

Abstract
Despite wide genetic, environmental and clinical heterogeneity in amyotrophic lateral sclerosis, a rapidly fatal neurodegenerative disease targeting motoneurons, neuroinflammation is a common finding. It is marked by local glial activation, T cell infiltration and systemic immune system activation. The immune system has a prominent role in the pathogenesis of various chronic diseases, hence some of them, including some types of cancer, are successfully targeted by immunotherapeutic approaches. However, various anti-inflammatory or immunosuppressive therapies in amyotrophic lateral sclerosis have failed. This prompted increased scrutiny over the immune-mediated processes underlying amyotrophic lateral sclerosis. Perhaps the biggest conundrum is that amyotrophic lateral sclerosis pathogenesis exhibits features of three otherwise distinct immune dysfunctions—excessive inflammation, autoimmunity and inefficient immune responses. Epidemiological and genome-wide association studies show only minimal overlap between amyotrophic lateral sclerosis and autoimmune diseases, so excessive inflammation is usually thought to be secondary to protein aggregation, mitochondrial damage or other stresses. In contrast, several recently characterized amyotrophic lateral sclerosis-linked mutations, including those in TBK1, OPTN, CYLD and C9orf72, could lead to inefficient immune responses and/or damage pile-up, suggesting that an innate immunodeficiency may also be a trigger and/or modifier of this disease. In such cases, non-selective immunosuppression would further restrict neuroprotective immune responses. Here we discuss multiple layers of immune-mediated neuroprotection and neurotoxicity in amyotrophic lateral sclerosis. Particular focus is placed on individual patient mutations that directly or indirectly affect the immune system, and the mechanisms by which these mutations influence disease progression. The topic of immunity in amyotrophic lateral sclerosis is timely and relevant, because it is one of the few common and potentially malleable denominators in this heterogenous disease. Importantly, amyotrophic lateral sclerosis progression has recently been intricately linked to patient T cell and monocyte profiles, as well as polymorphisms in cytokine and chemokine receptors. For this reason, precise patient stratification based on immunophenotyping will be crucial for efficient therapies.
Keywords: amyotrophic lateral sclerosis, neuroinflammation, neuroimmunity neurodegeneration, immunodeficiency
The immune system is an important driver of disease pathogenesis in amyotrophic lateral sclerosis. Here we explore diverse molecular, cellular and systemic processes leading to dysbalanced immunity in this disease. Notably, recent genetic evidence suggests that both excessive inflammation and inefficient immune responses may lead to motoneuron dysfunction and death.
Graphical Abstract
Graphical Abstract.
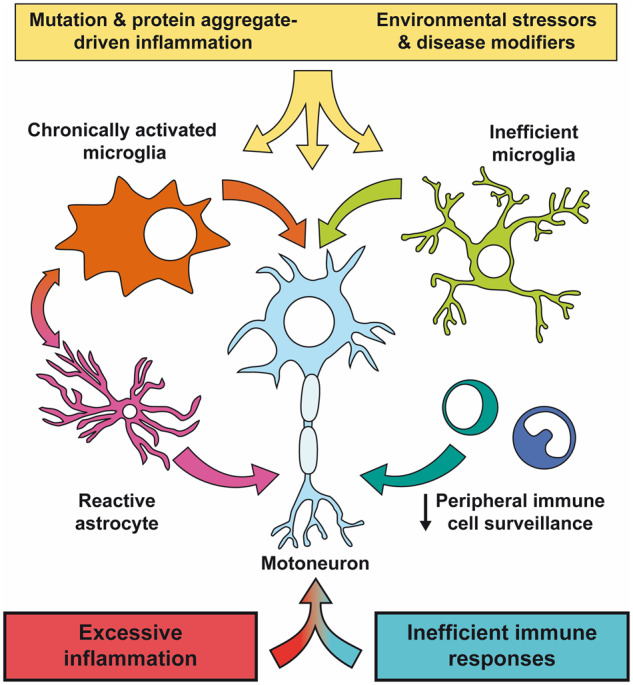
Introduction
Chronic inflammation is considered to be a key contributing factor to diseases as diverse as cancer, stroke, heart disease, obesity and depression. It is also an early and common hallmark of neurodegenerative diseases (Ransohoff, 2016). As the only immune cell subtype residing in the brain parenchyma, microglia are tasked with orchestrating inflammation against neurotropic infectious agents as well as clearing the debris in stroke and neurodegenerative diseases. Nevertheless, when Alois Alzheimer described reactive cells surrounding amyloid β plaques in the first case of his namesake disease in the early 1900s, the exact function of these cells was unclear (Alzheimer, 1911; Kettenmann et al., 2011). Microglia were subsequently identified by Pío del Río Hortega in 1919, who also described their phagocytic capacity and reactivity in response to external stimuli (Río-Hortega, 1919a, b; Sierra et al., 2016). However, it took another 70 years until it was recognized that activated microglia are key elements not only in Alzheimer’s disease but also in amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases (McGeer et al., 1987, 1988; McGeer and McGeer, 2011). Around the same time, the infiltration of T cells was first reported in ALS patients (Troost et al., 1989; Engelhardt et al., 1993). These findings strengthened the notion that glial activation and peripheral immune cell infiltration in the brain is neurotoxic and contributes to ALS, as it was previously reported for multiple sclerosis and spinal cord injury (DiSabato et al., 2016; Ransohoff, 2016). Hence, it came as both surprise and disappointment that a large number of ALS clinical trials found no protection provided by various anti-inflammatory agents, similar to trials in other neurodegenerative diseases and traumatic brain injury (Adapt-Fs, 2015; Russo and McGavern, 2016; Petrov et al., 2017). Multiple reasons for these failures likely exist. Firstly, despite the well-known neurotoxic effects of inflammation, non-specific downregulation of the immune system limits neuroprotective and neuroreparative functions of CD4 T cells and microglia (Schwartz and Shechter, 2010; Russo and McGavern, 2016). Secondly, ALS is a phenotypically and genetically heterogenous disease, so effective treatment necessitates stratified case monitoring (Goyal et al., 2020). Finally, immunological biomarkers are necessary for precise tracking of immune responses from preclinical to the end disease stages, thus guiding precision medicine.
This review discusses the complex role of inflammation and immunity during the course of ALS and details the functions of individual immune cell subsets during disease progression. We can nowadays firmly renounce the long-held dogma that the CNS remains safe by keeping the immune system off-limits (Schwartz and Deczkowska, 2016). Dural lymphatic vessels are now known to drain to deep cervical lymph nodes (Aspelund et al., 2015; Louveau et al., 2015). Furthermore, T cells, monocytes and other peripheral immune cells can directly access and/or indirectly influence the CNS via the choroid plexus (Baruch et al., 2014). Therefore, the longstanding perception of the brain as a strictly immunoprivileged organ is challenged by striking examples of active immune surveillance of the brain in both health and disease, suggesting that the main function of the immune system in the CNS is maintaining homeostasis (Medawar, 1948; Ziv et al., 2006; Aguzzi et al., 2013; Deczkowska et al., 2018a). However, in response to excessive or prolonged CNS damage, the protective functions of the immune system are thought to break down and lead to hyperinflammation, neuronal damage and unrelenting ALS progression. Moreover, in the recent years, a growing body of evidence has indicated that deregulation of immune response is not solely an aftermath of neuronal death, and that it may in many cases occur early in the course of ALS. This was corroborated by the longitudinal bioluminescence live imaging studies in mouse ALS models that revealed marked changes in activation of astrocytes and microglia in the very early presymptomatic stages of disease (Keller et al., 2011; Swarup et al., 2011a; Gravel et al., 2016). Furthermore, mutations in several genes directly linked to the immune response have recently been reported in ALS patients (Maruyama et al., 2010; CirulLi et al., 2015; Freischmidt et al., 2015; Dobson-Stone et al., 2020). Some of them could lead to inefficient immune responses rather than overt hyperinflammation, adding to the complexity of immune system dysfunctions in ALS. Therefore, we propose that the loss of immune homeostasis and/or early deregulation of immune responses may represent one of a key initial steps in disease pathogenesis leading to either excessive inflammation or an inefficient immune response (immunodeficiency), either of which could contribute to ALS progression. Due to the dual role of the immune system, patient stratification based on their immune profiles, and precise timing and/or specific targeting of excessive inflammation while maintaining the protective and regenerative immune functions, will likely be crucial for the efficient treatment of neurodegenerative diseases.
ALS is a systemic and immune-mediated disease
Commencing with Jean-Martin Charcot, considerable efforts have been made to correlate clinical symptoms to neuroanatomical findings, linking each neurodegenerative disease to discrete sets of damaged neurons (Kumar et al., 2011). However, most neurodegenerative diseases have systemic effects, clinically overlap and share common immunopathogenic mechanisms. In ALS, up to 50% of ALS patients have cognitive problems, some of which exhibit overt symptoms of frontotemporal dementia (Hardiman et al., 2017). Moreover, the same hexanucleotide repeat expansion in chromosome 9 open reading frame 72 gene (C9orf72), which is the most frequent genetic cause of ALS, may in some family members cause predominant frontotemporal dementia, suggesting that these disorders present the opposite ends of a clinical spectrum (Ling et al., 2013). The systemic aspect of ALS is perhaps best recognized in the seemingly pure motoric ALS cases, in which an accompanying inflammatory response is not restricted to the vicinity of motoneurons, and is detected in muscles, peripheral motor fibres, skin, liver and blood as well (Ono et al., 2001; Henkel et al., 2004; Chiu et al., 2009; Nardo et al., 2016; Gasco et al., 2017; Paré and Gros-Louis, 2017). An underlying immune component may contribute to delaying neurodegeneration in the individuals carrying highly penetrant familial mutations. Indeed, although late onset ALS is generally linked to an age-related deterioration of intrinsic motoneuron pathways (Khalil et al., 2018; Loeffler, 2019), immune senescence and proinflammatory skewing linked to the aging process (sometimes referred to as inflammaging) may also contribute to disease progression (Franceschi et al., 2000; Deleidi et al., 2015; Franceschi et al., 2018). Notably, once ALS symptoms appear, the progression towards the fatal outcome is extremely rapid, faster than in any other neurodegenerative disease, and in some cases, a rapid progression has been directly linked to immune parameters, which will be further discussed in subsequent chapters (Lopez-Lopez et al., 2014; Wosiski-Kuhn et al., 2019b).
ALS genetics and mechanisms
Since the discovery of the first ALS mutation in 1993 in superoxide dismutase 1 (SOD1), more than 50 different genes have been linked to ALS (Rosen et al., 1993; Renton et al., 2014; Brown and Al-Chalabi, 2017; Hardiman et al., 2017; Mejzini et al., 2019). Their exact roles in disease pathogenesis are still incompletely understood. The main proposed mechanisms include impaired proteostasis (enhanced protein aggregation and/or decreased degradation), dysregulated RNA metabolism, blockade of nucleocytoplasmic transport, mitochondrial dysfunction and impaired axonal transport. Thus, although inflammation is a common denominator in various ALS aetiologies, most ALS mutations do not directly target the immune system. However, proteins and/or peptides encoded by mutations in SOD1, TARDBP, FUS and C9orf72 gain propensity to form aggregates, which in turn trigger inflammatory responses in microglia (Fig. 1). Aggregates also form in sporadic ALS patients with unknown genetic background and contain wild type (WT) transactive response DNA-binding protein of 43 kDa (TDP-43, encoded by TARDBP). Notably, the crosstalk between protein aggregation and inflammation is bidirectional, and in inflammatory settings, nuclear TDP-43 gets phosphorylated, mislocalized and aggregated in the cytoplasm (Correia et al., 2015; Xue et al., 2018b). TDP-43 was also previously reported as an interacting partner and co-activator of p65, a subunit of nuclear factor of κB (NF-κB), the master transcription factor regulating proinflammatory factor production (Swarup et al., 2011b). Aggregation of ubiquilin 2 (encoded by UBQLN2 gene) stimulates TDP-43 co-aggregation and NF-κB activation (Picher-Martel et al., 2015). Another DNA/RNA-binding protein, fused in sarcoma (FUS) also acts as a co-activator of NF-κB, and expression of disease-associated FUS mutation (R521G) in astrocytes led to a tumour necrosis factor (TNF)-induced motoneuron death, which could be rescued by NF-κB inhibition (Uranishi et al., 2001; Kia et al., 2018). Moreover, viral infection causes mutant FUS aggregation (Shelkovnikova et al., 2019). In addition to these aggregate-prone proteins, numerous other genes mutated in ALS such as p62/SQSTM1, TBK1, OPTN, ALS2, CHMP2B, C9orf72 and CYLD were reported to disrupt proteostasis at the level of proteasomal or autophagosomal degradation, and could therefore indirectly facilitate aggregation and trigger inflammation (Fig. 1). Mitochondrial damage, endoplasmic reticulum stress response, and several other mechanisms also promote inflammation, and can at some point initiate a vicious cycle that leads to neurodegeneration. Therefore, inflammation, which is capable of exerting collateral damage, is linked to various aggregate-prone mutations, stress stimuli and/or defects in aggregate disposal.
Figure 1.
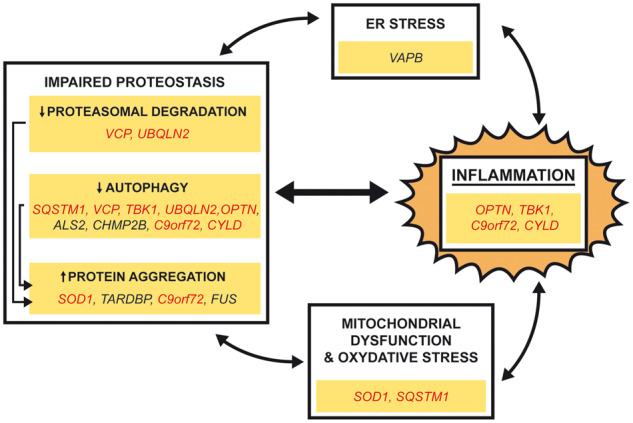
All roads lead to inflammation: genetic evidence. Mutations in a small subset of genes (OPTN, TBK1, CYLD and C9orf72) are directly linked with dysfunctional inflammatory responses. Most genes mutated in ALS disrupt proteostasis by increased protein aggregation, decreased proteasomal degradation or impaired autophagy. Aggregated proteins can in turn trigger inflammation. Inflammation can also be triggered by mutations in VAPB that causes endoplasmic reticulum stress or mutations in SOD1 and SQSTM1 that cause mitochondrial damage and oxidative stress. The link between inflammation and other proposed ALS-causing mechanisms is bidirectional: enhanced inflammation amplifies other pathogenic mechanism. Remark: genes marked in red are proposed act by more than one pathogenic mechanism.
The possibility that the immune system disbalance can also be an initial ALS trigger or a key disease modifier is only now taking hold. This has initially been dismissed because immune cell activation was reported to arise after the initial neuronal damage (Kano et al., 2012). Moreover, ALS was reported to be only slightly more frequent in patients with a prior diagnosis of autoimmune diseases (Turner et al., 2013), and recent genome-wide association studies found no strong association between ALS and autoimmune diseases (Broce et al., 2018). However, a subset of ALS-linked genes, such as TBK1, OPTN, CYLD and C9orf72, encode for multifunctional proteins involved in innate immune reactions. Moreover, polymorphisms in cytokine and chemokine receptors were shown to influence disease progression (Lopez-Lopez et al., 2014; Calvo et al., 2018; Wosiski-Kuhn et al., 2019b). Therefore, disbalanced immune responses leading to either excessive inflammation or inefficient immune response (immunodeficiency) may occur early during disease progression or serve as a disease modifier in ALS pathogenesis.
Glial cells as key contributors to ALS pathogenesis
The proposed passive role of glia as the glue that holds neurons together, has been firmly abandoned (Somjen, 1988; Ban et al., 2019). Glial cells actively maintain neuronal homeostasis but also take key roles in the neurodegenerative process (Ransohoff, 2016). Microglia represent the resident innate immune cells of the CNS, separated from peripheral populations during the early embryonic development, without a possibility of being replenished from bone marrow precursors (Ajami et al., 2007; Ginhoux and Prinz, 2015). They survey neuronal microenvironment with their remarkably motile processes and secrete various neurotrophic factors (Fig. 2A) (Davalos et al., 2005; Nimmerjahn et al., 2005; Kettenmann et al., 2013). The neighbouring healthy neurons mitigate microglial activation by negative co-stimulatory molecule CD200 and inhibitory chemokine fractalkine (CX3CL1), whereas astrocytes provide anti-inflammatory cytokine transforming growth factor β (TGF-β) (Fig. 2A) (Hellwig et al., 2013; Butovsky et al., 2014). Astrocytes provide major structural and trophic support to neurons, supplying them with nutrients, growth factors and maintaining the blood–brain barrier (BBB) (Verkhratsky et al., 2013; Liddelow and Barres, 2017). Although they are not considered to be immune cells per se, in response to proinflammatory cytokines from activated microglia they become reactive i.e. they proliferate and secrete proinflammatory cytokines (Welser-Alves et al., 2011; Liddelow et al., 2017).
Figure 2.

Acute neuroinflammation. (A) Microglia maintain homeostasis in the CNS by secreting neurotrophic factors (BDNF, GDNF, IGF-1). Microglial activation is suppressed by healthy neurons that provide negative co-stimulation via CD200 and CX3CL1, and astrocytes that secrete TGF-β. (B) Various PAMPs/DAMPs from dead/dying neurons and/or pathogens switch microglia from resting to proinflammatory state by binding to various pathogen and scavenger receptors. Such microglia clear cellular debris, produce proinflammatory factors (TNF, IL-1β, IL-6, ROS/RNS) and upregulate various receptors. (C) Upon damage resolution, the remaining neurons restore negative CD200 and CX3CL1 co-stimulation so microglia shift to an anti-inflammatory phenotype. Such microglia secrete anti-inflammatory cytokines IL-10 and IL-4, growth factors, and upregulate arginase 1, YM-1 and CD163, which leads to the resolution of inflammation and restoration of the resting state.
The data supporting a decisive role of non-neuronal cells in ALS are copious. Perhaps the strongest proof that motoneuron death in ALS is primarily non-cell autonomous came from analysing the mice carrying highly penetrant human SOD1 (hSOD1) mutations in neurons (Pramatarova et al., 2001; Lino et al., 2002). Unlike the ubiquitous expression of mutant hSOD1 in a mouse model, which leads to rapidly progressive disease and premature death, selective transgene expression in neurons or motoneurons fails to precipitate symptoms or death, strongly demonstrating that mutation-carrying glia contribute to neuronal pathology. Similarly, the disease could be alleviated in chimeric mutant hSOD1 mice when the diseased neurons were surrounded by healthy glia (Clement, 2003). Selective deletion of mutant hSOD1 transgene in motoneurons delays the disease onset, but provides no protection from disease progression and death, demonstrating that glial cells carrying the mutant hSOD1 transgene are sufficient to trigger motoneuron death (Boillee et al., 2006). Individual glial subsets from mutation-carrying mice have distinct neurotoxic potentials by themselves. Specifically, astrocytes carrying hSOD1G93A elicit motoneuron degeneration and microgliosis, whereas reconstitution of microglia-deficient (PU1−/−) mice with mSOD1G93A microglia did not result in motor weakness (Beers et al., 2006; Papadeas et al., 2011). These findings suggest that affected astrocytes can trigger motoneuron degeneration by themselves, while microglia require crosstalk with other diseased cells. In contrast, specific deletion of mutant hSOD1 transgene in the myeloid lineage (targeting both microglia and macrophages) or in astrocytes does not alter disease onset, but slows disease progression and increases life span (Boillee et al., 2006; Yamanaka et al., 2008). Experimental reconstitution with WT myeloid cells was also researched as a potential therapeutic intervention in microglia-deficient mutant hSOD1 mice (Beers et al., 2006). The reconstitution with the WT but not mutant SOD1 bone marrow extends the life span.
It was recently noted that glial activation differs between ALS models. In contrast to extensive activation in SOD1 models, mouse models carrying patient-derived mutations or haploinsufficiency of TANK-binding kinase 1 (TBK1) and optineurin (encoded by OPTN gene) lack overt microgliosis and astrocytosis (Ito et al., 2016; Gerbino et al., 2020). Similarly, in a model in which TDP-43 pathology can be reversibly induced in neurons, microglia are only subtly activated (Spiller et al., 2018). However, upon cessation of transgenic TDP-43 expression, microglia get robustly activated and clear the aggregated TDP-43 from the surviving neurons, suggesting that their function is inhibited by damaged neurons. Similar divergence in microglial phenotypes was found in ALS patients, whereby patients carrying SOD1 mutations had higher microglial activation than C9orf72 mutation carriers or sporadic ALS patients (Spiller et al., 2018). In contrast, another study found that C9orf72 mutation carriers had higher microglial activation than sporadic patients (Brettschneider et al., 2012), highlighting the considerable variability between patients.
Several ALS-related phenotypes have been recently reproduced in glial cells derived from patient-induced pluripotent stem cells, adding to the breadth of ALS models. Astrocytes derived from both sporadic and familial ALS patient-induced pluripotent stem cells have been shown to trigger necroptotic cell death of motoneurons in co-cultures (Re et al., 2014). Interestingly, SOD1 and TDP-43 were dispensable in this process, and death was triggered by soluble toxic factors. Another report showed that induced pluripotent stem cell-derived astrocytes from C9orf72 patients exerted motoneuron toxicity that correlated with elevated astrocytic oxidative stress, reduced antioxidant capacity and senescence (Birger et al., 2019). Interestingly, both necroptosis and oxidative stress have a potential to boost an inflammatory response, which can further increase astrocytic neurotoxicity. In contrast, motoneurons derived from TDP-43M337V patients showed increased cell-autonomous susceptibility to cell death, and co-culture with induced pluripotent stem cell-derived astrocytes—perhaps counterintuitively—increased their survival (Bilican et al., 2012; Serio et al., 2013). The observed discrepancies suggest a marked diversity in individual mutation-specific phenotypes. To conclude, the findings listed in this chapter, demonstrate that not only motoneurons but also glia can be intrinsically affected by the misfolded proteins, and that in ALS motoneurons in many cases die in a non-cell-autonomous manner. The particularities between the extent of glial activation in diverse genetic backgrounds are likely to be crucial for predicting prognosis, monitoring disease progression and determining personalized therapeutic approaches.
Acute versus chronic neuroinflammation
Damage sensing
Microglia are the first responders to damage in the CNS (Fig. 2B) (Kettenmann et al., 2011; Heneka et al., 2014; Roers et al., 2016). Pathogen-associated molecular patterns (PAMPs) such as microbial DNA, RNA, lipopolysaccharide (LPS), and endogenous damage-associated molecular patterns (DAMPs), such as aggregated proteins, high-mobility group box 1 (HMGB1) and heat shock proteins (HSP-60 and -70), bind to similar receptors to activate microglia. For example, LPS and ALS-specific aggregates of mutated SOD1 and TDP-43 bind to Toll-like receptors (TLR)-2 and 4 (Roberts et al., 2013; Zhao et al., 2015). Numerous other damage sensors might be important for microglial activation in neurodegenerative diseases including complement receptors, P2 purinergic receptors (for ATP), receptors for advanced glycation end products (RAGE), inflammasomes and triggering receptor expressed on myeloid cells 2 (TREM2) (Davalos et al., 2005; Lee et al., 2013; Heneka et al., 2014; Juranek et al., 2015; Debye et al., 2018; Deczkowska et al., 2018b). Importantly, glial responses to acute and chronic damage stimuli are very distinct.
Acute neuroinflammation
In response to acute injury, microglia change their resting ramified morphology to become ameboid and proliferate (Kettenmann et al., 2011). Activation also increases microglial phagocytic potential, production of reactive oxygen and nitrogen species (ROS/RNS) and a panel of proinflammatory factors such as cytokines TNF and interleukin 1β (IL-1β), chemokines and surface receptors (CD68) (Fig. 2B). Such proinflammatory polarization has commonly been described as classical or M1 phenotype, as first observed for macrophages in vitro, although most researchers nowadays avoid this term due to the fact that the in vivo polarization in neurodegenerative diseases is never so clear-cut. Productive proinflammatory responses clear cellular debris leading to resolution of the injury, but also trigger a reactive anti-inflammatory response, which in turn suppresses inflammation and promotes repair (Fig. 2C). Microglial neurotrophic and anti-inflammatory factors include insulin-like growth factor 1 (IGF-1), brain-derived neurotrophic factor (BDNF), IL-10, IL-4, arginase 1, and scavenger receptors CD163 and chitinase-like protein 3 (YM1) (Philips and Robberecht, 2011). Anti-inflammatory microglia are also known as alternatively activated or M2 microglia to be distinguished from the proinflammatory M1 microglia in the early phase of an acute inflammatory response.
Chronic neuroinflammation in ALS
In contrast to limited, acute and resolving inflammatory processes, the progressive nature of CNS damage in ALS precludes complete damage control and does not follow a simple proinflammatory/M1 to anti-inflammatory/M2 transition (Chiu et al., 2013). The initial acute phase is difficult to detect, suggesting that the primary damage is subtle and/or that the proinflammatory response seamlessly segues into an anti-inflammatory response (Fig 3A). Due to the difficulties of studying presymptomatic ALS in patients, most of the evidence on presymptomatic microglia comes from mouse models. Presymptomatic microglia in hSOD1 ALS mouse model express anti-inflammatory factors such as BDNF, IGF-1, IL-10, arginase 1, CD163 and YM1 (Appel et al., 2011; Chiu et al., 2013; Gravel et al., 2016). They also have an attenuated response to subsequent inflammatory insults, which is at least in part due to decreased TLR2 expression and activity (Gravel et al., 2016). The presymptomatic state is primarily maintained by secretion of IL-10 through autocrine signalling. An experimental increase in IL-10 production by microglia delays ALS onset slows progression and extends survival in the hSOD1 ALS mouse model. Furthermore, motoneurons in the presymptomatic stage are capable of providing negative co-stimulation via CX3CL1, which gets greatly reduced at disease onset (Zhang et al., 2018). For these reasons, presymptomatic microglia are thought to have a neuroprotective role.
Figure 3.

Chronic neuroinflammation. (A) The initial prolonged or repetitive damage in presymptomatic microglia elicits an anti-inflammatory phenotype. Neuronal negative co-stimulation with CD200 molecule and CX3CL1 is still active, and microglia show blunted proinflammatory response and neuroprotective properties: decreased TLR activity, increased production of anti-inflammatory cytokine IL-10, neurotrophic factors (BDNF, IGF-1) and scavenger receptors YM-1 and CD163. (B) Upon long-term chronic stimulation and/or repetitive hits microglia switch to highly proinflammatory state. Neurons in the symptomatic phase lose their ability to restrain microglial activation by negative co-stimulation, and various DAMPs (protein aggregates, debris, etc.) bind to their respective receptors (TLRs, TREM2, etc.) to activate downstream proinflammatory cascade, which results in profound changes in microglial transcriptional profiles (homeostatic genes are downregulated; proinflammatory genes are upregulated). Finally, proinflammatory microglia induce collateral neuronal damage, creating a vicious cycle that further amplifies inflammation.
The transcriptional profiles of chronically activated microglia markedly change over the course of ALS. With the onset of symptoms, microglia diminish their expression of anti-inflammatory and growth factors, and increase proinflammatory factor production, such as NADPH oxidase 2, and proinflammatory cytokines IL-1β, TNF and IL-6 (Fig. 3B) (Zhao et al., 2010; Beers et al., 2011; Liao et al., 2012). Notably, microglia in the most affected lumbar spinal cord regions are less responsive to inflammatory stimuli than healthy microglia, perhaps demonstrating exhaustion and/or desensitization (Nikodemova et al., 2014). A similar microglial phenotype has also been reported in Alzheimer’s disease and in models of experimental autoimmune encephalomyelitis, and was hence named disease-associated microglia (DAM) (Weissmann et al., 2016; Keren-Shaul et al., 2017; Krasemann et al., 2017; Kang et al., 2018; Deczkowska et al., 2018a, b). This phenotype is achieved through the TREM2 signalling pathway following chronic stimulation by neuronal debris, protein aggregates, etc. The DAM phenotype is characterized by the downregulation of homeostatic genes such as CX3CR1, P2RY12, TMEM119 and upregulation of APOE, CCL3, CLEC7A, CST7, CTSE, GPNMB, ITGAX, LGALS3, LILRB4 or LPL, which are linked to microglial activity, phagocytosis and inflammation (Keren-Shaul et al., 2017). The phagocytic DAM phenotype could be beneficial in plaque clearance in Alzheimer’s disease, whereas its role in ALS is less clear. Notably, DAM microglia are preferentially enriched around amyloid β plaques in Alzheimer’s disease, whereas their location in ALS was not reported. Since DAM do not express classic proinflammatory or anti-inflammatory markers and exhibit profound dysregulation of homeostatic genes (Keren-Shaul et al., 2017; Deczkowska et al., 2018b; Shi and Holtzman, 2018), they are distinct from M1 and M2 microglia. Of note, although DAM is a rather recent term, similar observations that microglia in ALS do not exhibit distinct textbook M1/M2 phenotypes have been reported previously (Chiu et al., 2013; Butovsky et al., 2014; Nikodemova et al., 2014). Therefore, symptomatic ALS microglia cannot be phenotypically categorized as classical proinflammatory or ‘M1’, which is associated with acute and not chronic inflammation. This paradigm shift in nomenclature is necessary to properly represent pathogenic microglia in ALS, which is crucial for development of targeted therapies.
With so many differentially expressed factors in chronically activated microglia, distinguishing the cause from the effect has been notoriously difficult, and the initial triggers for dysbalanced inflammatory reaction in ALS are still unclear. One plausible hypothesis is that because healthy microglia exhibit blunted inflammatory responses in comparison to macrophages, they are less effective in healing and repairing, and more prone to sustain chronic inflammation (Fig. 3B). Several reports have linked chronic inflammation in ALS to TGF-β (Butovsky et al., 2014; Cohen et al., 2014; Endo et al., 2015). TGF-β is mainly produced by astrocytes, and regulates several opposing immune functions such as promotion of anti-inflammatory functions and regulatory T cells (Treg), or supporting proinflammatory Th17 T cells and fibrosis, depending on the surrounding microenvironment and cytokine milieu (Katsuno et al., 2011; Butovsky et al., 2014; Endo et al., 2015). Activated microglia in mutant hSOD1 trigger differentiation of TGF-β-producing reactive astrocytes, suppress microglial neuroprotective functions and accelerate disease progression, whereas pharmacological blockade of TGF-β signalling extends survival (Iłzecka et al., 2002; Endo et al., 2015). Therefore, although TGF-β and perhaps other anti-inflammatory cytokines are initially beneficial, they are noxious on the long term (Cohen et al., 2014).
Chronic stimulation shuts down glial neuroprotective functions and elicits the production of a variety of proinflammatory factors, making late-stage microglia and astrocytes from mutant hSOD1 mice toxic to motoneurons (Haidet-Phillips et al., 2011; Liao et al., 2012; Re et al., 2014). Once generated, reactive astrocytes are sufficient to perpetuate the inflammatory reaction and/or neurotoxicity by themselves. This is partly explained by glutamate-mediated excitotoxic neuronal death due to lower levels of astrocytic excitatory amino acid transporter 2 (EAAT2) (Han and Whelan, 2010). Excitotoxicity is enhanced by proinflammatory cytokines such as TNF and motoneurons are exceptionally vulnerable to elevated glutamate levels. Aberrant populations of toxic microglia that express astrocytic markers and astrocytes expressing microglial markers have also been reported in ALS mouse models and patient samples (Díaz-Amarilla et al., 2011; Komine et al., 2018). Aberrant astrocytes develop at the onset of paralysis from Iba1+ microglia, express astrocytic markers such as GFAP, S100β and connexin 43, are detected around the dying motoneurons in a rat mutant hSOD1 ALS model, and increase in number with disease progression (Trias et al., 2013). The transdifferentiation processes leading to such aberrant cellular phenotypes are still unclear, but seem to be attractive therapeutic targets. Overall, ALS models demonstrate that a prolonged initial anti-inflammatory response is replaced by an excessive proinflammatory response towards the terminal stages of the disease, cautioning that therapeutic approaches will have to be stage-specific.
CNS macrophage and peripheral immune cell contribution to ALS pathogenesis
Growing evidence suggests that, in addition to microglia, several other innate immune cell subsets including macrophages, monocytes, dendritic cells (DC), natural killer (NK) cells, mastocytes and neutrophils are implicated in the ALS pathogenesis. Adaptive immune cells, in particular T cells play a key role as well, whereas the role of B cells and/or antibodies remains to be clarified. All these immune cell subsets likely exert a part or even most of their actions in the periphery as their access to the CNS is limited. Since ALS is a systemic disorder, regardless of their site of action, peripheral innate immune cells stand out as principal regulators of ALS pathogenesis.
Innate immunity
Similar to microglia, most CNS macrophages are tissue-resident long-lived cells derived from embryonic erythromyeloid precursors, with the exception of the choroid plexus macrophages, which partly originate from and are replenished by blood-derived monocytes (Prinz and Priller, 2014; Goldmann et al., 2016; Prinz et al., 2017). Their transcriptional profiles are more similar to microglia than to peripheral macrophages. We must bear in mind that to date it is still extremely challenging to distinguish the contribution of distinct myeloid cell types because of a substantial overlap in their activation markers. The same applies to DC and monocytes, which normally do not reside within the CNS, and their infiltration in ALS is still debated (Geissmann et al., 2008; Mrdjen et al., 2018). Some of the recent advances including transcriptional profiling, identification of a microglia-specific marker (transmembrane protein 119, Tmem119) and a potential monocyte marker not expressed on microglia (CD169), may help to distinguish innate immune cell types, but these approaches have not yet been validated and systematically implemented (Bennett et al., 2016; Zondler et al., 2016). Precise characterization of myeloid subsets will be crucial for understanding the extent of peripheral cell infiltration in ALS.
Several studies reported an increase in the monocyte chemoattractant CCL2 in the CNS and detected monocyte infiltration into spinal cords, suggesting that they promote disease progression and correlate to neuronal death in mutant hSOD1 ALS models (Henkel et al., 2004; Mantovani et al., 2009; Butovsky et al., 2012). This was not corroborated in several other studies, which claimed that monocyte infiltration is beneficial. For example, chimeric mice lacking the CCL2 receptor CCR2 in the bone marrow succumbed to ALS faster than the mice harbouring WT bone marrow, suggesting that monocyte presence and/or recruitment is protective (Beers et al., 2008). Similarly, experimentally induced increase in monocyte infiltration positively correlated with neuronal survival (Zondler et al., 2016). In contrast, parabiosis, single-cell mass spectrometry and other experiments have demonstrated that peripheral monocytes minimally infiltrate the CNS in ALS mouse models (Solomon et al., 2006; Ajami et al., 2007, 2018). The latter notion has been further supported by RNA sequencing of acutely isolated spinal cord immune cells/microglia from ALS models, which revealed that microglia do not share the molecular profiles of infiltrating monocytes, suggesting that without compromising BBB the infiltration of monocytes into CNS is limited (Chiu et al., 2013; Ajami et al., 2018). Taken together, it is still difficult to analyse the exact pattern and extent of monocyte infiltration in the brain and spinal cord in ALS models and patients. It is also notable that growing evidence suggests that endothelial cells are also affected in this disease, which in turn may cause progressive BBB alterations leading to infiltration of peripheral blood monocytes/macrophages. This has been confirmed in recent work by Garbuzova-Davis et al. (2017, 2019) as restoration of capillary integrity by delivery of human bone marrow stem cells reduced BBB permeability and delayed progression of disease in SOD1 mutant mice. Although the role and contribution of peripheral monocytes/macrophages in ALS-induced neurodegeneration remains elusive, these cells do have an important role in regulation of peripheral immunity. It is noteworthy that deregulation in peripheral blood monocyte immune profiles and function have been observed in sporadic ALS patients and may have some diagnostic and/or prognostic values. The role of monocytes in the regulation of peripheral immunity in ALS has been discussed in more details in the clinical chapter below, and some specific features of other peripheral innate subsets during ALS progression are listed in Box 1.
Box 1.
Innate immunity in ALS pathogenesis
Monocytes
The evidence on monocyte infiltration in ALS lesions is conflicting (Ajami et al., 2007; Butovsky et al., 2012; Zondler et al., 2016).
Familial and sporadic ALS patients have higher ratio of classical monocytes (CD14++CD16−) over non-classical monocytes (CD14++CD16++), which precedes disease onset; patients’ monocytes possess decreased velocity, increased adhesion and impaired phagocytic activity (Zondler et al., 2016).
ALS patient monocytes have a proinflammatory phenotype (IL-1β, IL-8, FosB, CXCL1 and CXCL2 mRNA upregulation) that correlates with disease progression (Zhang et al., 2005; Zhao et al., 2017).
Macrophages
CNS macrophages, found in the perivascular spaces of the BBB, meninges and choroid plexus, likely have overlapping roles with microglia in ALS pathogenesis (Prinz and Priller, 2014; Goldmann et al., 2016; Prinz et al., 2017).
Macrophage infiltration and complement secretion in peripheral nerves increase with disease progression (Chiu et al., 2009; Graber et al., 2010; Lincecum et al., 2010; Kano et al., 2012).
NK cells
NK cells increase with ALS progression in patients (Murdock et al., 2017).
NK cells infiltrate the motor cortex and the spinal cord of ALS patients and mouse models; NK cells kill motoneurons via NK2GD, reduce Treg infiltration and mediate microglial proinflammatory polarization via IFN-γ (Garofalo et al., 2020).
Dendritic cells
DC are reduced and dysfunctional in ALS patients’ blood (Rusconi et al., 2017).
DC increase in ALS patients’ spinal cords (Henkel et al., 2004; Sta et al., 2011).
Mast cells and neutrophils
Masts cells infiltrate skeletal muscles at the neuro-muscular junction and degranulate to help recruit neutrophils; neutrophils phagocytose degenerating axons and prevent reinnervation (Trias et al., 2018).
The drug masitinib targets these cells, reduces their infiltration and slows ALS progression in patients (Mora et al., 2019).
Adaptive immunity
T cells are present in negligible numbers in the CNS parenchyma, whereas slightly higher numbers are present in the meninges and the choroid plexus, with some entering the CSF through choroid plexus (Engelhardt and Ransohoff, 2005; Schwartz and Deczkowska, 2016). T cell infiltration to the CNS has for a long time been linked only to severe pathology, but it is now evident that CD4 T cells also exert important neuroprotective functions by improving cognition, and mediating CNS repair and recovery upon injury (Moalem et al., 1999; Hauben et al., 2001; Kipnis et al., 2012). The presence of CD4 and CD8 T cells in the ventral horns and CD4 T cells in the corticospinal tracts has been reported in ALS patient autopsies around the1990s, but it was not immediately clear if they were harmful or beneficial (Troost et al., 1989; Engelhardt et al., 1993). The beneficial effects of CD4 T cells, in particular Tregs, are now widely accepted (listed in Box 2). In contrast to T cells, the relevance of B cells during ALS is poorly understood, although immunoglobulins G (IgG) reportedly accumulate in motoneurons (Box 2) (Engelhardt and Appel, 1990). Both antigen-specific and Fc-mediated effects have been reported. Therefore, Bcell responses in ALS patients remain an interesting subject and their exact role remains to be defined.
Box 2.
Adaptive immunity in ALS pathogenesis
T cells
ALS patients have lower Treg numbers and function (Mantovani et al., 2009; Henkel et al., 2013; Beers et al., 2017; Sheean et al., 2018); at late disease stage, functional T cell defects and decreased numbers are seen in the peripheral immune compartments (Banerjee et al., 2008).
T cells in ALS patients and mouse models show proinflammatory Th1 and Th17 skewing at the late stages (Beers et al., 2011; Henkel et al., 2013; Saresella et al., 2013).
CD8 T cells specifically kill motoneurons in co-cultures, but CD8 T cell-deficient hSOD1 mice do not have shorter survival (Coque et al., 2019); CD8 T cells positively correlate to neuronal function in the peripheral nerves (Nardo et al., 2016).
Proposed CD4 T cell neuroprotective mechanisms include: (i) Tregs and/or Th2 cells suppress microglial and effector T cell activation; (ii) Tregs and/or Th2 cells support recruitment of peripheral monocytes to the CNS; (iii) autoimmune CD4 T cells release the blockade of the immune cell gateway to the CNS through the choroid plexus (Zhao et al., 2012; Ueno et al., 2013; Baruch et al., 2014; Kunis et al., 2015).
B cells and antibodies
B cells have no impact on disease progression in a mouse mutant hSOD1 model (Naor et al., 2009).
IgGs accumulate in motoneurons; IgG from ALS patients enter and stimulate motoneurons, astrocytes and microglia (Engelhardt and Appel, 1990; Appel et al., 1991; Pagani et al., 2011; Bataveljic et al., 2014; Miloševic et al., 2017).
Some antibodies recognize aggregated proteins and neurofilaments, but their pathogenicity is unclear (May et al., 2014; Malaspina et al., 2015).
Passive antibody transfer from ALS patients into mice increases serum and spinal cord cytokines (TNF, IL-6 and IL-10) and elicits motoneuron degeneration (Pullen et al., 2004; Obál et al., 2016).
The correlation of immune system activation to clinical phenotypes
Peripheral blood leukocytes are easily accessible and have been considered as possible ALS biomarkers. Decreased counts of CD4 T cells and increased counts of total leukocytes, neutrophils, CD16+ and CD16− monocytes and NK cells have been reported in ALS patients (Murdock et al., 2017). Importantly, the early changes in leukocyte numbers, in particular neutrophils and CD4 T cells, significantly correlated with disease progression measured by Amyotrophic Lateral Sclerosis Functional Rating Scale – Revised (ALSFRS-R) functional score. T cell data were corroborated in several other studies. For example, Treg numbers and function negatively correlate to disease severity i.e. low numbers are predictive for rapid disease progression and short survival (Fig. 4) (Henkel et al., 2013; Sheean et al., 2018), whereas there is moderate negative correlation between Th1 and Th17 T cells, ALSFRS-R and forced vital capacity (Jin et al., 2020). Another study observed a positive correlation between high baseline neutrophil-to-lymphocyte ratio and shorter survival, proposing that it can reflect the degree of neuroinflammation in ALS patients (Choi et al., 2020). A higher ratio of classical monocytes (CD14++CD16−) over non-classical monocytes (CD14++CD16++) is found in familial and sporadic ALS patients and precedes disease onset (Zondler et al., 2016). Other insights into ALS pathogenesis were provided by gene profiling studies. Zhang et al. showed that peripheral blood monocytes isolated from sporadic ALS patients strongly upregulate a cluster of LPS-TLR4 associated genes including those encoding for IL-1β, IL-8, FosB, CXCL1 and CXCL2, and that their level of activation correlates with disease severity (Zhang et al., 2005; Zhao et al., 2017). Surprisingly, they found that this upregulation correlated with elevated LPS plasma levels in sporadic ALS patients, suggesting that the molecular mechanisms of disease-associated deregulation of peripheral immunity may be more complex than initially thought. Importantly, immune profiles of peripheral monocytes at the mRNA level differ in rapidly and slowly progressing ALS patients, with rapidly progressing patients exhibiting more proinflammatory phenotypes (Zhao et al., 2017). In a recent study, TGF-β1 and TGF-β3 levels were also recently reported as potential markers of disease ALS severity, showing a significant negative correlation with ALSFRS-R (Duque et al., 2020). Therefore, the level of peripheral inflammation may predict disease progression.
Figure 4.

The timeline of ALS progression. (A) The progressive loss of neuroprotective immune responses over the course of ALS is depicted. (B) The potential prolonged and/or repetitive hits that affect ALS pathogenesis are shown. Inherited or sporadic mutations in ALS-associated genes could serve as primary hits that negatively affect to the initial anti-inflammatory phase, whereas secondary hit/s would set off the rapidly progressive stage marked with uncontrolled inflammation. (C) The disease progression is shaped by the immune factors. Various polymorphisms or other factors that result in lower-binding capacity of CX3CL1, higher levels of IL-6 and IL-6R, or reduced Treg numbers and function, are linked to rapid disease progression.
Some immune biomarkers associated with ALS have also been discovered in the CSF. For example, an increase in the level of macrophage-derived chitinases (CHIT1, CHI3L1 and CHI3L2), a signal of microglia/macrophage activation, was recently observed in ALS patients, and chitinase levels correlated with disease progression, severity, survival and other neurodegeneration markers such as phosphorylated neurofilament heavy chain (Steinacker et al., 2018; Thompson et al., 2018; Gille et al., 2019; Vu et al., 2020). A more detailed analysis of inflammatory factors, in both serum and CSF, and their correlation to disease severity is provided in Table 1.
Table 1.
Relevant immunological mediators and their correlation with clinical disease course
| Biomarker | Biofluid | Significance | References |
|---|---|---|---|
| TGF-β1 | Blood |
Higher levels with disease duration Negative correlation with ALSFRS-R |
Houi et al. (2002) and Duque et al. (2020) |
| TGF-β3 | Blood | Negative correlation with ALSFRS-R | Duque et al. (2020) |
| IL-2 | Blood |
Decrement with disease duration Poor survival |
Ehrhart et al. (2015) and Lu et al. (2016) |
| IL-4 | CSF | Slower disease progression; prevalent lower motor neuron phenotype | Furukawa et al. (2015) |
| IL-5 | Blood | Positive correlation with CK | Lu et al. (2016) |
| IL-6 |
Blood CSF and blood |
Positive correlation with CRP and end-stage disease Increment with respiratory dysfunction |
Moreau et al. (2005) and Lu et al. (2016) |
| IL-8 | Blood | Increment with disease duration | Ehrhart et al. (2015) |
| IL-10 |
Blood CSF |
Negative correlation with survival Slower disease progression; prevalent lower motor neuron phenotype |
Furukawa et al. (2015) and Olesen et al. (2020) |
| IL-13 | Blood | Negative correlation with ALSFRS-R and disease progression rate | Shi et al. (2007) |
| TNF | Blood |
Positive correlation with NfL Increment with respiratory dysfunction Negative correlation with survival |
Moreau et al. (2005), Lu et al. (2016) and Olesen et al. (2020) |
| Ferritin | Blood | Poor survival | Lu et al. (2016) |
| IFN-γ |
Blood CSF |
Increment with disease duration and progression rate Increment with disease duration and progression rate Shorter survival |
Liu et al. (2015) and Guo et al. (2017) |
| bFGF | CSF | Slower disease progression and longer disease duration | Guo et al. (2017) |
| VEGF | CSF | Slower disease progression and longer disease duration | Guo et al. (2017) |
| MIP-1α | CSF | Slower disease progression and longer disease duration | Guo et al. (2017) |
| CCL2 | CSF | Worse disease severity and faster progression | Guo et al. (2017) |
| TRAIL | Blood | Negative correlation with survival | Olesen et al. (2020) |
| CHIT1 | CSF | Positive correlation with rate of disease progression and level of NfL and NfH; negatively correlated with survival | Steinacker et al. (2018), Thompson et al. (2018), Gille et al. (2019), and Vu et al. (2020) |
|
CHI3L1 (YKL-40) CHI3L2 |
CSF | Positive correlation with rate of disease progression and level of NfH | Thompson et al. (2018) and Vu et al. (2020) |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale – Revised; bFGF = basic fibroblast growth factor; CK = creatine kinase; CRP = C-reactive protein; MIP-1α = macrophage inflammatory protein-1 alpha; NfL = neurofilament-light chain; NfH = neurofilament-heavy chain; TRAIL = TNF-related apoptosis-inducing ligand; VEGF = vascular endothelial growth factor.
It is notable that only few studies correlated immune parameters to specific ALS gene mutations. A study by Olesen et al. (2020) described differences in immune parameters and survival times in ALS subtypes. In particular, patients with sporadic ALS had higher CSF TNF compared with those with C9orf72 mutations, whereas patients with C9orf72 mutations had higher CSF IFN-α compared to those with SOD1 mutations or with sporadic ALS. The survival was negatively correlated with plasma IL-10, TNF and TNF-related apoptosis-inducing ligand (TRAIL), while in C9orf72 patients the levels of plasma IL-1β correlated negatively with survival. In addition, CHIT1 concentration was markedly increased in the CSF of symptomatic familial ALS patients, including C9orf72, SOD1, TARDBP and FUS mutation carriers, when compared to both healthy controls or asymptomatic mutation carriers (Oeckl et al., 2019). CHIT1 levels strongly correlated with axonal degeneration markers, suggesting that neuroinflammation is linked to the symptomatic phase of disease. Moreover, in a cohort of C9orf72 patients, a different protein profile, including neurofilament medium polypeptide (NEFM) and CHIT1, was observed in ALS and frontotemporal dementia patients, suggesting as these markers could distinguish between these two phenotypes (Barschke et al., 2020). Further research is needed to understand exactly which immune mediators are relevant for disease pathogenesis, and pinpoint other genetic variants with distinct immunological patterns.
How does immunity turn from friend to foe during ALS?
We have listed various immune cell subsets and neuroprotective and neurotoxic immune mechanisms that contribute to ALS pathogenesis. Extensive in vitro and in vivo evidence shows that inflammation can be secondary to prolonged and/or repetitive neuronal damage so this will be discussed first. After that, we will detail recent findings on immune system dysregulation as a potential initial trigger and/or a key disease modifier. This is in accordance with the recent mapping of several important immune-related genes and cellular pathways in ALS. Interestingly, two seemingly opposite immune defects were linked to these mutations: excessive inflammation and ineffective immune responses. However, the line between these two conditions is blurred because some immunodeficient states manifest as hyperinflammation due to the inability to contain pathogens (or even commensal bacteria) and/or clear tissue damage (Marks et al., 2010; Fodil et al., 2016). These processes comprise newer concepts in the pathogenesis of ALS, whose roles have only recently begun to be clarified.
Neuroinflammation secondary to prolonged and/or repetitive neuronal damage
As discussed earlier, typical ALS DAMPs arise from neuronal debris, but even prior to neuronal death, aggregated TDP-43 or SOD1 (mutant and/or oxidized WT), can be secreted or exocytosed by various CNS cells, and directly trigger inflammation (Urushitani et al., 2006; Ezzi et al., 2007; Zhao et al., 2010; Roberts et al., 2013; Zhao et al., 2015). The current model proposes that the predominant initial response of the immune system to aggregated proteins or similar stressors is neuroprotection, which at some still ill-defined point, when the damage can no longer be eliminated or contained, shifts to full-blown neuroinflammation and neurotoxicity, leading to an exacerbation of the primary damage (Fig. 4A). Data to support this neuroprotective to neurotoxic shift came largely from studies in mutant hSOD1 ALS models, in which the initial presymptomatic and early symptomatic phases are dominated by anti-inflammatory immune responses (anti-inflammatory microglia, Treg and Th2 CD4 T cells), whereas late symptomatic and terminal stages are dominated by proinflammatory immune responses (proinflammatory microglia and astrocytes, inflammatory monocytes, cytotoxic CD8 T cells, Th1 and Th17 CD4 T cells, NK cells and neutrophils) (Henkel et al., 2013; Zhao et al., 2013; Murdock et al., 2017; Sheean et al., 2018; Choi et al., 2020; Garofalo et al., 2020; Jin et al., 2020). As previously discussed, once that a hyperinflammatory state of microglia or astrocytes is established, these glial cells directly become toxic and can kill motoneurons (Roberts et al., 2013; Re et al., 2014; Zhao et al., 2015).
Although the exact turning point in this process remains unclear, it is reasonable to hypothesize that microglia and other immune cell subsets become neurotoxic upon facing prolonged and/or repetitive hits (Fig. 4B). The environmental factors that lead to ALS are poorly defined, except for aging, which is a common predisposing factor for all neurodegenerative diseases (Lucin and Wyss-Coray, 2009). Aged microglia exhibit enhanced and prolonged proinflammatory response to LPS challenge, accompanied with fractalkine receptor (CX3CR1) downregulation, which makes them refractory to neuronal immunosuppressive cues, whereas the aged choroid plexus decreases leukocyte entry from the blood, which reduces its neuroprotective potential (Godbout et al., 2005; Wynne et al., 2010; Baruch et al., 2014). Other environmental factors that may act as secondary hits or disease modifiers that could set off neurodegeneration include smoking, hormones, growth factors, head trauma, gut microbiome composition, peripheral nerve insults, and systemic and CNS inflammation (Nguyen et al., 2004; Bilic et al., 2006; Chen et al., 2007; Erny et al., 2015; Hancevic et al., 2019; Schram et al., 2019; Zhan and Fang, 2019).
Cytokines in systemic infections act as potential risk factors due to various mechanisms that allow microglia to ‘mirror’ systemic inflammation (Perry and Holmes, 2014). For this reason, chronic systemic LPS administration precipitates ALS symptoms in the mutant hSOD1 mouse model (Nguyen et al., 2004). Viral infections have also been proposed as potential risk factors as several studies detected viral genome elements in ALS patients’ spinal cords, mainly belonging to enteroviruses (Berger et al., 2000; Giraud et al., 2001; Xue et al., 2018a; Castanedo-Vazquez et al., 2019). Furthermore, a small fraction of patients infected with human immunodeficiency virus-1 (HIV-1) or human T cell leukaemia virus-1 (HTLV-1) progress to an ALS-like syndrome, which responds to antiretroviral therapy (Alfahad and Nath, 2013). ALS patients were reported to have significantly higher expression of human endogenous retroviral sequences in motoneurons compared to control subjects, and mice overexpressing endogenous retrovirus K develop an ALS-like syndrome (Douville et al., 2011; Li et al., 2015). Endogenous retrovirus K reactivation was found to be driven by neuroinflammatory cytokines TNF and IFN-γ, which act through IFN-stimulated responsive elements and NF-κB-binding sites in the viral promoter (Manghera et al., 2016). This allows for the possibility of a vicious cycle in which proinflammatory cytokines produced by microglia reactivate endogenous retrovirus K, which in turn supports chronic microglial activation and predisposes patients to ALS.
The gut microbiome is an important source of bioactive metabolites, and its imbalance can contribute to various diseases (Amedei and Boem, 2018; Di Gioia et al., 2020). It represents an important interface between the environment and the immune system, capable of affecting the CNS through microbial toxins or fermentation products, which can in turn trigger protein aggregation, proinflammatory cytokine secretion and microglial activation, among others (Erny et al., 2015). The link between gut microbiome and ALS is nowadays well-established in several animal models. SOD1 mice exhibit reduced colonic tight junction proteins and increased intestinal permeability prior to disease onset, and the levels and composition of commensal bacteria correlate with disease severity and exacerbation of the symptoms (Fang, 2016; Zhang et al., 2017; Blacher et al., 2019). Further confirmation came from C9orf72−/− mice in which reduced abundance of immune system-stimulating bacteria protected the mice from premature mortality and significantly attenuated their underlying systemic inflammation (Burberry et al., 2020). These findings suggest that in C9orf72−/− mice microbiome composition may be an important disease modifier for both onset and progression. Several studies have also advanced from preclinical to patients. Notably, a significant difference is observed in the global bacterial gene content in ALS patients compared with healthy controls (Rowin et al., 2017; Brenner et al., 2018; Blacher et al., 2019; Di Gioia et al., 2020). The alteration of gut microbiome and the other mechanisms listed in this section (prolonged hits from degenerating neurons, environmental factors, CNS trauma, systemic inflammation, etc.) could explain how initially protective microglial functions can, at a certain point, break down and lead to hyperinflammation, collateral neuronal damage and unrelenting ALS progression.
Excessive inflammation as a potential ALS trigger and/or disease modifier
A rare proof of principle that hyperactivated microglia can precede neuronal damage came from an experimental mouse model harbouring constitutively active IκB kinase β (IKKβ) in microglia, which led to a chronic NF-κB activation and gliosis, resulting in motoneuron death and a neurodegeneration similar to ALS (Frakes et al., 2014). Conversely, in a mutant hSOD1 model selective downregulation of NF-κB in microglia, but not in astrocytes, considerably prolonged the life span. However, immune system activation is complex and harmful processes in some cells or at some disease stages, can be beneficial in others. For example, astrocyte-specific NF-κB activation delayed the progressive phase of the disease by facilitating microglial transition to an anti-inflammatory state, enhancing autophagy and decreasing aggregated mutant hSOD1 (Ouali Alami et al., 2018). However, at later stages, it favoured a proinflammatory state of microglia and accelerated disease progression. These experiments opened the possibility that excessive inflammation through NF-κB pathway activation could be a primary pathogenic step in ALS.
The first ALS patient mutations proposed to be linked to excessive inflammation were reported in the OPTN gene encoding for an ubiquitin-binding adaptor protein optineurin (Maruyama et al., 2010). Due to its homology and competition to NF-κB essential modulator, optineurin was reported to act as a negative regulator of NF-κB signalling (Zhu et al., 2007; Maruyama et al., 2010; Akizuki et al., 2013). Microglial NF-κB activation was detected in autopsies of a homozygous OPTN mutation carrier and sporadic ALS patients (Sako et al., 2012). Unexpectedly though, extensive studies performed in various peripheral innate immune cell subsets and microglia from the OPTN−/− model or models lacking the ubiquitin-binding activity showed that optineurin is dispensable for NF-κB activation and production of proinflammatory cytokines (Gleason et al., 2011; Munitic et al., 2013; Pourcelot et al., 2016; Slowicka et al., 2016; Markovinovic et al., 2018). One group initially reported that OPTN−/− mice had a slight increase in proinflammatory cytokines, dysmyelination and deficits in vertical rearing activity, but other studies reported no neuroinflammation, dysmyelination and/or neurological defects (Ito et al., 2016; Slowicka et al., 2016; Dermentzaki et al., 2019). However, an ubiquitin-binding optineurin patient mutation (E478G) lentivirally delivered to the mouse motor cortex, triggered secretion of proinflammatory cytokines and neuronal death (Liu et al., 2018). Therefore, although most studies dismissed excessive inflammation as a mechanism of action of OPTN mutations in ALS, additional functional studies will be necessary to understand if individual OPTN mutations such as E478G act by distinct pathogenic mechanisms. Of note, optineurin is a multifunctional protein that also regulates the IFN-β pathway and autophagy, which will be discussed in the next chapter.
An observation that the C9orf72 gene has higher expression in microglia and other myeloid cells than in neurons, opened up the possibility that its loss- and/or gain-of-function may affect (neuro)inflammation (DeJesus-Hernandez et al., 2011; Gendron et al., 2013; O’Rourke, 2016). Gain of function of C9orf72 gene is observed either as toxicity linked to formation of RNA foci or as generation of dipeptide protein repeats. C9orf72−/− mice show defective lysosomal functions and increased production of proinflammatory cytokines in myeloid cells, splenomegaly, lymphadenopathy and production of self-reactive antibodies, indicating the appearance of autoinflammation and/or autoimmunity (Atanasio et al., 2016; O’Rourke, 2016). Due to the breadth of inflammatory markers expressed in these mice, we classified these mutations as potentially hyperinflammatory. It is notable though, that the initial trigger for hyperinflammation is unknown, and could in theory be defective lysosomal waste removal, which would in turn classify these mutations as an immunodeficiency, as discussed in the next chapter. This remains to be further investigated. However, neither whole body nor conditional C9orf72 loss in neurons or glia cause motoneuron degeneration in a mouse model (Koppers et al., 2015; O’Rourke, 2016). For this reason, it is unlikely that these models appropriately mimic human disease and that dysregulated inflammation is by itself sufficient to cause the disease in patients carrying C9orf72 mutations. Dipeptide protein repeat-mediated toxicity has been linked to inflammation as well. Several studies of C9orf72 BAC and poly-GA transgenic mouse models with widespread accumulation of dipeptide protein repeats showed appearance of either microgliosis and/or astrogliosis in the spinal cords and several brain regions (Zhang et al., 2015; Liu et al., 2016; Schludi et al., 2017). However, certain discrepancies in the extent and breadth of glial activation were reported in these otherwise similar models, necessitating further clarification. Nevertheless, the crosstalk of loss of function to other proposed mechanisms of action of C9orf72 mutations, which include toxicity of dipeptide repeats, RNA toxicity and impaired autophagy, could be necessary for disease appearance (Sellier et al., 2016; Sullivan et al., 2016).
A recent genome-wide association studies identified TNIP1 as a potential risk locus in Chinese ALS patients. The TNIP1 gene encodes for TNFAIP3-interacting protein 1, also known as A20-binding inhibitor of NF-κB (ABIN1) (Benyamin et al., 2017). ABIN1 has an ubiquitin-binding region that is highly homologous to optineurin and can shut down the NF-κB pathway via recruiting a deubiquitinating enzyme A20. Furthermore, polymorphisms in cytokine and chemokine receptors have been recently discovered as disease modifiers in ALS patients (Fig. 4C). Carriers of CX3CR1 249I allele, which has lower CX3CL1 binding capacity, have a substantially faster disease progression (Lopez-Lopez et al., 2014; Wosiski-Kuhn et al., 2019b). Similarly, carriers of the IL-6R Asp358Ala variant have higher IL-6 and IL-6R in the sera and CSF (Wosiski-Kuhn et al., 2019b). Notably, neither CX3CR1 nor IL-6R polymorphisms increase the risk of disease, but both lead to a rapid disease progression and shorter survival. This argues that immune system dysregulation linked to these polymorphisms is insufficient to cause the disease by itself but instead works together with other neurotoxic hits. Whether this is necessary for all ALS mutations affecting the immune system is still unknown; proofs of principle from mouse studies demonstrate that hyperactivated microglia can exert toxicity by itself (Frakes et al., 2014), but most of the other models discussed in this chapter were not cell-specific. Overall, several genes that directly regulate the immune system have been characterized, demonstrating that excessive responses to damage could in these particular cases be a primary cause or key disease modifier in ALS.
Immunodeficiency as a primary ALS trigger
Immunodeficiencies, subdivided into defects of innate or adaptive immunity, comprise inabilities to mount appropriate immune responses required for damage elimination. They are commonly linked to impaired responses to infection and are rarely discussed in the context of neurodegeneration. Here we propose that inefficient inflammatory factor secretion and/or clearance of neurodegenerative DAMPs reported for some ALS-causing mutations represent an innate immunodeficiency. An early proof of principle that innate immunodeficiency can accelerate ALS came from a chimeric mouse hSOD1 model that was (upon irradiation) reconstituted with WT or myeloid differentiation factor 88 (Myd88)-deficient bone marrow, replacing endogenous microglia. Myd88 is an adaptor to all TLRs except TLR3 and activates the NF-κB and mitogen-activated protein kinases (MAPK) pathways. Compared to microglial replacement with WT cells, microglial replacement with Myd88-deficient cells was associated with earlier disease onset and decreased survival (Kang and Rivest, 2007). Another example came from an experimental TIR-domain-containing adapter-inducing IFN-β (TRIF)-deficient mutant hSOD1 model (Komine et al., 2018). TRIF is an adaptor protein that primarily regulates IFN-β responses. It conveys signals from TLRs, but in contrast to MyD88, TRIF is activated via TLR3 or (together with MyD88) via TLR4, primarily resulting in TBK1 activation (O'Neill and Bowie, 2007). TBK1 is the primary kinase that phosphorylates transcription factor IFN regulatory factor 3 (IRF3) required for antiviral IFN-β production (Kielian, 2006). Whole-body TRIF deficiency substantially accelerated the development of ALS symptoms and shortened survival in the mutant hSOD1 model, whereas MyD88 deficiency had no effect (Komine et al., 2018). This suggests that IFN-β regulated responses delay pathology in that model.
Recently discovered ALS-linked mutations in TBK1 (Fig. 5) may also affect the IFN-β pathway and cause innate cell immunodeficiency (Maruyama et al., 2010; Cirulli et al., 2015; Freischmidt et al., 2015). While the role of TBK1 in neurodegeneration is rather new, neuroprotective effects of IFN-β in multiple sclerosis are well-known. It promotes microglial phagocytosis, regulates the switch from pro- to anti-inflammatory microglia, etc. (Cohen et al., 2014; Kocur et al., 2015). IFN-β also promotes neurite branching and autophagy in neurons, thus protecting against α-synuclein pathology and motor and cognitive defects in mice (Ejlerskov et al., 2015). In ALS patients bearing a TBK1 mutation, it was suggested that the TBK1-induced pathology was caused by a loss of function from haploinsufficiency as TBK1 mRNA and protein levels were reduced in comparison to sporadic ALS patients (Freischmidt et al., 2015; Pottier et al., 2015). In mice, TBK1 deficiency is lethal, whereas haploinsufficiency is insufficient to cause the disease by itself (Brenner et al., 2019; Gerbino et al., 2020). However, in a mutant hSOD1 model, haploinsufficiency of TBK1 or a loss-of-function mutation in TBK1R228H showed a biphasic effect on the disease course: accelerating disease onset, but afterwards suppressing the appearance of neurotoxic microgliosis and astrogliosis, and extending survival. Primary microglia from TBK1+/− SOD1G93A mice exhibited diminished production of proinflammatory cytokines in vitro (Brenner et al., 2019). TBK1R228H SOD1G93A mice led to diminished IRF3 activation and decreased induction of IFN-stimulated genes at the late stage of the disease. It was thus proposed that diminished inflammatory responses due to TBK1 insufficiency slow down the disease progression at late stages (Gerbino et al., 2020). Given that TBK1 also has a role in promoting autophagy, the authors proposed that the accelerated early-stage disease in TBK1+/− mutant hSOD1 ALS model resulted from defective autophagy (Brenner et al., 2019; Gerbino et al., 2020). However, this does not exclude the possibility that innate immunodeficiency resulting from diminished IFN-β production could contribute as well. Additional complexity might arise from the fact that more than 70 mutations in TBK1 have been found in ALS patients, which differently affect the kinase activity and may have different mechanisms of action (CirulLi et al., 2015; Freischmidt et al., 2015; Oakes et al., 2017; Ye et al., 2019).
Figure 5.

Hyperinflammation and immunodeficiency as primary triggers of motoneuron death in ALS. (A) Microglial skewing towards uncontrolled inflammation could be a repercussion of inadequate shut down of NF-κB signalling exerted by constitutive activation of IKKβ (*clinical correlate not yet reported), or mutations in proposed negative regulators OPTN and TNIP1. Overactivated NF-κB in reactive astrocytes elicits similar effects. Such excessive proinflammatory factor production from chronically activated glia ultimately causes motor neuron death. (B) The inability of microglia to optimally respond to damage is a potential trigger for neurodegeneration in ALS. ALS-linked loss-of-function mutations in OPTN and TBK1 lead to decreased production of IFN-β, dysregulated inflammatory responses, reduced phagocytic capacity of microglia and decreased neuronal autophagy. ALS-linked mutant CYLDM719V was proposed to impair autophagy flux and reduce NF-kB activation. Such inadequate response to damage finally leads to accumulation of various DAMPs (protein aggregates, ATP, HMGB1, etc.), thus sparking neurotoxic chronic inflammation. Loss of immunosurveillance by the adaptive immunity, in particular T cells, could contribute to dysregulated responses and neurotoxicity.
Insufficiency of optineurin also leads to diminished IFN-β responses in microglia (Fig. 5) (Markovinovic et al., 2018). Optineurin acts as an adaptor protein necessary for optimal TBK1 activation in various other innate immune cell subsets, suggesting that they likely act on the same axis in the pathogenesis of ALS (Munitic et al., 2013; Meena et al., 2016; Pourcelot et al., 2016). Moreover, previous findings linked optineurin deficiency with diminished acute immune response and neutrophil recruitment to the site of infection in mice (Chew et al., 2015; Smith et al., 2015), hence opening a possibility that inadequate first response to damage due to disruption in the optineurin-TBK1 axis could trigger neurodegeneration. In addition to diminished inflammatory response, ALS-linked mutations in optineurin also lead to diminished autophagy and mitophagy (Heo et al., 2015; Lazarou et al., 2015; Richter et al., 2016; Evans and Holzbaur, 2020). Interestingly, to become an autophagy adaptor, optineurin must be phosphorylated by TBK1. It is thus plausible that a breakdown in the crosstalk between autophagy and inflammation triggers disease (Markovinovic et al., 2017). The impaired autophagy itself could also lead to an accumulation of aggregated proteins, build-up of ROS from damaged mitochondria, and diminished elimination of bacteria and viruses. If these events are coupled by inadequate inflammatory response that cannot contain the damage, the likely final outcome is accumulation of PAMPs/DAMPs, chronic inflammation and neuronal death (Fig. 5).
Newly discovered missense mutation in the gene encoding for cylindromatosis (CYLD) protein, a deubiqutinase for K63-linked polyubiquitin chains that terminates NF-κB and TBK1 signalling, was recently linked to ALS (Dobson-Stone et al., 2020). ALS-linked mutant CYLDM719V exhibited increased deubiquitinase activity and subsequently reduced NF-κB activation in HEK293 cells (Fig. 5). The exact mechanism of action is still unclear since CYLDM719V also impairs autophagy by blocking autolysosome formation. This perhaps represents additional evidence that the combination of impaired autophagy and inadequate inflammatory response can promote motor neuron death, but further studies in primary cells and mouse models are needed to confirm that hypothesis.
Several lines of evidence have also suggested that dysfunction of adaptive immunity could contribute to ALS pathogenesis. Thymic involution and reduced T cell progenitor numbers are found in the mutant hSOD1 ALS mouse model, which results in reduced thymic output and restricted T cell repertoire in both mouse models and ALS patients (Fig. 5) (Seksenyan et al., 2010). Mutant hSOD1 ALS mice also mount diminished T cell response to vaccination in comparison to WT mice (Banerjee et al., 2008; Kunis et al., 2015). Furthermore, immunodeficiency could contribute to the aforementioned ALS-like syndrome associated with HIV infection (Sinha et al., 2004; Rowland, 2011). Therefore, although the data are still sparse, a possibility remains that adaptive immune system immunodeficiency could contribute to ALS pathogenesis too.
Remaining challenges for translational ALS research
Particularities of immune responses in individual patients (due to their genetic makeup and/or environmental cues), the dual roles of immune responses in ALS pathogenesis, and changes in these responses over the course of ALS, have rendered efficient targeting of pathogenic immune responses in ALS patients challenging. This can partially be attributed to the fact that historically most of the insight into the immune response in ALS came from the fast-progressing hSOD1 models, which do not necessarily replicate all the facets of the immune pathology observed in a wide spectrum of ALS patients (Wosiski-Kuhn et al., 2019a). The same reasons contributed to the failure of many clinical trials; for example, celeoxib, a COX2 inhibitor, successfully slowed the disease progression in the SOD1 mouse model, but showed no efficacy in ALS patients (Pompl et al., 2003; Cudkowicz et al., 2006). On the other hand, treatment with tocilizumab, a blocking antibody against IL-6 receptor, decreased inflammation in sporadic ALS patients with high levels of inflammation, whereas it increased inflammation in patients with initially lower level of inflammation (Fiala et al., 2013). This suggests that one of the major challenges facing efficient treatment will be accurate and time-dependent estimation of the level of immune system activation and inflammation.
In addition to evaluating peripheral inflammation, analysis of microgliosis and astrocytosis in patients can nowadays be done in real time with positron emission tomographic imaging using markers that target translocator protein, expressed primarily by activated microglia and astrocytes (Turner et al., 2004; Corcia et al., 2012; Alshikho et al., 2018). Increased uptake of translocator protein markers is seen in precentral gyri of ALS patients, colocalizes with cortical thinning and correlates with more severe clinical phenotype and markers of neurodegeneration (Turner et al., 2004; Alshikho et al., 2016; 2018). However, this method is still of limited use for monitoring ALS progression because the binding capacity seems to plateau shortly after the appearance of symptoms. Therefore, more precise readouts are highly sought-after. Hence, to efficiently target immunity in ALS, we need better diagnostic approaches for estimating inflammation, procedures to optimize duration and dosing for a desired therapeutic effect, and larger clinical trials with better patient stratification and appropriate controls (Berry et al., 2017; Wosiski-Kuhn et al., 2019a; Goyal et al., 2020).
Conclusion
ALS is an immune-mediated disease. Immune system dysregulation in ALS patients is present at multiple levels and many immune readouts including microglial activation, serum and CSF proteins, and blood cells are studied as potential therapeutic targets or biomarkers (Jeromin and Bowser, 2017; Beers and Appel, 2019). We have presented evidence that immune system dysregulation in ALS extends beyond excessive chronic inflammation, and that it may at some genetic backgrounds manifest as an inefficient immune response. We propose to classify the latter as an immunodeficiency, even though this term has been only rarely used in the context of ALS, and thus far only for T cell defects (Banerjee et al., 2008; Seksenyan et al., 2010). Immunodeficiency is an umbrella term for a variety of immune defects, ranging from adaptive to innate immunity, some of which lead to generally increased susceptibility to infection, whereas others are subtle and confer increased susceptibility to specific challenges. For example, haplodeficiency or dominant-negative effects of TBK1 mutations are linked to susceptibility to herpes simplex encephalitis in children, without other obvious immune failures (Herman et al., 2012). It may perhaps seem paradoxical that opposite immune dysfunctions such as excessive inflammation and immunodeficiency eventually lead to the same outcome. However, this is unsurprising because the inability to mount an appropriate immune response to eliminate cell debris, could lead to damage pile-up and thus trigger chronic inflammation. Indeed, an overlap between primary immunodeficiencies and inflammatory diseases has been reported for various inflammatory diseases such as inflammatory bowel disease, rheumatoid arthritis and others (Fodil et al., 2016; Melamed, 2016). Perhaps the best example is chronic inflammation in Crohn’s disease, which was initially regarded as an autoimmune disease, but is now thought to be an aftermath of defective production of immune factors, diminished immune cell recruitment and/or failed autophagy (Marks and Segal, 2008; Marks et al., 2010). Similar ineffective microglial activation has been reported for Alzheimer’s disease (Shi and Holtzman, 2018). Because of the wide plasticity of the immune system, manipulating immunity in ALS is a particularly attractive therapeutic approach. An experimental tyrosine-kinase inhibitor masitinib that targets signalling pathways in several innate immune cells showed promising results in a recent clinical trial (Mora et al., 2019), and various immunomodulatory transplantation approaches are being tested (haematopoietic stem cells, Tregs, mesenchymal stem cells) (Mazzini et al., 2003; Appel et al., 2008; Mazzini et al., 2018; Thonhoff et al., 2018). However, these approaches are in the early stages of development and currently do not target specific subsets of patients. Rapidly progressing disease has been linked to an aggressive immune phenotype, suggesting that groups of ALS patients could be distinguished by their immunological readouts (e.g. Tregs, IL-6, microglial activation, etc.) (Brettschneider et al., 2012; Beers et al., 2017; Wosiski-Kuhn et al., 2019b). Therefore, patient immune profiling will be crucial for selecting appropriate therapies. This will allow for overcoming the difficult task of dodging the Scylla and Charybdis of immune system activation in ALS—excessive inflammation and immunodeficiency—and possibly finding a sweet-spot for optimal immune responses for individual patients at specific disease stages.
Acknowledgements
We thank Dr. Christian Andrew Reynolds for critical revision of the manuscript. The authors would like to acknowledge networking support by the COST Action CA16122.
Funding
This work was supported by the Croatian Science Foundation (UIP-2013-11-7459 and IP-2018-01-8563), and University of Rijeka (18-211-1369) grants to I.M. and by the AGING Project for Department of Excellence at the Department of Translational Medicine (DIMET), Università del Piemonte Orientale, Novara, Italy to L.M. The Department of Biotechnology was equipped by European Regional Development Fund (ERDF) within the project ‘Research Infrastructure for Campus-based Laboratories at University of Rijeka’.
Competing interests
The authors report no competing interests.
Glossary
- ABIN1 =
A20-binding inhibitor of NF-κB
- ALS =
amyotrophic lateral sclerosis
- ALSFRS-R =
Amyotrophic Lateral Sclerosis Functional Rating Scale – Revised
- BBB =
blood–brain barrier
- BDNF =
brain-derived neurotrophic factor
- CD =
cluster of differentiation
- CHIT1 =
chitinase 1
- CHI3L1 =
chitinase 3-like 1
- CHI3L2 =
chitinase 3-like 2;CX3CL1 = fractalkine
- CX3CR =
CX3C chemokine receptor 1 or fractalkine receptor
- CYLD =
cylindromatosis
- C9orf72 =
chromosome 9 open reading frame 72 gene
- DAM =
disease-associated microglia
- DAMP =
damage-associated molecular patterns
- DC =
dendritic cells
- EAAT2 =
excitatory amino acid transporter 2
- FUS =
fused in sarcoma
- HMGB1 =
high-mobility group box 1
- hSOD1 =
human supeoxide dismutase 1
- HTLV-1 =
human T cell leukaemia virus-1
- IFN =
interferon
- IGF1 =
insulin-like growth factor 1
- IgG =
immunoglobulin G
- IL =
interleukin
- IKK =
IκB kinase
- IRF =
IFN regulatory factor
- LPS =
lipopolysaccharide
- MAPK =
mitogen-activated protein kinases
- MyD88 =
myeloid differentiation factor 88
- NEFM =
neurofilament medium polypeptide
- NK =
natural killer cells
- NF-κB =
nuclear factor of κB
- OPTN =
optineurin
- PAMP =
pathogen-associated molecular patterns
- RAGE =
receptors for advanced glycation end products
- RNS =
reactive nitrogen species
- ROS =
reactive oxygen species
- SOD1 =
superoxide dismutase 1
- SRSF3 =
serine/arginine-rich splicing factor 3
- TBK1 =
TANK-binding kinase 1
- TDP-43 =
transactive response DNA-binding protein of 43 kDa
- TGF =
transforming growth factor
- TLR =
Toll-like receptors
- Tmem119 =
transmembrane protein 119
- TNF =
tumour necrosis factor
- TRAIL =
TNF-related apoptosis-inducing ligand
- Treg =
regulatory T cells
- TREM2 =
triggering receptor expressed on myeloid cells 2
- TRIF =
TIR-domain-containing adapter-inducing IFN-β
- YM1 =
chitinase-like protein 3
- WT =
wild type
References
- ADAPT-FS Research Group Follow-up evaluation of cognitive function in the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial and its follow-up study. Alzheimers Dement 2015; 11: 216–25. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science 2013; 339: 156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 2007; 10: 1538–43. [DOI] [PubMed] [Google Scholar]
- Ajami B, Samusik N, Wieghofer P, Ho PP, Crotti A, Bjornson Z, et al. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat Neurosci 2018; 21: 541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, et al. Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J Neurochem 2013; 126: 699–704. [DOI] [PubMed] [Google Scholar]
- Alfahad T, Nath A. Retroviruses and amyotrophic lateral sclerosis. Antiviral Res 2013; 99: 180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshikho MJ, Zürcher NR, Loggia ML, Cernasov P, Chonde DB, Izquierdo Garcia D, et al. Glial activation colocalizes with structural abnormalities in amyotrophic lateral sclerosis. Neurology 2016; 87: 2554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshikho MJ, Zürcher NR, Loggia ML, Cernasov P, Reynolds B, Pijanowski O, et al. Integrated magnetic resonance imaging and [11 C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol 2018; 83: 1186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer A. Uber eigenartige Krankheitsfälle des späten Alters. Z f d g Neur u Psych 1911; 4: 356–85. [Google Scholar]
- Amedei A, Boem F. I’ve gut a feeling: microbiota impacting the conceptual and experimental perspectives of personalized medicine. Int J Mol Sci 2018; 19: 3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH, Engelhardt JI, García J, Stefani E. Immunoglobulins from animal models of motor neuron disease and from human amyotrophic lateral sclerosis patients passively transfer physiological abnormalities to the neuromuscular junction. Proc Natl Acad Sci U S A 1991; 88: 647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH, Engelhardt JI, Henkel JS, Siklos L, Beers DR, Yen AA, et al. Hematopoietic stem cell transplantation in patients with sporadic amyotrophic lateral sclerosis. Neurology 2008; 71: 1326–34. [DOI] [PubMed] [Google Scholar]
- Appel SH, Zhao W, Beers DR, Henkel JS. The microglial-motoneuron dialogue in ALS. Acta Myol 2011; 30: 4–8. [PMC free article] [PubMed] [Google Scholar]
- Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015; 212: 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee H-C, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 2016; 6: 23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban J, Sámano C, Mladinic M, Munitic I. Glia in amyotrophic lateral sclerosis and spinal cord injury: common therapeutic targets. Croat Med J 2019; 60: 109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Mosley RL, Reynolds AD, Dhar A, Jackson-Lewis V, Gordon PH, et al. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS One 2008; 3: e2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barschke P, Oeckl P, Steinacker P, Al Shweiki MR, Weishaupt JH, Landwehrmeyer GB, et al. Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J Neurol Neurosurg Psychiatry 2020; 91: 503–11. [DOI] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014; 346: 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataveljic D, Milosevic M, Radenovic L, Andjus P. Novel molecular biomarkers at the blood-brain barrier in ALS. Biomed Res Int 2014; 2014: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol 2019; 18: 211–20. [DOI] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2006; 103: 16021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A 2008; 105: 15558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A, et al. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun 2011; 25: 1025–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Zhao W, Wang J, Zhang X, Wen S, Neal D, et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017; 2: e89530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 2016; 113: E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyamin B, He J, Zhao Q, Gratten J, Garton F, Leo PJ, et al. Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat Commun 2017; 8: 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MM, Kopp N, Vital C, Redl B, Aymard M, Lina B. Detection and cellular localization of enterovirus RNA sequences in spinal cord of patients with ALS. Neurology 2000; 54: 20–5. [DOI] [PubMed] [Google Scholar]
- Berry JD, Paganoni S, Atassi N, Macklin EA, Goyal N, Rivner M, et al. Phase IIa trial of fingolimod for amyotrophic lateral sclerosis demonstrates acceptable acute safety and tolerability. Muscle Nerve 2017; 56: 1077–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilic E, Bilic E, Rudan I, Kusec V, Zurak N, Delimar D, et al. Comparison of the growth hormone, IGF-1 and insulin in cerebrospinal fluid and serum between patients with motor neuron disease and healthy controls. Eur J Neurol 2006; 13: 1340–5. [DOI] [PubMed] [Google Scholar]
- Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A 2012; 109: 5803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birger A, Ben-Dor I, Ottolenghi M, Turetsky T, Gil Y, Sweetat S, et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019; 50: 274–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacher E, Bashiardes S, Shapiro H, Rothschild D, Mor U, Dori-Bachash M, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019; 572: 474–80. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006; 312: 1389–92. [DOI] [PubMed] [Google Scholar]
- Brenner D, Hiergeist A, Adis C, Mayer B, Gessner A, Ludolph AC, et al. The fecal microbiome of ALS patients. Neurobiol Aging 2018; 61: 132–7. [DOI] [PubMed] [Google Scholar]
- Brenner D, Sieverding K, Bruno C, Lüningschrör P, Buck E, Mungwa S, et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med 2019; 216: 267–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, Lee VM-Y, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One 2012; 7: e39216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broce I, Karch CM, Wen N, Fan CC, Wang Y, Hong Tan C, et al. ; International FTD-Genomics Consortium. Immune-related genetic enrichment in frontotemporal dementia: an analysis of genome-wide association studies. PLoS Med 2018; 15: e1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med 2017; 377: 162–72. [DOI] [PubMed] [Google Scholar]
- Burberry A, Wells MF, Limone F, Couto A, Smith KS, Keaney J, et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 2020; 582: 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014; 17: 131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest 2012; 122: 3063–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo A, Moglia C, Canosa A, Cammarosano S, Ilardi A, Bertuzzo D, et al. Common polymorphisms of chemokine (C-X3-C motif) receptor 1 gene modify amyotrophic lateral sclerosis outcome: a population-based study. Muscle Nerve 2018; 57: 212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanedo-Vazquez D, Bosque-Varela P, Sainz-Pelayo A, Riancho J. Infectious agents and amyotrophic lateral sclerosis: another piece of the puzzle of motor neuron degeneration. J Neurol 2019; 266: 27–36. [DOI] [PubMed] [Google Scholar]
- Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. Am J Epidemiol 2007; 166: 810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew TS, O'Shea NR, Sewell GW, Oehlers SH, Mulvey CM, Crosier PS, et al. Optineurin deficiency contributes to impaired cytokine secretion and neutrophil recruitment in bacteria driven colitis. Dis Model Mech 2015; 8: 817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ETA, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 2013; 4: 385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Phatnani H, Kuligowski M, Tapia JC, Carrasco MA, Zhang M, et al. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci U S A 2009; 106: 20960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SJ, Hong YH, Kim SM, Shin JY, Suh YJ, Sung JJ. High neutrophil-to-lymphocyte ratio predicts short survival duration in amyotrophic lateral sclerosis. Sci Rep 2020; 10: 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. ; FALS Sequencing Consortium. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015; 347: 1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003; 302: 113–7. [DOI] [PubMed] [Google Scholar]
- Cohen M, Matcovitch O, David E, Barnett‐Itzhaki Z, Keren‐Shaul H, Blecher‐Gonen R, et al. Chronic exposure to TGFbeta1 regulates myeloid cell inflammatory response in an IRF7-dependent manner. EMBO J 2014; 33: 2906–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coque E, Salsac C, Espinosa-Carrasco G, Varga B, Degauque N, Cadoux M, et al. Cytotoxic CD8+ T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc Natl Acad Sci U S A 2019; 116: 2312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One 2012; 7: e52941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia AS, Patel P, Dutta K, Julien JP. Inflammation induces TDP-43 mislocalization and aggregation. PLoS One 2015; 10: e0140248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudkowicz ME, Shefner JM, Schoenfeld DA, Zhang H, Andreasson KI, Rothstein JD, et al. ; Northeast ALS Consortium. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol 2006; 60: 22–31. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005; 8: 752–8. [DOI] [PubMed] [Google Scholar]
- Debye B, Schmulling L, Zhou L, Rune G, Beyer C, Johann S. Neurodegeneration and NLRP3 inflammasome expression in the anterior thalamus of SOD1(G93A) ALS mice. Brain Pathol 2018; 28: 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Amit I, Schwartz M. Microglial immune checkpoint mechanisms. Nat Neurosci 2018. a; 21: 779–86. [DOI] [PubMed] [Google Scholar]
- Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 2018. b; 173: 1073–81. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleidi M, Jäggle M, Rubino G. Immune aging, dysmetabolism, and inflammation in neurological diseases. Front Neurosci 2015; 9: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermentzaki G, Politi KA, Lu L, Mishra V, Pérez-Torres EJ, Sosunov AA, et al. Deletion of Ripk3 prevents motor neuron death in vitro but not in vivo. eNeuro 2019; 6: ENEURO.0308-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gioia D, Bozzi Cionci N, Baffoni L, Amoruso A, Pane M, Mogna L, et al. A prospective longitudinal study on the microbiota composition in amyotrophic lateral sclerosis. BMC Med 2020; 18: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Amarilla P, Olivera-Bravo S, Trias E, Cragnolini A, Martinez-Palma L, Cassina P, et al. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2011; 108: 18126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem 2016; 139: 136–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone C, Hallup M, Shahheydari H, Ragagnin AMG, Chatterton Z, Carew-Jones F, et al. CYLD is a causative gene for frontotemporal dementia - amyotrophic lateral sclerosis. Brain 2020; 143: 783–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douville R, Liu J, Rothstein J, Nath A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol 2011; 69: 141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque T, Gromicho M, Pronto-Laborinho AC, de Carvalho M. Transforming growth factor-β plasma levels and its role in amyotrophic lateral sclerosis. Med Hypotheses 2020; 139: 109632. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Smith AJ, Kuzmin-Nichols N, Zesiewicz TA, Jahan I, Shytle RD, et al. Humoral factors in ALS patients during disease progression. J Neuroinflammation 2015; 12: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjørn M, Kuss M, et al. Lack of neuronal IFN-β-IFNAR causes lewy body- and Parkinson’s disease-like dementia. Cell 2015; 163: 324–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo F, Komine O, Fujimori-Tonou N, Katsuno M, Jin S, Watanabe S, et al. Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep 2015; 11: 592–604. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol 2005; 26: 485–95. [DOI] [PubMed] [Google Scholar]
- Engelhardt JI, Appel SH. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol 1990; 47: 1210–6. [DOI] [PubMed] [Google Scholar]
- Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 1993; 50: 30–6. [DOI] [PubMed] [Google Scholar]
- Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 2015; 18: 965–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CS, Holzbaur ELF. Lysosomal degradation of depolarized mitochondria is rate-limiting in OPTN-dependent neuronal mitophagy. Autophagy 2020; 16: 962–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzi SA, Urushitani M, Julien JP. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem 2007; 102: 170–8. [DOI] [PubMed] [Google Scholar]
- Fang X. Potential role of gut microbiota and tissue barriers in Parkinson’s disease and amyotrophic lateral sclerosis. Int J Neurosci 2016; 126: 771–6. [DOI] [PubMed] [Google Scholar]
- Fiala M, Mizwicki MT, Weitzman R, Magpantay L, Nishimoto N. Tocilizumab infusion therapy normalizes inflammation in sporadic ALS patients. Am J Neurodegener Dis 2013; 2: 129–39. [PMC free article] [PubMed] [Google Scholar]
- Fodil N, Langlais D, Gros P. Primary immunodeficiencies and inflammatory disease: a growing genetic intersection. Trends Immunol 2016; 37: 126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 2014; 81: 1009–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M, Valensin S, Olivieri F, DE Luca M, Ottaviani ENZO, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2000; 908: 244–54. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 2018; 14: 576–90. [DOI] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 2015; 18: 631–6. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Matsui N, Fujita K, Nodera H, Shimizu F, Miyamoto K, et al. CSF cytokine profile distinguishes multifocal motor neuropathy from progressive muscular atrophy. Neurol Neuroimmunol Neuroinflamm 2015; 2: e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova-Davis S, Kurien C, Haller E, Eve DJ, Navarro S, Steiner G, et al. Human bone marrow endothelial progenitor cell transplantation into symptomatic ALS mice delays disease progression and increases motor neuron survival by repairing blood-spinal cord barrier. Sci Rep 2019; 9: 5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova-Davis S, Kurien C, Thomson A, Falco D, Ahmad S, Staffetti J, et al. Endothelial and astrocytic support by human bone marrow stem cell grafts into symptomatic ALS mice towards blood-spinal cord barrier repair. Sci Rep 2017; 7: 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo S, Cocozza G, Porzia A, Inghilleri M, Raspa M, Scavizzi F, et al. Natural killer cells modulate motor neuron-immune cell cross talk in models of Amyotrophic Lateral Sclerosis. Nat Commun 2020; 11: 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasco S, Zaragoza P, García-Redondo A, Calvo AC, Osta R. Inflammatory and non-inflammatory monocytes as novel prognostic biomarkers of survival in SOD1G93A mouse model of Amyotrophic Lateral Sclerosis. PLoS One 2017; 12: e0184626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, et al. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol 2008; 86: 398–408. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang Y-J, Jansen-West K, Ash PEA, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 2013; 126: 829–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbino V, Kaunga E, Ye J, Canzio D, O’Keeffe S, Rudnick ND, et al. The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 2020; 106: 789–805.e5. [DOI] [PubMed] [Google Scholar]
- Gille B, De Schaepdryver M, Dedeene L, Goossens J, Claeys KG, Van Den Bosch L, et al. Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2019; 90: 1338–46. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol 2015; 7: a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud P, Beaulieux F, Ono S, Shimizu N, Chazot G, Lina B. Detection of enteroviral sequences from frozen spinal cord samples of Japanese ALS patients. Neurology 2001; 56: 1777–8. [DOI] [PubMed] [Google Scholar]
- Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J Biol Chem 2011; 286: 35663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, et al. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J 2005; 19: 1329–31. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 2016; 17: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal NA, Berry JD, Windebank A, Staff NP, Maragakis NJ, Berg LH, et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve 2020; 62: 156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber DJ, Hickey WF, Harris BT. Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J Neuroinflammation 2010; 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel M, Béland L-C, Soucy G, Abdelhamid E, Rahimian R, Gravel C, et al. IL-10 controls early microglial phenotypes and disease onset in ALS caused by misfolded superoxide dismutase 1. J Neurosci 2016; 36: 1031–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Yang X, Gao L, Zang D. Evaluating the levels of CSF and serum factors in ALS. Brain Behav 2017; 7: e00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 2011; 29: 824–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Whelan PJ. Tumor necrosis factor alpha enhances glutamatergic transmission onto spinal motoneurons. J Neurotrauma 2010; 27: 287–92. [DOI] [PubMed] [Google Scholar]
- Hancevic M, Bilic H, Sitas B, Pavlisa G, Borovecki F, Munitic I, et al. Attenuation of ALS progression during pregnancy-lessons to be learned or just a coincidence. Neurol Sci 2019; 40: 1275–8. [DOI] [PubMed] [Google Scholar]
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers 2017; 3: 17071. [DOI] [PubMed] [Google Scholar]
- Hauben E, Agranov E, Gothilf A, Nevo U, Cohen A, Smirnov I, et al. Posttraumatic therapeutic vaccination with modified myelin self-antigen prevents complete paralysis while avoiding autoimmune disease. J Clin Invest 2001; 108: 591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwig S, Heinrich A, Biber K. The brain’s best friend: microglial neurotoxicity revisited. Front Cell Neurosci 2013; 7: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol 2014; 14: 463–77. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 2013; 5: 64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, et al. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 2004; 55: 221–35. [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 2015; 60: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M, Ciancanelli M, Ou Y-H, Lorenzo L, Klaudel-Dreszler M, Pauwels E, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 2012; 209: 1567–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houi K, Kobayashi T, Kato S, Mochio S, Inoue K. Increased plasma TGF-beta1 in patients with amyotrophic lateral sclerosis. Acta Neurol Scand 2002; 106: 299–301. [DOI] [PubMed] [Google Scholar]
- Iłzecka J, Stelmasiak Z, Dobosz B. Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine 2002; 20: 239–43. [DOI] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016; 353: 603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeromin A, Bowser R. Biomarkers in neurodegenerative diseases. Adv Neurobiol 2017; 15: 491–528. [DOI] [PubMed] [Google Scholar]
- Jin M, Günther R, Akgün K, Hermann A, Ziemssen T. Peripheral proinflammatory Th1/Th17 immune cell shift is linked to disease severity in amyotrophic lateral sclerosis. Sci Rep 2020; 10: 5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juranek JK, Daffu GK, Wojtkiewicz J, Lacomis D, Kofler J, Schmidt AM. Receptor for advanced glycation end products and its inflammatory ligands are upregulated in amyotrophic lateral sclerosis. Front Cell Neurosci 2015; 9: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Rivest S. MyD88-deficient bone marrow cells accelerate onset and reduce survival in a mouse model of amyotrophic lateral sclerosis. J Cell Biol 2007; 179: 1219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, et al. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med 2018; 215: 2235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano O, Beers DR, Henkel JS, Appel SH. Peripheral nerve inflammation in ALS mice: cause or consequence. Neurology 2012; 78: 833–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Banno H, Suzuki K, Tanaka F, Sobue G. Transforming growth factor-β signaling in motor neuron diseases. Curr Mol Med 2011; 11: 48–56. [DOI] [PubMed] [Google Scholar]
- Keller AF, Gravel M, Kriz J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp Neurol 2011; 228: 69–79. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017; 169: 1276–90.e17. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011; 91: 461–553. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron 2013; 77: 10–8. [DOI] [PubMed] [Google Scholar]
- Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018; 14: 577–89. [DOI] [PubMed] [Google Scholar]
- Kia A, McAvoy K, Krishnamurthy K, Trotti D, Pasinelli P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018; 66: 1016–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res 2006; 83: 711–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipnis J, Gadani S, Derecki NC. Pro-cognitive properties of T cells. Nat Rev Immunol 2012; 12: 663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocur M, Schneider R, Pulm A-K, Bauer J, Kropp S, Gliem M, et al. IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol Commun 2015; 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komine O, Yamashita H, Fujimori-Tonou N, Koike M, Jin S, Moriwaki Y, et al. Innate immune adaptor TRIF deficiency accelerates disease progression of ALS mice with accumulation of aberrantly activated astrocytes. Cell Death Differ 2018; 25: 2130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppers M, Blokhuis AM, Westeneng H-J, Terpstra ML, Zundel CAC, Vieira de Sá R, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 2015; 78: 426–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017; 47: 566–81.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar DR, Aslinia F, Yale SH, Mazza JJ. Jean-Martin Charcot: the father of neurology. Clin Med Res 2011; 9: 46–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunis G, Baruch K, Miller O, Schwartz M. Immunization with a Myelin-derived antigen activates the brain’s choroid plexus for recruitment of immunoregulatory cells to the CNS and attenuates disease progression in a mouse model of ALS. J Neurosci 2015; 35: 6381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015; 524: 309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, et al. Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 2013; 10: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Lee M-H, Henderson L, Tyagi R, Bachani M, Steiner J, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 2015; 7: 307ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol 2012; 237: 147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity 2017; 46: 957–67. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017; 541: 481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincecum JM, Vieira FG, Wang MZ, Thompson K, De Zutter GS, Kidd J, et al. From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat Genet 2010; 42: 392–9. [DOI] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013; 79: 416–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lino MM, Schneider C, Caroni P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci 2002; 22: 4825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gao L, Zang D. Elevated levels of IFN-γ in CSF and serum of patients with amyotrophic lateral sclerosis. PLoS One 2015; 10: e0136937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 2016; 90: 521–34. [DOI] [PubMed] [Google Scholar]
- Liu Z, Li H, Hong C, Chen M, Yue T, Chen C, et al. ALS-associated E478G mutation in human OPTN (Optineurin) promotes inflammation and induces neuronal cell death. Front Immunol 2018; 9: 2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler DA. Influence of normal aging on brain autophagy: a complex scenario. Front Aging Neurosci 2019; 11: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez A, Gamez J, Syriani E, Morales M, Salvado M, Rodríguez MJ, et al. CX3CR1 is a modifying gene of survival and progression in amyotrophic lateral sclerosis. PLoS One 2014; 9: e96528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015; 523: 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C-H, Allen K, Oei F, Leoni E, Kuhle J, Tree T, et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm 2016; 3: e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 2009; 64: 110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina A, Puentes F, Amor S. Disease origin and progression in amyotrophic lateral sclerosis: an immunology perspective. Int Immunol 2015; 27: 117–29. [DOI] [PubMed] [Google Scholar]
- Manghera M, Ferguson-Parry J, Lin R, Douville RN. NF-κB and IRF1 induce endogenous retrovirus K expression via interferon-stimulated response elements in its 5’ long terminal repeat. J Virol 2016; 90: 9338–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M, et al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J Neuroimmunol 2009; 210: 73–9. [DOI] [PubMed] [Google Scholar]
- Markovinovic A, Cimbro R, Ljutic T, Kriz J, Rogelj B, Munitic I. Optineurin in amyotrophic lateral sclerosis: multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurobiol 2017; 154: 1–20. [DOI] [PubMed] [Google Scholar]
- Markovinovic A, Ljutic T, Beland LC, Munitic I. Optineurin insufficiency disbalances proinflammatory and anti-inflammatory factors by reducing microglial IFN-beta responses. Neuroscience 2018; 388: 139–51. [DOI] [PubMed] [Google Scholar]
- Marks DJ, Rahman FZ, Sewell GW, Segal AW. Crohn’s disease: an immune deficiency state. Clinic Rev Allerg Immunol 2010; 38: 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DJ, Segal AW. Innate immunity in inflammatory bowel disease: a disease hypothesis. J Pathol 2008; 214: 260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010; 465: 223–6. [DOI] [PubMed] [Google Scholar]
- May C, Nordhoff E, Casjens S, Turewicz M, Eisenacher M, Gold R, et al. Highly immunoreactive IgG antibodies directed against a set of twenty human proteins in the sera of patients with amyotrophic lateral sclerosis identified by protein array. PLoS One 2014; 9: e89596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzini L, Fagioli F, Boccaletti R, Mareschi K, Oliveri G, Olivieri C, et al. Stem cell therapy in amyotrophic lateral sclerosis: a methodological approach in humans. Amyotroph Lateral Scler Other Motor Neuron Disord 2003; 4: 158–61. [DOI] [PubMed] [Google Scholar]
- Mazzini L, Ferrari D, Andjus PR, Buzanska L, Cantello R, De Marchi F, et al. ; on behalf of BIONECA COST ACTION WG Neurology. Advances in stem cell therapy for amyotrophic lateral sclerosis. Expert Opin Biol Ther 2018; 18: 865–81. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, McGeer EG. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol 1988; 76: 550–7. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 1987; 79: 195–200. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. History of innate immunity in neurodegenerative disorders. Front Pharmacol 2011; 2: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol 1948; 29: 58–69. [PMC free article] [PubMed] [Google Scholar]
- Meena NP, Zhu G, Mittelstadt PR, Giardino Torchia ML, Pourcelot M, Arnoult D, et al. The TBK1-binding domain of optineurin promotes type I interferon responses. FEBS Lett 2016; 590: 1498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: where are we now. Front Neurosci 2019; 13: 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed I. Alzheimer’s disease of the immune system: a new variant of immune deficiency. Immunother Open Acc 2016; 2: 115. [Google Scholar]
- Miloševic M, Milicevic K, Bozic I, Lavrnja I, Stevanovic, I, Bijelic D, et al. Immunoglobulins G from sera of amyotrophic lateral sclerosis patients induce oxidative stress and upregulation of antioxidative system in BV-2 microglial cell line. Front Immunol 2017; 8: 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 1999; 5: 49–55. [DOI] [PubMed] [Google Scholar]
- Mora JS, Genge A, Chio A, Estol CJ, Chaverri D, Hernandez M, et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Frontotemporal Degener 2019; 21: 5–14. [DOI] [PubMed] [Google Scholar]
- Moreau C, Devos D, Brunaud-Danel V, Defebvre L, Perez T, Destee A, et al. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia. Neurology 2005; 65: 1958–60. [DOI] [PubMed] [Google Scholar]
- Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 2018; 48: 380–95.e6. [DOI] [PubMed] [Google Scholar]
- Munitic I, Giardino Torchia ML, Meena NP, Zhu G, Li CC, Ashwell JD. Optineurin insufficiency impairs IRF3 but not NF-κB activation in immune cells. J Immunol 2013; 191: 6231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdock BJ, Zhou T, Kashlan SR, Little RJ, Goutman SA, Feldman EL. Correlation of peripheral immunity with rapid amyotrophic lateral sclerosis progression. JAMA Neurol 2017; 74: 1446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naor S, Keren Z, Bronshtein T, Goren E, Machluf M, Melamed D. Development of ALS-like disease in SOD-1 mice deficient of B lymphocytes. J Neurol 2009; 256: 1228–35. [DOI] [PubMed] [Google Scholar]
- Nardo G, Trolese MC, de Vito G, Cecchi R, Riva N, Dina G, et al. Immune response in peripheral axons delays disease progression in SOD1G93A mice. J Neuroinflammation 2016; 13: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, D’Aigle T, Gowing G, Julien JP, Rivest S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci 2004; 24: 1340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikodemova M, Small AL, Smith SM, Mitchell GS, Watters JJ. Spinal but not cortical microglia acquire an atypical phenotype with high VEGF, galectin-3 and osteopontin, and blunted inflammatory responses in ALS rats. Neurobiol Dis 2014; 69: 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308: 1314–8. [DOI] [PubMed] [Google Scholar]
- O'Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007; 7: 353–64. [DOI] [PubMed] [Google Scholar]
- O’Rourke JG. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016; 351: 1324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain 2017; 10: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obál I, Klausz G, Mándi Y, Deli M, Siklós L, Engelhardt JI. Intraperitoneally administered IgG from patients with amyotrophic lateral sclerosis or from an immune-mediated goat model increase the levels of TNF-α, IL-6, and IL-10 in the spinal cord and serum of mice. J Neuroinflammation 2016; 13: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckl P, Weydt P, Steinacker P, Anderl-Straub S, Nordin F, Volk AE, et al. ; German Consortium for Frontotemporal Lobar Degeneration. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry 2019; 90: 4–10. [DOI] [PubMed] [Google Scholar]
- Olesen MN, Wuolikainen A, Nilsson AC, Wirenfeldt M, Forsberg K, Madsen JS, et al. Inflammatory profiles relate to survival in subtypes of amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm 2020; 7: e697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, Hu J, Shimizu N, Imai T, Nakagawa H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J Neurol Sci 2001; 187: 27–34. [DOI] [PubMed] [Google Scholar]
- Ouali Alami N, Schurr C, Olde Heuvel F, Tang L, Li Q, Tasdogan A, et al. NF‐κB activation in astrocytes drives a stage‐specific beneficial neuroimmunological response in ALS. EMBO J 2018; 37: e98697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani MR, Gonzalez LE, Uchitel OD. Autoimmunity in amyotrophic lateral sclerosis: past and present. Neurol Res Int 2011; 2011: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadeas ST, Kraig SE, O'Banion C, Lepore AC, Maragakis NJ. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A 2011; 108: 17803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré B, Gros-Louis F. Potential skin involvement in ALS: revisiting Charcot’s observation—a review of skin abnormalities in ALS. Rev Neurosci 2017; 28: 551–72. [DOI] [PubMed] [Google Scholar]
- Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol 2014; 10: 217–24. [DOI] [PubMed] [Google Scholar]
- Petrov D, Mansfield C, Moussy A, Hermine O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment. Front Aging Neurosci 2017; 9: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol 2011; 10: 253–63. [DOI] [PubMed] [Google Scholar]
- Picher-Martel V, Dutta K, Phaneuf D, Sobue G, Julien JP. Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol Brain 2015; 8: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompl PN, Ho L, Bianchi M, McManus T, Qin W, Pasinetti GM. A therapeutic role for cyclooxygenase-2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 2003; 17: 725–7. [DOI] [PubMed] [Google Scholar]
- Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 2015; 130: 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcelot M, Zemirli N, Silva Da Costa L, Loyant R, Garcin D, Vitour D, et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol 2016; 14: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci 2001; 21: 3369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol 2017; 18: 385–92. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 2014; 15: 300–12. [DOI] [PubMed] [Google Scholar]
- Pullen AH, Demestre M, Howard RS, Orrell RW. Passive transfer of purified IgG from patients with amyotrophic lateral sclerosis to mice results in degeneration of motor neurons accompanied by Ca2+ enhancement. Acta Neuropathol 2004; 107: 35–46. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science 2016; 353: 777–83. [DOI] [PubMed] [Google Scholar]
- Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014; 81: 1001–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014; 17: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 2016; 113: 4039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Río-Hortega P. El “Tercer Elemento” de los Centros Nerviosos I. La Microglia en Estado Normal. Bol Soc Esp Biol 1919. a; VIII: 67–82. [Google Scholar]
- Río-Hortega P. El “tercer elemento de los Centros Nerviosos” IV. Poder fagocitario y movilidad de la microglia. Bol Soc Esp Biol 1919. b; VIII: 154–66. [Google Scholar]
- Roberts K, Zeineddine R, Corcoran L, Li W, Campbell IL, Yerbury JJ. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia 2013; 61: 409–19. [DOI] [PubMed] [Google Scholar]
- Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity 2016; 44: 739–54. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362: 59–62. [DOI] [PubMed] [Google Scholar]
- Rowin J, Xia Y, Jung B, Sun J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol Rep 2017; 5: e13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LP. HIV-related neuromuscular diseases: nemaline myopathy, amyotrophic lateral sclerosis and bibrachial amyotrophic diplegia. Acta Myol 2011; 30: 29–31. [PMC free article] [PubMed] [Google Scholar]
- Rusconi M, Gerardi F, Santus W, Lizio A, Sansone VA, Lunetta C, et al. Inflammatory role of dendritic cells in amyotrophic lateral sclerosis revealed by an analysis of patients’ peripheral blood. Sci Rep 2017; 7: 7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science 2016; 353: 783–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako W, Ito H, Yoshida M, Koizumi H, Kamada M, Fujita K, et al. Nuclear factor kappa B expression in patients with sporadic amyotrophic lateral sclerosis and hereditary amyotrophic lateral sclerosis with optineurin mutations. NP 2012; 31: 418–23. [DOI] [PubMed] [Google Scholar]
- Saresella M, Piancone F, Tortorella P, Marventano I, Gatti A, Caputo D, et al. T helper-17 activation dominates the immunologic milieu of both amyotrophic lateral sclerosis and progressive multiple sclerosis. Clin Immunol 2013; 148: 79–88. [DOI] [PubMed] [Google Scholar]
- Schludi MH, Becker L, Garrett L, Gendron TF, Zhou Q, Schreiber F, et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol 2017; 134: 241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schram S, Chuang D, Schmidt G, Piponov H, Helder C, Kerns J, et al. Mutant SOD1 prevents normal functional recovery through enhanced glial activation and loss of motor neuron innervation after peripheral nerve injury. Neurobiol Dis 2019; 124: 469–78. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Deczkowska A. Neurological disease as a failure of brain-immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol 2016; 37: 668–79. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Shechter R. Systemic inflammatory cells fight off neurodegenerative disease. Nat Rev Neurol 2010; 6: 405–10. [DOI] [PubMed] [Google Scholar]
- Seksenyan A, Ron-Harel N, Azoulay D, Cahalon L, Cardon M, Rogeri P, et al. Thymic involution, a co-morbidity factor in amyotrophic lateral sclerosis. J Cell Mol Med 2010; 14: 2470–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Campanari M‐L, Julie Corbier C, Gaucherot A, Kolb‐Cheynel I, Oulad‐Abdelghani M, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 2016; 35: 1276–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A 2013; 110: 4697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheean RK, McKay FC, Cretney E, Bye CR, Perera ND, Tomas D, et al. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol 2018; 75: 681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, An H, Skelt L, Tregoning JS, Humphreys IR, Buchman VL. Antiviral immune response as a trigger of FUS proteinopathy in amyotrophic lateral sclerosis. Cell Rep 2019; 29: 4496–508.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi N, Kawano Y, Tateishi T, Kikuchi H, Osoegawa M, Ohyagi Y, et al. Increased IL-13-producing T cells in ALS: positive correlations with disease severity and progression rate. J Neuroimmunol 2007; 182: 232–5. [DOI] [PubMed] [Google Scholar]
- Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 2018; 18: 759–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, de Castro F, Del Río-Hortega J, Rafael Iglesias-Rozas J, Garrosa M, Kettenmann H. The “Big-Bang” for modern glial biology: translation and comments on Pío del Río-Hortega 1919 series of papers on microglia. Glia 2016; 64: 1801–40. [DOI] [PubMed] [Google Scholar]
- Sinha S, Mathews T, Arunodaya GR, Siddappa NB, Ranga U, Desai A, et al. HIV-1 clade-C-associated “ALS”-like disorder: first report from India. J Neurol Sci 2004; 224: 97–100. [DOI] [PubMed] [Google Scholar]
- Slowicka K, Vereecke L, Mc Guire C, Sze M, Maelfait J, Kolpe A, et al. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-kappaB signaling. Eur J Immunol 2016; 46: 971–80. [DOI] [PubMed] [Google Scholar]
- Smith AM, Sewell GW, Levine AP, Chew TS, Dunne J, O'Shea NR, et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology 2015; 144: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon JN, Lewis CA, Ajami B, Corbel SY, Rossi FM, Krieger C. Origin and distribution of bone marrow-derived cells in the central nervous system in a mouse model of amyotrophic lateral sclerosis. Glia 2006; 53: 744–53. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Nervenkitt: notes on the history of the concept of neuroglia. Glia 1988; 1: 2–9. [DOI] [PubMed] [Google Scholar]
- Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci 2018; 21: 329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sta M, Sylva-Steenland RMR, Casula M, de Jong JMBV, Troost D, Aronica E, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis 2011; 42: 211–20. [DOI] [PubMed] [Google Scholar]
- Steinacker P, Verde F, Fang L, Feneberg E, Oeckl P, Roeber S, et al. Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cord of amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J Neurol Neurosurg Psychiatry 2018; 89: 239–47. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun 2016; 4: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011. a; 134: 2610–26. [DOI] [PubMed] [Google Scholar]
- Swarup V, Phaneuf D, Dupré N, Petri S, Strong M, Kriz J, et al. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med 2011. b; 208: 2429–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AG, Gray E, Thézénas M-L, Charles PD, Evetts S, Hu MT, et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann Neurol 2018; 83: 258–68. [DOI] [PubMed] [Google Scholar]
- Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: a phase I, first-in-human study. Neurol Neuroimmunol Neuroinflamm 2018; 5: e465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trias E, Diaz-Amarilla P, Olivera-Bravo S, Isasi E, Drechsel DA, Lopez N, et al. Phenotypic transition of microglia into astrocyte-like cells associated with disease onset in a model of inherited ALS. Front Cell Neurosci 2013; 7: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trias E, King PH, Si Y, Kwon Y, Varela V, Ibarburu S, et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018; 3: e123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troost D, van den Oord JJ, de Jong JM, Swaab DF. Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin Neuropathol 1989; 8: 289–94. [PubMed] [Google Scholar]
- Turner MR, Cagnin A, Turkheimer FE, Miller CCJ, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 2004; 15: 601–9. [DOI] [PubMed] [Google Scholar]
- Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology 2013; 81: 1222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 2013; 16: 543–51. [DOI] [PubMed] [Google Scholar]
- Uranishi H, Tetsuka T, Yamashita M, Asamitsu K, Shimizu M, Itoh M, et al. Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-κB p65-mediated transcription as a coactivator. J Biol Chem 2001; 276: 13395–401. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 2006; 9: 108–18. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Rodriguez JJ, Parpura V. Astroglia in neurological diseases. Future Neurol 2013; 8: 149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu L, An J, Kovalik T, Gendron T, Petrucelli L, Bowser R. Cross-sectional and longitudinal measures of chitinase proteins in amyotrophic lateral sclerosis and expression of CHI3L1 in activated astrocytes. J Neurol Neurosurg Psychiatry 2020; 91: 350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann R, Hüttenrauch M, Kacprowski T, Bouter Y, Pradier L, Bayer TA, et al. Gene expression profiling in the APP/PS1KI mouse model of familial Alzheimer’s disease. J Alzheimers Dis 2016; 50: 397–409. [DOI] [PubMed] [Google Scholar]
- Welser-Alves JV, Crocker SJ, Milner R. A dual role for microglia in promoting tissue inhibitor of metalloproteinase (TIMP) expression in glial cells in response to neuroinflammatory stimuli. J Neuroinflammation 2011; 8: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosiski-Kuhn M, Lyon MS, Caress J, Milligan C. Inflammation, immunity, and amyotrophic lateral sclerosis: II. Immune-modulating therapies. Muscle Nerve 2019. a; 59: 23–33. [DOI] [PubMed] [Google Scholar]
- Wosiski-Kuhn M, Robinson M, Strupe J, Arounleut P, Martin M, Caress J, et al. IL6 receptor358Ala variant and trans-signaling are disease modifiers in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm 2019. b; 6: e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne AM, Henry CJ, Huang Y, Cleland A, Godbout JP. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav Immun 2010; 24: 1190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue YC, Feuer R, Cashman N, Luo H. Enteroviral infection: the forgotten link to amyotrophic lateral sclerosis. Front Mol Neurosci 2018. a; 11: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue YC, Ruller CM, Fung G, Mohamud Y, Deng H, Liu H, et al. Enteroviral infection leads to transactive response DNA-binding protein 43 pathology in vivo. Am J Pathol 2018. b; 188: 2853–62. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 2008; 11: 251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Cheung J, Gerbino V, Ahlsén G, Zimanyi C, Hirsh D, et al. Effects of ALS-associated TANK binding kinase 1 mutations on protein-protein interactions and kinase activity. Proc Natl Acad Sci U S A 2019; 116: 24517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Fang F. Smoking and amyotrophic lateral sclerosis: a Mendelian randomization study. Ann Neurol 2019; 85: 482–4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu Y, Liu X, Li S, Cheng C, Chen S, et al. Dynamic changes of CX3CL1/CX3CR1 axis during microglial activation and motor neuron loss in the spinal cord of ALS mouse model. Transl Neurodegener 2018; 7: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015; 525: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Gascon R, Miller RG, Gelinas DF, Mass J, Hadlock K, et al. Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol 2005; 159: 215–24. [DOI] [PubMed] [Google Scholar]
- Zhang Y-G, Wu S, Yi J, Xia Y, Jin D, Zhou J, et al. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin Ther 2017; 39: 322–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol 2013; 8: 888–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Bell S, Wang J, Wen S, Baloh RH, et al. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp Neurol 2015; 273: 24–35. [DOI] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, Julien J-P, et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010; 58: 231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Hooten KG, Sieglaff DH, Zhang A, Kalyana-Sundaram S, et al. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol 2017; 74: 677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Liao B, Henkel JS, Appel SH. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol Dis 2012; 48: 418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol 2007; 17: 1438–43. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 2006; 9: 268–75. [DOI] [PubMed] [Google Scholar]
- Zondler L, Müller K, Khalaji S, Bliederhäuser C, Ruf WP, Grozdanov V, et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol 2016; 132: 391–411. [DOI] [PubMed] [Google Scholar]