

Abstract
To aid in generating complex and diverse natural glycan libraries for functional glycomics, more efficient and reliable methods are needed to derivatize glycans. Here we present our development of a reversible, cleavable bifunctional linker 3-(methoxyamino)propylamine (MAPA). As the fluorenylmethyloxycarbonate (Fmoc) version (F-MAPA), it is highly fluorescent and efficiently derivatizes free reducing glycans to generate closed-ring derivatives that preserve the structural integrity of glycans. A library of glycans were derivatized and used to generate a covalent glycan microarray using N-hydroxysuccinimide derivatization. The array was successfully interrogated by a variety of lectins and antibodies, demonstrating the importance of closed-ring chemistry. The glycan derivatization was also performed at large scale using mg quantities of glycans and excess F-MAPA, and the reaction system was successfully recycled up to 5 times, without apparent decrease in conjugation efficiency. The MAPA-glycan is also easy to link to protein to generate neoglycoproteins with equivalent glycan densities. Importantly, the MAPA linker can be reversibly cleaved to regenerate free reducing glycans for detailed structural analysis (catch-and-release), often critical for functional studies of undefined glycans from natural sources. The high conjugation efficiency, bright fluorescence, and reversible cleavage of the linker enable access natural glycans for functional glycomics.
Graphic Abstract
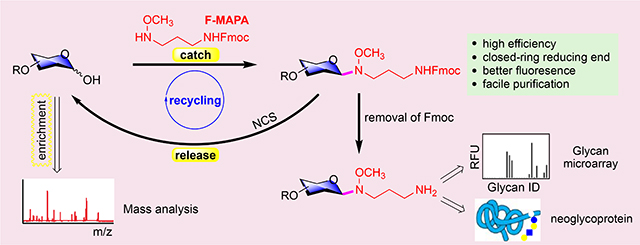
Complex carbohydrates (glycans) are essential constituents of all living organisms, occurring as both simple and complex structures in glycoproteins, proteoglycans, glycolipids, and as free glycans. Beyond their roles in energy storage and structural support, glycans via their interactions with glycan-binding proteins (GBPs) are important in numerous physiological and pathological processes, such as platelet clearance, cellular adhesion and migration, innate immune responses, fertilization, embryogenesis, pathogen infection, inflammation, and the development of autoimmune diseases and cancer1. Therefore, more and more therapeutic agents and diagnostic tools targeting on glycan-GBP interactions are under development2–7. Despite its well-recognized importance, glycomics, the structures and functions of glycans in biological systems, has lagged far behind genomics, transcriptomics and proteomics. This lag is mainly due to the unique structural complexity of glycans, the non-template driven synthesis of glycans and the indirect regulation of glycan synthesis by genes, which create unique technical challenges for glycan sequencing and synthesis, thereby hindering access to complex glycans for functional studies.
The ability to derivatize glycans to various supports, as in glycan microarray technologies and microbead presentations, has provided key insights into glycans recognition and studies to explore roles of glycans in cell adhesion and signaling, as well as recognition of glycans by viruses, antibodies, and various GBPs. These technologies require small quantities of glycans and permit rapid analysis of binding to hundreds of test glycans in relatively simple high-throughput assays formats8–12. The limitation of such technologies is the lack of diversity of synthetic glycans, which are limited because of the difficulties in glycan synthesis, as well as the questionable relevance of such limited repertoires of glycans to complex biological processes. One approach to circumvent these limitations is to acquire glycans from natural sources, e.g. organs, tissues, cells, bacteria, etc., which contain large glycomes of biological context and perhaps more relevant biological activities. However, while the isolation and purification of glycans from natural sources is a desirable strategy, there are many difficulties largely due to the lack of reversible and facile tagging methods for free, reducing glycans prepared from such sources.
To utilize glycans from natural sources, glycans need to be derivatized with functional tags to enable separation and purification by multi-dimensional chromatography, detection and quantification of purified glycans or sub-fractions, as well as subsequent immobilization on array surface and other applications13–17. Much effort has been devoted toward the development of linkers (or tags) for glycan derivatization18–22. Based on the chemistry of derivatization, these linkers can be classified into two categories, reductive amination and N-alkyl oxime ligation. Three representative linkers of these two categories are 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE), 2-amino-N-(2-amino-ethyl)-benzamide (AEAB) and 2-[(methylamino)oxy]ethylamine (AMNO) (Fig. 1a). DHPE and AEAB react with reducing glycans through reductive amination and open sugar ring of reducing end monosaccharide residue14, 23, which destroys the reducing end integrity of glycans, which may affect their binding and immunogenicity of glycans24, especially for small glycan epitopes. AMNO reacts with reducing glycans through N-alkyl oxime mediated selective ligation and forms closed-ring reducing end, which preserves the integrity of glycans25. Nevertheless, AMNO lacks a fluorophore, making it difficult to detect, separate and quantify. Thus, development of fluorescent linker, which can derivatize glycans efficiently and preserve closed-ring reducing end, is highly desired.
Figure 1.

Rationale for linker design.
Recently, Jiménez-Castells et al and Prudden et al developed fluorescent N-alkyl oxime linkers which can form closed-ring reducing end (Fig. 1a)26, 27. However, these two linkers required complex synthesis and gave moderate conjugation yields. Moreover, long reaction time under acidic conditions (72 h, pH 4.6) rendered them incompatible with labile modifications (eg. sulfation) and sialic acid residues typically found in glycans, thus substantially limiting their utility, especially for derivatization of natural glycans.
Here, we present a multifunctional fluorescent linker 3-(methoxyamino)propylamine (MAPA) with an Fmoc fluorescent addition, F-MAPA, for glycan derivatization, which can efficiently conjugate with glycans (synthetic and natural glycans) under mild conditions and form closed-ring reducing end (Fig. 1b). After conjugation with F-MAPA, the resulting Glycan-F-MAPA conjugates (GFMAPAs) can be purified by solid phase extraction (SPE), and Fmoc in the linker can be easily removed to generate an active alkyl amine enabling further manipulation and application of glycans for multiple purposes28–30. The linker can also be easily cleaved under mild conditions to regenerate free reducing glycans for detailed structure analysis by mass spectrometry (MS). The efficiency and reversibility of glycan derivatization as well as the enhanced detectability afforded by F-MAPA will promote novel studies of minor and major glycans in natural sources and their functional recognition by proteins.
RESULTS
Synthesis of MAPA.
We considered that N-alkyl oxime can stereoselectively couple with the hemiacetal of glycan reducing end to preserve the cyclic nature31, thus rendering development of fluorescent N-alkyl oxime linker highly attractive. To achieve high conjugation efficiency, the linker should be relatively small. In addition, its synthesis should be facile, efficient, scalable and cost-effective to enable general accessibility to research laboratories and potential industrial application. In these regards, linker F-MAPA was designed, which contains an N-alkyl oxime active motif and an Fmoc protected alkyl amine. Fmoc serves as a transient fluorophore for monitoring and quantification, as well as a hydrophobic tag for glycan enrichment, separation and purification. As shown in Fig. 2, F-MAPA (1) and its hydrochloride salt 2 were facilely and efficiently synthesized in 10 g scale.
Figure 2.

Synthesis of F-MAPA linker.
Glycan and Linker Conjugation Condition Optimization.
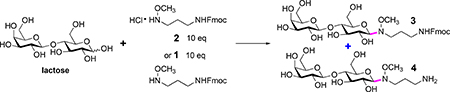
The conjugation of N-alkyl oxime with reducing glycans was generally conducted in pH 4.5 aqueous buffer. Nevertheless, due to poor solubility of 1 and 2 in water, combinations of aqueous buffer with methanol and dimethyl sulfoxide (DMSO), DMSO, DMSO/HOAc were tested as solvent for the conjugation of lactose and 2 (Table 1. Entry 1–4). When MeOH/NaOAc buffer and DMSO/HOAc were used as solvent, 40% conversions were given and conjugation product 3 was provided exclusively with 33% and 31% yields respectively (entry 1 & 4). DMSO/NaOAc buffer gave higher conversion and yield, but a mixture of 3 and 4 were generated (entry 2). The low conversion in DMSO (entry 3) might be attributed to its low polarity, which suppressed the dissociation of protonated N-alkyl oxime, thereby blocking ligation of N-alkyl oxime to reducing end hemiacetal of glycans. Thus, unprotonated 1 or 2 with base additive were screened (entry 5–7). Indeed, conversions and yields were substantially increased in all three scenarios. Nevertheless, 1 or 2 with N,N-diisopropylethylamine (DIPEA) gave a mixture of 3 and 4 (entry 5 & 6), whereas 2 with NaOAc provided 4 exclusively (entry 7). According to the results above, DMSO/HOAc provides 3 exclusively and base increases conversion and yield. Therefore, DMSO/HOAc was chosen as solvent, and 1 or 2 with base additive was used (entry 8–12). When 1 was used as linker, 3 was provided exclusively with 89% yield (entry 8), whereas addition of base led to formation of a mixture of 3 and 4 (entry 9 & 10). For 2, addition of base gave higher yields and 3 was provided exclusively (entry 11 & 12 vs entry 4). When conjugation temperature was increased to 65 °C, reaction time was substantially reduced to 2 hours and 3 was provided with high yields (entry 13–15). Nevertheless, in consideration of conjugation efficiency, convenient linker handling and storage, the combination of 2 (white powder) and NaOAc were chosen. Conjugation conditions were set to DMSO/HOAc as solvent and heating at 65 °C for 2 hours (entry 15), which had been demonstrated to be compatible with natural glycans in previous works14, 32, 33.
Table 1.
Glycan and linker conjugation condition optimization.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | linker | solvent | base | temp. | time | conversiona | yieldb |
|
| 3 | 4 | |||||||
| 1 | 2 | MeOH/NaOAc buffer (pH 4.5), 1/1, v/v | 40 °C | 48 h | 40% | 33% | 0% | |
| 2 | 2 | DMSO/NaOAc buffer (pH 4.5), 1/1, v/v | 40 °C | 48 h | 50% | 24% | 21% | |
| 3 | 2 | DMSO | 40 °C | 48 h | < 5% | ND | ND | |
| 4 | 2 | DMSO/HOAc 7/3, v/v | 40 °C | 48 h | 40% | 31% | 0% | |
| 5 | 1 | DMSO | 40 °C | 48 h | 80% | 53% | 17% | |
| 6 | 2 | DMSO | DIPEA, 3 eq | 40 °C | 48 h | 75% | 11% | 52% |
| 7 | 2 | DMSO | NaOAc, 3 eq | 40 °C | 12 h | 100% | 0% | 91% |
| 8 | 1 | DMSO/HOAc 7/3, v/v | 40 °C | 36 h | > 95% | 89% | 0% | |
| 9 | 1 | DMSO/HOAc 7/3, v/v | DIPEA 10 eq | 40 °C | 36 h | > 95% | 35% | 50% |
| 10 | 1 | DMSO/HOAc 7/3, v/v | NaOAc 10 eq | 40 °C | 36 h | > 95% | 52% | 31% |
| 11 | 2 | DMSO/HOAc 7/3, v/v | DIPEA 10 eq | 40 °C | 36 h | > 95% | 87% | 0% |
| 12 | 2 | DMSO/HOAc 7/3, v/v | NaOAc 10 eq | 40 °C | 36 h | > 95% | 88% | 0% |
| 13 | 1 | DMSO/HOAc 7/3, v/v | 65 °C | 2 h | 100% | 90% | 0% | |
| 14 | 2 | DMSO/HOAc 7/3, v/v | DIPEA 10 eq | 65 °C | 2 h | 100% | 88% | 0% |
| 15 | 2 | DMSO/HOAc 7/3, v/v | NaOAc 10 eq | 65 °C | 2 h | 100% | 91% | 0% |
Based on lactose (5 mg)
Isolated yield
ND: not determined; eq: equivalent.
Derivatization of Reducing Glycans.
To demonstrate the utility of F-MAPA linker, 24 commercially available blood group glycans were derivatized with 2 in micro-gram scale. 5-Methoxyanthranilic acid (5MA) was reported to promote ligation of N-alkyl oxime and free reducing glycans34, 35. To assure complete conversion of glycans, 100 equivalents of 2 were used and 0.2 equivalent of 5MA was added. The reaction mixture was shaken at 65°C for 2 hours, then 10 volume of ethyl acetate were added to precipitate GFMAPAs. The precipitates were collected and purified by C-18 SPE to provide GFMAPAs with high yields (Fig. 3). The 24 blood group glycans were also derivatized with AEAB for comparison and further evaluation of F-MAPA linker in microarray analysis.
Figure 3.

Derivatization of blood group glycans with F-MAPA.
Microarray Analysis of F-MAPA- and AEAB-labeled Glycans.
Fmoc was found to be a good hydrophobic tag for reverse phase purification of GFMAPAs. We tried to print GFMAPAs directly on nitrocellulose and polystyrene slides, but unfortunately, GFMAPAs do not attach to either slide, although a wide range of concentrations were tested (from 50 μM up to 1 mM). Thus, we removed the Fmoc by piperidine to give MAPA-labeled glycans in quantitative yield, which provided active alkyl amine and allowed printing on NHS-activated slides. MAPA- and AEAB-labeled glycans were printed on NHS-activated slides at the same concentration (50 μM). Probe 1a-24a were AEAB-labeled blood group glycans with open-ring reducing ends. Probe 1b-24b were MAPA-labeled blood group glycans with closed-ring reducing ends. Probe 1-5 were H antigens, 6-7 were A antigens, 8-14 were B antigens, and 15-24 were Lewis antigens and analog. Probe 25a is biotinylated hydrazide and probe 25b is sodium phosphate. Quality of the array was validated by specificity well defined plant lectins, and then the array was interrogated by anti-blood group antibodies.
Aleuria aurantia lectin (AAL) and Ulex europaeus agglutinin I (UEA-I) are fucose binding lectins. AAL recognizes terminal fucose residues and core fucose residues36, whereas UEA-I is specific for α1–2-linked fucose of terminal Fucα1–2Galβ1–4GlcNAc/Glc determinants37. As expected, both AEAB- and MAPA-labeled blood group H glycans (1-5) and Lewis antigens (15-24) exhibited strong binding with AAL (Fig. 4a), whereas blood group A glycans (6 & 7) and blood group B glycans (9-13) generally did not bind AAL. Interestingly, although both AEAB- and MAPA-labeled 8 and 14 bound to AAL, the binding of AEAB-labeled 8b and 14b with AAL was much stronger, which might result from increased accessibility of fucose residues by AAL due to open-ring reducing end. In agreement with previous reports, only H2 and H5 glycans (1, 2 & 4), Ley (17 & 18) and 2,2’-FL (24) were recognized by UEA-I (Fig. 4b), and the binding of 17a is much weaker than that of 17b. Griffonia simplicifolia I-B4 isolectin (GSL-I B4) and Dolichos biflorus lectin (DBA) recognize terminal Galα-38 and GalNAcα1–3GalNAc/Gal39, 40 respectively. Therefore, only blood group B glycans (8-14) were bound by GSL-I B4 (Fig. 4c), and blood group A glycans (6&7) were bound by DBA (Fig. 4d). Altogether, the lectin studies demonstrated that all probes were well printed on the array, and that overall, the lectin binding patterns of AEAB- and MAPA-labeled glycans were similar or almost the same.
Figure 4.

Recognition of blood group antigens by plant lectins and anti-blood group antibodies.
To further investigate the influence of linker on glycan recognition by GPBs, the glycan microarray was interrogated by human dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), and monoclonal anti-CD15, anti-SLex/SLea, anti-Lea antibodies. DC-SIGN bound to H2, H5 (1, 2, 4) and Lewis glycans (15-18 & 21-23), which corroborated previous studies on the glycan specificity of DC-SIGN41–43. Notably, glycan 14 and 24 exhibited strong binding, whereas glycan 8 were not bound by DC-SIGN (Fig. 4e), which suggested that 2,2’-FL was a new binding motif of DC-SIGN. Moreover, DC-SIGN bound to MAPA-labeled Lex trisaccharide 15b but not AEAB-labeled Lex trisaccharide 15a, indicating the importance of preserving intact closed-ring reducing end for glycan recognition. Similarly, anti-CD15 antibody and anti-SLex/SLea antibody bound to MAPA-labeled 15b and 19b respectively, but not AEAB-labeled 15a or 19a (Fig. 4f & 4g). In addition, MAPA-labeled 21b exhibited strong binding, but AEAB-labeled 21a was weakly bound by the anti-Lea antibody (Fig. 4h). These preference towards MAPA-labeled glycans further highlight the importance of preserving intact closed-ring reducing ends for glycan recognition by GBPs.
Reaction System Recycling and Cleavage of Linker to Regenerate Free Reducing Glycans.
Given the high conjugation efficiency and facile purification procedure, we envisioned that F-MAPA could be applied in large scale glycan derivatization and the reaction system could be recycled. As a demonstration, LNnT was derivatized with F-MAPA in preparative scale. LNnT (20 mg) was dissolved in DMSO/HOAc, then 100 equivalents of F-MAPA and 0.2 equivalent of 5MA were added. After stirring at 65°C for 4 hours, LNnT was completely consumed. Then, 10 volumes of ethyl acetate were added to precipitate LNnT-F-MAPA conjugate. The precipitate was collected by centrifugation and purified by C-18 SPE to give LNnT-F-MAPA with 86% yield. The supernatant was concentrated to remove ethyl acetate, and 20 mg LNnT was added once again (Fig. 5a). The mixture was stirred at 65°C for another 4 hours and LNnT-F-MAPA was generated with 83% yield. The reaction system was recycled for 5 times and there was no apparent deterioration in the conjugation efficiency, with the yield of LNnT-F-MAPA ranging from 86% in the first cycle to 78% in the sixth cycle (Fig. 5b). In addition, after 6 cycles, linker F-MAPA was also recovered with 80% yield. The successful recycling of reaction system and recovery of linker render linker F-MAPA hold great potential for industrial-scale application.
Figure 5.

a) Recycling of reaction system. b) Conjugation efficiency of F-MAPA in recycling. c) cleavage of linker to regenerate free reducing glycans.
F-MAPA/MAPA tag can increase MS sensitivity compared with unmodified glycans, nevertheless it also complicates permethylation of glycans and limits more detailed structural analysis by MS, which is critical for the functional study of undefined glycans from natural sources. Anomeric N-alkyl oxime can be hydrolyzed under acidic conditions44, 45, however, the challenge was to establish conditions to cleave the N-alkyl oxime linker without affecting the labile fucose and sialic acid residues. N-chlorosuccinimide (NCS) was reported to cleave anomeric N-alkyl oxime with high yield, nevertheless, only protected mono- or di-saccharides were tested46. Therefore, NCS was explored for the cleavage of F-MAPA linker. LNnT-F-MAPA and LNnT-MAPA were treated with NCS in water. Importantly, both F-MAPA and MAPA were cleanly cleaved by NCS in 2 hours and LNnT (confirmed by MS and NMR) was regenerated with 82% and 81% yield, respectively (Fig.5c and Fig. S1). To further demonstrate the robustness of NCS mediated cleavage of N-alkyl oxime linker, fucosylated and sialylated 20b was incubated with NCS in water for 2 hours. The F-MAPA linker was selectively removed and reducing glycan 20 was provided in 80% yield without affecting labile sialic acid and fucose residues (Fig. S2). The ability to add and remove F-MAPA in a facile manner brings versatility to F-MAPA-derivatized glycans, facilitating both glycomics studies and functional glycan recognition.
Synthesis of Neoglycoprotein.
After removal of Fmoc, an active alkyl amine is generated, which enables further manipulation and application of MAPA-labeled glycans. As an example, LNnT-MAPA was applied in the synthesis of neoglycoprotein. LNnT-MAPA was first conjugated to NHS-activated suberic acid spacer, then the LNnT-suberate conjugate was coupled to alkyl amine side chain of lysine residues on bovine serum albumin (BSA) to form neoglycoprotein (Fig. 6a). Since the conjugation of LNnT-MAPA and NHS-activated suberic acid was quantitative, LNnT-suberate was conjugated with BSA at different molar ratios to further evaluate the conjugation of LNnT-MAPA with proteins. As expected, the average number of glycan conjugated on per BSA is dependent on the ratio of LNnT-MAPA to BSA. At 10:1 ratio, ∼4–5 LNnT/BSA were conjugated; at 25:1 ratio, ∼10–11 LNnT/BSA were conjugated; at 50:1 ratio, ∼16–17 LNnT/BSA were conjugated (Fig. 6b, 6c and S3).
Figure 6.

Synthesis of neoglycoprotein. a) Synthetic scheme. b) Mass spectra of LNnT-BSA conjugates. c) Average number of glycan on BSA.
Derivatization of Natural Glycans.
To further demonstrate the utility of F-MAPA linker, same amount of crude egg yolk N-glycans (released by ORNG method47, 1.5 mg) were conjugated with F-MAPA versus AEAB. F-MAPA- and AEAB-labeled egg yolk N-glycans were quantified according to their fluorescence, and F-MAPA gave slightly higher yield (0.201 μmol vs 0.193 μmol, Fig. 7a). F-MAPA- and AEAB-labeled N-glycans were normalized to the same concentration and injected into HPLC (NH2 HILIC column). As shown in Fig. 7b, the fluorescence intensity of F-MAPA was substantially higher than that of AEAB. In addition, according to the fluorescent standard curves of F-MAPA -, AEAB- and anthranilamide (2-AB)-labeled LNnT, F-MAPA was 18-fold brighter than AEAB and 16-fold brighter than 2-AB (Fig. S4), which could enable more sensitive detection and quantification during isolation, purification and LC/MS analysis of F-MAPA-labeled natural glycans, thereby facilitating the development of natural glycan microarray. Then, F-MAPA-labeled egg yolk N-glycans were treated by NCS. F-MAPA was selectively removed and reducing egg yolk N-glycans were regenerated. This catch-and release chemoselectively isolated reducing egg yolk N-glycans from the crude mixture, which suggested that F-MAPA could potentially be used for the enrichment of reducing glycans in biological samples (urine, blood, human milk, etc.).
Figure 7.

a) Derivatization of crude egg yolk N-glycans with F-MAPA and AEAB. b) HPLC profiles of F-MAPA- and AEAB-labeled egg yolk N-glycans.
DISCUSSION
Our results demonstrate that we have successfully developed a highly efficient, fluorescent and reversible tagging approach for reducing glycans that promotes their detection, separation, and analysis, as well as providing a linker to covalently coupling glycans to other molecules, including proteins and biotin. The fluorescent linker F-MAPA is easy to synthesize and its fluorescence is more sensitive than conventional fluorophores by many folds, such as 2-AB. The catch-and-release advantages of F-MAPA, as well as the reversibility of tagging, provides an unprecedented opportunity to detect and functionally assess minor quantities of glycans from natural sources. The availability of glycans from natural sources using F-MAPA as a linker will enable both glycan analytical and purification approaches and enlarge the availability of glycans to explore for functional recognition.
Despite the well-recognized importance of glycans in numerous physiopathological processes and medicine, functional glycomics study has been impeded by limited access to complex glycans. Tremendous progress has been achieved in the synthesis of glycans48–52, and several defined glycan microarrays with large glycan libraries have been developed. Nevertheless, the synthetic approach still lags far behind the explosive need for more diverse and biologically relevant glycans for biomedical research. Synthesis of modified mammalian glycans (phosphorylation, sulfation, etc.), plant and microbial glycans is still challenging, due to a lack of general chemical glycosylation protocols53, 54 and limited types of glycosyltransferases available at current stage55, 56. More importantly, synthetic approach is target-driven, but often the actual biological target is unknown. On the other hand, glycans from a natural source (tissue, bacteria, cells, organisms) contain the entire glycome of interest and are directly implicated in the biological issues probed.
The power of natural glycan microarrays has been highlighted in several reports32, 33, 57, 58, and one of the key aspects for development of natural glycan microarrays is efficient and reliable method to derivatize glycans. The F-MAPA linker described here can efficiently derivatize glycans and facilitate glycan-F-MAPA conjugate separation/purification. Synthesis of the linker was facile, efficient and cost-effective, with 10 g of F-MAPA being synthesized in one batch in a few days. Considering operational simplicity of the synthetic design, the synthesis can easily be reproduced and scaled up in other research laboratories. The linker will also be available to the research community via our center with cost for shipping and handling to assure general accessibility.
To demonstrate the utility of F-MAPA, blood group glycans were derivatized with F-MAPA in microgram scale and printed on NHS-activated slides. The resulting glycan microarray was validated by specificity well defined plant lectins. The lectin binding patterns of AEAB- and MAPA-labeled glycans were similar or almost the same. Then, the array was interrogated by anti-blood group antibodies. 2,2’-FL was identified as a new binding motif of human DC-SIGN. The binding preference towards MAPA labeled 15b, 19b, and 21b by DC-SIGN and anti-Lewis antibodies highlighted the importance of preserving intact closed-ring reducing end for glycan recognition by GBPs.
Given the high conjugation efficiency and facile purification procedure of F-MAPA mediated glycan derivatization, LNnT was conjugated with F-MAPA in preparative scale and the reaction system was recycled for 5 times. The yield of LNnT-F-MAPA conjugate ranged from 86% in the first cycle to 78% in the sixth cycle, without apparent deterioration in the conjugation efficiency. In addition, after 6 cycles, the linker F-MAPA was also recovered with 80% yield. After removal of Fmoc, active alkyl amine is generated, enabling further manipulation and application of MAPA-labeled glycans. LNnT-MAPA was coupled to BSA to synthesize neoglycoproteins. The average number of glycans conjugated on per BSA is dependent on the ratio of LNnT-MAPA to BSA. The successful recycling of the reaction system and synthesis of neoglycoproteins render F-MAPA hold great potential for industrial synthesis of neoglycoprotein therapeutics and vaccines.
To further investigate the scope of F-MAPA-mediated glycan derivatization, same amount of crude egg yolk N-glycans (released by bleach) were conjugated with F-MAPA and AEAB separately. F-MAPA gave slightly higher yield (0.201 μmol vs 0.193 μmol) than AEAB, and the fluorescence of F-MAPA was 18-fold brighter than that of AEAB, which could enable efficient derivatization, more sensitive detection and quantification of F-MAPA-labeled natural glycans, thereby facilitating the development of natural glycan microarray. Upon treatment of F-MAPA-labeled glycans with NCS, F-MAPA was removed and reducing egg yolk N-glycans were regenerated, thereby enabling detailed glycan structural analysis by MS. This catch-and-release chemoselectively isolated reducing egg yolk N-glycans from the crude mixture, which suggested that F-MAPA could potentially be used for the enrichment of reducing glycans in biological samples (urine, blood, human milk, etc.).
CONCLUSIONS
Natural glycans from biological sources contain the whole glycome of interest and are more biologically relevant. Expanding the glycan microarray technology with natural glycans represents the most attractive strategy to access complex and diverse glycan libraries for functional glycomics. For this approach to be effective in practice, an efficient and reliable method to derivatize glycans is needed. We developed a multifunctional fluorescent linker F-MAPA, which can efficiently derivatize reducing glycans under mild conditions and preserve the structure integrity. F-MAPA can also be facilely cleaved under mild conditions to regenerate free reducing glycans for detailed structure analysis by MS, which is the key for functional glycomics of undefined natural glycan microarrays. This catch-and-release approach also allows selectively enrichment of free reducing glycans from biological samples, which has vast potential applications in the bioanalysis and diagnosis. The facile and scalable synthesis of F-MAPA, high conjugation efficiency and operational simplicity of F-MAPA mediated glycan derivatization make the linker accessible to general research laboratories and allow derivatization of glycans by biologists, thereby enabling general accessibility of glycan microarrays and customized development of glycan microarrays by researchers themselves. Moreover, the successful recycling of reaction system and synthesis of neoglycoprotein render F-MAPA hold great potential for its industrial application in the synthesis of neoglycoprotein therapeutics and vaccines.
METHODS
Synthesis of F-MAPA.
To a stirred solution of 3-aminopropionaldehyde diethylacetal (7.36 g, 50 mmol, 1 eq) and N,N-diisopropylethylamine (DIPEA, 12.93 g, 100 mmol, 2 eq) in dichloromethane (DCM, 250 mL), Fmoc-Cl (13.58 g, 52.5 mmol, 1.05 eq in 30 mL DCM) was added dropwise at 0 °C. After completion of addition, the mixture was allowed to warm to rt and stirred overnight. The reaction mixture was washed with 1 M HCl, dried over Na2SO4, and concentrated to provide Fmoc-3-amino propionaldehyde diethylacetal with quantitative yield, which was sufficiently pure and used directly.
Fmoc-3-amino propionaldehyde diethylacetal was dissolved in 150 mL DCM, then 4 M HCl in dioxane (30 mL) was added at −78°C and stirred for 1 h. The mixture was diluted with DCM (150 mL) and washed with aqueous Na2CO3 (1 M, 200 mL). After phase separation and extraction of the aqueous phase with DCM (100 mL), the combined organic phases were dried over Na2SO4, concentrated under reduced pressure and purified by flash chromatography (silica gel column, hexane/ethyl acetate, from 0 to 35%) to afford Fmoc-3-amino propionaldehyde as a white solid (12.85 g, 87% yield). 1H NMR (400 MHz, CDCl3): δ 9.80 (s, 1 H), 7.76 (d, J = 7.6 Hz, 2 H), 7.57 (d, J = 7.6 Hz, 2 H), 7.40 (m, 2 H), 7.31 (m, 2 H), 5.21 (s, 1 H), 4.39 (d, J = 6.8 Hz, 2 H), 4.20 (t, J = 6.8 Hz, 1 H), 3.49 (dd, J = 12.0, 6.0 Hz, 2 H), 2.72 (t, J = 6.0 Hz, 2 H); 13C NMR (100 MHz, CDCl3) δ 201.1, 156.3, 143.8, 141.3, 127.6, 127.0, 125.0, 119.9, 66.6, 47.2 44.0, 34.4.
To a solution of Fmoc-3-amino propionaldehyde (12.85 g, 43.5 mmol, 1 eq) and CH3ONH3Cl (5.45 g, 65.25 mmol, 1.5 eq) in DCM (150 mL), DIPEA (16.83 g, 130.5 mmol, 3 eq) was added. The mixture was stirred overnight at rt, then concentrated. The residue was dissolved in DCM/ HOAc (4/1, v/v, 200 mL), then NaBH3CN (4.11 g, 65.25 mmol, 1.5 eq) was added at 0°C in several portions and stirred for 1 h. The reaction mixture was concentrated and the residue was purified by flash chromatography (silica gel column, hexane/ethyl acetate, from 0 to 50%) to afford F-MAPA (1) as a colorless oil (10.51 g, 74% yield). 1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 7.6 Hz, 2 H), 7.59 (d, J = 7.6 Hz, 2 H), 7.40 (dd, J = 7.6, 7.6 Hz, 2 H), 7.31 (dd, J = 7.6, 7.6 Hz, 2 H), 5.20 (s, 1 H), 4.41 (d, J = 6.8 Hz, 2 H), 4.21 (t, J = 6.8 Hz, 1 H), 3.54 (s, 1 H), 3.30 (t, J = 6.4 Hz, 2 H), 2.97 (t, J = 6.4 Hz, 2 H), 1.73 (tt, J = 6.4, 6.4 Hz, 2 H); 13C NMR (100 MHz, CDCl3) δ 156.5, 144.0, 141.3, 127.6, 127.0, 125.0, 119.9, 66.5, 61.9, 49.5, 47.3, 39.4, 27.5. ESI HRMS, calcd for C19H23N2O3 [M+H]+ 327.1703, found 327.1711.
To a solution of F-MAPA (10.51 g, 32.19 mmol) in dioxane (50 mL), 4 M HCl in dioxane (10 mL) was added dropwise at 0°C. Then, cold Et2O was added until no more precipitate formed. The precipitate was collected and washed with cold Et2O to afford F-MAPA hydrochloride salt 2 as a white powder (10.51 g, 90% yield). 1H NMR (400 MHz, MeOD): δ 7.77 (d, J = 7.6 Hz, 2 H), 7.62 (d, J = 7.6 Hz, 2 H), 7.37 (dd, J = 7.6, 7.6 Hz, 2 H), 7.29 (dd, J = 7.6, 7.6 Hz, 2 H), 4.37 (d, J = 6.8 Hz, 2 H), 4.17 (t, J = 6.8 Hz, 1 H), 3.91 (s, 1 H), 3.28 (t, J = 7.6 Hz, 2 H), 2.97 (t, J = 6.4 Hz, 2 H), 1.89 (tt, J = 7.6, 6.4 Hz, 2 H); 13C NMR (100 MHz, MeOD) δ 157.7, 143.8, 141.2, 127.4, 126.7, 124.7, 119.6, 66.3, 60.9, 47.1, 46.5, 37.2, 24.1.
Conjugation of Free Oligosaccharides with 2 and AEAB.
0.35 M 2, 1 M NaOAc (with 2 mM 5MA), 0.35 M AEAB and 1M NaBH3CN in DMSO/AcOH (7:3 v/v) were freshly prepared. To 100 μg glycan, 100 eq 2, 100 eq NaOAc and 0.2 eq 5MA were added. The mixture was vortexed and heated at 65°C on shaker for 2 h. Then, 10 volume of ethyl acetate was added, vortexed and froze at −20°C for 20 min to precipitate the glycan-F-MAPA conjugate. The precipitate was collected by centrifugation and dried in a Centra-vap evaporator. The dried sample was dissolved in 150 μL water and loaded on a C18 SPE cartridge (50 mg). The cartridge was washed with 5 × 1 mL of water and the glycan-F-MAPA conjugate was eluted by 4 × 0.6 mL 30% acetonitrile (ACN). Fractions were combined and lyophilized. glycan-F-MAPA was quantified by fluorometer (Ex 265 nm, Em 315 nm).
After quantification, glycan-F-MAPA was treated with 5% piperidine in water (500 μL) for 30 min, MALDI analysis showed complete removal of Fmoc. The mixture was washed with chloroform for 3 times and the aqueous phase was loaded on C18 SPE cartridge (50 mg). The cartridge was eluted with 4 × 0.6 mL of water. Fractions were collected and lyophilized to provide glycan-F-MAPA conjugate with quantitative yield.
To 100 μg glycan, 200 eq AEAB and 600 eq NaBH3CN were added. The mixture was vortexed and heated at 65°C on shaker for 2 h. Then, 10 volume of ACN was added, vortexed and froze at −20°C for 20 min to precipitate glycan-AEAB conjugate. The precipitate was collected by centrifugation. The sample was dissolved in 150 μL 85% ACN and loaded on a NH2 SPE cartridge (100 mg). The cartridge was washed with 6 × 1 mL of 85% ACN and the glycan-AEAB conjugate was eluted by 4 × 0.6 mL 100 mM NH4HCO3 aqueous solution. Fractions containing glycan-AEAB (monitored by UV Lamp at 365 nm) was collected and lyophilized. The sample was reconstituted in 150 water and loaded on a C18 SPE cartridge (50 mg). The cartridge was washed with 5 × 0.3 mL of water and the glycan-AEAB conjugate was eluted by 4 × 0.6 mL 50% ACN. Fractions containing glycan-AEAB were collected and lyophilized. Glycan-AEAB was quantified by fluorometer (Ex 330 nm, Em 420 nm).
Microarray Printing.
MAPA- and AEAB-labeled glycans were reconstituted in phosphate buffer (100 mM sodium phosphate, pH 8.5) at final concentration of 50 mM and were arrayed onto Nexterion H NHS functionalized slides (Schott AG, Louisville, KY) using a sciFLEXARRAYER S11 from Scienion AG (Berlin, Germany). Each probe was arrayed in 4 replicates. The average drop volume was within 5% variation of the target volume of 330 pL. After printing, slides were incubated at room temperature in a humidity cabinet set at 80% relative humidity overnight. The slides were then blocked with 50 mM ethanolamine in 100 mM sodium tetraborate buffer (pH 9) for 1 hour and washed briefly with 1x phosphate buffered saline with 0.05% polysorbate 20 (Tween-20). This was followed by brief rinsing with ultrapure water before drying by centrifugation. The slides were stored desiccated at −20°C until use.
Microarray Analysis.
After rehydration with TSM buffer (20 mM Tris–HCl, 150 mM sodium chloride, 0.2 mM calcium chloride, and 0.2 mM magnesium chloride), the microarray slides were probed with biotinylated lectins and anti-blood group antibodies. The bound lectins were detected with SA-Cy5 (0.5 μg/mL). Recombinant human DC-SIGN Fc chimera (10 μg/mL) was detected with Alexa Fluor 488 labelled goat anti-human IgG (H+L, 5 μg/mL). Rat anti-human cutaneous lymphocyte antigen antibody (10 μg/mL) was detected with Alexa Fluor 488 labelled goat anti-rat IgM (5 μg/mL). Blood Group Lewis a Antibody (7LE, 10 μg/mL) was detected with Alexa Fluor 488 labelled goat anti-mouse IgG (5 μg/mL). Slides were scanned with a Genepix 4300A microarray scanner from Molecular Devices (Sunnyvale, CA). For cyanine5, Alexa594 and Alexa488, settings of 649 nm (Ex) and 670 nm (Em), 594 nm (Ex) and 645 nm (Em), and 495 nm (Ex) and 519 nm (Em) were used respectively. Scanned images were quantified using Genepix Pro 7 software.
Reaction System Recycling and Linker Recovery.
To a solution of LNnT (20 mg, 28.29 μmol, 1 eq) in DMSO/HOAc (7/3, 11 mL), F-MAPA (923.39 mg, 2.83 mmol, 100 eq) and 5MA (1 mg, 5.66 μmol, 0.2 eq) were added. The mixture was stirred at 65°C for 4 hours. LNnT was completely consumed, monitored by thin layer chromatography (TLC) analysis. Then, 10 volume of ethyl acetate (120 mL) was added to the reaction mixture. Ethyl acetate was mixture thoroughly with the reaction mixture, then frozen at −20°C for 20 min to precipitate LNnT-F-MAPA conjugate. The precipitate was collected by centrifugation and purified by C18 SPE cartridge (2 g) to give 24.7 mg LNnT-F-MAPA with 86% yield. The supernatant was concentrated to remove ethyl acetate, and 20 mg LNnT was added to the residue. The mixture was stirred at 65°C for another 4 hours, then 120 mL ethyl acetate was added to the mixture. Ethyl acetate was mixture thoroughly with the reaction mixture, then frozen at −20°C for 20 min to precipitate LNnT-F-MAPA conjugate. The precipitate was collected by centrifugation and purified by C18 SPE cartridge (2 g) to give 23.83 mg LNnT-F-MAPA with 83% yield. Once again, the supernatant was concentrated to remove ethyl acetate, and 20 mg LNnT was added to the residue. The reaction system was recycled for 5 times with the yield of LNnT-F-MAPA ranging from 86% in the first cycle to 78% in the sixth cycle.
After 6 cycles, the supernatant was concentrated under reduced pressure to remove acetic acid. Ethyl acetate and 1 M Na2CO3 aqueous solution were added to the residue. After phase separation and extraction of the aqueous phase with ethyl acetate, the combined organic phases were dried over Na2SO4, concentrated under reduced pressure and purified by flash chromatography (silica gel column, hexane/ethyl acetate, from 0 to 50%) to provide 738.4 mg F-MAPA with 80% recovery.
Synthesis of Neoglycoprotein.
To a stirred solution of diN-hydroxysuccinimidyl suberate (41.8 mg, 113.6 μmol, 15 eq) in DMF (1.5 mL), a solution of LNnT-MAPA (6 mg, 7.57 μmol, 1 eq) and triethylamine (TEA, 114.9 mg, 1.136 mmol, 150 eq) in 1. 5 mL DMF was added dropwise at 0 °C. The reaction mixture was stirred at room temperature for 3 h. TLC analysis showed complete conversion of LNnT-MAPA. The reaction was then concentrated under high vacuum; the residue was reconstituted in 3 mL water, extracted with ethyl acetate (3 mL × 3). The aqueous layer was lyophilized to give NHS activated LNnT as a white powder. MALDI TOF-MS: m/z calcd for C42H70N4O26Na [M+Na]+ 1046.43, found 1046.46. The NHS activated LNnT was conjugated to BSA (5 mg/mL) at a molar ratio of 50:1 (25:1, 10:1) in 3 × PBS buffer (pH 7.4). The solution was incubated overnight at room temperature. Then the resulting solution was ultrafiltered and washed with 1 × PBS buffer using Amicon Centrifugal Filter Devices (Ultracel 10 000). The glycoprotein solution was lyophilized to give a white solid. Glycoproteins were analyzed by Bruker ultrafleXtreme MALDI TOF/TOF mass spectrometer.
Derivatization of Egg Yolk N-Glycans.
As described in blood glycans derivatization, same amount of crude egg yolk N-glycans (released by ORNG Method, 1.5 mg) were conjugated with F-MAPA and AEAB separately.
Cleavage of F-MAPA/MAPA linker.
LNnT-F-MAPA (10.15 mg, 10 μmol, 1 eq) and LNnT-MAPA (7.94 mg, 10 μmol) were dissolved in water (2 mL) separately, then 4 mg (30 μmol, 3 eq) and 6.68 mg (50 μmol, 5 eq) NCS were added respectively. The mixtures were stirred at 50°C for 2 h and starting material disappeared completely (monitored by MALDI). Then 2 μL and 4 μL formic acid were added, respectively, to quench the reaction. The mixtures were lyophilized and purified by P2 bio-gel filtration to afford 5.8 mg (82% yield) and 5.7 mg (81% yield) LNnT respectively.
The glycan derivative 20-F-MAPA (1.57 mg, 1 μmol, 1 eq) was dissolved in water (100 μL), then NCS (3 μmol, 3 eq) was added. The mixture was stirred at 50°C for 2 h and starting material disappeared completely (monitored by MALDI). Then formic acid (3 μmol, 3 eq) was added to quench the reaction. The mixture was lyophilized and purified by P2 bio-gel filtration to afford 1.0 mg 20 with 80% yield.
Release of F-MAPA-labeled egg yolk N-glycans was performed as described above. F-MAPA-labeled egg yolk N-glycans (0.201 μmol) were dissolved in water (200 μL), then NCS (0.6 μmol, 3 eq) was added. The mixture was shaken at 50°C for 2 h, then washed with chloroform (3 × 0.5 mL). The aqueous phase was detected with fluorometer (Ex 265 nm, Em 315 nM) and the fluorescence disappeared, which suggested that the F-MAPA was completely cleaved. The aqueous phase was concentrated and purified by P2 bio-gel filtration to afford free reducing egg yolk N-glycans.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant P41GM103694 to R.D.C.
Footnotes
The authors declare no competing financial interest.
REFERENCE
- (1).Varki A (2016) Biological roles of glycans, Glycobiology 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, et al. (2017) Crizanlizumab for the prevention of pain crises in sickle cell disease, N. Engl. J. Med. 376, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhou Q (2018) Recent Progress in Clinical Development of Therapeutic Antibodies Targeting Glycan-Binding Proteins, Curr. Drug Targets 19, 1491–1497. [DOI] [PubMed] [Google Scholar]
- (4).Gonzalez PS, O’Prey J, Cardaci S, Barthet VJA, Sakamaki J. i., Beaumatin F, Roseweir A, Gay DM, Mackay G, Malviya G, et al. (2018) Mannose impairs tumour growth and enhances chemotherapy, Nature (London, U. K.) 563, 719–723. [DOI] [PubMed] [Google Scholar]
- (5).Xu Y, Chandarajoti K, Zhang X, Pagadala V, Dou W, Hoppensteadt DM, Sparkenbaugh EM, Cooley B, Daily S, Key NS, et al. (2017) Synthetic oligosaccharides can replace animal-sourced low-molecular weight heparins, Sci. Transl. Med 9, eaan5954/5951–eaan5954/5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Giddens JP, Lomino JV, DiLillo DJ, Ravetch JV, and Wang L-X (2018) Site-selective chemoenzymatic glycoengineering of Fab and Fc glycans of a therapeutic antibody, Proc. Natl. Acad. Sci. U. S. A 115, 12023–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xiao H, Woods EC, Vukojicic P, and Bertozzi CR (2016) Precision glycocalyx editing as a strategy for cancer immunotherapy, Proceedings of the National Academy of Sciences, 201608069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wang D, Liu S, Trummer BJ, Deng C, and Wang A (2002) Carbohydrate microarrays for the recognition of cross-reactive molecular markers of microbes and host cells, Nat. Biotechnol 20, 275–281. [DOI] [PubMed] [Google Scholar]
- (9).Fukui S, Feizi T, Galustian C, Lawson AM, and Chai W (2002) Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions, Nat. Biotechnol 20, 1011–1017. [DOI] [PubMed] [Google Scholar]
- (10).Feizi T, Fazio F, Chai W, and Wong C-H (2003) Carbohydrate microarrays - a new set of technologies at the frontiers of glycomics, Curr. Opin. Struct. Biol 13, 637–645. [DOI] [PubMed] [Google Scholar]
- (11).Cummings Richard D., and Pierce JM (2014) The Challenge and Promise of Glycomics, Chemistry & Biology 21, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Geissner A, and Seeberger PH (2016) Glycan Arrays: From Basic Biochemical Research to Bioanalytical and Biomedical Applications, Annu. Rev. Anal. Chem 9, 223–247. [DOI] [PubMed] [Google Scholar]
- (13).Xia B, Kawar ZS, Ju T, Alvarez RA, Sachdev GP, and Cummings RD (2005) Versatile fluorescent derivatization of glycans for glycomic analysis, Nat. Methods 2, 845–850. [DOI] [PubMed] [Google Scholar]
- (14).Song X, Xia B, Stowell SR, Lasanajak Y, Smith DF, and Cummings RD (2009) Novel Fluorescent Glycan Microarray Strategy Reveals Ligands for Galectins, Chemistry & Biology 16, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhu Y, Liu X, Zhang Y, Wang Z, Lasanajak Y, and Song X (2018) Anthranilic Acid as a Versatile Fluorescent Tag and Linker for Functional Glycomics, Bioconjugate Chem 29, 3847–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Song X, Heimburg-Molinaro J, Cummings RD, and Smith DF (2014) Chemistry of natural glycan microarrays, Curr. Opin. Chem. Biol 18, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Song X, Heimburg-Molinaro J, Smith DF, and Cummings RD (2015) Glycan microarrays of fluorescently-tagged natural glycans, Glycoconjugate J. 32, 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Munneke S, Prevost JRC, Painter GF, Stocker BL, and Timmer MSM (2015) The Rapid and Facile Synthesis of Oxyamine Linkers for the Preparation of Hydrolytically Stable Glycoconjugates, Organic Letters 17, 624–627. [DOI] [PubMed] [Google Scholar]
- (19).Hjuler CT, Maolanon NN, Sauer J, Stougaard J, Thygesen MB, and Jensen KJ (2017) Preparation of glycoconjugates from unprotected carbohydrates for protein-binding studies, Nature Protocols 12, 2411. [DOI] [PubMed] [Google Scholar]
- (20).Chen N, and Xie J (2016) N–O linkage in carbohydrates and glycoconjugates, Organic & Biomolecular Chemistry 14, 11028–11047. [DOI] [PubMed] [Google Scholar]
- (21).Chen G-S, and Pohl NL (2008) Synthesis of Fluorous Tags for Incorporation of Reducing Sugars into a Quantitative Microarray Platform, Organic Letters 10, 785–788. [DOI] [PubMed] [Google Scholar]
- (22).Liu Y, Feizi T, Campanero-Rhodes MA, Childs RA, Zhang Y, Mulloy B, Evans PG, Osborn HMI, Otto D, Crocker PR, et al. (2007) Neoglycolipid Probes Prepared via Oxime Ligation for Microarray Analysis of Oligosaccharide-Protein Interactions, Chemistry & Biology 14, 847–859. [DOI] [PubMed] [Google Scholar]
- (23).Feizi T, and Childs RA (1994) [19] Neoglycolipids: Probes in structure /function assignments to oligosaccharides, In Methods in Enzymology, pp 205–217, Academic Press. [DOI] [PubMed] [Google Scholar]
- (24).Prasanphanich NS, Song X, Heimburg-Molinaro J, Luyai AE, Lasanajak Y, Cutler CE, Smith DF, and Cummings RD (2015) Intact Reducing Glycan Promotes the Specific Immune Response to Lacto-N-neotetraose-BSA Neoglycoconjugates, Bioconjugate Chem 26, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bohorov O, Andersson-Sand H, Hoffmann J, and Blixt O (2006) Arraying glycomics: a novel bi-functional spacer for one-step microscale derivatization of free reducing glycans, Glycobiology 16, 21C–27C. [DOI] [PubMed] [Google Scholar]
- (26).Prudden AR, Liu L, Capicciotti CJ, Wolfert MA, Wang S, Gao Z, Meng L, Moremen KW, and Boons G-J (2017) Synthesis of asymmetrical multiantennary human milk oligosaccharides, Proc. Natl. Acad. Sci. U. S. A 114, 6954–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Jimenez-Castells C, Stanton R, Yan S, Kosma P, and Wilson IBH (2016) Development of a multifunctional aminoxy-based fluorescent linker for glycan immobilization and analysis, Glycobiology 26, 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Purohit S, Li T, Guan W, Song X, Song J, Tian Y, Li L, Sharma A, Dun B, Mysona D, et al. (2018) Multiplex glycan bead array for high throughput and high content analyses of glycan binding proteins, Nature Communications 9, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Palmioli A, and La Ferla B (2018) Glycofunctionalization of Poly(lactic-co-glycolic acid) Polymers: Building Blocks for the Generation of Defined Sugar-Coated Nanoparticles, Organic Letters 20, 3509–3512. [DOI] [PubMed] [Google Scholar]
- (30).Munneke S, Dangerfield EM, Stocker BL, and Timmer MSM (2017) The versatility of N-alkyl-methoxyamine bi-functional linkers for the preparation of glycoconjugates, Glycoconjugate Journal 34, 633–642. [DOI] [PubMed] [Google Scholar]
- (31).Peri F, Dumy P, and Mutter M (1998) Chemo- and stereoselective glycosylation of hydroxylamino derivatives: a versatile approach to glycoconjugates, Tetrahedron 54, 12269–12278. [Google Scholar]
- (32).Song X, Lasanajak Y, Xia B, Heimburg-Molinaro J, Rhea JM, Ju H, Zhao C, Molinaro RJ, Cummings RD, and Smith DF (2010) Shotgun glycomics: a microarray strategy for functional glycomics, Nature Methods 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Byrd-Leotis L, Liu R, Bradley KC, Lasanajak Y, Cummings SF, Song X, Heimburg-Molinaro J, Galloway SE, Culhane MR, Smith DF, et al. (2014) Shotgun glycomics of pig lung identifies natural endogenous receptors for influenza viruses, Proceedings of the National Academy of Sciences 111, E2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Dirksen A, Hackeng TM, and Dawson PE (2006) Nucleophilic Catalysis of Oxime Ligation, Angewandte Chemie International Edition 45, 7581–7584. [DOI] [PubMed] [Google Scholar]
- (35).Loskot SA, Zhang J, and Langenhan JM (2013) Nucleophilic Catalysis of MeON-Neoglycoside Formation by Aniline Derivatives, The Journal of Organic Chemistry 78, 12189–12193. [DOI] [PubMed] [Google Scholar]
- (36).Kochibe N, and Furukawa K (1980) Purification and properties of a novel fucose-specific hemagglutinin of Aleuria aurantia, Biochemistry 19, 2841–2846. [DOI] [PubMed] [Google Scholar]
- (37).Matsumoto I, and Osawa T (1969) Purification and characterization of an anti-H(O) phytohemagglutinin of Ulex europeus, Biochim Biophys Acta 194, 180–189. [DOI] [PubMed] [Google Scholar]
- (38).Peters BP, and Goldstein IJ (1979) The use of fluorescein-conjugated Bandeiraea simplicifolia B4-isolectin as a histochemical reagent for the detection of α-D-galactopyranosyl groups. Their occurrence in basement membranes, Exp. Cell Res 120, 321–334. [DOI] [PubMed] [Google Scholar]
- (39).Etzler ME, and Kabat EA (1970) Purification and characterization of a lectin (plant hemagglutinin) with blood group A specificity from Dolichos biflorus, Biochemistry 9, 869–877. [DOI] [PubMed] [Google Scholar]
- (40).Hammarstrom S, Murphy LA, Goldstein IJ, and Etzler ME (1977) Carbohydrate binding specificity of four N-acetyl-D-galactosamine-”specific” lectins: Helix pomatia A hemagglutinin, soy bean agglutinin, lima bean lectin, and Dolichos biflorus lectin, Biochemistry 16, 2750–2755. [DOI] [PubMed] [Google Scholar]
- (41).Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CMJE, Geijtenbeek TBH, and van Kooyk Y (2003) Cutting Edge: Carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells, J. Immunol 170, 1635–1639. [DOI] [PubMed] [Google Scholar]
- (42).van Die I, van Vliet SJ, Nyame AK, Cummings RD, Bank CMC, Appelmelk B, Geijtenbeek TBH, and van Kooyk Y (2003) The dendritic cell-specific C-type lectin DC-SIGN is a receptor for Schistosoma mansoni egg antigens and recognizes the glycan antigen Lewis x, Glycobiology 13, 471–478. [DOI] [PubMed] [Google Scholar]
- (43).Noll AJ, Yu Y, Lasanajak Y, Duska-McEwen G, Buck RH, Smith DF, and Cummings RD (2016) Human DC-SIGN binds specific human milk glycans, Biochem. J 473, 1343–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gudmundsdottir AV, Paul CE, and Nitz M (2009) Stability studies of hydrazide and hydroxylamine-based glycoconjugates in aqueous solution, Carbohydr. Res 344, 278–284. [DOI] [PubMed] [Google Scholar]
- (45).Huang S, Liao J, Zhao Q, Chai X, Wang B, Yu S, and Wu Q (2013) A New Method for Selective Deprotection of Anomeric N,O-Dimethylhydroxylamine Promoted by TMSCl, J. Carbohydr. Chem 32, 158–168. [Google Scholar]
- (46).Dasgupta S, and Nitz M (2011) Use of N,O-Dimethylhydroxylamine As an Anomeric Protecting Group in Carbohydrate Synthesis, J. Org. Chem 76, 1918–1921. [DOI] [PubMed] [Google Scholar]
- (47).Song X, Ju H, Lasanajak Y, Kudelka MR, Smith DF, and Cummings RD (2016) Oxidative release of natural glycans for functional glycomics, Nature Methods 13, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Plante OJ, Palmacci ER, and Seeberger PH (2001) Automated solid-phase synthesis of oligosaccharides, Science (Washington, DC, U. S.) 291, 1523–1527. [DOI] [PubMed] [Google Scholar]
- (49).Hahm HS, Schlegel MK, Hurevich M, Eller S, Schuhmacher F, Hofmann J, Pagel K, and Seeberger PH (2017) Automated glycan assembly using the Glyconeer 2.1 synthesizer, Proc. Natl. Acad. Sci. U. S. A 114, E3385–E3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhang J, Chen C, Gadi MR, Gibbons C, Guo Y, Cao X, Edmunds G, Wang S, Liu D, Yu J, et al. (2018) Machine-Driven Enzymatic Oligosaccharide Synthesis by Using a Peptide Synthesizer, Angew. Chem., Int. Ed 57, 16638–16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Li T, Liu L, Wei N, Yang J-Y, Chapla DG, Moremen KW, and Boons G-J (2019) An automated platform for the enzyme-mediated assembly of complex oligosaccharides, Nature Chemistry 11, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Krasnova L, and Wong C-H (2019) Oligosaccharide Synthesis and Translational Innovation, J. Am. Chem. Soc 141, 3735–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Crich D (2010) Mechanism of a Chemical Glycosylation Reaction, Acc. Chem. Res 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- (54).Zhu X, and Schmidt RR (2009) New principles for glycoside-bond formation, Angew. Chem., Int. Ed 48, 1900–1934. [DOI] [PubMed] [Google Scholar]
- (55).Yu H, and Chen X (2016) One-pot multienzyme (OPME) systems for chemoenzymatic synthesis of carbohydrates, Org. Biomol. Chem 14, 2809–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Moremen KW, Ramiah A, Stuart M, Steel J, Meng L, Forouhar F, Moniz HA, Gahlay G, Gao Z, Chapla D, et al. (2017) Expression system for structural and functional studies of human glycosylation enzymes, Nature Chemical Biology 14, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yu Y, Lasanajak Y, Song X, Hu L, Ramani S, Mickum ML, Ashline DJ, Venkataram Prasad BV, Estes MK, Reinhold VN, et al. (2014) Human Milk Contains Novel Glycans That Are Potential Decoy Receptors for Neonatal Rotaviruses, Mol. Cell. Proteomics 13, 2944–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Byrd-Leotis L, Jia N, Dutta S, Trost JF, Gao C, Cummings SF, Braulke T, Müller-Loennies S, Heimburg-Molinaro J, Steinhauer DA, et al. (2019) Influenza binds phosphorylated glycans from human lung, Science Advances 5, eaav2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.