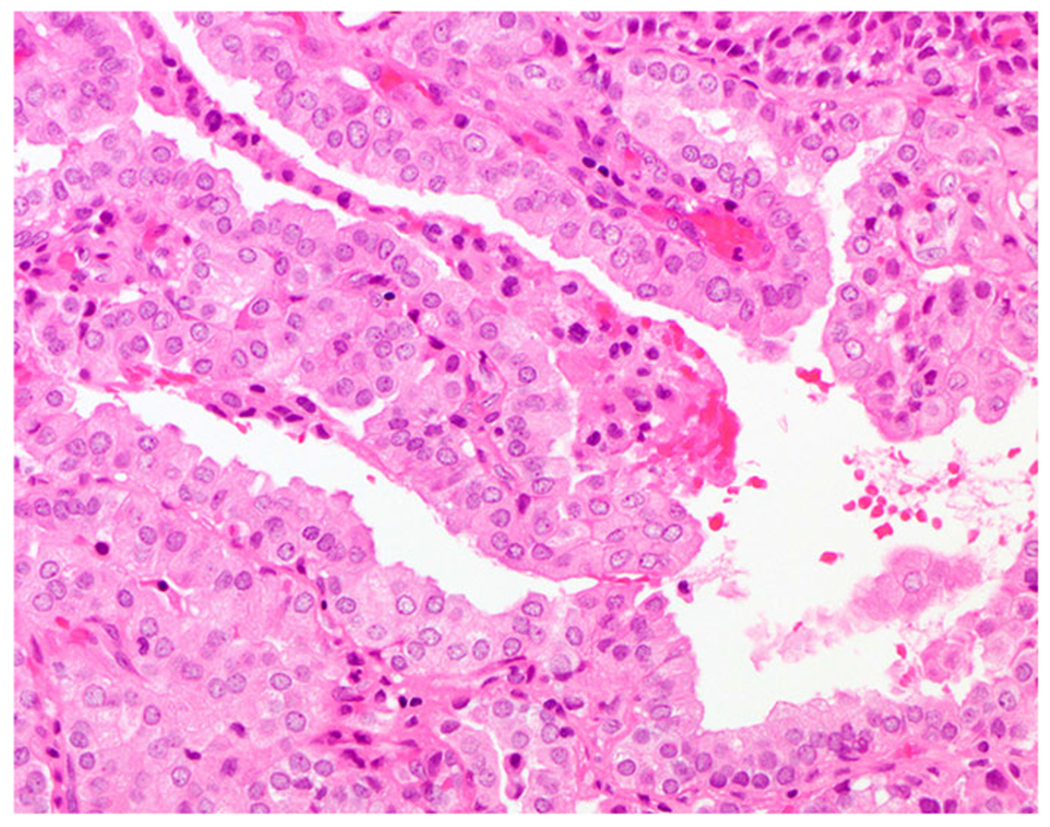
Tumor cells were oncocytic with finely stippled nuclei and occasional nuclear grooves, forming microfollicles and papillae.
SNaPshot mutational profiling
SNaPshot targeted mutational NGS analysis was successfully performed in nine cases. In cases 2 and 9, the data output was not interpretable due to nucleic acid quality. In genes that were covered by the assay, all NRTC showed a low mutation burden. Variants were detected in four (44%) cases, including a pathogenic TERT promotor mutation c.−124C>T (C228T) in two cases (cases 1 and 5) and a likely pathogenic frameshift deletion in MEN1 (ENSP00000308975.6:p.Pro55Leuf-sTer64) in case 1, who had neither personal nor family history of multiple neuroendocrine neoplasia or any neuroendocrine tumor. In addition, two variants of uncertain significance were also noted: a splicing region variant in GNA11 (ENST00000078429.4:c.735+7C>G) in case 7 and a non-binding site missense variant in IDH1 (ENSP00000260985.2:p.Ala51Gly) in case 10. No alterations were detected in cases 3, 4, 6, 8, 11.
Treatment and outcome
As summarized in Table 1, all patients initially received total thyroidectomy and radioactive iodine (RAI) treatment. Nine (82%) patients had disease persistence or recurrence, including all cases with RBPMS-NTRK3, TPR-NTRK1, SQSTM1-NTRK3, and EML4-NTRK3 fusions and three of the four (75%) cases with ETV6-NTRK3 fusion. The SQSTM1-NTRK1 fusion case (case 6) and one ETV6-NTRK3 fusion case (case 3) showed no evidence of disease at 44 and 39 months after the initial treatment, respectively. Over a median (range) follow-up of 44 (11–471) months, no deaths occurred. Three patients developed lung metastases at recurrence, bringing the overall lung metastatic rate to 55% (Table 1). Brain metastasis occurred more than three decades after thyroidectomy in case 1. No other distant organ was involved. NTRK inhibitor therapy was given in cases 1, 9 and 11. In case 1, which tested positive for the pTERT C228T mutation, the MEN1 frameshift mutation and the EML4-NTRK3 fusion prior to therapy, there was a 69.7% decrease in disease burden following 34 cycles of entrectinib (400 mg daily). In Case 9, a TPR-NTRK1 fusion tumor, the disease burden decreased by 33% compared with his baseline after 2 cycles of larotrectinib (100 mg twice per day) and remained stable for the subsequent 15 months leading to the study endpoint. Case 11, with a primary thyroid SC harboring ETV6-NTRK3 fusion, experienced complete radiologic resolution after four cycles of larotrectinib (100 mg twice per day). The patient has remained free of any detectable disease for the subsequent 21 months until the study endpoint. Based on the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, most adverse events were grade 1 to 2 and clinically non-serious (case 1: fatigue, exertional dyspnea, gait disturbance, anemia, leukopenia, serum creatinine elevation; case 9: hoarseness, neck stiffness, hot flashes; fatigue, constipation, elevation of hepatic enzymes aspartate transaminase and alanine transaminase; case 11: instability, hypophosphatemia). Case 1 had dose reduction from 600 to 400 mg daily during the first cycle due to grade 2 gait disturbance; he also had transient grade 3 orthostatic hypotension during the 15th treatment cycle, which lasted for two weeks and subsequently improved. No grade 4 adverse event or death was noted.
Discussion
NTRK-related thyroid cancers are rare tumors (1); however, their identification has recently gained high clinical relevance since anti-NTRK therapeutic agents have become available. Our series demonstrated a wide morphologic spectrum including predominantly PTC with remarkably varied growth patterns and SC. Based upon these findings, there may be significant clinical benefit to performing NTRK status testing in subset of thyroid carcinomas with unusual morphologies, particularly those that present with locally advanced disease with frequently observed histologic findings of multinodularity and extensive lymphovascular spread, as noted in the current study, and those lacking BRAF V600E, available with rapid molecular testing or immunohistochemistry [28]. While our subjects were mainly adults, similar histologic characteristics had also been noted by Prasad et al. in seven pediatric NRTC [5], and thus seemed to apply to both age populations.
In our series, histologic tumor patterns are variable, but they each showed correlation with underlying gene rearrangements. Except for case 11 (the SC), the other three ETV6-NTRK3 fusion cases were histologically similar with a predominantly follicular architecture. The same morphology was also reported by Prasad et al. in three pediatric ETV6-NTRK3 fusion thyroid carcinomas [5]. As for TPR-NTRK1 fusion thyroid carcinoma, both of our cases were composed of packeted papillae with glomeruloid structures. Interestingly, one solid-patterned and one follicular NRTC had been previously reported with TPR-NTRK1 fusion [5, 8]. Case 8, an SQSTM1-NTRK3 fusion NRTC, was a PTC with high-grade features, including insular growth and elevated mitotic rate. The only previously published SQSTM1-NTRK3 thyroid tumor was a classical type PTC [4]. RBPMS-NTRK3 and SQSTM1-NTRK1 fusion thyroid carcinomas were first time reported here, although both fusions have been noted in tumors of other organs [11].
Previously, Musholt et al. reported frequent pT4 disease (40%), lymph node metastases (67%), and tumor recurrence (60%) in a predominantly adult cohort of 15 sporadic NTRK1-rearranged thyroid tumors [10]. Of note, their series demonstrated a much lower rate of distant metastasis (7%) and higher tumor-related mortality (27%) with similar follow-up time [10] compared with our cases (55% and 0%, respectively). Since the current series included mostly NTRK3- rearranged cases, this raises an interesting question of whether there might be a behavioral difference between NTRK3-rearranged and NTRKI-rearranged thyroid carcinomas. Currently, the literature lacks sufficient data, and this remains an interesting topic for future research as more cases appear.
Concurrent TERT promotor mutation was previously undocumented in NRTC. pTERT C228T mutation was detected in an intracranial metastatic NRTC harboring EML4-NTRK3 (case 1) and a locally recurrent tumor harboring RBPMS-NTRK3 (case 5). Since molecular testing was performed in post-RAI recurrent tumors in both cases, it is unclear whether the pTERT C228T mutation had been present in the primary tumor versus representing an acquired change after RAI treatment. TERT promotor C228T mutation is a well characterized oncogenic mutation that restores the telomerase reverse transcriptase (TERT) activity and facilitates uncontrolled cell proliferation in melanomas, gliomas and carcinomas of the thyroid, bladder, kidney, and gastrointestinal tract [29]. Of note, case 1, with additional pTERT C228T mutation and frameshift MEN1 deletion, received entrectinib and had a 69.7% decrease in disease burden after 34 treatment cycles. In this case, neither the pTERT C228T mutation nor the frameshift MEN1 deletion conferred entrectinib resistance. More clinical experience with pTERT mutant NRTC is needed to understand its impact on the therapeutic sensitivity and other behavioral aspects of NRTC.
It is noteworthy that case 11, a primary thyroid SC, had been initially diagnosed as PTC with oncocytic and signet ring features until further workup revealed a SC immuno-profile. Primary thyroid SC represent an extremely rare phenotype for NRTC. In our series, the ratio of PTC and SC among all NRTC was 10:1. Histologically, although the nuclei of SC were elongated and often grooved, the chromatin appearance was finely stippled rather than clear, and the prominent central nucleoli would be unusual for PTC. Additional helpful morphologic clues included the micro-cystic/tubulopapillary architecture and abundant intracellular and extracellular eosinophilic secretion. As recently reviewed by Desai et al., there have been 11 primary thyroid SC documented in the literature, most of which were locally advanced tumors with similar morphology to case 11 [20]. Thyroid SC appeared to be clinically aggressive, with three (27%) previously reported tumor-related deaths. Larotrectinib has demonstrated 70–90% response rate for NRTC, mammary SC and salivary gland SC in published clinical trials [16]. Indeed, case 11 has shown a long-lasting response (21 months) to larotrectinib with complete resolution of radiologically detectable tumor.
In the literature, sensitivity to entrectinib and larotrectinib has been best described in ETV6-NTRK3 fusion tumors, and a smaller number of cases carrying CTRC-NTRK1, EML4-NTRK3, LMNA-NTRK1, PDE4DIP-NTRK1, PPL-NTRK1, STRN-NTRK2, TPM3-NTRK1, IRF2BP2-NTRK1, TPM4-NTRK3, TPR-NTRK1, TRIM63-NTRK1, and SQSTM1-NTRK1 have also been reportedly responsive [11, 30]. These tumors seem to respond with very little adverse symptomatology, with CTCAE grade 3 or 4 treatment-related adverse events occurring in <15% [16, 17]. In the current series, three cases with the EML4-NTRK3, TPR-NTRK1 and ETV6-NTRK3 fusions, all with an intact kinase domain, received anti-NTRK therapy with partial response and mild side effects. Point mutations in the kinase domain and bypassing BRAF and KRAS mutations are recently characterized causes of resistance to larotrectinib and entrectinib, and none were detected in our cases in pre-treatment testing. It was unclear why tumor shrinkage plateaued in cases 1 and 9 (at −69.7% and −33%, respectively) while case 11 had long-lasting complete resolution since no posttreatment genetic characterization of residual tumor was performed.
Okamura et al. reported several genetic co-alterations in NTRK-rearranged tumors of various organs, including other tyrosine kinases (FGFR, EGFR, KIT, DDR2, KDR, FLT1/4, PDGFRA/B, and JAK1/2/3), cell cycle-related proteins (CDKNA2A, CDKN2C, CCND1/2, CDK4/6, and RB1), MAPK signaling proteins (BRAF, HRAS, NRAS, NF1, RAF1), PI3K signaling proteins (PTEN, NF2, STK11, PIK3CA, and MTOR), MDM2, ATM, and TP53 [11]. None of these genetic changes were detected in our cases. Instead, we observed a novel and likely pathogenic frameshift deletion in MEN1, p.Pro55LeufsTer64, and two cases with pTERT activating mutation C228T. The MEN1 deletion was detected with a variant allele frequency of 55% in a sample with 70% tumor without paired normal tissue testing. It was thus difficult to determine whether the finding represented a germline versus somatic change. Nevertheless, since the patient had neither a personal nor family history of multiple neuroendocrine neoplasia or any endocrine malignancy, we speculate that it is likely an acquired mutation. In addition, there were two previously unreported changes, a splicing site SNV in GNA11 and a nonbinding site missense mutation in IDH1, of which the clinical significance is unknown. It should be noted that since we did not perform paired testing of normal tissue, the possibility of these being rare germline variants cannot be excluded.
Of note, case 8, an NTRK-rearranged PTC with high-grade features, showed no additional genetic changes other than the SQSTM1-NTRK3 fusion. Tumors designated as “PTC with high-grade features” are considered more biologically consequential than classical PTC but with features precluding a diagnosis of PDTC [31]. The current literature is scarce on fusion-driven PDTC, on which two recent studies have provided some interesting genetic insights [32, 33]. Duan et al. employed an 18-gene thyroid-focused NGS panel in 41 PDTC and found nine cases carrying rearrangements of RET (6 cases), NTRK1, PPARG, and ALK (1 case each) [32]. The NTRK1-rearranged PDTC reportedly showed combined PTC and PDTC morphological components, arguably features of which were seen in our case, and had no other genetic alteration based on the panel [32]. Also reported in the study was another NTRK1- rearranged tumor with combined anaplastic thyroid carcinoma and PTC morphology and a concurrent unspecified BRAF mutation [32]. In another study, Landa et al. performed a 341-gene sequencing panel in 84 PDTC and found 11 cases harboring various oncogenic gene fusions including PAX8-PPARG, CCDC6-RET, NCOA4-RET, STRN-ALK, EML4-ALK, and CCDC149-ALK [33]. All the 11 fusion-positive PDTC showed less than five concurrent mutations, mainly pTERT C228T (4 cases) and TP53 mutations (2 cases) [33]. Due to the scarcity of data, it remains unclear whether fusion-driven PDTC represent a genetically and prognostically meaningful ‘progression’ from PTC that carry the same fusion, particularly since the Turin criteria were developed based on mostly non-fusion-related tumors. Nevertheless, the combined morphology found in Duan’s case [32] was suggestive of progression from NTRK1-rearranged PTC to PDTC, which might presumably have been caused by some unusual molecular mechanism that happened to be outside the scope of the sequencing method. Further research is needed to elucidate the clinical and molecular natural history of NRTC.
In summary, our data demonstrate that NRTC are phenotypically diverse and encompass a unique subtype of thyroid carcinomas which show morphologic overlap with other more common thyroid carcinomas, particularly PTC, but also SC. SC, although NTRK driven and respond to NTRK directed therapy, are phenotypically different, including divergent immunohistochemistry. All studied cases show an unusual multinodular histologic appearance and a propensity for early extensive lymphovascular spread, which may serve in the morphologic recognition of NRTC to trigger confirmatory laboratory testing. Clinically, NRTC are characterized by high recurrence rate and prevalent distant metastases, which generally lead to a protracted clinical course with overall low mortality with NTRK inhibitor therapy. This study also demonstrates the genetic landscape of NRTC with multiple fusion combinations and otherwise low mutation burden. We propose an encompassing terminology of NTRK-rearranged thyroid carcinomas (NRTC) for a histologic spectrum of tumors with NTRK fusion-driven pathogenesis with overlapping clinical features and unique pathologic features as noted.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Solomon JP, Linkov I, Rosado A, Mullaney K, Rosen EY, Frosina D, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997;57:1690–4. [PubMed] [Google Scholar]
- 4.Iyama K, Matsuse M, Mitsutake N, Rogounovitch T, Saenko V, Suzuki K, et al. Identification of three novel fusion oncogenes, SQSTM1/NTRK3, AFAP1L2/RET, and PPFIBP2/RET, in thyroid cancers of young patients in Fukushima. Thyroid. 2017;27:811–8. [DOI] [PubMed] [Google Scholar]
- 5.Prasad ML, Vyas M, Horne MJ, Virk RK, Morotti R, Liu Z, et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in northeast United States. Cancer. 2016;122:1097–107. [DOI] [PubMed] [Google Scholar]
- 6.Leeman-Neill RJ, Kelly LM, Liu P, Brenner AV, Little MP, Bogdanova TI, et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer. 2014;120:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabes HM, Demidchik EP, Sidorow JD, Lengfelder E, Beimfohr C, Hoelzel D, et al. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clin Cancer Res. 2000;6:1093–103. [PubMed] [Google Scholar]
- 8.Ricarte-Filho JC, Li S, Garcia-Rendueles ME, Montero-Conde C, Voza F, Knauf JA, et al. Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J Clin Investig. 2013;123:4935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bongarzone I, Vigneri P, Mariani L, Collini P, Pilotti S, Pierotti MA. RET/NTRK1 rearrangements in thyroid gland tumors of the papillary carcinoma family: correlation with clinicopathological features. Clin Cancer Res. 1998;4:223–8. [PubMed] [Google Scholar]
- 10.Musholt TJ, Musholt PB, Khaladj N, Schulz D, Scheumann GF, Klempnauer J. Prognostic significance of RET and NTRK1 rearrangements in sporadic papillary thyroid carcinoma. Surgery. 2000;128:984–93. [DOI] [PubMed] [Google Scholar]
- 11.Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol. 2018;20:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang J, Cai W, Feng D, Teng H, Mao F, Jiang Y, et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J Pathol. 2018;244:215–26. [DOI] [PubMed] [Google Scholar]
- 13.Pfeifer A, Rusinek D, Zebracka-Gala J, Czarniecka A, Chmielik E, Zembala-Nozynska E, et al. Novel TG-FGFR1 and TRIM33-NTRK1 transcript fusions in papillary thyroid carcinoma. Genes Chromosom Cancer. 2019;58:558–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabanillas ME, Ryder M, Jimenez C. Targeted therapy for advanced thyroid cancer: kinase inhibitors and beyond. Endocr Rev. 2019;40:1573–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19:705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gatalica Z, Xiu J, Swensen J, Vranic S. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol. 2019;32:147–53. [DOI] [PubMed] [Google Scholar]
- 19.Hechtman JF, Benayed R, Hyman DM, Drilon A, Zehir A, Frosina D, et al. Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am J Surg Pathol. 2017;41:1547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desai MA, Mehrad M, Ely KA, Bishop JA, Netterville J, Aulino JM, et al. Secretory carcinoma of the thyroid gland: report of a highly aggressive case clinically mimicking undifferentiated carcinoma and review of the literature. Head Neck Pathol. 2019;13:562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dogan S, Wang L, Ptashkin RN, Dawson RR, Shah JP, Sherman EJ, et al. Mammary analog secretory carcinoma of the thyroid gland: a primary thyroid adenocarcinoma harboring ETV6-NTRK3 fusion. Mod Pathol. 2016;29:985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl J Med. 2018;378:731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK Fusion-positive solid tumors. Cancer Discov. 2017;7: 963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med. 2019;25:1422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2:146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Routhier CA, Mochel MC, Lynch K, Dias-Santagata D, Louis DN, Hoang MP, et al. Comparison of 2 monoclonal antibodies for immunohistochemical detection of BRAF V600E mutation in malignant melanoma, pulmonary carcinoma, gastrointestinal carcinoma, thyroid carcinoma, and gliomas. Hum Pathol. 2013;44: 2563–70. [DOI] [PubMed] [Google Scholar]
- 29.Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. [DOI] [PubMed] [Google Scholar]
- 30.Xu T, Wang H, Huang X, Li W, Huang Q, Yan Y, et al. Gene fusion in malignant glioma: an emerging target for next-generation personalized treatment. Transl Oncol. 2018;11:609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sadow PM, Faquin WC. Poorly differentiated thyroid carcinoma: an incubating entity. Front Endocrinol (Lausanne). 2012;3:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan H, Li Y, Hu P, Gao J, Ying J, Xu W, et al. Mutational profiling of poorly differentiated and anaplastic thyroid carcinoma by the use of targeted next-generation sequencing. Histopathology. 2019;75:890–9. [DOI] [PubMed] [Google Scholar]
- 33.Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Investig. 2016;126:1052–66. [DOI] [PMC free article] [PubMed] [Google Scholar]