

Abstract
Purpose
Corneal injury that occurs after burning with alkali initiates wound-healing processes, including inflammation, neovascularization, and fibrosis. Excessive reactions to injury can reduce corneal transparency and thereby compromise vision. The NADPH oxidase (Nox) enzyme complex is known to be involved in cell signaling for wound-healing angiogenesis, but its role in corneal neovascularization has been little studied.
Methods
The center corneas of wild-type and Nox4 knockout (KO) mice were injured with 3 µL 1 M NaOH, while the contralateral corneas remained untouched. On day 7, mRNA expression levels of NADPH oxidase isoforms, the proangiogenic factors VEGF-A and TGFβ1, and proinflammatory genes ICAM-1 and VCAM-1 were determined. Corneal neovascularization and fibrosis were visualized using PECAM-1 antibody and picrosirius red staining, respectively, on the same day.
Results
Expressions of both Nox2 and Nox4 gene isoforms as well as the above genes were markedly increased in the injured corneas at 7 days. Injured corneas showed neovascularization and fibrosis as well as an increase in clinical opacity score. All responses stimulated by alkali burn were abrogated in Nox4 KO mice.
Conclusions
Nox4 could be a new target to treat pathologic corneal wound-healing responses and such targeting might prevent blindness caused by burn injuries.
Keywords: NADPH oxidase, Nox4, corneal injury, neovascularization, corneal fibrosis
Eye injuries due to chemical burns are true ophthalmic emergencies that account for up to 18% of ocular trauma, and pediatric patients frequently have severe vision loss due to ocular burns.1 First-line treatment for ocular chemical burns includes antioxidant vitamin C (ascorbic acid) and topical steroids.2 This reduces ocular inflammation and scar formation and promotes wound-healing responses. However, serious injuries often require long courses of steroid treatment, which can increase risk of infection, ocular hypertension, and glaucoma,3 and therefore steroids are tapered beyond 10 to 14 days. Chronic epithelial defects may require transplantation of amniotic membrane, limbal stem cells (from patient's fellow eye), or corneal donor tissue to allow recovery after severe chemical burns. However, surgical success is greatly influenced by the severity of the burn.4 A recent clinical trial shows that amniotic membrane transplantation only accelerates the healing process in patients with moderate ocular burns compared to conventional steroid therapy.5 Long-term corneal scarring with associated poor vision is still likely after severe alkali burns. Thus, alternative treatments that are safe, are cost-effective, and can reduce corneal scarring are sought for improved management of corneal burn injury.
NADPH oxidases are known to be involved in inflammatory responses to injury and fibrosis.6–9 We and others have also shown key roles for Nox isoforms in retinal angiogenesis and neovascularization10–12 and similar roles for NADPH oxidase 2 (Nox2) in wound healing and neovascularization following chemical burns in the cornea.13 In this study, we sought to clarify the role of the Nox4 isoform in neovascularization and wound healing after an alkali burn injury, using Nox4 knockout mice.14
Methods
Alkali Burn of Cornea in Mice
All surgical procedures were performed under animal care guidelines comparable to those published by ARVO and the Institute for Laboratory Animal Research (Guide for the Care and Use of Laboratory Animals) and guidelines of St. Vincent's Hospital Animal Ethics Committee (Ethics no. AEC 004/14). Wild-type (WT) C57BL/6J mice and Nox4 knockout mice (Nox4 KO) on a C57BL/6J background were kindly provided by Professor Karl-Heinz Krause,14 University of Geneva and bred at the BRC mouse facility (Fitzroy, Victoria, Australia). The Nox4 genotype was confirmed with conventional genotyping (check primers see Supplementary Fig. S1; Nox4 WT forward primer, 50AAGAGAAACTCCTCTGCTGTGAA-30; reverse primer, 50CGCACTGGAACCCCTGAGAAAGG-30; Nox4KO forward primer, 50AAGAGAAACTCCTCTGCTGTGAA-30; reverse primer, GTTCTAATTCCATCAGAAGCTTATCG-30; Sigma-Aldrich, Sydney, New South Wales, Australia). Mice aged between 8 and 9 weeks were used. Animals were anaesthetized with intraperitoneal injection of a tribromoethanol (250 mg/kg) supplemented with a topical corneal anesthetic agent (oxybuprocaine hydrochloride 0.4%). Corneal injury was caused by placing a drop of NaOH solution (1 N, 3 µL) to the center of the cornea for 10 seconds by a trained operator masked to the genotype of mice. The cornea was then rinsed off with 10 mL saline solution (0.9% sodium chloride). The fellow eye was not treated and used as a control to the alkali-treated eye. Mice were euthanized and eyes were harvested on days 7 and 14 postinjury for endpoint analysis. From previous pilot studies, it was found that a change in gene expression of acute inflammatory markers subsided before the injury was fully developed, so the earlier time point (day 7) was chosen for mRNA and proinflammatory and proangiogenic markers (growth factors and adhesion molecules) studies, whereas day 14 was chosen to study the reparative phase when chronic inflammation, neovascularization, and the early fibrotic response occur.
Corneal Flat-Mount Staining
Corneas from WT and Nox4 KO mice were fixed with 4% paraformaldehyde at 4°C for at least 1 hour. Enzymatic antigen retrieval was performed with proteinase K (Dako, Glostrup, Denmark) for 5 minutes at room temperature. Flat mounts were then blocked with total protein block (Dako) for 30 minutes at room temperature and then stained with rat anti-mouse CD31 conjugated with FITC for endothelial cells (1:150, 0.5 mg/mL; BD Biosciences Pharmingen, Sydney, NSW) overnight at 4°C. The wholemounts were then mounted in fluorescent mounting media (Dako) and images were captured using fluorescence microscopy (Zeiss AxioImager.2 microscope; Zeiss, Sydney, NSW). The degree of neovascularization (CD31+ area) was then quantified with ImageJ and expressed as percentage of the corneal area (% NV). A ratio of percentage NV between control and alkali-treated eye was then determined.
Gene Expression Detected by Real-Time PCR
Total corneal RNA was purified using commercial kits in accordance with the manufacturer's instructions (RNeasy Mini Kit; Qiagen, Victoria, Australia). Briefly, a single cornea was lysed and homogenized in the lysate, and RNA was purified using a column system. RNA was reverse-transcribed to cDNA (100 ng) using a high-capacity cDNA reverse transcription kit (cat. no. 4374996; Life Technologies, Melbourne, Victoria). Real-time PCR reactions were performed (7300 real-time PCR systems; Life Technologies) using a TaqMan Universal PCR master mix and commercially available probe and primer sets (TaqMan Gene Expression Assay; Life Technologies) for mouse Nox1 (Mm00549170_m1), Nox2 (Mm00432775_m1), Nox4 (Mm00479246_m1), VEGF-A (Mm00437304_m1), TGFβ1 (Mm00441724_m1), ICAM-1 (Mm00516023_m1), and VCAM-1 (Mm00449197_m1). Mouse GAPDH (4352339E-0912025) was used as a reference gene. Gene expression changes in the corneas of the cauterized eyes were normalized using values from untreated fellow eyes.
Picrosirius Red Staining
Paraffin-embedded cross sections (5 µm) were stained with 0.1% picric acid (Sigma-Aldrich) for 1 hour to identify total collagen. Pictures were taken with an Olympus (Notting Hill, Victoria) DP71 microscope using polarized and nonpolarized light filters.
Clinical Opacity Score Assessment
Assessment of injury was performed 14 days posttreatment with NaOH. Briefly, examination of the mouse eyes, under a microscope, was performed and corneal neovascularization was recorded based on neovascularization area and corneal opacity. Scores from three observers were recorded. Corneal opacity was scored using a scale of 0 to 4 used by Yoeruek et al.15 Grade 0 means completely clear cornea; grade 1 means slightly hazy, with iris and pupils easily detectable; grade 2 means slightly opaque, but iris and pupils still detectable; grade 3 means opaque with hard detection of pupils. Finally, grade 4 means completely opaque with no pupils observable. Representative in vivo images were taken using a digital camera (Cannon, Tokyo, Japan), also to be used for corneal neovascularization measurement.
Data and Statistics
All data were expressed as mean ± SEM. The mean data were analyzed by Student's t-test or one-way ANOVA followed by post hoc Tukey analysis (GraphPad Prism 6.0; GraphPad Software, La Jolla, CA, USA). A value of P < 0.05 was regarded as statistically significant.
Results
Effects of Alkali Burn on NADPH Oxidase Isoforms in Mouse Cornea
Corneal RNAs were extracted at day 7 and Nox1, Nox2, and Nox4 gene expression levels were determined using RT-PCR and gene-specific primers and probes (Nox5 is not expressed in rodents). As shown in Figure 1, Nox4 and Nox2 mRNA expression increased in NaOH-treated corneas compared with untreated corneas, whereas Nox1 expression was unaltered in this model. Thus, Nox2 and Nox4 are two isoforms of NADPH oxidase that might be involved in corneal alkali burn injury.
Figure 1.

Expression of NADPH oxidase isoforms in mouse alkali-burned cornea. Right eyes of C57BL/6 wild-type and Nox4 knockout mice (8–9 weeks old) were treated with 3 µL NaOH (see Methods). Values (mean ± SEM; n = 3–4) are represented as a fold change from control; *P < 0.05 from control following a Students’ paired t-test. NS, not significant.
Nox4-Deficient, Alkali-Burned Corneas Showed Less Neovascularization
Corneas were photographed on day 14 after full response to alkali burn injury, in order to quantify the areas of neovascularization. To do that, total area of cornea (100%) and the avascular area were outlined using ImageJ software. Neovascularization area was calculated by excluding the avascular area from the total area. Induction of corneal neovascularization of treated eyes was expressed relative to untreated corneas in both WT and Nox4 KO groups. Macroscopic observation of mouse eyes in Figure 2A shows the appearance of blood vessels emerging from the limbus across the cornea after alkali burn in WT (Fig. 2A II) and Nox4 KO groups (Fig. 2A IV) compared with their untreated controls (Fig. 2A I and III, respectively). Interestingly, there was 50% less neovascularization after alkali burn injury in Nox4 KO cornea (Figs. 2A IV, 2B) compared with WT cornea (Figs. 2A II, 2B).
Figure 2.
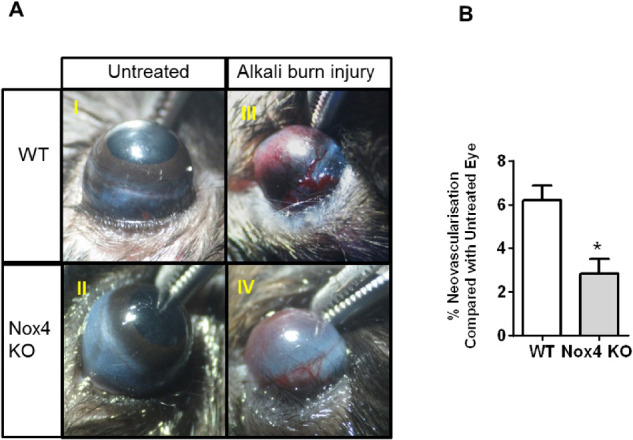
Progression of neovascularization in cornea after alkali burn injury. (A) Representative corneal images at day 14 of WT and Nox4 KO untreated eyes (I and III, respectively) and WT and Nox4 KO eyes (II and IV, respectively) after treatment with 1 M NaOH. (B) Quantitation of blood vessel area from limbal cells to the cornea before and after alkali burn injury using ImageJ shows a marked reduction in corneal neovascularization area in the absence of Nox4 gene after NaOH treatment compared with WT treated control. Values (mean ± SEM; n = 5) are represented as a percentage of neovascularization; *P < 0.0001 from WT-treated eye following a Student's paired t-test.
To characterize the neovascularization further, neovessels in flat mounts were stained and quantified at day 7 after alkali burn injury. Wholemount staining of corneas in WT and Nox4 KO using PECAM-1 antibody again demonstrated a stark difference in neovascularization between WT and Nox4 KO groups (Fig. 3A II and IV, respectively). Quantitative analyses showed a marked reduction of the neovascularization ratio in the absence of Nox4 compared to WT after NaOH treatment (Fig. 3B), suggesting that Nox4 plays a critical role in corneal neovascularization.
Figure 3.
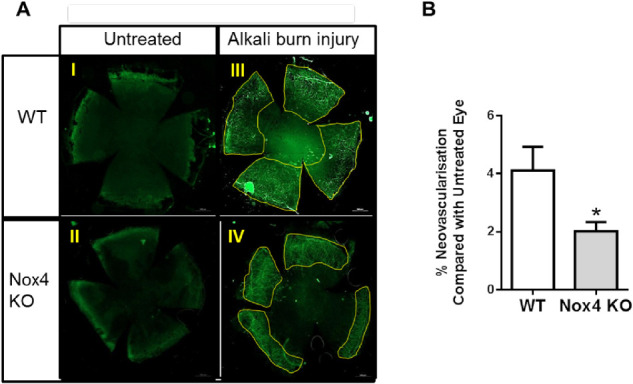
Progression of neovascularization as shown by PECAM-1 staining on corneal flat mounts after alkali burn injury. (A) Representative images of corneal flat-mount immunostaining using PECAM-1 antibody (1:150; BD) at day 7 of WT and Nox4 KO untreated controls (I and III, respectively) and NaOH-treated WT and Nox4 KO corneas (II and IV, respectively). (B) Quantitation of flat-mount corneal neovascularization ratio before and after alkali burn injury using ImageJ shows a marked reduction in blood vessels growing in the Nox4 KO compared with WT corneas after injury. Values (mean ± SEM; n = 5–6) are represented as a percentage of neovascularization; *P < 0.0001 from untreated WT cornea following a Student's paired t-test. Scale bar: 500 µm.
Induction of Proangiogenic Factors Diminished in the Absence of Nox4
Real time PCR was performed to examine the expression of proangiogenic factors in the presence and absence of the Nox4 gene. As shown in Figures 4A and 4B, both TGFβ1 and VEGF mRNA expression were upregulated in WT-derived cornea after alkali burn injury compared to untreated cornea. In the absence of Nox4, there was a dramatic reduction of both proangiogenic factors compared with WT-injured corneas. These results suggest that targeting Nox4 could reduce angiogenesis in the cornea after injury.
Figure 4.

Expression of proangiogenic factors after corneal alkali burn injury. RNA isolated from WT and Nox4 KO corneas 7 days after NaOH treatment. Injury markedly increased the expression of proangiogenic factors: (A) VEGF and (B) TGFβ1 in WT corneas. The upregulation of proangiogenic factors was reduced in the absence of Nox4. Values (mean ± SEM; n = 4) are expressed as a fold change compared with untreated WT control; *P < 0.05 from WT without NaOH treatment; #P < 0.05 between WT and Nox4 KO with NaOH treatment. Data were analyzed with one-way ANOVA followed by post hoc Tukey analysis.
Inhibition of Nox4 Reduced Collagen Synthesis in Cornea After Alkali Burn
Corneal stroma contains many highly organized collagen fibrils,16,17 which increase in the fibrotic process, and collagen is a well-established marker of fibrosis.18 Here, we stained the collagen in corneal stroma using picrosirius red. A complication can arise because muscle, in addition to collagen fibers, can be stained with picrosirius red, so we used a polarization method where only collagen is detected.19 Using nonpolarized light (Fig. 5A), an increase in red staining in the cornea after alkali burn in both WT (II) and Nox4 knockout (IV) groups was observed, but such staining was clearly reduced in the absence of Nox4 (IV) when compared with WT (II). Switching to a polarized filter (Fig. 5B), an increase of collagen fibers was observed after NaOH treatment in WT and Nox4 KO corneas (II, IV, respectively) compared with untreated corneas in each group (I, III, respectively). Again, there were less collagen fibers after NaOH in the Nox4 KO group (IV) compared to the WT-treated group (II). Total collagen content was significantly reduced in Nox4 KO cornea after alkali burn injury compared to the WT (Fig. 5C). These results indicate that lack of Nox4 reduces collagen synthesis after alkali burn injury.
Figure 5.

Collagen fibers in mouse cornea detected by picrosirius red polarization method. Collagen staining of WT and Nox4 KO corneas shows an increase of collagen fibers after NaOH treatment (II and IV, respectively) compared with untreated control (I and III, respectively) using (A) nonpolarized light (4×) and (B) polarized light (10×). (C) Quantitation shows a significant reduction of collagen ratio after NaOH treatment in Nox4 KO-derived compared with WT-derived cornea. Values (mean ± SEM; n = 4–5) are represented as a fold change from WT control; *P < 0.05 from WT following a Student's paired t-test. Scale bar: 50 µm.
Nox4 Deficiency Improved the Opacity Score in the Burned Mouse Cornea
Opacity score is used in the clinic to grade the clarity of cornea after injury (Fig. 6A). Using this system, corneal opacity score was increased by corneal burn in the WT group (Fig. 6B), but it was lower in the Nox4 KO group, confirming that inhibition of Nox4 improves corneal opacity score.
Figure 6.
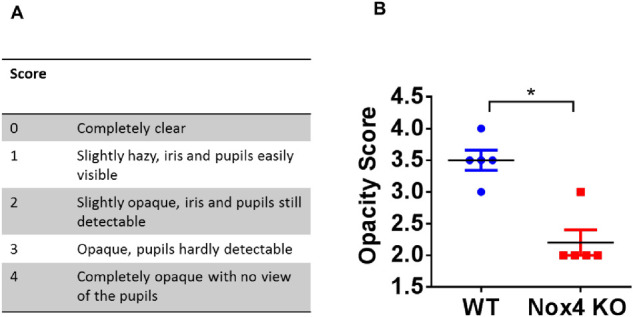
Nox4 deficiency reduced clinical corneal opacity score. Table (A) shows the scoring system used in this study. Graph (B) represents the marked difference in corneal opacity score at day 14 after alkali burn. The opacity score after NaOH treatment is markedly reduced in Nox4 KO cornea compared with WT cornea. Values (mean ± SEM; n = 5) are represented as a fold change from WT control; *P < 0.001 from NaOH treated WT following a Student's paired t-test.
Nox4 Deficiency Reduced the Upregulation of Proinflammatory Factors in Alkali Burned Cornea
Nox2 is involved in inflammatory responses in many different organs,20–23 including the eye.24 Chemokines such as ICAM-1 and VCAM-1 are released in inflamed tissues.25 To demonstrate the functional importance of Nox4 on inflammation, we assessed the expression of proinflammatory factors Nox2, ICAM-1, and VCAM-1 after alkali burn in the presence and absence of Nox4. As shown in Figure 7, there was a massive increase in Nox2 (Fig. 7A), ICAM-1 (Fig. 7B), and VCAM-1 (Fig. 7C) gene expressions after alkali burn injury, but deficiency of Nox4 considerably reduced the expression of all these genes (Figs. 7A–C), indicating that targeting Nox4 could reduce inflammation in alkali burn injury.
Figure 7.

Expression of proinflammatory factors in alkali-burned corneas. Proinflammatory genes: (A) Nox2, (B) ICAM-1, and (C) VCAM-1 were upregulated in cornea 7 days postinjury, and all were reduced in the absence of Nox4. Values (mean ± SEM; n = 4) are expressed as a fold change compared with WT control; *P < 0.05 from WT without NaOH treatment; #P < 0.05 between WT and Nox4 KO with NaOH treatment. Data were analyzed with one-way ANOVA followed by post hoc Tukey analysis.
Discussion
In this experimental model of alkali burn injury to the cornea, we found that both Nox2 and Nox4 mRNA expression were upregulated. Inflammation, neovascularization, fibrosis, and opacity scores were all increased in WT corneas after alkali burn injury. In addition, there was a significant induction of proinflammatory factors (Nox2, ICAM-1, and VCAM-1), as well as proangiogenic factors (VEGF and TGFβ1) after alkali burn injury in WT corneas. However, all these gene expressions were markedly reduced in alkali-burned Nox4-deficient corneas. Taken together, it is clear that targeting Nox4 might well be useful for the reduction of inflammation, neovascularization, and fibrosis in the cornea as well as improving corneal opacity score after alkali burn injury.
Corneal injury leads to production of reactive oxygen species (ROS) and release of cytokines to trigger the healing process.26 Kubota et al.27 first showed that alkali burn increased ROS production, and this was partly responsible for corneal neovascularization, but the source of ROS was not identified. Gu et al.28 confirmed that alkali burn injury increases protein expression of both Nox2 and Nox4 in the cornea. They also found that nonselective inhibitors of NADPH oxidase like apocynin and diphenyliodonium (DPI) are capable of attenuating burn-induced neovascularization and inflammation, supporting the notion that inhibiting Nox could be a therapeutic target for ocular chemical burn injuries. However, it is important to note that apocynin not only can be a nonspecific inhibitor of Nox2 activity (but not Nox4) under prescribed circumstances (mainly in vitro cells) but can also act in vivo as a nonspecific antioxidant.6 In addition, DPI binds to the flavin moiety of some NADPH oxidases, thus suppressing many flavin-containing enzymes, including, for example, nitric oxide (NO) synthases, so using these toxic inhibitors in vivo does not strengthen the conclusion that NADPH oxidase is involved. Finally, the use of 3-nitrotyrosine as an indicator of oxidative stress is a very nonspecific measure of just one oxidation product, and this relies also on production of sufficient quantities of NO, not just superoxide anion. Interestingly, in Figure 2 of that study, it appears that apocynin did not reduce 3-nitrotyrosine levels, which is not consistent with their conclusion that apocynin blocks NADPH oxidase.
NADPH oxidase isoforms are clearly involved in retinal neovascularization,10–12,29–31 but the relative contributions of Nox isoforms to corneal neovascularization have not been identified. We showed a marked increase of both Nox2 and Nox4 expression after alkali burn, while Nox1 expression did not change (Fig. 3), suggesting that Nox2 and Nox4 are the primary isoforms involved in this model. Although Nox1 expression does not change with burn injury, its activity could be altered, which needs further investigation.
To our knowledge, no studies have previously investigated the role of Nox4 in corneal neovascularization. It has been previously shown that in alkali-burned cornea, the expression of TGFβ1 increases,32 which promotes the wound-healing process, and it has been well established that TGFβ1 induces Nox4 expression in a variety of cells.33–38 TGFβ1 also triggers inflammation-associated chemokine expression such as ICAM-1 and VCAM-1.39 Similarly, in our study, we found that treating the cornea with NaOH increased the inflammation pathway by increasing the expression of ICAM-1 and VCAM-1. The infiltrating inflammatory cells highly express the Nox2 isoform, and this may explain the increase of the Nox2 gene after injury. Here, we showed that inhibition of Nox4 dramatically reduced the upregulation of ICAM-1 and VCAM-1, as well as Nox2, without affecting Nox2 baseline expression (hence, not compromising immunity). Thus, targeting Nox4 could moderate inflammatory processes after alkali burn injury of the cornea. Although the induction of Nox2 gene is considered highly inflammatory, targeting this gene directly might also compromise the immune system and enhance the severity of autoimmune40,41 and hyperinflammatory diseases.42,43 Therefore, Nox4 would appear to be a preferred target in this model.
Inflammation of the cornea also triggers the neovascularization process.27,32 Neovascularization can be activated by angiogenic factors such as VEGF-A and TGFβ1,44,45 and both VEGF-A and TGFβ1 expression increased after alkali burn.32 Consistent with this, we also found VEGF-A and TGFβ1 were upregulated 7 days after alkali burn in WT cornea. Different techniques have been used to reduce the upregulation of either VEGF-A or TGFβ1 after alkali injury,46,47 and we know that Nox4 is involved in VEGF-induced48,49 and TGFβ1-induced signaling.34,38 Thus, upregulation of both proangiogenic genes was markedly reduced in Nox4-deficient compared to WT cornea (Fig. 6). We suggest targeting Nox4 might then obviate targeting these signaling factors individually.
Release of TGFβ1 after injury induces the production of extracellular proteins such as collagen, but excessive production of collagen can compromise normal tissue function through fibrosis and scar formation.32,50,51 Nox4 has a pivotal role in TGFβ-induced fibrotic responses.9,14,35,52–54 We and others showed that Nox4 induces collagen production in vitro and in vivo via stimulation by TGFβ1.9,55,56 Inhibition of Nox4, by expressing a dominant negative form of Nox4 (Adv-Nox4DNADPH) or an adenovirus carrying siRNA targeting Nox4 (Adv-Nox4i), abrogated the production of collagen in cardiac fibroblasts.9 Another study showed that interstitial fibrosis increased in ureteral obstruction in Nox4 KO mice compared with WT control.57 These results support our finding that induction of collagen synthesis in WT-derived cornea after alkali burn injury is reduced in the absence of Nox4 (Fig. 5).
Excessive production of collagen can reduce corneal transparency.17 In our study, corneas lost transparency by 14 days after alkali burn in WT. Blocking TGF signaling using Smad7 has been shown to reduce fibrosis and improve opacity score,32 and consistent with this, in our study, corneas appeared less opaque, fibrosis was reduced, and opacity score improved in the absence of Nox4 (Fig. 6). Thus, targeting Nox4 can also improve the opacity score and reduce vision loss.
Interestingly, the details of these mechanisms of response to injury vary a little between different models of corneal injury. Chan et al.13 used acidic silver nitrate to inflict an injury to the corneal surface, and this results in an upregulation of Nox2 mRNA and a downregulation of the Nox4 gene over 7 days. In that study, Nox2 upregulation was found to be associated with inflammation and neovascularization in the burnt corneas. In the current study, we use basic NaOH to produce a more severe burn to the eye and show that both Nox2 and Nox4 are induced in the cornea at day 7. It is worth noting that the nitrate injury is associated with an induction of the proangiogenic VEGF-A but not profibrotic factor TGFβ1.13 In contrast, NaOH injury stimulates the expression of both VEGF and TGFβ1 in the burnt corneas, consistent with recent findings by Gu et al.28 In this regard, both Nox2 and Nox4 appear to regulate the initial inflammation and neovascular responses in the corneas up to day 7 following chemical insults. The induction of TGFβ1 is ablated in burnt corneas from Nox4 KO mice, which actually show a lower degree of opacity and collagen accumulation in the burnt corneas when compared to the WT. Thus, Nox4 may also regulate corneal scarring at the later stage of wound healing through induction of TGFβ1. Therefore, inhibiting Nox4 and Nox2 may well have a competitive edge for the treatment of alkali injury due to its additional profibrotic activity. Nevertheless, further studies are needed to clarify this aspect with selective inhibitors for Nox2 and Nox4.
In summary, we showed that Nox4 has a potential role in the corneal healing process after alkali burn injury and that Nox4 may be a target for limiting corneal neovascularization, inflammation, and fibrosis, as well as opacity score. These findings demonstrate a new therapeutic target that could be of value to reduce aberrant wound-healing responses. Recently, a pyrazolopyridinedione derivative, GKT137831, has been shown to inhibit Nox4 in liver fibrosis58 and pulmonary hypoxia models.59 Studies using GKT137831 to inhibit Nox4-derived corneal neovascularization and fibrosis after alkali burn injury could also be of value.
Supplementary Material
Acknowledgments
The authors thank Mark Daniell for his suggestions on clinical context of this work and reviewing the manuscript.
Supported by King Abdul Aziz University, Jeddah, Saudi Arabi (NYH); Heart Foundation Postdoctoral Fellowship (PF11M6093) (HMP); and NHMRC Principal Research Fellowship and the Wiseman Trust (gift to University of Melbourne) (GJD). Studies were also supported by the National Heart Foundation of Australia Grant-in-Aid (G12M6726) and by grants from the Ophthalmic Research Institute of Australia grant and the Eye Research Australia Foundation. The Centre for Eye Research Australia received Operational Infrastructure Support from the Victorian State Government.
Disclosure: N.Y. Hakami, None; G.J. Dusting, None; E.C. Chan, None; M.H. Shah, None; H.M. Peshavariya, None
References
- 1. Singh P, Tyagi M, Kumar Y, Gupta KK, Sharma PD. Ocular chemical injuries and their management. Oman J Ophthalmol. 2013; 6: 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brodovsky SC, McCarty CA, Snibson G, et al.. Management of alkali burns: an 11-year retrospective review. Ophthalmology. 2000; 107: 1829–1835. [DOI] [PubMed] [Google Scholar]
- 3. Eslani M, Baradaran-Rafii A, Movahedan A, Djalilian AR. The ocular surface chemical burns. J Ophthalmol. 2014; 2014: 196827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burcu A, Yalniz-Akkaya Z, Ozdemir MF, Erdem E, Onat MM, Ornek F. Surgical rehabilitation following ocular chemical injury. Cutan Ocul Toxicol. 2014; 33: 42–48. [DOI] [PubMed] [Google Scholar]
- 5. Tandon R, Gupta N, Kalaivani M, Sharma N, Titiyal JS, Vajpayee RB. Amniotic membrane transplantation as an adjunct to medical therapy in acute ocular burns. Br J Ophthalmol. 2011; 95: 199–204. [DOI] [PubMed] [Google Scholar]
- 6. Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011; 63: 218–242. [DOI] [PubMed] [Google Scholar]
- 7. Brown KD, Shah MH, Liu GS, Chan EC, Crowston JG, Peshavariya HM. Transforming growth factor beta1-induced NADPH oxidase-4 expression and fibrotic response in conjunctival fibroblasts. Invest Ophthalmol Vis Sci. 2017; 58: 3011–3017. [DOI] [PubMed] [Google Scholar]
- 8. Bedard K, Krause KH.. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007; 87: 245–313. [DOI] [PubMed] [Google Scholar]
- 9. Chan EC, Peshavariya HM, Liu GS, Jiang F, Lim SY, Dusting GJ. Nox4 modulates collagen production stimulated by transforming growth factor beta1 in vivo and in vitro. Biochem Biophys Res Commun. 2013; 430: 918–925. [DOI] [PubMed] [Google Scholar]
- 10. Wilkinson-Berka JL, Deliyanti D, Rana I, et al.. NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy. Antioxidants Redox Signal. 2014; 20: 2726–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan EC, Liu GS, Dusting GJ. Redox mechanisms in pathological angiogenesis in the retina: roles for NADPH oxidase. Curr Pharm Des. 2015; 21: 5988–5998. [DOI] [PubMed] [Google Scholar]
- 12. Chan EC, van Wijngaarden P, Liu GS, Jiang F, Peshavariya H, Dusting GJ. Involvement of Nox2 NADPH oxidase in retinal neovascularization. Invest Ophthalmol Vis Sci. 2013; 54: 7061–7067. [DOI] [PubMed] [Google Scholar]
- 13. Chan EC, van Wijngaarden P, Chan E, et al.. NADPH oxidase 2 plays a role in experimental corneal neovascularization. Clin Sci (Lond). 2016; 130: 683–696. [DOI] [PubMed] [Google Scholar]
- 14. Carnesecchi S, Deffert C, Donati Y, et al.. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxidants Redox Signal. 2011; 15: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoeruek E, Ziemssen F, Henke‐Fahle S, et al.. Safety, penetration and efficacy of topically applied bevacizumab: evaluation of eyedrops in corneal neovascularization after chemical burn. Acta Ophthalmol. 2008; 86: 322–328. [DOI] [PubMed] [Google Scholar]
- 16. DelMonte DW, Kim T.. Anatomy and physiology of the cornea. J Cataract Refract Surg. 2011; 37: 588–598. [DOI] [PubMed] [Google Scholar]
- 17. Hassell JR, Birk DE.. The molecular basis of corneal transparency. Exp Eye Res. 2010; 91: 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leask A. Potential therapeutic targets for cardiac fibrosis TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010; 106: 1675–1680. [DOI] [PubMed] [Google Scholar]
- 19. Rich L, Whittaker P. Collagen and picrosirius red staining: a polarized light assessment of fibrillar hue and spatial distribution. Braz J Morphol Sci. 2005; 22: 97–104. [Google Scholar]
- 20. Cannizzo B, Quesada I, Militello R, et al.. Tempol attenuates atherosclerosis associated with metabolic syndrome via decreased vascular inflammation and NADPH-2 oxidase expression. Free Radical Res. 2014; 48: 526–533. [DOI] [PubMed] [Google Scholar]
- 21. Gandhirajan RK, Meng S, Chandramoorthy HC, et al.. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest. 2013; 123: 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vlahos R, Stambas J, Bozinovski S, Broughton B, Drummond GR, Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011; 7: e1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen H, Kim GS, Okami N, Narasimhan P, Chan PH. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis. 2011; 42: 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Al-Shabrawey M, Rojas M, Sanders T, et al.. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2008; 49: 3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mollà M, Gironella M, Miquel R, et al.. Relative roles of ICAM-1 and VCAM-1 in the pathogenesis of experimental radiation-induced intestinal inflammation. Int J Radiat Oncol Biol Phys. 2003; 57: 264–273. [DOI] [PubMed] [Google Scholar]
- 26. Shoham A, Hadziahmetovic M, Dunaief JL, Mydlarski MB, Schipper HM. Oxidative stress in diseases of the human cornea. Free Radical Biol Med. 2008; 45: 1047–1055. [DOI] [PubMed] [Google Scholar]
- 27. Kubota M, Shimmura S, Kubota S, et al.. Hydrogen and N-acetyl-L-cysteine rescue oxidative stress-induced angiogenesis in a mouse corneal alkali-burn model. Invest Ophthalmol Vis Sci. 2011; 52: 427–433. [DOI] [PubMed] [Google Scholar]
- 28. Gu XJ, Liu X, Chen YY, et al.. Involvement of NADPH oxidases in alkali burn-induced corneal injury. Int J Mol Med. 2016; 38: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saito Y, Geisen P, Uppal A, Hartnett ME. Inhibition of NAD (P) H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis. 2007; 13: 840. [PMC free article] [PubMed] [Google Scholar]
- 30. Al-Shabrawey M, Bartoli M, El-Remessy AB, et al.. Role of NADPH oxidase and Stat3 in statin-mediated protection against diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008; 49: 3231–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li J, Wang JJ, Yu Q, Chen K, Mahadev K, Zhang SX. Inhibition of reactive oxygen species by lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice role of NADPH oxidase 4. Diabetes. 2010; 59: 1528–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saika S, Ikeda K, Yamanaka O, et al.. Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. Am J Pathol. 2005; 166: 1405–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carmona-Cuenca I, Roncero C, Sancho P, et al.. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol. 2008; 49: 965–976. [DOI] [PubMed] [Google Scholar]
- 34. Boudreau HE, Casterline BW, Rada B, Korzeniowska A, Leto TL. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radical Biol Med. 2012; 53: 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hecker L, Vittal R, Jones T, et al.. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009; 15: 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sturrock A, Cahill B, Norman K, et al.. Transforming growth factor-β1 induces Nox4 NAD (P) H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006; 290: L661–L673. [DOI] [PubMed] [Google Scholar]
- 37. Michaeloudes C, Sukkar MB, Khorasani NM, Bhavsar PK, Chung KF. TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2011; 300: L295–L304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cucoranu I, Clempus R, Dikalova A, et al.. NAD (P) H oxidase 4 mediates transforming growth factor-β1–induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005; 97: 900–907. [DOI] [PubMed] [Google Scholar]
- 39. Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4–and TGF-β1–dependent pathway. Arterioscler Thromb Vasc Biol. 2009; 29: 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. George‐Chandy A, Nordström I, Nygren E, et al.. Th17 development and autoimmune arthritis in the absence of reactive oxygen species. Eur J Immunol. 2008; 38: 1118–1126. [DOI] [PubMed] [Google Scholar]
- 41. Hultqvist M, Olofsson P, Holmberg J, Bäckström BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci USA. 2004; 101: 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Björkman L, Dahlgren C, Karlsson A, Brown KL, Bylund J. Phagocyte‐derived reactive oxygen species as suppressors of inflammatory disease. Arthritis Rheum. 2008; 58: 2931–2935. [DOI] [PubMed] [Google Scholar]
- 43. Ferguson PJ, Lokuta MA, El‐Shanti HI, Muhle L, Bing X, Huttenlocher A. Neutrophil dysfunction in a family with a SAPHO syndrome–like phenotype. Arthritis Rheum. 2008; 58: 3264–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ucuzian AA, Gassman AA, East AT, Greisler HP. Molecular mediators of angiogenesis. J Burn Care Res. 2010; 31: 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saika S. TGFβ pathobiology in the eye. Lab Invest. 2006; 86: 106–115. [DOI] [PubMed] [Google Scholar]
- 46. Chen M, Matsuda H, Wang L, et al.. Pretranscriptional regulation of Tgf-β1 by PI polyamide prevents scarring and accelerates wound healing of the cornea after exposure to alkali. Mol Ther. 2010; 18: 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim SW, Ha BJ, Kim EK, Tchah H. The effect of topical bevacizumab on corneal neovascularization. Ophthalmology. 2008; 115: e33–e38. [DOI] [PubMed] [Google Scholar]
- 48. Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol. 2007; 27: 2319–2324. [DOI] [PubMed] [Google Scholar]
- 49. Meng D, Mei A, Liu J, et al.. NADPH oxidase 4 mediates insulin-stimulated HIF-1α and VEGF expression, and angiogenesis in vitro. PLoS One. 2012; 7: e48393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eckes B, Zigrino P, Kessler D, et al.. Fibroblast-matrix interactions in wound healing and fibrosis. Matrix Biol. 2000; 19: 325–332. [DOI] [PubMed] [Google Scholar]
- 51. Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003; 200: 500–503. [DOI] [PubMed] [Google Scholar]
- 52. Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal. 2013; 25: 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sampson N, Berger P, Zenzmaier C. Therapeutic targeting of redox signaling in myofibroblast differentiation and age-related fibrotic disease. Oxid Med Cell Longev. 2012; 2012: 458276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD (P) H oxidases. Kidney Int. 2011; 79: 944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010; 65: 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Crestani B, Besnard V, Boczkowski J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2011; 43: 1086–1089. [DOI] [PubMed] [Google Scholar]
- 57. Khodo SN, Dizin E, Sossauer G, et al.. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol. 2012; 23: 1967–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aoyama T, Paik YH, Watanabe S, et al.. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012; 56: 2316–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Green DE, Murphy TC, Kang BY, et al.. The Nox4 inhibitor, GKT137831, attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol. 2012; 47: 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.