

Abstract
The genetic underpinnings of late-onset degenerative disease have typically been determined by screening families for the segregation of genetic variants with the disease trait in affected, but not unaffected, individuals. However, instances of intrafamilial etiological heterogeneity, where pathogenic variants in a culprit gene are not shared among all affected family members, continue to emerge and confound gene-discovery and genetic counselling efforts. Discordant intrafamilial cases lacking a mutation shared by other affected family members are described as disease phenocopies. This description often results in an over-simplified acceptance of an environmental cause of disease in the phenocopy cases, while the role of intrafamilial genetic heterogeneity, shared de novo mutations or epigenetic aberrations in such families is often ignored. On a related note, it is now evident that the same disease-associated variant can be present in individuals exhibiting clinically distinct phenotypes, thereby genetically uniting seemingly unrelated syndromes to form a spectrum of disease. Herein, we discuss the intricacies of determining complex degenerative disease aetiology and suggest alternative mechanisms of disease transmission that may account for the apparent missing heritability of disease.
Keywords: phenocopy, pleiotropy, inheritance, aetiology, neurodegeneration
This review addresses the pitfalls of classifying cases of apparently discordant disease aetiology as phenocopies, offers alternative explanations for hereditary disease transmission and discusses the complex roles of pleiotropic genes in degenerative disease.
Graphical Abstract
Graphical Abstract.
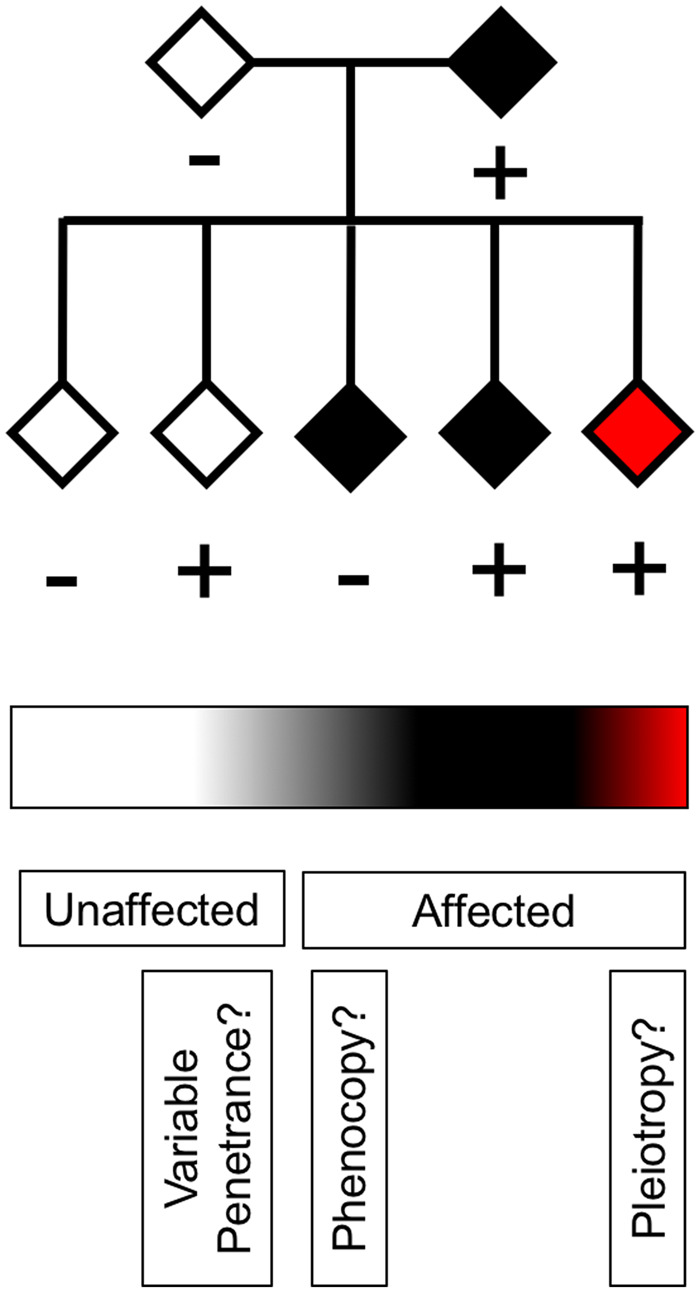
Introduction
Advances in the field of molecular genetics have facilitated the identification of disease-causing or associated genetic variants in many late-onset degenerative conditions. This is typically achieved by identifying segregating genetic variants present in affected cases, but absent from healthy, unaffected family members of similar or older in age. A causal link between genotype and disease phenotype can be most readily identified in cases of highly penetrant and monogenic diseases, e.g. Huntington disease. However, in genetically complex diseases such as Parkinson disease and amyotrophic lateral sclerosis (ALS), variability of disease penetrance and manifestation, both within and across families, can hinder the identification of a co-segregating genetic variant(s) with a disease phenotype.
As the era of next-generation sequencing forged ahead and more pedigrees were studied, intrafamilial cases of presumed discordant disease aetiology continued to emerge. Assuming accurate familial relationship reporting, affected cases that lack a known disease-associated variant(s) that co-segregates with the clinical phenotype in other affected family members are commonly referred to as phenocopies. The classical definition of phenocopy is the manifestation of a phenotype due to an environmental condition that mimics a phenotype produced by a gene. The term has come to be used more loosely with regard to familial disease, allowing any affected family member that does not share a mutation present in other affected family members to be identified as a phenocopy.
Classifying intrafamilial cases of apparent discordant disease aetiology as phenocopies can be problematic for gene-discovery efforts. Such classification can lead to the erroneous conclusion that a benign variant is disease-associated if affected individuals lacking the variant are readily accepted as phenocopies. A pathogenic variant may also be missed if apparent phenocopies are not considered as such. When two or more genes or loci contend for causing a phenotype in a family, the most parsimonious aetiology is the one with the least phenocopies (Deng et al., 2019). Combined with our limited understanding of genetic variants of low effect and incomplete penetrance, genes implicated in more than one condition (i.e. pleiotropic genes), and unconventional modes of disease inheritance, it can be extremely difficult to identify a genetic cause of disease within a family. Ultimately, the segregation of genotype with phenotype can only be strictly determined through statistical testing of a large number of similar cases and by experimental validation, a task made difficult when studying rare disease phenotypes of late-onset with incomplete penetrance and particularly in small families.
Here, we provide examples of families we have studied in our laboratory and use Parkinson disease and ALS as exemplars of genetically complex late-onset neurodegenerative diseases to examine the concepts of intrafamilial genetic heterogeneity, sharing of de novo mutations (DNMs) or epimutations, and pleiotropy to provide a deeper understanding of phenocopy beyond assuming the cause to be purely environmental.
Unmasking phenocopies
Mendelian inheritance of disease is common among the majority of Parkinson disease and ALS pedigrees harbouring mutations in single genes, but there are occasions where disease inheritance does not appear to follow Mendelian patterns. Intrafamilial genetic heterogeneity, where an observed phenotype within a family is caused by more than one genetic variant, can give rise to this apparent disconnect between genotype and phenotype. Approximately 10% of Parkinson disease cases are inherited, due to mutations in genes such as SNCA, LRRK2, PRKN, PINK1, VPS35, TMEM230 and others (Polymeropoulos, 1997; Lucking et al., 2000; Valente, 2004; Zimprich et al., 2004; Vilarino-Guell et al., 2011; Zimprich et al., 2011; Deng et al., 2016). However, Parkinson disease can also occur after ingestion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a well-characterized toxin of dopaminergic neurons (Betarbet and Greenamyre, 2007). Using the classical definition of phenocopy, Parkinson disease cases caused by exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine are phenocopies, although not all reported Parkinson disease phenocopies are the result of a known environmental exposure (Delamarre and Meissner, 2017). A review of all Parkinson disease pedigrees published between 1997 and 2009 with a SNCA, LRRK2, PRKN or PINK1 mutation concluded that 5% of all affected individuals, that is 27 patients from 23 families out of a cohort of 537 patients from 160 families, were phenocopies (Klein et al., 2011). Intriguingly, four of the presumed phenocopies were found to carry a Parkinson disease-associated mutation that was different from that detected in the proband, demonstrating that intrafamilial genetic heterogeneity can account for apparent disease phenocopies.
Familial amyotrophic lateral sclerosis accounts for ∼10% of all ALS cases, with disease-causing mutations in SOD1, FUS and C9orf72 identified in ∼60% of probands of familial amyotrophic lateral sclerosis families (Ajroud-Driss and Siddique, 2015; Taylor et al., 2016; Al-Chalabi et al., 2017). While the occurrence of intrafamilial genetic heterogeneity is presumed to be rare in ALS pedigrees, a report describes 5 of 97 families with mutations in more than one ALS-associated gene (van Blitterswijk et al., 2012). Here we describe a family we have studied, Family 379, in which three individuals with a clinical diagnosis of ALS do not carry the TAR DNA-binding protein 43 p. G298S mutation identified in 10 other affected family members (Fig. 1 and Table 1). Genotyping of the three TAR DNA-binding protein 43 p. G298S-negative individuals for mutations in other known ALS-causing genes revealed a heterozygous superoxide dismutase 1 (SOD1) p. D91A mutation in individual III-6 that is not present in any other affected family members. Such an individual may have inherited the discordant disease-causing mutation from the ‘married in’ parent who is part of a distinct kindred or it could be a DNM. Whatever the source of the mutation, this case is further demonstration of intrafamilial genetic heterogeneity accounting for an apparent phenocopy.
Figure 1.

Family 379. Unaffected individuals are represented by white diamonds. For simplicity and clarity, multiple unaffected individuals within a sibship are represented by a single diamond and spouses are omitted. All affected individuals in this pedigree have a clinical diagnosis of ALS. Affected individuals for which DNA samples were unavailable are represented by grey diamonds. Affected family members with a TAR DNA-binding protein 43 p. G298S mutation are represented by black diamonds. Affected individual III-6 (yellow diamond) harbours a heterozygous SOD1 p. D91A mutation. Affected individuals IV-30 and V-3 (red diamonds) lack a mutation in known ALS-associated genes. Individual IV-25 is a true obligate carrier of TAR DNA-binding protein 43 p. G298S. All other indicated obligate carriers, represented by a black circle within a diamond, are possible obligate carriers since DNA samples were unavailable to confirm the presence of a mutation or the affected individuals in subsequent generations lack a known mutation.
Table 1.
Clinical characteristics of family 379
| All affected (n = 16) | TARDBP p. G298S (n = 10) | SOD1 p. D91A (n = 1) | V-3 | IV-30 | ||
|---|---|---|---|---|---|---|
| Age of onset (years) | Average ± SD | 52.0 ± 12.6 | 52.9 ± 12.4 | 64 | 33 | 46 |
| Range | 33–73 | 36–71 | ||||
| Disease duration (months) | Average ± SD | 39.5 ± 621 | 11.7 ± 7.8 | 254 | 35 | 80 |
| Range | 4–254 | 4–27 | ||||
Tabulation of age of disease onset and duration based on affected status and genotype of family 379. Invasive ventilation or death, which ever occurred first, marked the disease endpoint.
SD: standard deviation; SOD1: superoxide dismutase 1; TARDBP: TAR DNA-binding protein 43.
Consideration must also be given to the incidence of more than one disease-relevant mutation in an individual. Historically, genetic screening of cases often ceased once a single disease-relevant mutation was identified and, therefore, the frequency and consequence of inheriting multiple causative genes for ALS is largely unknown. An oligogenic presentation may belie the variable penetrance of a particular genetic variant and the variability of disease course. With more comprehensive next-generation sequencing strategies now available, several small studies have shown that harbouring more than one causative variant resulted in a significantly earlier age of symptom onset (Cady et al., 2015; Naruse et al., 2019) or faster disease course (Cooper-Knock et al., 2017). Genetic background or individual variants may also serve as disease modifiers and an additional ‘second hit’ variant might be required in order for disease to manifest, or to induce a specific disease phenotype. The SOD1-D91A mutation is a useful illustration of the relevance of genetic background to disease manifestation. This is typically a tolerated heterozygous polymorphism in northern Swedish and Finnish populations, but causes ALS when homozygous (Andersen et al., 1995). Heterozygous SOD1 p. D91A is sufficient to cause ALS in other populations (Al-Chalabi, 1998; Parton et al., 2002; Pasinelli and Brown, 2006), demonstrating that the genomic background of northern Swedish and Finnish populations might confer protection against the deleterious effects of heterozygous SOD1 p. D91A. The factors, such as gene dose or other more complex gene–gene or gene–environmental factors, responsible for the variability of disease penetrance and phenotype manifestation in heterozygous and homozygous carriers are not fully understood.
Validation of the direct effect of a genetic variant on a human phenotype is often an arduous task that requires examination at both the gene and variant levels to determine whether toxic gain-of-function, loss-of-function, dominant negative or a combination of mechanisms are involved (MacArthur et al., 2014). At the gene level, this involves examination of the expression of a candidate disease gene in disease-relevant tissue or its interactions with other proteins previously implicated in the disease pathway, or a de novo mechanism. At the variant level, the pathogenic mechanism by which the candidate variant adopts a toxic property or alters the function of the wild type gene or gene product is examined. Acquiring disease-relevant human tissue in the premorbid condition or during the process of disease progression in neurodegenerative conditions is obviously impractical. Thus, genetic models, where the gene of interest is knocked out, or the comparable mutation knocked in or over-expressed can provide useful insight, and also introduce a host of confounding factors that can obscure the pathogenesis. Indeed, it is the exception rather than the rule that model systems accurately and/or completely recapitulate a human disease in its clinical, molecular and pathological phenotypes. Presence of the culprit gene product in pathology of relevant tissue links the culprit gene and its product with the pathology, but does not prove mechanism of pathogenesis.
Testing for validated disease-causing mutations in cases presenting with apparently discordant disease aetiology is a rational approach, but negative results, as obtained in individuals IV-30 and V-3 in Family 379, do not exclude the possibility that disease is caused by a rare, and/or novel, mutation(s) that may, or may not, be expressed in all tissue or cells of an individual’s genome. DNMs provide an alternative to environmental exposure for phenocopy disease aetiology. DNMs can arise in germline or somatic tissue due to endogenous or exogenous causes, such as inefficient DNA repair or ionizing radiation, respectively (Acuna-Hidalgo et al., 2016). This may lead to varying degrees of mosaicism depending on the stage of development in which the DNM was acquired, causing different cells to express distinct complements of genetic variants. Rare DNMs have been identified in ALS and Parkinson disease patients both in novel genes (Chesi et al., 2013; Kun-Rodrigues et al., 2015; Steinberg et al., 2015) and previously implicated genes (Alexander et al., 2002; Puschmann et al., 2009; DeJesus-Hernandez et al., 2010; Chio et al., 2011; Zou et al., 2013; Calvo et al., 2014; Leblond et al., 2016; Williams et al., 2018). It is difficult to determine with certainty whether DNMs in genes not previously implicated in disease are causal since they may be unique to that individual. Patterns and rates of DNMs in ALS patients do not appear to differ from the general population as was noted in 82 new patient–parent trios when combined with datasets of all previously published ALS trios (173 trios in total) (van Doormaal et al., 2017). In general, these studies suggest that DNMs may account for rare cases of ALS but do not appear to play a major role in disease pathogenesis. Nevertheless, DNMs should be considered when studying families with cases of discordant disease aetiology.
The apparent discordance between genotype and phenotype could also be explained by the acquisition of an epimutation, i.e. an epigenetic aberration that alters the transcriptional regulation of a gene (Hesson et al., 2010; Sloane et al., 2016). Epigenetic modifications have been proposed as disease modifiers in a range of neurodegenerative disorders as previously reviewed (Belzil et al., 2016; Hwang et al., 2017; Miranda-Morales et al., 2017). The role of epimutations in disease manifestation and whether they can be inherited by subsequent generations is discussed later in this review.
Linking what is shared with what causes disease
It is worthwhile to consider unconventional modes of inheritance to identify what might be shared among family members that causes a shared phenotype when cases of presumed discordant disease aetiology arise. Sharing of a phenotype among siblings could occur through transmission of DNMs arising at the time of fertilization or during postzygotic early cell division of the parent. The sooner the DNM arises after fertilization, the greater the chance that both somatic and germline tissue will be affected, i.e. gonosomal mosaicism (Campbell et al., 2015). While a parent exhibiting gonosomal mosaicism may lack or show variable expression of a phenotype depending on which somatic tissue harbours the DNM, they may transmit a germline DNM to their offspring. Consequently, in the offspring the DNM could be ubiquitously expressed and a different phenotype from the parent may appear. Although the recurrence of the same mutation in siblings arising from parental germline DNMs is rare (Rahbari et al., 2016), it is another potential avenue for the emergence of a shared disease phenotype among siblings.
Similar to DNMs, epimutations may also be distributed in somatic and/or germline tissue. Epimutations are either primary or secondary depending on the absence or presence of a co-segregating genetic variant, respectively. Secondary epimutations display clear patterns of Mendelian inheritance in which the co-segregating genetic variant re-establishes the epimutation in each generation. It is proposed, but less widely accepted, that inheritance of a primary epimutation may account for the shared disease phenotype observed among affected siblings in a family (Chong et al., 2007; Daxinger and Whitelaw, 2012; Heard and Martienssen, 2014; Yan, 2014; Blake and Watson, 2016; Sharma, 2017).
Human studies exploring how epigenetic inheritance may drive a phenotype are limited by the fact that humans are outbred, making it difficult to ascertain whether an inherited epigenetic modification is truly primary and not the result of an inherited genetic variant re-establishing the epigenetic modification, i.e. secondary (Chong et al., 2007). A number of criteria must be met to demonstrate the occurrence of epigenetic inheritance, including ruling out other modes of inheritance, identifying the epigenetic factor(s) involved and demonstrating that it results in an inherited phenotype in subsequent generations (Horsthemke, 2018). Studies exploring how epigenetic inheritance drives phenotypes in offspring have been more fruitful in inbred animal studies where confounding genetic influences are reduced and tightly controlled experimentation is facilitated. While the epigenome of both sperm and ova are susceptible to environmental influences, paternal models of epigenetic inheritance are often studied to minimize confounding factors imparted in utero in maternal models (Donkin and Barres, 2018). Paternal exposure to dietary changes, psychological stress or toxins are common paradigms used to demonstrate environmental influences driving phenotypic changes in offspring (Boskovic and Rando, 2018; Sharma, 2019). It becomes increasingly difficult to investigate the impact of an epimutation on degenerative disease phenotypes as environmental causes are generally unknown, and even if a putative epimutation were to be identified, a reliable technique must be employed to introduce it in a model organism. Furthermore, late-onset degenerative disease phenotypes provide possibly decades of confounding influences that limit our ability to trace the cascade of events that might drive manifestation of a phenotype. It remains to be determined whether any shared epimutation drives degenerative disease pathogenesis among family members.
The mechanisms underlying epigenetic information transfer across generations are not fully established but have been proposed to involve DNA methylation, histone modifications and small RNAs (Boskovic and Rando, 2018). DNA methylation at CpG sites is the most well-characterized epigenetic modification that regulates gene expression. Most methylation in germ cells is erased upon fertilization to generate a totipotent state. Imprinted genes and metastable epialleles are exceptions and can escape epigenetic reprogramming. In such cases, DNA methylation states can persist across generations, thus providing a mechanism of transmission of information across generations independent of DNA sequence. Imprinted genes play an important role in neurodevelopment, and while ALS is a late-onset neurodegenerative disorder, evidence from ALS mouse models suggests that neurodevelopmental processes may be disturbed, and ultimately contribute to disease pathogenesis in later life (Bories et al., 2007; Pambo-Pambo et al., 2009; Chang and Martin, 2011; Quinlan et al., 2011; Filipchuk and Durand, 2012; Martin et al., 2013; Milan et al., 2014).
During spermatogenesis, most histones are replaced by protamines that facilitate DNA compaction. The remaining histones and newly acquired protamines can undergo post-translational modification and regulate gene expression (Bohacek and Mansuy, 2016). A study proposes that environmentally induced liver injury gives rise to a soluble factor in serum that modulates histone methylation of PPARγ chromatin in sperm and impacts the hepatic wound healing response in subsequent generations (Zeybel et al., 2012). Environmental stress and toxins have also been shown to influence the expression of RNA species in mammals (Sharma, 2019). For example, microinjection of small RNAs purified from the sperm of male mice exposed to a maternal stress and separation paradigm into control zygotes resulted in a behavioural and metabolic phenotype as the mice developed (Gapp et al., 2014). These data raise the possibility that environmentally induced changes in germline methylation profiles, histone modifications and small RNA species may alter embryonic development and offspring susceptibility to a neurodegenerative disease phenotype later in life that may or may not be passed on to future generations.
Another conceptual explanation for the apparent discordance of genotype and disease phenotype is the incidence of chimerism. A mutation may be apparently lacking in an affected individual, but it is only definitively lacking from the cells from which the DNA was derived. Chimerism arises when individuals harbour more than one genome that is variably expressed in cells throughout the body (Rinkevich, 2001). Although chimerism has been reported in humans (Mayr et al., 1979; Sheets et al., 2018), we are unaware of presumed phenocopy cases that can be attributed to this phenomenon. Genotyping disease-relevant tissue in all affected family members would be desirable for determining whether a pathogenic mutation is present, but this is impractical, particularly in neurodegenerative diseases. Also, depending on the sensitivity threshold of the sequencing assay, the degree of chimerism may preclude identification of a genetic variant if only a few vulnerable, although crucial, cells harbour the mutation and are responsible for disease pathogenesis.
In summary, the absence of a genetic mutation or epimutation in blood does not rule out the possibility that other cells and/or tissues harbour the mutation. Non-Mendelian inheritance of a neurodegenerative disease phenotype, in which the shared inherited phenotype among siblings may arise due to DNMs or epimutations via germline mosaicism or chimerism is a possibility and should be considered in efforts to determine disease aetiology. Although less well studied and understood, non-genetic modes of inheritance remain possibilities for the emergence of a shared phenotype in offspring where a genetic explanation is apparently lacking.
Multi-tasking genes and variants
Pleiotropy is a phenomenon whereby a single genetic locus has an impact on multiple phenotypic traits. The genetic locus can vary from an individual nucleotide to vast regions of a chromosome. The breadth of this description necessitates that context drives further refinement. Three categorical forms of pleiotropy are described to encapsulate the phenomenon (Solovieff et al., 2013). Biological pleiotropy implicates a locus in the manifestation of more than one phenotypic trait. Mediated pleiotropy arises from the impact of a primary phenotypic trait on a second phenotypic trait, indirectly associating a genetic locus with a second phenotypic trait. Spurious pleiotropy occurs due to the inability to distinguish a non-associated genetic locus from the manifestation of a phenotypic trait, thus implicating this locus in error. Consider the example of attempting to distinguish between biological and spurious pleiotropy for copy number variations. This is challenging because it may be unclear whether a single locus affects multiple traits or whether different loci within the region affect different traits. For clarity, we will define the genetic locus as a gene and the phenotypic trait as the clinical diagnosis of a degenerative disease. Below, we outline the complexity of pleiotropy and the necessity of being cautious in describing genes as pleiotropic. Understanding the pleiotropic nature of genes may redefine apparently sporadic disease and shed new light upon disease phenocopies.
A hexanucleotide repeat expansion (HRE) mutation in C9orf72 is the most common known genetic cause of frontotemporal dementia (FTD) and ALS (Majounie et al., 2012; Rohrer et al., 2015). The discovery of the C9orf72 HRE firmly established that FTD and ALS may be considered parts of a disease spectrum (Swinnen and Robberecht, 2014). Mutation carriers are typically identified if they have an HRE >30 repeats, although this threshold is theoretical and recent work supports earlier studies indicating that intermediate HREs of 24–30 repeats are also pathogenic (Iacoangeli et al., 2019). It is possible that an individual that has reached the average age of disease ascertainment could harbour an HRE of (G4C2)>30 but present as healthy, with ALS alone, with FTD alone or with ALS/FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Majounie et al., 2012; Xi et al., 2015). This incomplete penetrance indicates that factors in addition to C9orf72 genotype may be involved. These factors may act to cause, delay or prevent disease, and/or have a protective or deleterious effect on neuronal subtypes. To complicate matters, evidence has emerged that the length of HREs can vary across tissues and cells (Beck et al., 2013; Dols-Icardo et al., 2014; van Blitterswijk et al., 2013). HRE size is routinely determined using DNA extracted from white blood cells, which may not reflect the HRE size within the CNS. Indeed, the HRE size may not be homogenous across the CNS. This is challenged by a study showing the size of the hexanucleotide repeat was consistent across spinal cord levels in ALS cases lacking a C9orf72 HRE, but the possibility of somatic mosaicism of expanded alleles still remains (Ross et al., 2019). One may hypothesize that HREs could cause disease through the same molecular mechanism, yet clinical disease manifestation might depend on the neurons or glia harbouring the HRE. This calls into question whether C9orf72 is pleiotropic, or whether a deeper understanding of individual biological attributes of affected cell types and systems would allow for the identification of individual causes for distinct diseases. Notably, a study described somatic recombination of APP-gene products that resulted in the incorporation of ‘genomic cDNAs’ into the genome (Lee et al., 2018). These genomic cDNAs are numerous, diverse and specifically identified in neurons. A number of Alzheimer disease-associated mutations were found in genomic cDNAs of individual cells from sporadic Alzheimer disease patients. These data highlight the effect even a few cells might play in driving disease pathogenesis, although further validation and confirmation will be required to determine the extent of the role this phenomenon might play.
The pleiotropic nature of C9orf72 may also arise due to epigenetic variation, rather than sequence variation. Differential methylation of CpG islands in the gene promoter, the HRE, or associated histones has been suggested as a disease-modifying feature of C9orf72-associated diseases. Thus far, a consensus has not been reached concerning the mutant gene methylation profile, or indeed, the mechanism(s) by which it may impact disease.
Similar to C9orf72, ATXN2 provides an example of a pleiotropic phenomenon arising due to variability of an intragenic locus. ATXN2 contains a CAG trinucleotide repeat that encodes for polyglutamine. More than 34 repeats have been shown to cause spinocerebellar ataxia type 2 (Imbert et al., 1996; Pulst et al., 1996), while individuals with 24–33 repeats have demonstrated an increased risk of developing ALS (Elden et al., 2010). The mechanism(s) by which the differing trinucleotide repeat lengths, and/or their products, manifest as different phenotypic traits remain unclear.
Pleiotropy can be more easily recognized when assessing the impact of single nucleotide variants. We report here a Puerto Rican family, Family 1174, with individuals displaying either ALS, Paget disease of bone, probable Alzheimer disease or spastic paraparesis (Fig. 2). Some, but not all, individuals in this family with ALS harbour a Sequestosome 1(SQSTM1) p. P392L mutation, as do the individuals with Paget disease of bone and probable Alzheimer disease. Without prior knowledge of the pathogenicity of the mutation (Fecto, 2011; Rubino et al., 2012; Le Ber et al., 2013; Teyssou et al., 2013; Kwok et al., 2014; Rea et al., 2014), its disease-associated role in this family might be challenged due to its association with distinct phenotypes and its absence from affected individuals. A number of questions arise. Is SQSTM1 p. P392L causing three distinct diseases in three first cousins? If so, it should be considered pleiotropic. The genome aggregation database reports an allele frequency of 0.127% (45/35 440) for this variant in the Latino population and 0.09171% (259/282 398) for all populations. This frequency raises doubts as to whether SQSTM1 p. P392L is implicated in three degenerative diseases in this family. While two ALS-affected individuals are known to share the mutation, (II-2 and III-11), an apparent ALS phenocopy (III-22) weakens the argument for SQSTM1 p. P392L causing ALS in this family. Alternatively, if individual III-22 is accepted as an ALS phenocopy, a gene–environment interaction where different environmental factors act as triggers to cause distinct phenotypes in individuals with the mutation might explain the variety of disease phenotypes. SQSTM1 is a known pleiotropic gene given that it is implicated in multiple degenerative diseases, including ALS, FTD, ALS/FTD, vacuolar myopathy and Paget disease of bone (Laurin et al., 2002; Fecto, 2011; Bucelli et al., 2015). Similarly, mutations in CHCHD10 have been reported in individuals with ALS, FTD, myopathy and spinal muscular atrophy-Jokela type, among others, making it pleiotropic (Bannwarth et al., 2014; Ajroud-Driss et al., 2015; Penttila et al., 2015; Jiao et al., 2016). It is important to note that while the same gene might be involved, individual mutations may dictate disease pathogenesis, therefore implicating intragenic variability in apparent pleiotropy.
Figure 2.
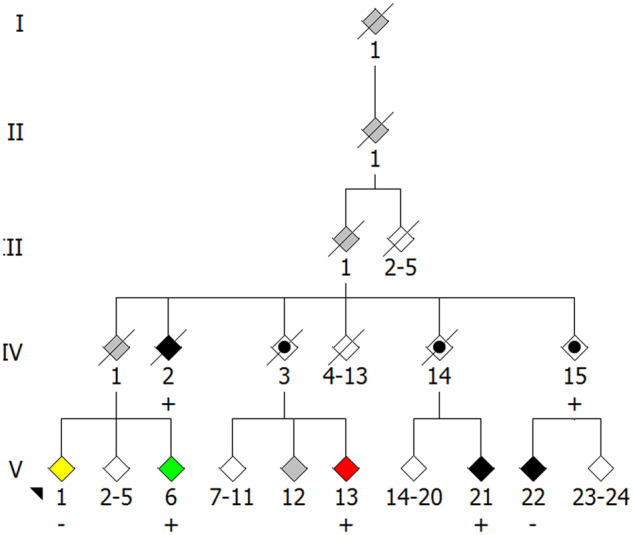
Family 1174. Unaffected individuals are represented by white diamonds. For simplicity and clarity, multiple unaffected individuals within a sibship are represented by a single diamond and spouses are omitted. Individuals affected with ALS for which DNA samples were unavailable are represented by grey diamonds. Individuals affected with ALS for which DNA samples are available are represented by black diamonds. Individual V-1, represented by a yellow diamond, presented with bilateral leg weakness at 45 years of age and has a clinical diagnosis of spastic paraparesis with lower extremity weakness. Their condition has been stable for 30 years. Individual V-6, represented by a green diamond, has a clinical diagnosis of probable Alzheimer Disease. Individual V-13, represented by a red diamond, has a clinical diagnosis of Paget Disease of Bone. SQSTM1 p. P392L-positive individuals are indicated by a ‘+’ sign. SQSTM1 p. P392L-negative individuals are indicated by a ‘−’ sign. Possible obligate carriers are represented by a black circle within a diamond.
In addition to the role of pathogenic variants in SQSTM1 contributing to degenerative disease aetiology, SQSTM1/p62-positive neuronal inclusions are a pathological hallmark of ALS, even in the absence of a mutation in the gene itself. Among other functions, SQSTM1/p62 is an adaptor protein that mediates autophagic degradation of ubiquitinated proteins. SQSTM1/p62 is a component of a group of proteins independently implicated in ALS pathogenesis that play roles in protein degradation pathways. Included in this group are UBQLN2,Valosin containing protein, OPTN and TBK1 (Johnson et al., 2010; Maruyama et al., 2010; Deng et al., 2011; Cirulli et al., 2015). UBQLN4 and CYLD, which have been implicated in ALS in single, multi-generational families also feature in this group (Dobson-Stone et al., 2013, 2020; Edens et al., 2017). SQSTM1/p62, TBK1, optineurin and CYLD all participate closely in autophagy, but the extent of their interaction in various cell types and under various conditions is yet to be elucidated. Investigation remains to be carried out to determine the consequence of putative mutations or variants of unknown significance in this wider group of proteins that might disrupt the function of their counterparts or other important components of protein degradation pathways, ultimately leading to a cell type-specific degenerative disease phenotype.
VCP, hnRNPA1 and hnRNPA2B1 mutations have been identified in individuals displaying a complex phenotype with features of inclusion body myopathy, Paget disease of bone, FTD and ALS (Watts et al., 2004; Johnson et al., 2010; Kim et al., 2013). These clinical manifestations can present in a single patient as multisystem proteinopathy. Mutations in these genes are also present in individuals with a clinical diagnosis of each disease independently. It remains to be determined how other cell types susceptible to degeneration in multisystem proteinopathy might be protected in individuals displaying only one of the diseases caused by these mutations. There are likely modifying factors that may act at the molecular, cellular, physiological, system or environmental level, making them difficult to identify.
Epistatic interactions between nuclear and mitochondrial DNA-encoded proteins may be one such modifier (Tranah, 2011; Dunham-Snary and Ballinger, 2015). Although a report suggests biparental inheritance of mitochondrial DNA is possible in humans (Luo et al., 2018), discounting any DNMs, siblings will typically share mitochondrial DNA inherited from their mother, while their nuclear DNA will vary. A typically benign variant might become pathogenic in concert with a mitochondrial population that is impaired due to a mismatch between nuclear and mitochondrial-encoded proteins. Alternatively, there might be a protective effect in an individual with a pathogenic variant if that ‘mismatch’ turned out to beneficial. Mitochondrial DNA heteroplasmy might also amplify those deleterious or protective effects.
Expression quantitative trait loci (eQTLs) may also underlie the variability of phenotype manifestation. Sequence variation at specific loci can account for expression level differences of apparently independent genes in different cell types. eQTLs may become relevant to disease manifestation if they modulate expression of a gene in a relevant pathway or cell type. It can be difficult to disentangle benign and pathogenic effects of eQTLs due to the vast number of genes and cell types that might be involved in a disease pathway. Furthermore, the recurring challenge of obtaining data from the human CNS can hinder examination of the impact of eQTLs. If relevant data can be obtained, techniques such as Mendelian randomization, where genetic variants can be used as instrumental variables to test the causative effect of gene expression on phenotype manifestation, can be employed to identify eQTLs that impact upon disease manifestation (Zhu et al., 2016; Storm et al., 2020).
In summary, the scope of the pleiotropy of genetic loci associated with degenerative disease continues to broaden. With greater understanding of disease aetiology, the commonalities of degenerative diseases are becoming more apparent. While a single gene may be implicated in multiple degenerative diseases, it is important to distinguish true pleiotropy from intragenic variation accounting for the manifestation of multiple phenotypic traits. Disease phenocopies can obscure recognition of the pleiotropic effects of genes as they amplify the disconnect between genotype and phenotype among family members. Pleiotropy and phenocopy may not occur in isolation, thus both possibilities should be considered. Lastly, we may be unaware of cell type-specific phenotypes that arise due to pleiotropy, which may prevent recognition of heritable disease phenotypes. Consequently, disease-associated variants may be missed, and familial disease falsely identified as sporadic, if the complete spectrum of phenotypes is not recognized. For this reason, we endorse the proposal to use caution with the terms familial and sporadic, since they can serve to obscure disease aetiology (Al-Chalabi, 2017).
Conclusion
This review highlights the complexities of determining late-onset degenerative disease aetiology. Exceptions to patterns of Mendelian inheritance in which genotype and phenotype appear uncorrelated continue to emerge and must be examined carefully. It is essential that assumptions are not made in classifying affected individuals as phenocopies, since intrafamilial genetic heterogeneity, shared DNMs or epimutations and pleiotropy may underlie the apparent discordant disease etiologies (Fig. 3). Genetic counselling for late-onset disorders is often difficult due to the variable penetrance and pathogenicity of mutations. It cannot be overstated that the presence of an identified familial mutation does not guarantee that disease will manifest, while absence of a familial mutation does not eliminate risk of developing disease. Although presymptomatic genetic screening can be helpful for purposes such as reproductive planning, the certainty that disease will manifest cannot be guaranteed. Finally, the continuing emergence of pleiotropic genes involved in degenerative disease provides an excellent opportunity to study the underlying mechanisms that govern different cells and tissues and how disease manifests. Ultimately, this understanding will allow development of rational and effective interventions that will prevent, halt and cure disease.
Figure 3.
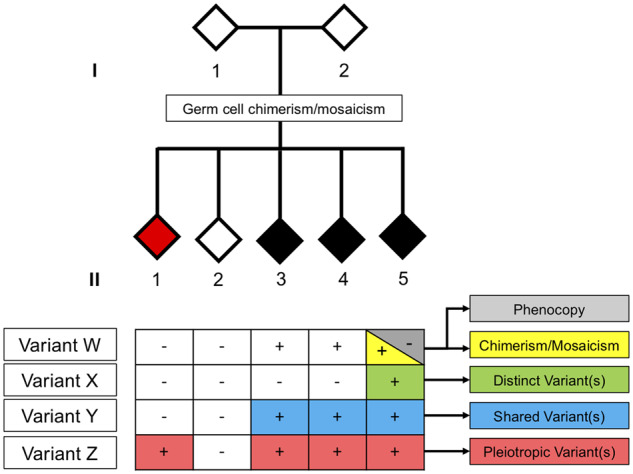
A hypothetical pedigree depicting scenarios to consider when encountering cases of presumed discordant disease aetiology. We indicate three siblings with a clinically similar phenotype (black). Two of the three affected individuals (II-3 and II-4) harbour a candidate disease-causing variant W, whereas all others tested negative for variant W in blood. While the phenotype in individual II-5 may be the result of an environmental trigger (i.e. classical phenocopy), the apparent disconnect between genotype and phenotype may also be explained by intrafamilial genetic heterogeneity (variants W and X), chimerism, mosaicism or identification of a distinct shared disease-causing variant Y. Alternatively, variant Z may have pleiotropic functions, and therefore cause a clinically distinct phenotype (red and black). In situations where both parents appear healthy and/or test negative for the suspected disease-causing variant, one must consider the potential role of germline chimerism or germline mosaicism via DNMs or epimutations.
Acknowledgements
We wish to thank the families described in this review for their willingness to participate in our research studies. We would also like to thank members of the Siddique laboratory that participated in genotyping DNA samples collected from the pedigrees discussed.
Funding
This study would not have been possible without generous funding from the Foglia Family Fund for ALS Research and over the years from the National Institute of Neurological Disorders and Stroke (NS046535 and NS099638), the Les Turner ALS Foundation/Herbert C. Wenske Professorship, the Vena Schaff ALS Fund, the Herbert C. Wenske Fund and the Les Turner ALS Foundation.
Competing interests
The authors report no competing interests.
Data availability
All data are available upon request to the corresponding author.
Glossary
- ALS =
amyotrophic lateral sclerosis
- DNM =
de novo mutation
- eQTLs =
expression quantitative trait loci
- FTD =
frontotemporal dementia
- HRE =
hexanucleotide repeat expansion
- SOD1 =
superoxide dismutase 1
- SQSTM1 =
Sequestosome 1
References
- Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol 2016; 17: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajroud-Driss S, Fecto F, Ajroud K, Lalani I, Calvo SE, Mootha VK, et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 2015; 16: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta 2015; 1852: 679–84. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A. Perspective: don't keep it in the family. Nature 2017; 550: S112. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor. Hum Mol Genet 1998; 7: 2045–50. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 2017; 13: 96–104. [DOI] [PubMed] [Google Scholar]
- Alexander MD, Traynor BJ, Miller N, Corr B, Frost E, McQuaid S, et al. “True” sporadic ALS associated with a novel SOD-1 mutation. Ann Neurol 2002; 52: 680–3. [DOI] [PubMed] [Google Scholar]
- Andersen PM, Nilsson P, Ala-Hurula V, Keränen M-L, Tarvainen I, Haltia T, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet 1995; 10: 61–6. [DOI] [PubMed] [Google Scholar]
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014; 137: 2329–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 2013; 92: 345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzil VV, Katzman RB, Petrucelli L. ALS and FTD: an epigenetic perspective. Acta Neuropathol 2016; 132: 487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Greenamyre JT. Parkinson's disease: animal models. Handb Clin Neurol 2007; 83: 265–87. [DOI] [PubMed] [Google Scholar]
- Blake GE, Watson ED. Unravelling the complex mechanisms of transgenerational epigenetic inheritance. Curr Opin Chem Biol 2016; 33: 101–7. [DOI] [PubMed] [Google Scholar]
- Bohacek J, Mansuy IM. Epigenetic risk factors for diseases: a transgenerational perspective In: Dietmar S, Elisabeth B, editors. Epigenetics and neuroendocrinology: clinical focus on psychiatry, Vol. 2 Cham: Springer International Publishing; 2016. p. 79–119. [Google Scholar]
- Bories C, Amendola J, Lamotte d'Incamps B, Durand J. Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 2007; 25: 451–9. [DOI] [PubMed] [Google Scholar]
- Boskovic A, Rando OJ. Transgenerational epigenetic inheritance. Annu Rev Genet 2018; 52: 21–41. [DOI] [PubMed] [Google Scholar]
- Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 2015; 85: 665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol 2015; 77: 100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo A, et al. De novo nonsense mutation of the FUS gene in an apparently familial amyotrophic lateral sclerosis case. Neurobiol Aging 2014; 35: 1513.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet 2015; 31: 382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Martin LJ. Glycine receptor channels in spinal motoneurons are abnormal in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 2011; 31: 2815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi A, Staahl BT, Jovičić A, Couthouis J, Fasolino M, Raphael AR, et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci 2013; 16: 851–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A, et al. A de novo missense mutation of the FUS gene in a “true” sporadic ALS case. Neurobiol Aging 2011; 32: 553.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Youngson NA, Whitelaw E. Heritable germline epimutation is not the same as transgenerational epigenetic inheritance. Nat Genet 2007; 39: 574–5; author reply 75–6. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015; 347: 1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J, Robins H, Niedermoser I, Wyles M, Heath PR, Higginbottom A, et al. Targeted genetic screen in amyotrophic lateral sclerosis reveals novel genetic variants with synergistic effect on clinical phenotype. Front Mol Neurosci 2017; 10: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet 2012; 13: 153–62. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Kocerha J, Finch N, Crook R, Baker M, Desaro P, et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat 2010; 31: E1377–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarre A, Meissner WG. Epidemiology, environmental risk factors and genetics of Parkinson's disease. Presse Med 2017; 46: 175–81. [DOI] [PubMed] [Google Scholar]
- Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011; 477: 211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H-X, Pericak-Vance MA, Siddique T. Reply to ‘TMEM230 variants in Parkinson’s disease' and ‘doubts about TMEM230 as a gene for parkinsonism’. Nat Genet 2019; 51: 369–71. [DOI] [PubMed] [Google Scholar]
- Deng H-X, Shi Y, Yang Y, Ahmeti KB, Miller N, Huang C, et al. Identification of TMEM230 mutations in familial Parkinson's disease. Nat Genet 2016; 48: 733–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone C, Luty AA, Thompson EM, Blumbergs P, Brooks WS, Short CL, et al. Frontotemporal dementia–amyotrophic lateral sclerosis syndrome locus on chromosome 16p12.1-q12.2: genetic, clinical and neuropathological analysis. Acta Neuropathol 2013; 125: 523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone C, Hallupp M, Shahheydari H, Ragagnin AMG, Chatterton Z, Carew-Jones F, et al. CYLD is a causative gene for frontotemporal dementia–amyotrophic lateral sclerosis. Brain 2020; 143: 783–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dols-Icardo O, García-Redondo A, Rojas-García R, Sánchez-Valle R, Noguera A, Gómez-Tortosa E, et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet 2014; 23: 749–54. [DOI] [PubMed] [Google Scholar]
- Donkin I, Barres R. Sperm epigenetics and influence of environmental factors. Mol Metab 2018; 14: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Snary KJ, Ballinger SW. GENETICS. Mitochondrial-nuclear DNA mismatch matters. Science 2015; 349: 1449–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens BM, Yan J, Miller N, Deng H-X, Siddique T, Ma YC, et al. A novel ALS-associated variant in UBQLN4 regulates motor axon morphogenesis. Elife 2017; 6: e25453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466: 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecto F. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011; 68: 1440–6. [DOI] [PubMed] [Google Scholar]
- Filipchuk AA, Durand J. Postnatal dendritic development in lumbar motoneurons in mutant superoxide dismutase 1 mouse model of amyotrophic lateral sclerosis. Neuroscience 2012; 209: 144–54. [DOI] [PubMed] [Google Scholar]
- Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J, et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci 2014; 17: 667–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 2014; 157: 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesson LB, Hitchins MP, Ward RL. Epimutations and cancer predisposition: importance and mechanisms. Curr Opin Genet Dev 2010; 20: 290–8. [DOI] [PubMed] [Google Scholar]
- Horsthemke B. A critical view on transgenerational epigenetic inheritance in humans. Nat Commun 2018; 9: 2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JY, Aromolaran KA, Zukin RS. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat Rev Neurosci 2017; 18: 347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S, et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol Commun 2019; 7: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier J-M, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 1996; 14: 285–91. [DOI] [PubMed] [Google Scholar]
- Jiao B, Xiao T, Hou L, Gu X, Zhou Y, Zhou L, et al. High prevalence of CHCHD10 mutation in patients with frontotemporal dementia from China. Brain 2016; 139: e21. [DOI] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010; 68: 857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang Y-D, Scarborough EA, Moore J, Diaz Z, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495: 467–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Chuang R, Marras C, Lang AE. The curious case of phenocopies in families with genetic Parkinson's disease. Mov Disord 2011; 26: 1793–802. [DOI] [PubMed] [Google Scholar]
- Kun-Rodrigues C, Ganos C, Guerreiro R, Schneider SA, Schulte C, Lesage S, et al. A systematic screening to identify de novo mutations causing sporadic early-onset Parkinson's disease. Hum Mol Genet 2015; 24: 6711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CT, Morris A, de Belleroche JS. Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur J Hum Genet 2014; 22: 492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding Sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70: 1582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, Nicolas G, et al. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol 2013; 70: 1403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Webber A, Gan-Or Z, Moore F, Dagher A, Dion PA, et al. De novo FUS P525L mutation in Juvenile amyotrophic lateral sclerosis with dysphonia and diplopia. Neurol Genet 2016; 2: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-H, Siddoway B, Kaeser GE, Segota I, Rivera R, Romanow WJ, et al. Somatic APP gene recombination in Alzheimer's disease and normal neurons. Nature 2018; 563: 639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000; 342: 1560–7. [DOI] [PubMed] [Google Scholar]
- Luo S, Valencia CA, Zhang J, Lee N-C, Slone J, Gui B, et al. Biparental inheritance of mitochondrial DNA in humans. Proc Natl Acad Sci USA 2018; 115: 13039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014; 508: 469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012; 11: 323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Cazenave W, Cattaert D, Branchereau P. Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 2013; 54: 116–26. [DOI] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010; 465: 223–6. [DOI] [PubMed] [Google Scholar]
- Mayr WR, Pausch V, Schnedl W. Human chimaera detectable only by investigation of her progeny. Nature 1979; 277: 210–11. [DOI] [PubMed] [Google Scholar]
- Milan L, Barrière G, De Deurwaerdère P, Cazalets J-R, Bertrand SS. Monoaminergic control of spinal locomotor networks in SOD1G93A newborn mice. Front Neural Circuits 2014; 8: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Morales E, Meier K, Sandoval-Carrillo A, Salas-Pacheco J, Vázquez-Cárdenas P, Arias-Carrión O, et al. Implications of DNA methylation in Parkinson's disease. Front Mol Neurosci 2017; 10: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse H, Ishiura H, Mitsui J, Takahashi Y, Matsukawa T, Tanaka M, et al. Burden of rare variants in causative genes for amyotrophic lateral sclerosis (ALS) accelerates age at onset of ALS. J Neurol Neurosurg Psychiatry 2019; 90: 537–42. [DOI] [PubMed] [Google Scholar]
- Pambo-Pambo A, Durand J, Gueritaud JP. Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-low) mice. J Neurophysiol 2009; 102: 3627–42. [DOI] [PubMed] [Google Scholar]
- Parton MJ, Broom W, Andersen PM, Al-Chalabi A, Nigel Leigh P, Powell JF, et al. D90A-SOD1 mediated amyotrophic lateral sclerosis: a single founder for all cases with evidence for a cis-acting disease modifier in the recessive haplotype. Hum Mutat 2002; 20: 473. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006; 7: 710–23. [DOI] [PubMed] [Google Scholar]
- Penttila S, et al. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann Neurol 2015; 77: 163–72. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997; 276: 2045–7. [DOI] [PubMed] [Google Scholar]
- Pulst S-M, Nechiporuk A, Nechiporuk T, Gispert S, Chen X-N, Lopes-Cendes I, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996; 14: 269–76. [DOI] [PubMed] [Google Scholar]
- Puschmann A, Ross OA, Vilariño-Güell C, Lincoln SJ, Kachergus JM, Cobb SA, et al. A Swedish family with de novo alpha-synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat Disord 2009; 15: 627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ. Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol 2011; 589: 2245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, et al. Timing, rates and spectra of human germline mutation. Nat Genet 2016; 48: 126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations—bridging Paget disease of bone and ALS/FTLD. Exp Cell Res 2014; 325: 27–37. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72: 257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich B. Human natural chimerism: an acquired character or a vestige of evolution? Hum Immunol 2001; 62: 651–7. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 2015; 14: 291–301. [DOI] [PubMed] [Google Scholar]
- Ross JP, Leblond CS, Catoire H, Volkening K, Strong M, Zinman L, et al. Somatic expansion of the C9orf72 hexanucleotide repeat does not occur in ALS spinal cord tissues. Neurol Genet 2019; 5: e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012; 79: 1556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A. Transgenerational epigenetics: integrating soma to germline communication with gametic inheritance. Mech Ageing Dev 2017; 163: 15–22. [DOI] [PubMed] [Google Scholar]
- Sharma U. Paternal contributions to offspring health: role of sperm small RNAs in intergenerational transmission of epigenetic information. Front Cell Dev Biol 2019; 7: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets KM, Baird ML, Heinig J, Davis D, Sabatini M, Starr DB, et al. A case of chimerism-induced paternity confusion: what ART practitioners can do to prevent future calamity for families. J Assist Reprod Genet 2018; 35: 345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane MA, Ward RL, Hesson LB. Defining the criteria for identifying constitutional epimutations. Clin Epigenet 2016; 8: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet 2013; 14: 483–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg KM, Yu B, Koboldt DC, Mardis ER, Pamphlett R. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep 2015; 5: 9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm CS, Kia DA, Almramhi M, Wood NW. Using Mendelian randomization to understand and develop treatments for neurodegenerative disease. Brain Commun 2020; 2: fcaa031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014; 10: 661–70. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature 2016; 539: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teyssou E, Takeda T, Lebon V, Boillée S, Doukouré B, Bataillon G, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 2013; 125: 511–22. [DOI] [PubMed] [Google Scholar]
- Tranah GJ. Mitochondrial-nuclear epistasis: implications for human aging and longevity. Ageing Res Rev 2011; 10: 238–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158–60. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk M, van Es MA, Hennekam EAM, Dooijes D, van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012; 21: 3776–84. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013; 12: 978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doormaal PTC, Ticozzi N, Weishaupt JH, Kenna K, Diekstra FP, Verde F, et al. The role of de novo mutations in the development of amyotrophic lateral sclerosis. Hum Mutat 2017; 38: 1534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011; 89: 162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant Valosin-containing protein. Nat Genet 2004; 36: 377–81. [DOI] [PubMed] [Google Scholar]
- Williams ES, Barrett MJ, Dhamija R, Toran L, Chambers C, Mahadevan MS, et al. Phase determination using chromosomal microarray and fluorescence in situ hybridization in a patient with early onset Parkinson disease and two deletions in PRKN. Mol Genet Genomic Med 2018; 6: 457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z, van Blitterswijk M, Zhang M, McGoldrick P, McLean JR, Yunusova Y, et al. Jump from pre-mutation to pathologic expansion in C9orf72. Am J Hum Genet 2015; 96: 962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W. Potential roles of noncoding RNAs in environmental epigenetic transgenerational inheritance. Mol Cell Endocrinol 2014; 398: 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med 2012; 18: 1369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016; 48: 481–7. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011; 89: 168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004; 44: 601–7. [DOI] [PubMed] [Google Scholar]
- Zou ZY, Cui L-Y, Sun Q, Li X-G, Liu M-S, Xu Y, et al. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol Aging 2013; 34: 1312.e1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available upon request to the corresponding author.