

Abstract
Objective
The purpose of this study was to investigate whether somatic nonsynonymous variants in tumor tissue can potentially be identified in circulating cell‐free DNA (cfDNA) of head and neck oropharyngeal squamous cell carcinoma (OPSCC) patients using next‐generation sequencing and can predict recurrence or persistence disease.
Methods
A total of 22 OPSCC patients with tumor tissue and respective plasma samples were included in this study. Matching cfDNA and tumor tissues were processed, and DNA sequencing was conducted using the MiSeq platform. Variants were identified using Biomedical Genomic Workbench and Genialis's online data analysis platform for Swift Biosciences' Accel‐amplicon panels.
Results
Among 11 nonresponders, 6 matched mutations were detected in 5 patients suggesting a predictive factor for patients with likelihood of recurrence. The matched variants and their allele frequencies identified in the nonresponder group were (tumor DNA/cfDNA in %): TP53 G325fs (27/0.62), TP53 R282W (48/1.74), TP53 R273C (39/2.17), FBXW7 R505G (30/0.6), FBXW7 R505L (31/0.65), and TP53 Q331H (56.5/0.52). Interestingly, the matched somatic mutations were only detected in patients who did not respond to therapy or had persistent disease.
Conclusions
Somatic nonsynonymous variants in tumor tissue can potentially be identified in cfDNA of OPSCC patients using NGS. The likelihood of variant detection in cfDNA is greater in nonresponders, especially in human papillomavirus‐negative nonresponders, rendering it beneficial as a less invasive detection method for disease persistence/recurrence and prognosis.
Level of evidence
Cohort study.
Keywords: cfDNA, oropharynx, recurrence, TP53
Somatic nonsynonymous variants in tumor tissue can potentially be identified in cell‐free DNA (cfDNA) of head and neck oropharyngeal squamous cell carcinoma patients using next‐generation sequencing (NGS). The likelihood of variant detection in cfDNA is greater in nonresponders, especially in human papillomavirus‐negative nonresponders, rendering it beneficial as a less invasive detection method for disease persistence/recurrence and prognosis.
1. INTRODUCTION
Head and neck squamous cell carcinomas (HNSCC) encompass all cancers arising in the mucosa of the oral cavity, pharynx, and larynx. HNSCC is the sixth leading common malignancy worldwide with high rates of mortality and morbidity. 1 HNSCC accounts for more than 650 000 cases and 330 000 deaths worldwide, annually. In the United States alone, it is estimated that approximately 53 000 Americans develop HNSCC annually and about 10 800 deaths occur due to tobacco, alcohol use, and human papillomavirus (HPV) infection. 2 , 3 , 4 Unfortunately, the 5‐year mortality rate stands at approximately 50% despite advances in multimodal therapy over the past two decades for the non‐HPV associated cancers. Furthermore, the prognosis of patients with recurrent or metastatic HNSCC is generally poor. The median survival in most series is 6 to 12 months depending upon patient and disease‐related factors. Thus, there is a need for better detection measures to determine persistent or recurrent disease that could potentially lead to improved survival as a result of salvage surgery.
Circulating cell‐free DNA (cfDNA) is extracellular DNA found in circulating blood. The presence of cancer‐specific genomic alterations allows the differentiation between circular tumor DNA (ctDNA) and DNA from normal healthy cells. Similar to DNA from apoptotic cells, cfDNA consist of a range of multiples of 180 bp, consistent with the unit size of nucleosomes. 5 , 6 , 7 Recent studies have identified increased levels of circulating DNA fragments in patients with cancer. 8 , 9 , 10 , 11 , 12 , 13 Recent advances have also aided in identifying epigenetic and genetic characteristics of HNSCC in multiple data sets. 14 Leveraging information from these genetic alterations and applying it in the context of ctDNA carrying information on tumor‐related genetic and epigenetic changes may help identify recurrences or persistent disease.
Our primary goal was to determine if somatic nonsynonymous variants could be identified in cfDNA of OPSCC patients. Furthermore, we also investigated the extent to which the mutations found in the ctDNA correlated to those altered in paired tumor tissues. Finally, we elucidated if variants determined in the ctDNA correlated with response to treatment, recurrence, or persistent disease.
2. MATERIALS AND METHODS
2.1. Study design
An Institutional Review Board approved retrospective study was conducted on 22 patients with advanced squamous cell carcinoma of the oropharynx treated at Louisiana State University Health Shreveport. HPV status was determined based on p16 immunohistochemistry (IHC) on tumor samples. Patients were selected based on the following criteria: (a) diagnosis of OPSCC (HPV+ and HPV−) between the years 2008 and 2014 and staging according to the American Joint Committee on Cancer TNM staging eighth edition; (b) available plasma specimens and matched flash frozen tumors; (c) stage III or IV with at least N1 disease; and (d) free of distant metastases, M0. Patients were included in the study only if 2 years follow‐up data were available. Patients were divided into either disease‐free or nonresponders to standard therapy based on radiographic data and clinical follow‐up 2 years posttreatment (Table 1, 2 ). No statistically significant difference in patient demographics was observed between the disease‐free and nonresponder groups suggesting a Gaussian distribution in patient samples. The patients were treated using standard therapy based on National Comprehensive Cancer Network guidelines for OPSCC (https://www.nccn.org). The time to recurrence or persistence for the nonresponder was determined to be 0 to 11.7 months with an average of 2.6 months and median of 0 months. For follow‐up, the mean value accounting for both the groups was 38.6 months with the mean at 30.6 months and a range of 5.9 to 98 months.
TABLE 1.
Expected compared to detected allele frequency with Q‐Seq HDx quantitative multiplex DNA reference standard
| Gene | AA | CHR | POS | REF | ALT | Expected allele frequency | Detected allele frequency (N = 4) | SE |
|---|---|---|---|---|---|---|---|---|
| EGFR | G719S | 7 | 55241707 | G | A | 24.5 | 25.9 | 1.55 |
| PIK3CA | H1047R | 3 | 178952085 | A | G | 17.5 | 18.2 | 1.95 |
| KRAS | G13D | 12 | 25398281 | C | T | 15.0 | 16.9 | 1.93 |
| NRAS | Q61K | 1 | 115256530 | G | T | 12.5 | 13.5 | 1.94 |
| BRAF | V600E | 7 | 140453136 | A | T | 10.5 | 9.2 | 0.30 |
| KIT | D816V | 4 | 55599321 | A | T | 10.0 | 9.7 | 1.20 |
| PIK3CA | E545K | 3 | 178936091 | G | A | 9.0 | 8.4 | 0.76 |
| KRAS | G12D | 12 | 25398284 | C | T | 6.0 | 5.6 | 0.90 |
| EGFR | L858R | 7 | 55259515 | T | G | 3.0 | 2.8 | 0.71 |
| EGFR | ΔR746‐A750 | 7 | 55242465‐55242479 | Del 15 bp | 2.0 | 1.5 | 0.35 | |
| EGFR | T790M | 7 | 55249071 | C | T | 1.0 | 1.0 | 0.50 |
Note: The detected allele frequency was called by Biomedical Genomic Workbench.
TABLE 2.
Patient clinicodemographic characteristics
| Disease‐free (n = 11) | Non‐responder (n = 11) | P value | |
|---|---|---|---|
| Age | .85 | ||
| Age at diagnosis, median (IQR) | 55.00 (46.00, 63.00) | 55 (47.50, 59.00) | |
| Age at diagnosis, mean (SD) | 52.36 (10.71) | 54 (8.52) | |
| Race | 1.00 | ||
| Caucasian | 7 (64%) | 8 (73%) | |
| Black | 4 (36%) | 3 (27%) | |
| Gender | .31 | ||
| Male | 10 (91%) | 7 (64%) | |
| Female | 1 (9%) | 4 (36%) | |
| Current smoker | .18 | ||
| No | 6 (55%) | 2 (18%) | |
| Yes | 5 (45%) | 9 (82%) | |
| Tobacco use | .58 | ||
| Pack‐years, median (IQR) | 18.00 (10.00, 38.00) | 20 (15.00, 39.00) | |
| Pack‐years, mean (SD) | 27.05 (22.39) | 28 (20.28) | |
| Patient with over 10 pack‐years | 1.00 | ||
| No | 1 (9%) | 2 (18%) | |
| Yes | 10 (91%) | 9 (82%) | |
| Alcohol use | .18 | ||
| No | 9 (82%) | 5 (45%) | |
| Yes | 2 (18%) | 6 (55%) | |
| HPV status | .0861 | ||
| Negative | 3 (27%) | 8 (73%) | |
| Positive | 8 (73%) | 3 (27%) | |
| Tumor stage a | .22 | ||
| T1 | 1 (9%) | ||
| T2 | 5 (45%) | 2 (18%) | |
| T3 | 1 (9%) | ||
| T4 | 4 (36%) | 9 (82%) | |
| Nodal stage | 1.00 | ||
| N1 | 1 (9%) | 2 (18%) | |
| N2 | 10 (91%) | 9 (82%) | |
| Squamous cell carcinoma pathology grade | 0.79 | ||
| G2, moderately differentiated | 7 (64%) | 6 (55%) | |
| G3, poorly differentiated | 4 (36%) | 3 (27%) | |
| B3, poorly differentiated, basaloid features | 22 (18%) |
TNM staging according to AJCC eighth edition.
2.2. Sample preparation and DNA extraction
All specimens were collected at diagnosis and before any treatment. Blood cells and plasma were separated by centrifugation at 2000 rpm for 10 minutes and stored locally at a −80°C freezer within 10 minutes after collection. Circulating cfDNA was extracted from plasma using Zymo's Quick‐cfDNA Serum and Plasma Kit according to the manufacturer's protocol. Briefly, about 1.5 to 3 mL of plasma was mixed with S&P 5× Digestion Buffer and digested by incubating with Proteinase K solution at 55°C over 30 minutes. Postdigestion, the DNA lysate was added with two volumes of S&P DNA Binding Buffer and then transferred to a Zymo‐Spin III‐S Column. After centrifuging at 1000g for 2 minutes, the flow‐through was discarded. The column was washed with S&P DNA Prep Buffer once and followed with S&P DNA Wash Buffer twice. The cfDNA was eluted with DNA Elution Buffer. The size distribution of extracted cfDNA was examined with Agilent Tapestation 2200 with high‐sensitivity D1000 tape system. In some samples, non‐cfDNA contamination was removed by Zymo's Select‐A‐Size DNA Clean & Concentrator kit. The size distribution of extracted cfDNA is shown on Figure 1.Tissue DNA was extracted from flash frozen tissue with NucleoSpin Tissue kit (MACHEREY‐NAGEL GmbH & Co. KG). Briefly, 25 mg of tissue was ground and digested by proteinase K at 56°C overnight and then lysed at 70°C for 10 minutes. After grinding and washing in the column, DNA was eluted. The quality of genomic DNA from tissue samples was evaluated by Agilent Tape station 2200 with genomic tape system. Irrespective of the concentration of cfDNA obtained from patients, 200 ng of cfDNA was used for all analyses.
FIGURE 1.
Cell‐free DNA (cfDNA) size distribution. cfDNA was extracted from plasma and analyzed using Agilent 2200 to determine fragment size and concentration
2.3. DNA sequencing
Genomic DNA was quantified by Thermo Fisher's Qubit 2.0 with Qubit dsDNA HS Assay Kit. CfDNA was quantified by quantative real‐time polymerase chain reaction (qPCR) using Alu primers. Library was prepared with Swift Biosciences's Accel‐Amplicon 56G Oncology Panel v2. Briefly, 20 ng DNA were amplified with multiplex PCR using gene‐specific primers. The PCR product was size selected and cleaned up with SPRI select beads (Beckman Coulter) and dual‐indexed with Illumina's sequencing adapters. After second size selection and cleanup, the library was quantified by qPCR using KAPA Library Quantification Kits (Roche). A pair‐end DNA sequencing was conducted on Illumina's MiSeq platform with MiSeq Reagent Kits v2 (2 × 150) and adapter‐trimmed fastq files were generated. The average coverage is set about 1500× for tissue DNA and over 10 000× for cfDNA.
2.4. Detection of mutations in cfDNA
To identify the variants in our DNA samples by NGS, we prepared the library with the Accel‐Amplicon 56G Oncology Panel v2 from Swift Biosciences. This panel has hotspot coverage of 56 clinical oncology‐related genes and most common mutations in head and neck cancer. 15 This panel uses a 263‐amplicon design covering 16 000 mutations. Furthermore, the panel requires the input to be as low as 10 ng. The average amplicon size is about 138 bp compatible with the size limitation of our cfDNA. Also, over 95% on‐target specificity and uniformity strengthen the reliability of this panel. First, we tested the panel with A Q‐Seq HDx quantitative multiplex DNA reference standard (HD701). This standard DNA contains 6 genes and 11 variants with expected allele frequency from 1% to 24.5%. The test results showed that the detected allele frequency of 11 variants is similar to the expected allele frequency (Table 1 ), indicating that the panel will generate reliable results and meet our standards.
2.5. Bioinformatics analysis of sequencing data
Demultiplex fastq files were analyzed by two work flows, somatic cancer work flow for targeted amplicon sequencing in Biomedical Genomics Workbench (BGW) (Qiagen) and Swift Bio Accel‐Amplicon panel pipelines of Genialis (Genialis).In somatic workflow of BGW, paired‐end reads were imported into BGW and failed reads were excluded according to Illumina pipeline 1.8 or later. The primers were trimmed out according to primer list of Swift Biosciences. Reads were aligned with the hg19 human reference genome. A ready‐to‐use workflow for targeted amplicon sequencing was selected. The default setting was applied. The QC reports and variant calls in the targeting regions were generated according to bed files for Accel‐Amplicon 56G Oncology Panel v2. A minimum frequency for variant detection was set at 5% for tumor tissue and 0.5% for cfDNA sequencing data, respectively. Through Swift Bio Accel‐Amplicon panel pipeline for Accel‐Amplicon 56G Oncology Panel v2, fastq data were aligned with hg19 human reference genome. Variants were called by GATK haplotype caller and LoFreg. Reported variant calls were further filtered by read count, read coverage, frequency, forward/reverse balance, homopolymer status, and non‐synonymous status. Amino acid change was determined. The Oncoprint of ctDNA was prepared with D3Oncoprint software (Alida Palmisano, Yingdong Zhao and Richard Simon. JCO Clinical Cancer Informatics, 2018. nr. 2, p. 1‐9).
2.6. Statistical analysis
Participants were divided into two subgroups: (a) “disease‐free,” reflecting those without relapse or persistence; and (b) “non‐responders,” referring to those who experience persistence or recurrence. Sociodemographic characteristics, tumor characteristics, HPV status, and treatment variables were measured. Continuous variables are presented as mean (SD), whereas categorical variables are presented as the number (percentage) of patients. P values for continuous variables correspond to analysis of variances. P values for categorical variables with fewer than two patients in a given category correspond to Fisher's exact test. P values for categorical variables with at least two patients correspond to Pearson's chi‐square tests. Data management and analyses were performed with STATA/SE statistical software, version 13 (Stata Corp).
3. RESULTS
3.1. Mutations in cfDNA
To validate liquid biopsies in OPSCC patients, we sought to isolate tumor DNA and cfDNA from blood samples obtained from HNSCC patients and matched them to their corresponding tumor samples. In the current study, 22 patients with advanced‐stage squamous cell carcinoma of the oropharynx were selected. As shown in Table 2, there were no statistically significant differences in patient demographics such as age at diagnosis, race, gender, smoking status, and alcohol between disease‐free and nonresponders groups. We also evaluated the viral status within the cohort and the percentage of nonresponder was different between HPV‐positive and HPV‐negative groups. Consistent with TCGA data, more nonresponders were observed in the HPV‐negative group than that in the HPV‐positive group (Table 2). Additionally, the tumor characteristics such as primary site, tumor, and nodal stage were also non‐significant between the two groups (Table 2).
3.2. Identification of mutations in tumor
We used the panel to prepare the sequencing library for all tumor tissue and matched plasma DNA samples. The quality of sequencing data was evaluated by FastQC and coverage analysis was performed by BGW. The average sequencing depth for 22 tissue DNA samples is over 1600×. The average percentage of aligned reads on target of all tissue DNA samples was greater than 95%. The base coverage uniformity at 0.2× ranged from 93% to 98% with a mean of 96. The mean sequencing depth for matched 22 cfDNA samples were over 12 000×. The average percentage of aligned reads on the target of cfDNA samples was 90.5%. The mean base coverage uniformity at 0.2× was 92%.
The sequencing data from 22 tissue samples were analyzed with BGW and verified by Genialis's Workflow for Swift Biosciences's Accel‐Amplicon Panels. Both germline and somatic mutations determined in the tumor tissue samples are presented in Table 3. A total of 12 patients with nonsynonymous somatic mutations were determined among 22 patients. The most frequently occurred somatic mutant gene was TP53, which was detected in nine patients. Among nine patients, one was an HPV‐positive disease‐free, two were HPV‐negative disease‐free, and six were HPV‐negative nonresponders. There was a significant difference in the TP53 mutation distribution between HPV‐positive and HPV‐negative group (P = .0075 by Fisher's exact test). Somatic mutant FBXW7 (F‐box and WD repeat domain containing‐7) and PTEN was detected in two patients, respectively. The somatic mutated PIK3CA, HRAS, and CDKN2A were found in one patient for each gene. All these mutations have previously been reported in head and neck cancer and are consistent with the TCGA data. Among 12 patients, 3 patients have 2 or 3 somatic mutations. One HPV‐negative nonresponder exhibited two somatic mutations in the TP53 gene, suggesting strengthening dysfunctions of the gene. Further, we detected polymorphisms or germline mutations in all 22 patients. The most frequent germline polymorphism genes in 22 patients were TP53 and kinase insert domain Receptor (KDR). The polymorphism of TP53 (P72R) occurred in 16 of 22 patients. The KDR polymorphism including Q472H and V297I was detected in 15 patients. Other observed germline mutations were HINF1A (11/22), PIK3CA (4/22), KIT (2/22), APC (2/22), and CDKN2A (2/22).
TABLE 3.
Germline and somatic mutations determined in the tumor tissue samples
| ID | Chr | Region | Type | Reference | Allele | Frequency | Read count | Read coverage | Gene name | Amino acid change |
|---|---|---|---|---|---|---|---|---|---|---|
| HPV‐positive responder | ||||||||||
| 517 | ||||||||||
| 3 | 178927410 | SNV | A | G | 55.3 | 3854 | 6983 | PIK3CA | Ile391Met | |
| 17 | 7579472 | SNV | G | C | 99.7 | 3901 | 3910 | TP53 | Pro72Arg | |
| 558 | ||||||||||
| 4 | 55972974 | SNV | T | A | 99.8 | 1113 | 1115 | KDR | Gln472His | |
| 4 | 55979558 | SNV | C | T | 99.8 | 938 | 940 | KDR | Val297Ile | |
| 17 | 7579472 | SNV | G | C | 99.9 | 3901 | 1351 | TP53 | Pro72Arg | |
| 738 | ||||||||||
| 4 | 55972974 | SNV | T | A | 50.1 | 584 | 1162 | KDR | Gln472His | |
| 4 | 55979558 | SNV | C | T | 46.6 | 410 | 884 | KDR | Val297Ile | |
| 12 | 121432117 | SNV | G | C | 58.6 | 34 | 56 | HNF1A | Gly226Ala | |
| 17 | 7579472 | SNV | G | C | 100 | 1364 | 1364 | TP53 | Pro72Arg | |
| 744 | ||||||||||
| 4 | 153247289 | SNV | G | C | 30.5 | 621 | 2026 | FBXW7 | Arg505Gly | |
| 9 | 21971160 | SNV | G | C | 54.4 | 506 | 779 | CDKN2A | Arg81Gly | |
| 12 | 121432117 | SNV | G | C | 43.8 | 27 | 80 | HNF1A | Gly226Ala | |
| 17 | 7574012 | SNV | C | T | 19.4 | 239 | 1226 | TP53 | Glu339Lys | |
| 17 | 7579472 | SNV | G | C | 99.9 | 2209 | 2211 | TP53 | Pro72Arg | |
| 749 | ||||||||||
| 3 | 178936082 | SNV | G | A | 16.3 | 1415 | 8656 | PIK3CA | Glu542Lys | |
| 4 | 55972974 | SNV | T | A | 49.1 | 1113 | 2268 | KDR | Gln472His | |
| 17 | 7579472 | SNV | G | C | 52.5 | 951 | 1814 | TP53 | Pro72Arg | |
| 756 | ||||||||||
| 4 | 55979558 | SNV | C | T | 100 | 983 | 983 | KDR | Val297Ile | |
| 780 | ||||||||||
| 3 | 178927410 | SNV | A | G | 48.8 | 1564 | 3207 | PIK3CA | Ile391Met | |
| 4 | 55593464 | SNV | A | C | 50.1 | 1489 | 2980 | KIT | Met541Leu | |
| 12 | 121432117 | SNV | G | C | 92.9 | 128 | 137 | HNF1A | Gly226Ala | |
| 17 | 7579472 | SNV | G | C | 99.9 | 2131 | 2133 | TP53 | Pro72Arg | |
| C3‐32 | ||||||||||
| 3 | 178927410 | SNV | A | G | 23.7 | 3367 | 14253 | PIK3CA | Ile391Met | |
| 4 | 55979558 | SNV | C | T | 46.3 | 1315 | 2845 | KDR | Val297Ile | |
| 10 | 89692905 | SNV | G | A | 20 | 546 | 2800 | PTEN | Arg130Gln | |
| 17 | 7579548 | SNV | G | A | 48.4 | 2261 | 4666 | TP53 | Pro47Ser | |
| HPV‐positive nonresponder | ||||||||||
| 668 | ||||||||||
| 10 | 89720779̂89720780 | Insertion | ‐ | A | 41.5 | 1847 | 4456 | PTEN | Asn311fs | |
| 12 | 121432117 | SNV | G | C | 51.7 | 58 | 113 | HNF1A | Gly226Ala | |
| 17 | 7579472 | SNV | G | C | 51.9 | 1684 | 3260 | TP53 | Pro72Arg | |
| 789 | ||||||||||
| 3 | 178927410 | SNV | A | G | 56 | 4137 | 7385 | PIK3CA | Ile391Met | |
| 4 | 55979558 | SNV | C | T | 48 | 1237 | 2580 | KDR | Val297Ile | |
| 12 | 121432117 | SNV | G | C | 93.8 | 145 | 153 | HNF1A | Gly226Ala | |
| 17 | 7579472 | SNV | G | C | 45.2 | 1231 | 2746 | TP53 | Pro72Arg | |
| 807 | ||||||||||
| 4 | 55593464 | SNV | A | C | 49.7 | 2092 | 4225 | KIT | Met541Leu | |
| 4 | 55972974 | SNV | T | A | 52.1 | 1326 | 2550 | KDR | Gln472His | |
| 12 | 121432117 | SNV | G | C | 96.3 | 162 | 159 | HNF1A | Gly226Ala | |
| 17 | 7579472 | SNV | G | C | 99.9 | 3057 | 3059 | TP53 | Pro72Arg | |
| HPV‐negative responder | ||||||||||
| 723 | ||||||||||
| 4 | 55979558 | SNV | C | T | 46.9 | 714 | 1521 | KDR | Val297Ile | |
| 5 | 112173899 | SNV | C | T | 100 | 1382 | 1382 | APC | Pro870Ser | |
| 766 | ||||||||||
| 4 | 55972974 | SNV | T | A | 38.7 | 859 | 2213 | KDR | Gln472His | |
| 17 | 7578382 | SNV | G | T | 69.5 | 2047 | 2910 | TP53 | Ser183* | |
| 17 | 7579472 | SNV | G | C | 17.3 | 259 | 1520 | TP53 | Pro72Arg | |
| C3‐15 | ||||||||||
| 12 | 121432117 | SNV | G | C | 44.3 | 85 | 192 | HNF1A | Gly226Ala | |
| 17 | 7578263 | SNV | G | A | 90.3 | 6664 | 7347 | TP53 | Arg196* | |
| 17 | 7579472 | SNV | G | C | 5.5 | 158 | 2702 | TP53 | Pro72Arg | |
| HPV‐negative nonresponder | ||||||||||
| 460 | ||||||||||
| 11 | 534289 | SNV | C | A | 36.9 | 1956 | 5296 | HRAS | Gly12Cys | |
| 12 | 121432117 | SNV | G | C | 42.7 | 58 | 132 | HNF1A | Gly226Ala | |
| 17 | 7576872 | Deletion | C | ‐ | 27 | 889 | 3260 | TP53 | Gly325fs | |
| 17 | 7577574 | SNV | T | C | 22.3 | 294 | 1317 | TP53 | Tyr236Cys | |
| 17 | 7579472 | SNV | G | C | 99.8 | 2134 | 2136 | TP53 | Pro72Arg | |
| 499 | ||||||||||
| 4 | 55979558 | SNV | C | T | 48.3 | 417 | 859 | KDR | Val297Ile | |
| 9 | 21971017 | SNV | G | C | 49.8 | 879 | 1771 | CDKN2A | Pro114Arg | |
| 12 | 121432117 | SNV | G | C | 96.9 | 120 | 123 | HNF1A | Gly226Ala | |
| 17 | 7577094 | SNV | G | A | 48.1 | 1119 | 2331 | TP53 | Arg282Trp | |
| 17 | 7579472 | SNV | G | C | 25 | 257 | 1064 | TP53 | Pro72Arg | |
| 606 | ||||||||||
| 4 | 55972974 | SNV | T | A | 43.9 | 664 | 1509 | KDR | Gln472His | |
| 17 | 7577046 | SNV | C | A | 5.9 | 158 | 2744 | TP53 | Glu298* | |
| 662 | ||||||||||
| 4 | 55972974 | SNV | T | A | 44.6 | 765 | 1714 | KDR | Gln472His | |
| 17 | 7579442 | Deletion | G | ‐ | 21.4 | 358 | 1672 | TP53 | Pro82fs | |
| 767 | ||||||||||
| 9 | 21970979 | SNV | C | A | 52 | 3112 | 5963 | CDKN2A | Ala127Ser | |
| 817 | ||||||||||
| 4 | 55972946 | SNV | A | G | 33.4 | 370 | 1111 | KDR | Cys482Arg | |
| 4 | 55972974 | SNV | T | A | 33.9 | 375 | 1109 | KDR | Gln472His | |
| 17 | 7576853 | SNV | C | A | 56.5 | 1646 | 2906 | TP53 | Gln331His | |
| 17 | 7579472 | SNV | G | C | 80.5 | 914 | 1153 | TP53 | Pro72Arg | |
| 836 | ||||||||||
| 4 | 55972974 | SNV | T | A | 51.3 | 924 | 1798 | KDR | Gln472His | |
| 4 | 153247288 | SNV | C | A | 30.9 | 762 | 2467 | FBXW7 | Arg505Leu | |
| 12 | 121432117 | SNV | G | C | 68.9 | 58 | 84 | HNF1A | Gly226Ala | |
| 17 | 7577121 | SNV | G | A | 38.7 | 1460 | 3773 | TP53 | Arg273Cys | |
| 17 | 7579472 | SNV | G | C | 99.9 | 1839 | 1841 | TP53 | Pro72Arg | |
| C3‐12 | ||||||||||
| 4 | 55972974 | SNV | T | A | 52.7 | 1130 | 2148 | KDR | Gln472His | |
| 5 | 112175711 | SNV | G | A | 45 | 1220 | 2706 | APC | Ala1474Thr | |
| 12 | 121432117 | SNV | G | C | 54.8 | 78 | 143 | HNF1A | Gly226Ala | |
| 17 | 7579472 | SNV | G | C | 99.8 | 2060 | 2064 | TP53 | Pro72Arg | |
Note: Samples were segregated based on human papillomavirus (HPV) status and response to treatment.
3.3. Validation of liquid biopsies
To further validate liquid biopsies in OPSCC patients, we determined if somatic mutations could be detected in cfDNA. We sequenced cfDNA samples with the same library preparation as we did for tissue samples (Figure 1 ). To get rid of false‐positive variants, we combined two variant callers, loFreg and BGW, to identify the variants with allele frequency over 1% according to instruction of Accel‐Amplicon 56G Oncology Panel v2. We also excluded potential random errors and recurrent background noise by quadruple analyzing standard DNA samples. Among 22 cfDNA samples, somatic variants were detected in 11 samples. The variant number for each patient varied from 1 to 6 variants. The variant gene distribution over the patient's cfDNA samples is shown in Figure 2. The variants in EGFR genes were detected in six patients. Variants in TP53 and APC were detected in three patients, respectively. CDKN2A, Eerbb2, and PIK3CA were observed in two patients, respectively.
FIGURE 2.
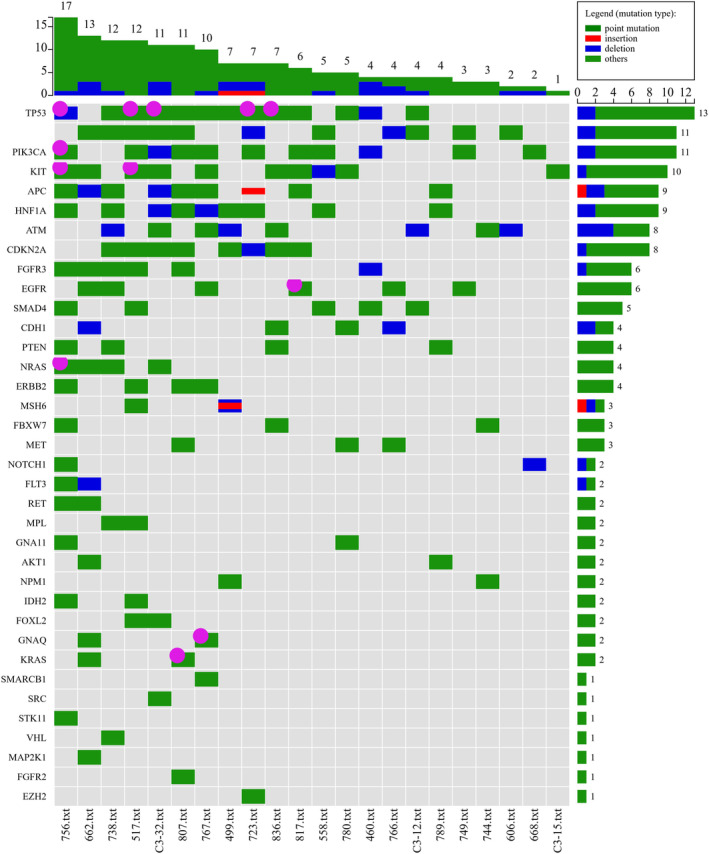
Oncoprint for variants detected from cell‐free DNA (cfDNA). Variants were picked up as described in materials and methods. Frequency number of patient's cfDNA are shown in top panel. The right panel lists frequency number of variant from each gene. The solid dots in dark pink indicate the curated variant according to Clinic Interpretation of Variants in Cancer (CIViC), FDA‐approved drug (target mutation), My Cancer Genome, and/ or OncoKB, respectively
To investigate if cfDNA sequencing can detect the somatic mutations noted in the original tumor tissue, somatic mutations with allele frequency over 0.5% were identified by two callers and are listed in Table 4. Analysis of cfDNA revealed that five patients carried tumor sample bearded somatic mutations (Table 4 ), in which, four patients were HPV‐negative nonresponders, and one was HPV‐positive disease free. Among somatic mutations detected in tumor DNA samples, four TP53 mutants, two FBXW7 mutants, and one CDNK2A mutant were observed in cfDNA samples. The detected TP53 mutants were higher in HPV‐negative patients than in HPV‐positive patients (P = .0451 by one‐tailed Fisher's exact test). The high incidence of TP53 mutants in cfDNA samples among HPV‐negative patients was correlated with a high incidence of detected TP53 mutations in tissue samples of HPV‐negative patients. It suggests that TP53 mutations could be an important characteristic of HPV‐negative OPSCC patients.
TABLE 4.
Matching mutation detected in both tissue DNA and cell‐free DNA (cfDNA)
| Sample ID | Chr | Region | Type | Tumor DNA | ctDNA | Gene | AA Change |
|---|---|---|---|---|---|---|---|
| Freq. >5 | Freq. >0.5% | ||||||
| cf460 | 17 | 7576872 | Deletion | 27.0 | 0.62 | TP53 | G325fs |
| cf499 | 17 | 7577094 | SNV | 48.1 | 1.74 | TP53 | R282W |
| cf744 | 4 | 153247289 | SNV | 30.5 | 0.6 | FBXW7 | R505G |
| cf817 | 17 | 7576853 | SNV | 56.5 | 0.52 | TP53 | Q331H |
| cf836 | 4 | 153247288 | SNV | 30.9 | 0.65 | FBXW7 | R505L |
| 17 | 7577121 | SNV | 38.7 | 2.18 | TP53 | R273C |
3.4. Survival status between disease‐free and nonresponder groups
Kaplan Meier survival analysis between disease‐free and nonresponders is depicted in Figure 3A. Based on the survival curve, the median survival in nonresponder was 18.6 months and undefined in disease‐free group as all patients survived at the end time point of this study. Considering that the distribution of HPV‐positive and HPV‐negative patients was different between the disease‐free and nonresponders group (Table 1, 2), we investigated survival status between HPV‐positive and HPV‐negative patients. The survival analysis showed that HPV‐positive patients had much better survival than HPV‐negative patients did (Figure 3B ). Two death cases were reported in the HPV‐positive group and six death cases occurred in the HPV‐negative group. Therefore, the median survival in HPV‐positive patients had undefined time compared to 25.05 months in HPV‐negative patients. To see if the detection of tissue‐matched ctDNA helps assess the disease progression and further treatment, we analyzed the disease‐free survival of ctDNA‐positive and ctDNA‐negative patients (Figure 3C ). The average disease‐free survival was 12.3 months in ctDNA‐positive patients and 37.0 months in ctDNA‐negative patients. Interestingly, among four ctDNA‐positive and HPV‐negative patients, three patients recurred and one patient had 8 months of disease‐free follow‐up.
FIGURE 3.
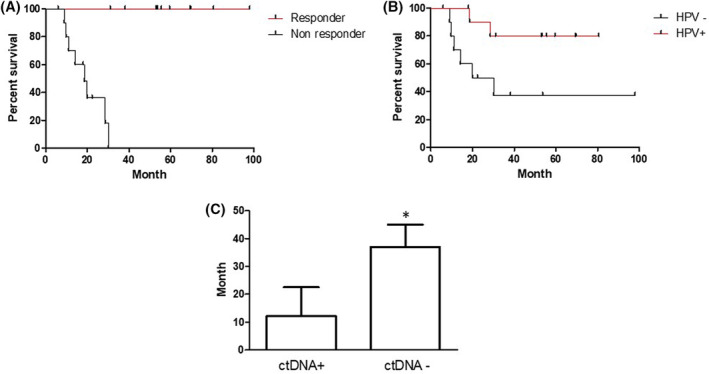
A, Kaplan‐Meier curves illustrating overall survival outcomes by response status for head and neck oropharyngeal squamous cell carcinoma (OPSCC) patients. Log‐rank (Mantel‐Cox) test, P < .0001. B. Kaplan‐Meier curves illustrating overall survival outcomes by human papillomavirus (HPV) status for OPSCC patients. Log‐rank (Mantel‐Cox) test, P < .05. C, Disease‐free survival by cell‐free DNA (cfDNA) detection. *P value = .0668 for one‐sided T test
4. DISCUSSION
Personalized pharmacogenomics strategies for treating HNSCC patients require the detection and modulation of potent mutations that could predict response to therapy, affect drug responses and disease progression. While genotyping tumor tissues can help clinical decisions to a certain extent, conventional biopsies are temporally and spatially restricted, due to the brief insight they provide of a single region of heterogeneous tumor tissue. In addition, due to the nonexistence of consistent biomarkers currently, tissue biopsies involving cytopathology or histopathology have been the backbone for diagnosis and clinical management of head and neck cancers.
The ctDNA is a promising and emerging biomarker from significant prior studies. However, because ctDNA is extremely diluted in cfDNA by multiple factors. It has been a challenging work to true ctDNA at a low‐frequency level. Several methods have been developed recently. For detection of unknown or new tumor‐related variants in the clinic, the limit of detection of allele frequency of several commercial available cfDNA sequencing kit is set at 0.5% to 1% in order to keep the sensitivity and reduce the false‐positive errors. In this study, cfDNA was sequenced with the Swift Biosciences's Accel‐Amplicon 56G Oncology Panel v2. This kit is one of the first commercially available kits on the market for cfDNA sequencing. Though the kit is not molecular barcoding based, it has a limit of detection of allele frequency as low as 1% and overall detection sensitivity ranged 94% to 100%. The amplicon‐based kit has high specificity with on‐target reads over 95%. In the analysis of the tumor tissue bearded variants in cfDNA samples, we expanded the detection limit of allele frequency down to 0.5%. One reason to lower the detection limit is these variants were known variants in tumor tissue. Second, detected low‐frequency variant with Accel‐Amplicon 56G Oncology kit was verified for sample samples by ERASE‐Seq (Elimination of Recurrent Artifacts and Stochastic Errors) method. 16 For newly detected somatic variants in cfDNA only, we followed the instruction of the manufacturer of Accel‐Amplicon 56G Oncology Panel v2. The limit of detection was set up at 1%. Considering a large number of potential false‐positive variants at low‐frequency level, the variant called by both Lofreg and BGW caller was picked up. These variants were further screened out for random and recurrent errors by quadruple analysis of standard DNA and available cfDNA samples.
We evaluated the ctDNA in OPSCC patient subgroups responding to therapy in comparison to nonresponders. While aspects of data from this study were in accordance with prior studies, indicating oncogenic mutations in TP53 and FBXW7 gene in tumor tissues, 17 this study has distinctively identified correlations of the nonsynonymous mutations in ctDNA between disease‐free and nonresponders groups in OPSCC. Among 11 nonresponders, matched mutations were detected in 4 patients suggesting a predictive factor for patients with the likelihood of recurrence. FBXW7 is a critical tumor suppressor and one of the most commonly deregulated ubiquitin‐proteasome system proteins in human cancer. A wide variety of FBXW7 substrates have been identified including oncoproteins such as cyclin E, c‐Myc, Mcl‐1, mTOR, Jun, Notch and AURKA that undergo proteasome‐mediated degradation. 18 Furthermore, recent studies have highlighted the importance of FBXW7 in promoting mitotic cell death and help prevent premature escape from mitosis through mitotic slippage. 19 Mitotic slippage is a process that can cause chemoresistance and tumor relapse. Therefore, further studies are warranted in identifying the role of FBXW7 in HNSCC and more importantly in non‐responding patients. Interestingly, there exist a cross talk between the FBXW7 and p53 axis. FBXW7 interacts with p53 and leads to its polyubiquitylation and proteasomal degradation. Interestingly, an increase in FBXW7 reduced the survival of patients with wild type p53 but not in patients carrying a p53 mutation or deletion suggesting that the tumorigenic effect of FBXW7 amplification is dependent on the degradation of a functional p53.
FBXW7 sensitizes cancer cells to radiation via stabilizing p53 leading to cell cycle arrest and apoptosis. 20 Furthermore, Tripathi et al demonstrated that TP53 mutant cell lines with unaltered FBXW7 status, when treated with doxorubicin to test their drug resistance attenuated its ability to form clones and to invade. 21 Taken together, DNA damage, FBXW7‐mediated degradation of p53 allows cell proliferation recovery and could have a potentially negative effect on the outcome of treatment of cancer disease.
4.1. HPV‐negative nonresponder mutations
While mutations in TP53 have been prominently associated with oncogenesis, the significant observation in this present study is the differential expression of these mutations between disease‐free and nonresponders groups to therapy, with specific differentiation between HPV‐positive and HPV‐negative cases. 22 As previously reported elsewhere, the HPV‐negative patients showed a higher incidence of TP53 mutations as what HPV‐positive patients did in this study. The noteworthy presence of the hotspot DNA‐binding surface structural mutations TP53 R282W (48/1.74) and TP53 R273C (39/2.17) in the nonresponder group indicates the effects of a highly destabilized and dysfunctional p53 mechanism in this patient group. TP53 R282W causes substantial destabilization of the mutant protein with a loss of thermodynamic stability of more than >3 kcal/mol, consequently causing changes in the folding of the mutant protein, making it highly destabilized and nonfunctional. TP53 R273C mutation affects the DNA contact functionality of the p53 protein, as ARG‐273 is vital in making key interactions with the phosphate backbone of the target DNA. 23 Thus, this mutation leads to a critical loss of contact with the target DNA. The presence of the TP53 R282W (48/1.74) and TP53 R273C (39/2.17) mutational effects in the nonresponder groups could implicate the destabilized p53 mechanism to be a factor for therapeutic failure in some of these patients. Furthermore, the presence of the truncating mutation TP53 G325fs (27/0.62), which dysregulates p53 functionality to promote tumorigenesis instead of halting it, in the nonresponder groups implicates the consequence of a tumorigenic mutant p53 may be significant in the nonresponder groups in comparison to the responder group. Consistent with TCGA data, more nonresponders were observed in the HPV‐negative group than in the HPV‐positive group. Also, a substantially higher percentage of matched TP53 mutations occurred in 80% of the nonresponder patients. Furthermore, our study also determined TP53 mutations in 100% of the nonresponder HPV‐negative HNSCC patients in our cohort.
Nevertheless, our studies suggest that ctDNA could potentially be a specific, noninvasive liquid biomarker to assess recurrence in advanced OPSCC patients, especially in HPV‐negative patients.
ACKNOWLEDGMENTS
Cherie‐Ann Nathan is currently an Associate Editor for Laryngoscope Investigate Otolaryngology Journal. The project was funded by an intramural grant from the Feist‐Weiller Cancer Center, Shreveport, Louisiana. We thank the Feist‐Weiller Cancer Center at LSU Health Shreveport for providing us with funding support.
Khandelwal AR, Greer AH, Hamiter M, et al. Comparing cell‐free circulating tumor DNA mutational profiles of disease‐free and nonresponders patients with oropharyngeal squamous cell carcinoma. Laryngoscope Investigative Otolaryngology. 2020;5:868–878. 10.1002/lio2.447
Alok R. Khandelwal, Adam H. Greer, and Mickie Hamiter contributed equally to this study.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Warnakulasuriya S. Causes of oral cancer—an appraisal of controversies. Br Dent J. 2009;207(10):471‐475. [DOI] [PubMed] [Google Scholar]
- 3. Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294‐4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chung CH, Gillison ML. Human papillomavirus in head and neck cancer: its role in pathogenesis and clinical implications. Clin Cancer Res. 2009;15(22):6758‐6762. [DOI] [PubMed] [Google Scholar]
- 5. Sidransky D. Nucleic acid‐based methods for the detection of cancer. Science. 1997;278(5340):1054‐1059. [DOI] [PubMed] [Google Scholar]
- 6. Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole‐genome sequencing. Sci Transl Med. 2012;4(162):162ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aarthy R, Mani S, Velusami S, Sundarsingh S, Rajkumar T. Role of circulating cell‐free DNA in cancers. Mol Diagn Ther. 2015;19(6):339‐350. [DOI] [PubMed] [Google Scholar]
- 8. Lochowska BA, Nowak D, Bialasiewicz P. Cell‐free tumour DNA as a diagnostic and prognostic biomarker in non‐small cell lung carcinoma. Adv Respir Med. 2019;87(2):118‐122. [DOI] [PubMed] [Google Scholar]
- 9. Kato S, Schwaederlé MC, Fanta PT, et al. Genomic assessment of blood‐derived circulating tumor DNA in patients with colorectal cancers: correlation with tissue sequencing, therapeutic response, and survival. JCO Precis Oncol. 2019;(3):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mithraprabhu S, Morley R, Khong T, et al. Monitoring tumour burden and therapeutic response through analysis of circulating tumour DNA and extracellular RNA in multiple myeloma patients. Leukemia. 2019;33:2022‐2033. [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto Y, Uemura M, Nakano K, et al. Increased level and fragmentation of plasma circulating cell‐free DNA are diagnostic and prognostic markers for renal cell carcinoma. Oncotarget. 2018;9(29):20467‐20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hamfjord J, Guren TK, Dajani O, et al. Total circulating cell‐free DNA as a prognostic biomarker in metastatic colorectal cancer prior to first‐line oxaliplatin‐based chemotherapy. Ann Oncol. 2019;30:1088‐1095. [DOI] [PubMed] [Google Scholar]
- 13. Wang Y, Zhao C, Chang L, et al. Circulating tumor DNA analyses predict progressive disease and indicate trastuzumab‐resistant mechanism in advanced gastric cancer. EBioMedicine. 2019;43:261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Choudhury JH, Ghosh SK. Promoter hypermethylation profiling identifies subtypes of head and neck Cancer with distinct viral, environmental, genetic and survival characteristics. PLoS One. 2015;10(6):e0129808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stadler ME, Patel MR, Couch ME, Hayes DN. Molecular biology of head and neck cancer: risks and pathways. Hematol Oncol Clin North Am. 2008;22(6):1099‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamps‐Hughes N, McUsic A, Kurihara L, et al. ERASE‐Seq: leveraging replicate measurements to enhance ultralow frequency variant detection in NGS data. PLoS One. 2018;13(4):e0195272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richter KT, Kschonsak YT, Vodicska B, Hoffmann I. FBXO45‐MYCBP2 regulates mitotic cell fate by targeting FBXW7 for degradation. Cell Death Differ. 2019;27:758–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galindo‐Moreno M, Giráldez S, Limón‐Mortés MC, et al. SCF(FBXW7)‐mediated degradation of p53 promotes cell recovery after UV‐induced DNA damage. FASEB J. 2019;33(10):11420‐11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tripathi V, Kaur E, Kharat SS, et al. Abrogation of FBW7alpha‐dependent p53 degradation enhances p53's function as a tumor suppressor. J Biol Chem. 2019;294(36):13224‐13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou G, Liu Z, Myers JN. TP53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. J Cell Biochem. 2016;117(12):2682‐2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kamaraj B, Bogaerts A. Structure and function of p53‐DNA complexes with inactivation and rescue mutations: a molecular dynamics simulation study. PLoS One. 2015;10(8):e0134638. [DOI] [PMC free article] [PubMed] [Google Scholar]