

Abstract
Polycomb group (PcG) proteins are evolutionary conserved chromatin-modifying complexes, essential for the regulation of developmental and cell-identity genes. Polycomb-mediated transcriptional regulation is provided by two multi-protein complexes known as Polycomb repressive complex 1 (PRC1) and PRC2. Recent studies positioned PRC1 as a foremost executer of Polycomb-mediated transcriptional control. Mammalian PRC1 complexes can form multiple sub-complexes that vary in their core and accessory subunit composition, leading to fascinating and diverse transcriptional regulatory mechanisms employed by PRC1 complexes. These mechanisms include PRC1-catalytic activity towards monoubiquitination of histone H2AK119, a well-established hallmark of PRC1 complexes whose importance has been long debated. In this review, we will emphasize the central roles that PRC1-catalytic activity plays in transcriptional repression and discuss the recent evidence supporting a role for PRC1 complexes in gene activation.
Keywords: Polycomb, PRC1, PRC2, histone modification, catalytic activity, H2AK119ub, transcriptional activation
Graphical Abstract
Mammalian PRC1 complex composition is highly diverse, resulting in a variety of molecular mechanisms by which they regulate gene expression. Here, we discuss the molecular mechanisms underlying PRC1-mediated repression, focusing on the role of the PRC1 catalytic activity, H2AK119 mono-ubiquitination, and review the evidence linking PRC1 to gene activation.
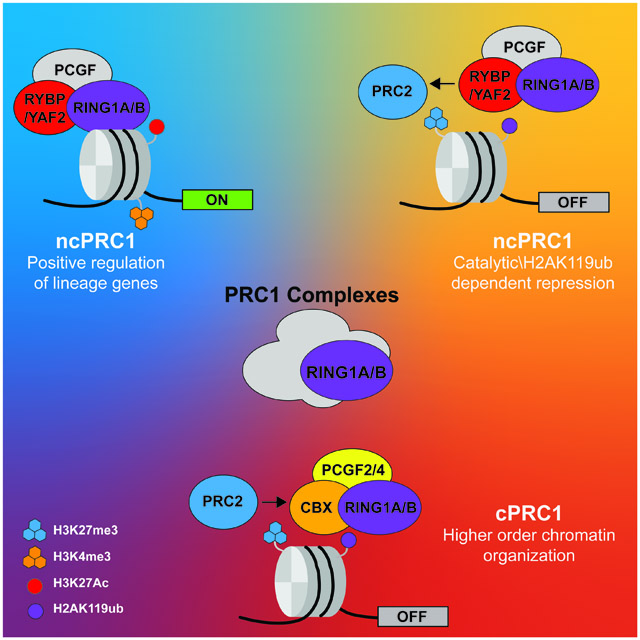
1. Introduction
Transcriptional regulation is a key process underlying the formation and maintenance of cellular diversity and tissue patterns. Transcriptional regulation is a complex process achieved by multiple mechanisms, including transcription factors binding, modification of DNA and histones, and large-scale chromatin architecture organization.[1] Among these transcriptional regulatory mechanisms, the Polycomb group (PcG) proteins have emerged as a paradigm for epigenetic regulation and transcriptional repression. PcG proteins are evolutionary conserved chromatin modifiers,[2] initially discovered in Drosophila melanogaster as critical repressors of homeotic genes, controlling body segmentation.[3] Mutations in different PcG genes result in similar defects in the fly body plan, suggestive of a common pathway/function.[3, 4] Later, extensive biochemical studies revealed that PcG proteins function as two multi-subunit complexes, Polycomb repressive complex 1 (PRC1) and 2 (PRC2).[5] PRC2 consists of the core subunits EED, SUZ12 and EZH1/2, and catalyzes mono-, di- or tri-methylation on lysine 27 of histone H3 (H3K27me1, 2, or 3, respectively).[6, 7] PRC1 complex composition is more heterogeneous and PRC1 can form several complexes (Fig. 1), all of which containing the E3 ligase RING1A or RING1B that catalyzes monoubiquitination on lysine 119 of histone H2A (H2AK119ub).[8-10] Together, PRC1 and PRC2 localization to chromatin results in the formation of Polycomb domains, characterized by compacted chromatin demarcated by H2AK119ub and H3K27me3.[11, 12] Historically, Polycomb complexes were proposed to function through a simplistic hierarchical model, where PRC2 recruitment and H3K27me3 deposition is recognized by so-called canonical PRC1 (cPRC1) complexes, leading cPRC1 recruitment, deposition of H2AK119 monoubiquitination and chromatin compaction.[9, 13-15] Research progress during the past decade brought to our attention the diversity of mammalian Polycomb complexes, with an emphasis on variant types of PRC1 complexes, better known as non-canonical PRC1 (ncPRC1) complexes.[8, 11] This diversity has revealed previously unknown and surprising functions of ncPRC1 complexes as regulators of gene expression,[16] including a reversed Polycomb recruitment model governed by PRC1 complexes[17, 18-20] and the identification of PRC1 complexes at active genes.[21-24] In this review, we will focus on PRC1 complexes and explore the known and unknowns in their modulation of gene expression.
Fig. 1. Schematic representation of PRC1 complexes.

PRC1 complexes are divided into two main groups, canonical PRC1 (cPRC1) complexes and non-canonical PRC1 (ncPRC1) complexes. Both groups contain core subunits, present in all sub-complexes of a subtype, which together with other accessory proteins define the complete composition of each sub-complex. (A) At their core, cPRC1 complexes each contain the E3-ligase subunit RING1A or RING1B that catalyzes the monoubiquitination of histone H2AK119, together with either PCGF2 or PCGF4. The additions of a CBX protein and PHC protein define a cPRC1 complex that can recognize the PRC2-mediated H3K27me3 histone mark. (B) In ncPRC1 complexes, CBX and PHC proteins are replaced by RYBP or YAF2 proteins, and the core complex is not restricted to PCGF2/4 and can be formed with any other PCGF1–6 protein.
2. Canonical PRC1 complexes mediate gene repression by multiple mechanisms
Mammalian PRC1 complexes can be divided into two main groups, cPRC1 and ncPRC1 complexes, depending on the presence or absence of a chromobox domain family (CBX) protein, respectively (Fig. 1a).[8, 11] cPRC1 complexes consist of a catalytic subunit, RING1A or RING1B, PCGF2 (MEL18) or PCGF4 (BMI1), one of three PHC subunits (PHC1 - 3), and one of five CBX subunits (CBX2, 4, 6, 7, or 8).[25] The CBX proteins provide the first molecular link between PRC1 and PRC2 complexes, based on their ability to bind the PRC2-mediated H3K27me3 modification via their chromobox domain.[13, 15, 26] Moreover, PRC2 loss of function or manipulation of H3K27me3 levels affects PRC1 binding, further demonstrating the strong interplay between Polycomb complexes.[7, 13, 27] The observed interdependence of PRC1 and PRC2 has led to the hypothesis that PRC2 is recruited to chromatin first, and H3K27me3 serves as a cue to recruit PRC1. This hypothesis is well-known as the canonical Polycomb recruitment model.
In line with the recruitment of cPRC1 complexes to chromatin by PRC2-mediated H3K27me3, similar roles have been identified for PRC2 complex and cPRC1 proteins in regulating the Drosophila body plan, early mouse embryonic development, and the control of cell proliferation and survival.[28, 29, 30] Among these roles, cPRC1 proteins are mostly recognized for their role in repression of the Cdkn2a locus, encoding the cell-cycle inhibitor INK4A and the pro-apoptotic protein ARF.[31, 32] Ablation of the cPRC1 subunits BMI1, CBX7, or CBX8 in several cell types results in de-repression of the Cdkn2a locus, decreased cell proliferation, and premature senescence.[32, 33] Overexpression of BMI1 or CBX8 in murine embryonic fibroblasts leads to immortalization of these cells.[29, 32] Finally, deletion of Cdkn2a in mice where RING1B has been ablated partially rescues early developmental arrest in mouse embryos, supporting the hypothesis that cPRC1 promotes proliferation and survival during gastrulation by repressing Cdkn2a.[30] While these studies highlight essential roles for cPRC1 complexes in cell proliferation/survival and early tissue development, recent studies in embryonic stem cells have shown that cPRC1 complexes have low contribution to transcriptional repression.[34, 35] Separate roles of cPRC1 and ncPRC1 in somatic stem cells have been recently characterized in neonatal skin epidermis, highlighting both cPRC1-depedent and ncPRC1-dependent essential functions in skin epithelium development.[21, 36] However, such roles remain to be elucidated in other tissues and other developmental stages.
The aforementioned prominent roles of cPRC1 complexes as repressors have led to extensive research focusing on the molecular mechanisms underlying PRC1 repression. Recent gain- and loss-of-function studies of PRC1 complexes with monitoring H2AK119ub by chromatin immunoprecipitation (ChIP) have shown that cPRC1 complexes have a smaller contribution to H2AK119ub levels relative to ncPRC1 complexes,[20, 35] suggesting that cPRC1 complexes execute other mechanisms to mediate transcriptional repression. Earlier studies of PRC1 function have shown that PRC1 complexes can mediate chromatin compaction, and that this function is conserved from fruit flies to mammals.[14, 37] The chromatin compaction region is conserved among PRC1 complexes of multiple organisms but can be found on different complex subunits, depending on the organism. In mammals, the compaction function is attributed to the CBX2 subunit. In Drosophila and other non-mammals, chromatin compaction function has been attributed to BMI1, MEL18, CBX6 and CBX8.[38] Mutations in the mammalian CBX2 basic region result in upregulation of gene expression and homeotic transformations on the anterior-posterior axis. These findings propose nucleosome compaction as a key mechanism for gene silencing during the establishment of body patterning.[39] In addition, the basic region of CBX2 is linked to another repressive mechanism, related to PRC1 localization in condensed nuclear foci called PcG bodies.[40] In mouse ES cells, CBX2 is localized to these nuclear condensates and is required for the assembly of PRC1-CBX2 condensates.[41] Point mutations in the internal domain of CBX2 that disrupt phase separation also impair PcG bodies formation.[41, 42] Interestingly, phase separation is driven by the basic region of CBX2,[41, 42] which also mediates chromatin compaction and essential for axial patterning,[38, 39] suggesting that phase separation mediates repression side by side with chromatin compaction during development. Finally, cPRC1 complexes can mediate transcriptional repression by sub-nuclear clustering. The sterile alpha motif (SAM) domain of the cPRC1 component, PHC2, is able to polymerize, thus enabling interactions between different PRC1 complexes bound to distant genomic areas, supporting the formation of high-order chromatin structures.[43] Mutations in the SAM domain polymerization prevent the formation of PRC1 clusters and result in homeotic transformations, directly linking the function of cPRC1 polymerization domains to gene repression.[44]
Collectively, these studies demonstrate that cPRC1 subunits are important for chromatin compaction and large-scale chromatin organization, allowing efficient repression of unwanted genes, and proper patterning and tissue development. That been said, and although Polycomb-occupied promoters are typically more compact than promoters lacking Polycomb complexes,[45] the loss of PRC1 or PRC2 in ES cells has minor effect on chromatin accessibility,[46] suggesting that other mechanisms that restrict promoter accessibility are independent of Polycomb activity.
3. Recruitment of Non-canonical PRC1 complexes involves both general and context-specific mechanisms.
The canonical Polycomb recruitment model, governed by PRC2-mediated recruitment of the PRC1 complex, has been recently challenged by the discovery of ncPRC1 complexes that can be recruited to chromatin and establish H2AK119ub independently of PRC2.[8, 20, 47, 48] These findings are further reinforced by observations that the genome-wide distribution of PRC1 and PRC2 does not fully overlap [8, 21, 49] and that the global levels of H2AK119ub are maintained in mouse embryonic stem cells and mouse epithelial stem cells lacking the core PRC2 subunits.[21, 48, 50, 51]
Mammalian ncPRC1 complexes are defined by the absence of CBX proteins that can bind H3K27me3, the histone mark deposited by PRC2. Instead of CBX and PHC subunits, ncPRC1 complexes contain RYBP or its homolog YAF2, together with a catalytic subunit RING1A or RING1B and one of six PCGF1-6 subunits (ncPRC1.1-ncPRC1.6) (Fig. 1b).[8]
Several mechanisms have been proposed for PRC2-independent recruitment of PRC1 complexes. The high prevalence of Polycomb complexes at unmethylated CpG islands, mostly around promoter regions, suggests a key role for CpG islands in Polycomb complex recruitment. The molecular link between Polycomb recruitment and CpG islands is mainly attributed to KDM2B, a H3K36 histone lysine demethylase that binds to unmethylated CpG islands through its CxxC-zinc finger domain.[52] Interestingly, the majority of PRC1/RING1B-bound promoter regions are also bound by KDM2B in several systems, including mouse embryonic stem cells and mouse epidermal stem cells.[21, 35, 53] Initial biochemical studies in cancer cells have identified that KDM2B is associated with PRC1 complex components, including RING1B, RYBP, and BCOR.[54] Further dissection of PRC1 sub-complexes demonstrated that KDM2B forms a PRC1 complex with PCGF1/PRC1 (ncPRC1.1),[8, 55] and contributes to generic recruitment of the PRC1 complex to unmethylated CpG islands.[53, 55, 56] In addition, artificial tethering of KDM2B is sufficient for the recruitment of PRC1 and de novo deposition of H2AK119ub.[20] However, despite the practically indistinctive binding of KDM2B to all unmethylated CpG islands, PRC1 occupancy is mostly enriched at repressed sites marked by PRC2-mediated H3K27me3.[53, 55, 56] Moreover, loss of KDM2B or PCGF1 does not lead to complete loss of PRC1 binding or H2AK119ub deposition,[20, 34, 35, 53, 55] suggesting additional mechanisms that guide and/or stabilize PRC1 complex binding to their targets. Such additional mechanism includes the PCGF6-containing ncPRC1.6 complexes. ncPRC1.6 complexes are enriched at PRC1-bound sites in ES cells[34, 35, 57] and maintains RING1B binding and H2AK119ub deposition in the absence of PCGF1 or PCGF2/4 at ncPRC1.6 bound loci,[34, 35] suggesting for alternative non-canonical binding mechanism employed by ncPRC1.6 complexes. This genomic recruitment relies on the presence of ncPRC1.6 accessory subunits with DNA-binding activity, such as E2F6, MGA, and MAX transcription factors. Indeed, PCGF6-bound loci are enriched for MGA/MAX and E2F6 DNA-binding motifs and functional studies have demonstrated that MGA/MAX heterodimers are required for global ncPRC1.6 binding, while loss of E2F6 resulted in locus-specific loss of ncPRC1.6 binding. [34, 57, 58] Additional context-dependent recruitment mechanisms of PRC1 complexes have been shown to include sequence-specific transcription factors such as REST and RUNX1.[59] Together, these mechanisms direct both generic and context-specific recruitment of PRC1 complexes to their genomic targets.
4. H2AK119 monoubiquitination plays a critical role in Polycomb domain formation
Genome-wide studies have demonstrated that ncPRC1 complexes are bound at genomic loci together with cPRC1 and PRC2 complexes or at loci lacking cPRC1 and PRC2 complexes [8, 21, 35, 60, 61] suggesting intricate and diverse mechanisms behind ncPRC1 complexes functions. If ncPRC1 complexes recruitment is independent of PRC2, what is the rationale for colocalization between ncPRC1 and PRC2 complexes? A possible answer lies within the recently proposed reversed Polycomb recruitment model.[17] According to this alternative model, ncPRC1 complex recruitment and H2AK119ub deposition drives PRC2 recruitment, resulting in Polycomb domain formation and transcriptional repression of unwanted genes. Indeed, artificial recruitment of PRC1 to genomic regions normally devoid of Polycomb complexes leads to H2AK119 monoubiquitination followed by PRC2 recruitment and H3K27me3 deposition.[18-20] The molecular link between PRC1 and PRC2 complexes in this model relies on the interaction between PRC1-mediated H2AK119ub and the PRC2 subunit JARID2. JARID2 recognizes nucleosomes containing H2AK119ub via its ubiquitin interacting motif and facilitates PRC2 complex localization and H3K27me3 deposition on PRC1-marked nucleosomes.[18, 62] In line with these observations, loss of PRC1-catalytic activity toward H2AK119 monoubiquitination leads to marked reduction in PRC2 subunits binding and deposition of H3K27me3.[21, 63] Because ncPRC1 complexes, rather than cPRC1 complexes, are proficient for H2AK119 monoubiquitination,[8, 20, 64] the reversed Polycomb recruitment model therefore positions ncPRC1 complexes and H2AK119 monoubiquitination as critical components of Polycomb domain formation and transcriptional repression.
5. The role of PRC1-catalytic activity and H2AK119ub in transcriptional repression is context-dependent.
Histone modifications are thought to be important features regulating gene expression. Yet, the functional significance of many histone modifications, including PRC1-mediated H2AK119ub, is poorly understood. Early studies proposed that PRC1-mediated H2AK119ub interferes with transcriptional elongation.[65] However, initial studies testing whether PRC1 catalytic activity is necessary for gene repression in ES cells yielded conflicting results,[63, 66, 67] leaving the functional role of PRC1 catalytic activity and the H2AK119ub histone mark highly controversial. Arguing against a functional role, studies in PRC1-mutant flies have reported that both catalytic-inactive PRC1 clones and mutated histone H2A/H2Av residues in Drosophila maintain the repression of classical Polycomb targets that are typically de-repressed upon loss of PRC1 or PRC2 complexes.[68] Similarly, catalytic-inactive RING1B mutants have been shown to be largely sufficient for maintenance of gene repression in ES cells and to promote early mouse embryonic development, otherwise impaired in the absence of RING1B.[30, 63, 67] However, at later stages of development, catalytic-inactive PRC1 mutant flies and catalytic-inactive RING1B mutant mice exhibit developmental defects and lethality,[63, 68] suggesting that PRC1 catalytic activity is essential but may function in a gene-specific or cell type-specific manner. Indeed, in neuronal progenitor cells, catalytic-dependent repression of developmental genes has been shown to be unique to poised genes marked by H3K27ac in neurogenic neuronal progenitor cells, while these genes are repressed by PRC1 in a catalytic-independent manner in non-neurogenic neuronal progenitor cells. [69] Studies of catalytically inactive PRC1 mutant complexes in the skin epidermis have also demonstrated a requirement for catalytic-dependent PRC1 activity in gene repression in vivo. Comparison between PRC1 catalytic-inactive and PRC1-null epidermal progenitors demonstrates that hundreds of PRC1-dependent genes that are de-repressed upon global loss of PRC1 function were also de-repressed upon the loss of PRC1 catalytic activity. [21] Importantly, loss of PRC1 catalytic activity in vivo leads to de-repression of Merkel-lineage commitment transcription factors and ectopic formation of mechanosensory Merkel cells.[21, 70, 71] Taken together, these studies provide compelling evidence of essential roles for PRC1 catalytic activity in Polycomb-mediated gene repression.
The specific functions of PRC1 catalytic activity in Polycomb-mediated transcriptional repression are currently unclear. As suggested by the reversed PRC1-mediated recruitment model, one possibility is that PRC1 catalytic activity and H2AK119ub are required to facilitate the efficient recruitment of PRC2. This notion is supported by several studies showing a reduction in chromatin-bound PRC2 subunits and lower H3K27me3 deposition in catalytic-inactive PRC1 mutants.[21, 63, 68, 72] Such a reduction in PRC2-mediated H3K27me3, can lead to impaired binding of cPRC1 complexes that contribute to Polycomb-mediated transcriptional repression by mediating long-range interactions between Polycomb domains.[73, 74] On this note, it is important to mention that PRC2 complexes can be divided into two main subtypes,[75] PRC2.1 complexes that contain PCL proteins and bind to unmethylated CpG islands[76, 77] and PRC2.2 complexes that contain the JARID2 subunit that recognizes H2AK119ub to promote PRC2 recruitment.[18, 62, 76, 78] Therefore, it would be interesting to test whether specific loci enriched for PRC2.2 are more susceptible to loss of PRC1-mediated H2AK119ub compared with loci enriched for PRC2.1-type complexes, as suggested by recent studies in mouse ES cells[79]. Alternatively, H2AK119ub may also contribute to PRC1-mediated transcriptional repression in a PRC2/cPRC1-independent fashion through direct interference with RNA polymerase II activity[65] or through H2AK119ub-dependent interactions with reader proteins, other than JARID2, that possess repressive functions.[80] PRC2/cPRC1-independent roles for ncPRC1 complexes and H2AK119ub are also supported by the observation that the cPRC1 complex contribution to H2AK119ub and transcriptional repression is minimal relative to that of ncPRC1 complexes.[35] Likewise, the transcriptional changes and in vivo phenotypes associated with loss of PRC1 or its catalytic activity are in many cases more pronounced compared with the loss of cPRC1 or PRC2 (Fig. 2a).[21, 35, 36, 50, 70, 72, 81] Remarkably, for a subset of PRC2-independent genes, loss of PRC1-catalytic activity results in gene de-repression without altering RING1B binding, suggesting a direct role for H2AK119ub in gene repression (Fig. 2b).[21] Therefore, these studies provide examples of essential roles of PRC1-catalytic activity, and most likely H2AK119ub, in Polycomb-mediated transcriptional repression. However, dissection of the molecular mechanisms downstream of PRC1 catalytic activity is required to elucidate whether gene repression is PRC2/cPRC1-dependent or -independent.
Fig. 2. PRC1-catalytic activity is essential for PRC1-mediated transcriptional repression.

(A) Venn diagram showing the overlap in significantly upregulated genes in epidermal progenitors for the indicated genotypes that are marked by H2AK119ub and H3K27me3 in epidermal progenitors. (B) Integrative Genomics Viewer (IGV) ChIP-seq and RNA-seq browser views for the indicated markers and genotypes in epidermal progenitors. Note that some genes co-marked by H2AK119ub and H3K27me3 are de-repressed only upon loss of PRC1-catalytic or -global activities, but not upon loss of PRC2 activity. In addition, note that loss of PRC1-catalytic activity does not impair RING1B binding to those genes in epidermal progenitors. Data was obtained from the Gene Expression Omnibus (GEO) database (GSE112460) and was analyzed as previously described.[21]
6. ncPRC1 complexes can facilitate gene activation.
Although the Polycomb complexes are historically viewed as key transcriptional repressors,[5] early studies in Drosophila have shown that PcG subunits binding to several Polycomb response elements (PREs) nearby homeotic genes persist even when these genes are actively transcribed.[82, 83] While most of the PREs near homeotic genes demonstrated repressor functions in transgenic assays, some PREs were required for proper gene activation.[82, 84] Recently, several genome-wide chromatin profiling studies have mapped PRC1 core components and associating proteins to active genes in multiple mammalian cell types, including ES cells,[23, 60] neural progenitors,[23] epidermal progenitors,[21] quiescent lymphocytes,[24] leukemic cells,[22] and breast cancer cells.[85] In most of these cases, PRC1-bound loci are devoid of PRC2 subunits or the H3K27me3 mark, suggesting for PRC2-independent functions at those active genes. [21-23] Typically, PRC1-bound active genes are enriched for ncPRC1 complexes rather than cPRC1 complexes, and are devoid of or marked by low levels of H2AK119ub, whereas PRC1-bound silent genes contain cPRC1 and/or ncPRC1 complexes (Fig. 3a).[21, 23, 34, 36, 60, 61, 86] The regulatory function of ncPRC1 complexes at active genes is poorly understood. In some cases, however, accumulating evidence suggests that ncPRC1 complexes function to promote the expression of actively transcribed target genes, rather than repressing it, as loss of PRC1 binding and function leads to the downregulation of those target genes (Fig. 3b).[21, 24, 36, 87] In addition, reporter assay experiments have demonstrated that the recruitment of specific ncPRC1 complexes containing PCGF3 or PCGF5 (ncPRC1.3 and ncPRC1.5, respectively) leads to increased reporter activity,[34, 87] supporting a role for ncPRC1 complexes in gene activation. Paradoxically, ncPRC1 complexes are more proficient in H2AK119 monoubiquitination than cPRC1 complexes, while ncPRC1-bound active genes display low levels of H2AK119ub.[21, 34, 36, 87] Thus, molecular mechanisms underlying ncPRC1-mediated regulation of actively transcribed genes should favor accessory subunit composition or additional interacting partners that reduce PRC1-catalytic activity toward H2AK119 monoubiquitination. Indeed, in quiescent lymphocytes, the Aurora B kinase cooperates with RING1B to promote gene expression by modulating PRC1 activity and H2AK119ub deposition through diverse mechanisms. On one hand, Aurora B phosphorylates the E2-conjugating enzyme UBE2D3 and inhibits its catalytic activity required for RING1B E3 ligase activity, thereby leading to reduced H2AK119ub deposition.[24] On the other hand, Aurora B phosphorylates histone H3S28 as well as the USP16 histone de-ubiquitinase, leading to enhanced USP16 binding and removal of the H2AK119ub histone mark.[24] A somewhat similar mechanism was also reported in the murine brain, where ncPRC1.3/ncPRC1.5 forms a complex with AUTS2 that recruits the casein kinase 2 (CK2) subunits CSNK2A1 and CSNK2A2.[87] In turn, CK2 phosphorylates RING1B at serine 168 (S168) to inhibit the E3 ligase activity of RING1B.[87] Finally, functional studies done in the developing epidermis and in epidermal progenitors have demonstrated that PRC1-catalytic activity is dispensable for promoting the expression of PRC1-dependent genes (Fig. 3b).[21] These studies therefore suggest that while PRC1-catalytic activity plays essential roles in Polycomb-mediated transcriptional repression, it may be dispensable for PRC1 function in promoting the expression of active genes.
Fig. 3. ncPRC1 core subunits are bound to both silent and active genes.

(A) Expression analysis of genes located near the indicated PRC1 subunits in control epidermal progenitors. Note higher level of expression in genes bound by ncPRC1 subunits, compared with genes bound by cRPC1 subunits. Gene expression data is presented as FPKM values in box-and-whisker boxplots. Midline, median; box limits, 25th percentile (lower quartile) and 75th percentile (upper quartile); upper whisker, 75th-95th percentile; lower whisker, 5th-25th percentile. (B) Integrative Genomics Viewer (IGV) ChIP-seq and RNA-seq browser views for the indicated markers and genotypes in epidermal progenitors. Note that in active gene bound by ncPRC1 subunits, loss of PRC1-global activity results in downregulation of gene expression, whereas loss of PRC1-catalytic activity or PRC2 does not affect gene expression. Data was obtained from the Gene Expression Omnibus (GEO) database (GSE112460) and was analyzed as previously described.[21]
In addition to the suppression of PRC1-catalytic activity and displacement of H2AK119ub deposition from active genes, PRC1-dependent transcriptional activation probably requires additional mechanisms. PRC1 plays an important role in regulating the genomic architecture, securing proper topological promoter-promoter and promoter-enhancer interactions.[74] Recently, the three-dimensional organization of enhancer-promoter interactions mediated by PRC1 has been shown to be essential for the activation of Meis2 in midbrain during embryonic development.[88] It is noteworthy that ncPRC1.3 and ncPRC1.5 complexes function to repress Meis2 at the distal forelimb bud,[89] suggesting that specific interactions with corepressors or coactivators might determine the switch between gene repression and activation by PRC1. Indeed, in the central nervous system, PRC1 interaction with the tissue-specific transcription factor AUTS2 mediates the recruitment of P300 histone acetyltransferase, which in turn activates transcription.[87] Future studies of tissue-specific PRC1-interacting partners, as well as the dynamic changes in PRC1 accessory subunits composition during cell differentiation, are expected to shed light on the mechanisms by which PRC1 complexes contribute to gene activation.
7. Concluding remarks and future perspectives
Here we describe some of the recent and exciting findings on PRC1 complexes, stressing the complexity behind Polycomb systems and highlighting our incomplete understanding of the diverse molecular mechanisms by which PRC1 complexes regulate gene expression. PRC1-catalytic activity has recently emerged as a central feature required for Polycomb-mediated transcriptional repression; yet we still do not entirely understand the specific function of PRC1-mediated H2AK119ub in this process. It is currently unclear whether this histone mark has a direct repressive role, or whether it mostly facilitates the recruitment of PRC2 complexes to chromatin. In addition to H2AK119 monoubiquitination, it is possible that PRC1-catalytic activity represses transcription by modifying histone H2A.Z variant[90] or does so indirectly through the modification of RING1A/B itself and/or other protein substrates.[91] Finally, the presence of ncPRC1 complexes at active genes is surprising and fascinating, given the view of Polycomb complexes as classical transcriptional repressors. Remarkably, it seems that ncPRC1 complexes are more abundant at active genes compared with their relevant occupancy at silent genes. In some cases, PRC1 complexes attenuate the expression of those active genes, whereas in other cases PRC1 complexes promote the expression of their target genes.[21] Given the overwhelming diversity in PRC1 complexes and their accessory subunit composition, understanding the dynamic changes in PRC1 complexes composition during transition from silent to active states is warranted. Careful and in-depth dissection of these open questions should increase our knowledge and provide novel insights into the precise functions and mechanisms behind PRC1-mediated transcriptional control.
Acknowledgements
I.C. was supported by a Training Program in Stem Cell Biology fellowship from the New York State Department of Health (NYSTEM-C32561GG). C.B. is a Merksamer Fund scholar. Research in the laboratory of E.E was supported by the Tisch Cancer Institute P30 Cancer Support Grant, the NIH/NIAMS under award number R01 AR069078, and the NIH/NIDCD under award number R01 DC017400. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
References
- [1].Lelli KM, Slattery M, Mann RS, Annu Rev Genet 2012, 46, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G, Cell 2017, 171, 34. [DOI] [PubMed] [Google Scholar]
- [3].Lewis EB, Nature 1978, 276, 565. [DOI] [PubMed] [Google Scholar]
- [4].Jurgens G, Nature 1985, 316, 153 [Google Scholar]; Struhl G, Nature 1981, 293, 36. [DOI] [PubMed] [Google Scholar]
- [5].Simon JA, Kingston RE, Mol Cell 2013, 49, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Margueron R, Reinberg D, Nature 2011, 469, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D, Genes Dev 2002, 16, 2893. [DOI] [PMC free article] [PubMed] [Google Scholar]; Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V, Cell 2002, 111, 185. [DOI] [PubMed] [Google Scholar]
- [7].Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y, Science 2002, 298, 1039. [DOI] [PubMed] [Google Scholar]
- [8].Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D, Mol Cell 2012, 45, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y, Nature 2004, 431, 873. [DOI] [PubMed] [Google Scholar]
- [10].de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, Koseki H, Brockdorff N, Dev Cell 2004, 7, 663. [DOI] [PubMed] [Google Scholar]
- [11].Di Croce L, Helin K, Nat Struct Mol Biol 2013, 20, 1147. [DOI] [PubMed] [Google Scholar]
- [12].Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE, PLoS Genet 2008, 4, e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS, Mol Cell 2004, 14, 637. [DOI] [PubMed] [Google Scholar]
- [14].Francis NJ, Kingston RE, Woodcock CL, Science 2004, 306, 1574. [DOI] [PubMed] [Google Scholar]
- [15].Min J, Zhang Y, Xu RM, Genes Dev 2003, 17, 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Miroshnikova YA, Cohen I, Ezhkova E, Wickstrom SA, Curr Opin Genet Dev 2019, 55, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]; Blackledge NP, Rose NR, Klose RJ, Nat Rev Mol Cell Biol 2015, 16, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]; Aranda S, Mas G, Di Croce L, Sci Adv 2015, 1, e1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schwartz YB, Pirrotta V, Cell Rep 2014, 8, 321. [DOI] [PubMed] [Google Scholar]; Comet I, Helin K, Nat Struct Mol Biol 2014, 21, 573. [DOI] [PubMed] [Google Scholar]
- [18].Kalb R, Latwiel S, Baymaz HI, Jansen PW, Muller CW, Vermeulen M, Muller J, Nat Struct Mol Biol 2014, 21, 569. [DOI] [PubMed] [Google Scholar]
- [19].Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, Heger A, Brockdorff N, Cell Rep 2014, 7, 1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LL, Ito S, Cooper S, Kondo K, Koseki Y, Ishikura T, Long HK, Sheahan TW, Brockdorff N, Kessler BM, Koseki H, Klose RJ, Cell 2014, 157, 1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cohen I, Zhao D, Bar C, Valdes VJ, Dauber-Decker KL, Nguyen MB, Nakayama M, Rendl M, Bickmore WA, Koseki H, Zheng D, Ezhkova E, Cell Stem Cell 2018, 22, 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].van den Boom V, Maat H, Geugien M, Rodriguez Lopez A, Sotoca AM, Jaques J, Brouwers-Vos AZ, Fusetti F, Groen RW, Yuan H, Martens AC, Stunnenberg HG, Vellenga E, Martens JH, Schuringa JJ, Cell Rep 2016, 14, 332. [DOI] [PubMed] [Google Scholar]
- [23].Kloet SL, Makowski MM, Baymaz HI, van Voorthuijsen L, Karemaker ID, Santanach A, Jansen P, Di Croce L, Vermeulen M, Nat Struct Mol Biol 2016, 23, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frangini A, Sjoberg M, Roman-Trufero M, Dharmalingam G, Haberle V, Bartke T, Lenhard B, Malumbres M, Vidal M, Dillon N, Mol Cell 2013, 51, 647. [DOI] [PubMed] [Google Scholar]
- [25].Simon JA, Kingston RE, Nat Rev Mol Cell Biol 2009, 10, 697. [DOI] [PubMed] [Google Scholar]; Levine SS, Weiss A, Erdjument-Bromage H, Shao Z, Tempst P, Kingston RE, Mol Cell Biol 2002, 22, 6070. [DOI] [PMC free article] [PubMed] [Google Scholar]; Francis NJ, Saurin AJ, Shao Z, Kingston RE, Mol Cell 2001, 8, 545. [DOI] [PubMed] [Google Scholar]; Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, Kingston RE, Cell 1999, 98, 37. [DOI] [PubMed] [Google Scholar]
- [26].Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S, Genes Dev 2003, 17, 1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R, Science 2007, 318, 447. [DOI] [PubMed] [Google Scholar]; Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R, Nature 2006, 441, 349. [DOI] [PubMed] [Google Scholar]
- [28].Sauvageau M, Sauvageau G, PLoS Biol 2008, 6, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]; O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T, Mol Cell Biol 2001, 21, 4330. [DOI] [PMC free article] [PubMed] [Google Scholar]; Faust C, Lawson KA, Schork NJ, Thiel B, Magnuson T, Development 1998, 125, 4495. [DOI] [PubMed] [Google Scholar]
- [29].Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, Hansen KH, Helin K, Genes Dev 2007, 21, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Voncken JW, Roelen BA, Roefs M, de Vries S, Verhoeven E, Marino S, Deschamps J, van Lohuizen M, Proc Natl Acad Sci U S A 2003, 100, 2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Popov N, Gil J, Epigenetics 2010, 5, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M, Nature 1999, 397, 164. [DOI] [PubMed] [Google Scholar]
- [33].Dietrich N, Bracken AP, Trinh E, Schjerling CK, Koseki H, Rappsilber J, Helin K, Hansen KH, EMBO J 2007, 26, 1637. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gil J, Bernard D, Martinez D, Beach D, Nat Cell Biol 2004, 6, 67. [DOI] [PubMed] [Google Scholar]
- [34].Scelfo A, Fernandez-Perez D, Tamburri S, Zanotti M, Lavarone E, Soldi M, Bonaldi T, Ferrari KJ, Pasini D, Mol Cell 2019, 74, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fursova NA, Blackledge NP, Nakayama M, Ito S, Koseki Y, Farcas AM, King HW, Koseki H, Klose RJ, Mol Cell 2019, 74, 1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cohen I, Zhao D, Menon G, Nakayama M, Koseki H, Zheng D, Ezhkova E, Genes Dev 2019, 33, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lavigne M, Francis NJ, King IF, Kingston RE, Mol Cell 2004, 13, 415. [DOI] [PubMed] [Google Scholar]
- [38].Grau DJ, Chapman BA, Garlick JD, Borowsky M, Francis NJ, Kingston RE, Genes Dev 2011, 25, 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lau MS, Schwartz MG, Kundu S, Savol AJ, Wang PI, Marr SK, Grau DJ, Schorderet P, Sadreyev RI, Tabin CJ, Kingston RE, Science 2017, 355, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Saurin AJ, Shiels C, Williamson J, Satijn DP, Otte AP, Sheer D, Freemont PS, J Cell Biol 1998, 142, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]; Satijn DP, Gunster MJ, van der Vlag J, Hamer KM, Schul W, Alkema MJ, Saurin AJ, Freemont PS, van Driel R, Otte AP, Mol Cell Biol 1997, 17, 4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tatavosian R, Kent S, Brown K, Yao T, Duc HN, Huynh TN, Zhen CY, Ma B, Wang H, Ren X, J Biol Chem 2019, 294, 1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, Kingston RE, Genes Dev 2019, 33, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim CA, Gingery M, Pilpa RM, Bowie JU, Nat Struct Biol 2002, 9, 453. [DOI] [PubMed] [Google Scholar]
- [44].Isono K, Endo TA, Ku M, Yamada D, Suzuki R, Sharif J, Ishikura T, Toyoda T, Bernstein BE, Koseki H, Dev Cell 2013, 26, 565. [DOI] [PubMed] [Google Scholar]
- [45].Beck S, Lee BK, Rhee C, Song J, Woo AJ, Kim J, Nat Commun 2014, 5, 5490. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bell O, Schwaiger M, Oakeley EJ, Lienert F, Beisel C, Stadler MB, Schubeler D, Nat Struct Mol Biol 2010, 17, 894. [DOI] [PubMed] [Google Scholar]
- [46].King HW, Fursova NA, Blackledge NP, Klose RJ, Genome Res 2018, 28, 1494. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hodges HC, Stanton BZ, Cermakova K, Chang CY, Miller EL, Kirkland JG, Ku WL, Veverka V, Zhao K, Crabtree GR, Nat Struct Mol Biol 2018, 25, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lagarou A, Mohd-Sarip A, Moshkin YM, Chalkley GE, Bezstarosti K, Demmers JA, Verrijzer CP, Genes Dev 2008, 22, 2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, Wutz A, Vidal M, Elderkin S, Brockdorff N, Cell 2012, 148, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Loubiere V, Delest A, Thomas A, Bonev B, Schuettengruber B, Sati S, Martinez AM, Cavalli G, Nat Genet 2016, 48, 1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bar C, Cohen I, Zhao D, Pothula V, Litskevitch A, Koseki H, Zheng D, Ezhkova E, Cell Rep 2019, 28, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A, Genes Dev 2010, 24, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ, Mol Cell 2010, 38, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]; He J, Kallin EM, Tsukada Y, Zhang Y, Nat Struct Mol Biol 2008, 15, 1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].He J, Shen L, Wan M, Taranova O, Wu H, Zhang Y, Nat Cell Biol 2013, 15, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sanchez C, Sanchez I, Demmers JA, Rodriguez P, Strouboulis J, Vidal M, Mol Cell Proteomics 2007, 6, 820. [DOI] [PubMed] [Google Scholar]; Gearhart MD, Corcoran CM, Wamstad JA, Bardwell VJ, Mol Cell Biol 2006, 26, 6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Farcas AM, Blackledge NP, Sudbery I, Long HK, McGouran JF, Rose NR, Lee S, Sims D, Cerase A, Sheahan TW, Koseki H, Brockdorff N, Ponting CP, Kessler BM, Klose RJ, Elife 2012, 1, e00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wu X, Johansen JV, Helin K, Mol Cell 2013, 49, 1134. [DOI] [PubMed] [Google Scholar]
- [57].Endoh M, Endo TA, Shinga J, Hayashi K, Farcas A, Ma KW, Ito S, Sharif J, Endoh T, Onaga N, Nakayama M, Ishikura T, Masui O, Kessler BM, Suda T, Ohara O, Okuda A, Klose R, Koseki H, Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Stielow B, Finkernagel F, Stiewe T, Nist A, Suske G, PLoS Genet 2018, 14, e1007193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yu M, Mazor T, Huang H, Huang HT, Kathrein KL, Woo AJ, Chouinard CR, Labadorf A, Akie TE, Moran TB, Xie H, Zacharek S, Taniuchi I, Roeder RG, Kim CF, Zon LI, Fraenkel E, Cantor AB, Mol Cell 2012, 45, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dietrich N, Lerdrup M, Landt E, Agrawal-Singh S, Bak M, Tommerup N, Rappsilber J, Sodersten E, Hansen K, PLoS Genet 2012, 8, e1002494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Morey L, Santanach A, Blanco E, Aloia L, Nora EP, Bruneau BG, Di Croce L, Cell Stem Cell 2015, 17, 300. [DOI] [PubMed] [Google Scholar]
- [61].Morey L, Aloia L, Cozzuto L, Benitah SA, Di Croce L, Cell Rep 2013, 3, 60. [DOI] [PubMed] [Google Scholar]
- [62].Cooper S, Grijzenhout A, Underwood E, Ancelin K, Zhang T, Nesterova TB, Anil-Kirmizitas B, Bassett A, Kooistra SM, Agger K, Helin K, Heard E, Brockdorff N, Nat Commun 2016, 7, 13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Illingworth RS, Moffat M, Mann AR, Read D, Hunter CJ, Pradeepa MM, Adams IR, Bickmore WA, Genes Dev 2015, 29, 1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rose NR, King HW, Blackledge NP, Fursova NA, Ember KJ, Fischer R, Kessler BM, Klose RJ, Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG, Mol Cell 2008, 29, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]; Stock JK, Giadrossi S, Casanova M, Brookes E, Vidal M, Koseki H, Brockdorff N, Fisher AG, Pombo A, Nat Cell Biol 2007, 9, 1428. [DOI] [PubMed] [Google Scholar]
- [66].Endoh M, Endo TA, Endoh T, Isono K, Sharif J, Ohara O, Toyoda T, Ito T, Eskeland R, Bickmore WA, Vidal M, Bernstein BE, Koseki H, PLoS Genet 2012, 8, e1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, Gilbert N, Fan Y, Skoultchi AI, Wutz A, Bickmore WA, Mol Cell 2010, 38, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pengelly AR, Kalb R, Finkl K, Muller J, Genes Dev 2015, 29, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tsuboi M, Kishi Y, Yokozeki W, Koseki H, Hirabayashi Y, Gotoh Y, Dev Cell 2018, 47, 758. [DOI] [PubMed] [Google Scholar]
- [70].Dauber KL, Perdigoto CN, Valdes VJ, Santoriello FJ, Cohen I, Ezhkova E, J Invest Dermatol 2016, 136, 1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bardot ES, Valdes VJ, Zhang J, Perdigoto CN, Nicolis S, Hearn SA, Silva JM, Ezhkova E, EMBO J 2013, 32, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Blackledge NP, Fursova NA, Kelley JR, Huseyin MK, Feldmann A, Klose RJ, bioRxiv 2019, 667667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, Dekker J, Kingston RE, Mol Cell 2017, 65, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Schoenfelder S, Sugar R, Dimond A, Javierre BM, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, Segonds-Pichon A, Jurkowski W, Wingett SW, Tabbada K, Andrews S, Herman B, LeProust E, Osborne CS, Koseki H, Fraser P, Luscombe NM, Elderkin S, Nat Genet 2015, 47, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Laugesen A, Hojfeldt JW, Helin K, Mol Cell 2019, 74, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Marasca F, Bodega B, Orlando V, Bioessays 2018, 40, e1700137. [DOI] [PubMed] [Google Scholar]
- [76].Healy E, Mucha M, Glancy E, Fitzpatrick DJ, Conway E, Neikes HK, Monger C, Van Mierlo G, Baltissen MP, Koseki Y, Vermeulen M, Koseki H, Bracken AP, Mol Cell 2019. [DOI] [PubMed] [Google Scholar]
- [77].Conway E, Jerman E, Healy E, Ito S, Holoch D, Oliviero G, Deevy O, Glancy E, Fitzpatrick DJ, Mucha M, Watson A, Rice AM, Chammas P, Huang C, Pratt-Kelly I, Koseki Y, Nakayama M, Ishikura T, Streubel G, Wynne K, Hokamp K, McLysaght A, Ciferri C, Di Croce L, Cagney G, Margueron R, Koseki H, Bracken AP, Mol Cell 2018, 70, 408. [DOI] [PubMed] [Google Scholar]; Beringer M, Pisano P, Di Carlo V, Blanco E, Chammas P, Vizan P, Gutierrez A, Aranda S, Payer B, Wierer M, Di Croce L, Mol Cell 2016, 64, 645. [DOI] [PubMed] [Google Scholar]
- [78].Holoch D, Margueron R, Trends Biochem Sci 2017, 42, 531. [DOI] [PubMed] [Google Scholar]
- [79].Tamburri S, Lavarone E, Fernandez-Perez D, Conway E, Zanotti M, Manganaro D, Pasini D, Mol Cell 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhang Z, Jones AE, Wu W, Kim J, Kang Y, Bi X, Gu Y, Popov IK, Renfrow MB, Vassylyeva MN, Vassylyev DG, Giles KE, Chen D, Kumar A, Fan Y, Tong Y, Liu CF, An W, Chang C, Luo J, Chow LT, Wang H, Proc Natl Acad Sci U S A 2017, 114, E7949. [DOI] [PMC free article] [PubMed] [Google Scholar]; Qin W, Wolf P, Liu N, Link S, Smets M, La Mastra F, Forne I, Pichler G, Horl D, Fellinger K, Spada F, Bonapace IM, Imhof A, Harz H, Leonhardt H, Cell Res 2015, 25, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Chiacchiera F, Rossi A, Jammula S, Zanotti M, Pasini D, EMBO J 2016, 35, 2301. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chiacchiera F, Rossi A, Jammula S, Piunti A, Scelfo A, Ordonez-Moran P, Huelsken J, Koseki H, Pasini D, Cell Stem Cell 2016, 18, 91. [DOI] [PubMed] [Google Scholar]
- [82].Park SY, Schwartz YB, Kahn TG, Asker D, Pirrotta V, Mech Dev 2012, 128, 536. [DOI] [PubMed] [Google Scholar]
- [83].Langlais KK, Brown JL, Kassis JA, PLoS One 2012, 7, e48765. [DOI] [PMC free article] [PubMed] [Google Scholar]; Papp B, Muller J, Genes Dev 2006, 20, 2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ahmad K, Spens AE, PLoS Genet 2019, 15, e1007877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chan HL, Beckedorff F, Zhang Y, Garcia-Huidobro J, Jiang H, Colaprico A, Bilbao D, Figueroa ME, LaCava J, Shiekhattar R, Morey L, Nat Commun 2018, 9, 3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pivetti S, Fernandez-Perez D, D'Ambrosio A, Barbieri CM, Manganaro D, Rossi A, Barnabei L, Zanotti M, Scelfo A, Chiacchiera F, Pasini D, Sci Adv 2019, 5, eaav1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, Reinberg D, Nature 2014, 516, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kondo T, Ito S, Koseki H, Trends Biochem Sci 2016, 41, 9. [DOI] [PubMed] [Google Scholar]; Kondo T, Isono K, Kondo K, Endo TA, Itohara S, Vidal M, Koseki H, Dev Cell 2014, 28, 94. [DOI] [PubMed] [Google Scholar]
- [89].Yakushiji-Kaminatsui N, Kondo T, Hironaka KI, Sharif J, Endo TA, Nakayama M, Masui O, Koseki Y, Kondo K, Ohara O, Vidal M, Morishita Y, Koseki H, Development 2018, 145. [DOI] [PubMed] [Google Scholar]
- [90].Surface LE, Fields PA, Subramanian V, Behmer R, Udeshi N, Peach SE, Carr SA, Jaffe JD, Boyer LA, Cell Rep 2016, 14, 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A, Mol Cell 2006, 24, 701. [DOI] [PubMed] [Google Scholar]