

Graphical abstract
Keywords: Regulatory network, Breast cancer, Multi-omics, Integrative network-based approach, GWASs
Abstract
Although genome-wide association studies (GWASs) have successfully identified thousands of risk variants for human complex diseases, understanding the biological function and molecular mechanisms of the associated SNPs involved in complex diseases is challenging. Here we developed a framework named integrative multi-omics network-based approach (IMNA), aiming to identify potential key genes in regulatory networks by integrating molecular interactions across multiple biological scales, including GWAS signals, gene expression-based signatures, chromatin interactions and protein interactions from the network topology. We applied this approach to breast cancer, and prioritized key genes involved in regulatory networks. We also developed an abnormal gene expression score (AGES) signature based on the gene expression deviation of the top 20 rank-ordered genes in breast cancer. The AGES values are associated with genetic variants, tumor properties and patient survival outcomes. Among the top 20 genes, RNASEH2A was identified as a new candidate gene for breast cancer. Thus, our integrative network-based approach provides a genetic-driven framework to unveil tissue-specific interactions from multiple biological scales and reveal potential key regulatory genes for breast cancer. This approach can also be applied in other complex diseases such as ovarian cancer to unravel underlying mechanisms and help for developing therapeutic targets.
1. Introduction
Genome-wide association studies (GWASs) have identified thousands of risk single nucleotide polymorphisms (SNPs) for human complex diseases [1], [2], [3]. However, understanding the biological function and molecular mechanisms of the associated SNPs involved in complex diseases is challenging. Breast cancer represents a typical example of such status quo. Breast cancer is one of the most common diagnosed cancers and leading causes of cancer-related death in females [4]. GWASs have identified over 7000 SNPs for breast cancer based on the results collected from the NHGRI GWAS Catalog (release on 2020-08-26) [1] and published GWASs [5], [6]. Most of these SNPs are located within non-coding genomic regions and their functions need to be interpreted. The genetic information flow from genome to traits goes through various intermediate molecular layers, including genome, epigenome, transcriptome, proteome, and metabolome. Different type of molecules works together as an interactive system to affect biological processes [7], [8]. Therefore, integration of multiple omics data might offer an unprecedented opportunity to illuminate the complex pathogenesis [9]. For example, Zhang et al. used datasets from multiple biological scales to construct regulatory networks and identified TYROBP as a key gene in late-onset Alzheimer's disease [10]. Therefore, integrating GWASs findings with multiple layers of functional data might provide new insight into the complex pathogenesis of breast cancer.
There are increasing evidence showing that genetic variants can perturb the regulatory network and consequently contribute to the changing of traits [11], [12]. Incorporating multi-omic data into network analyses has the advantage of constructing regulatory networks by combining biological signals from different scales [11], [12], [13], [14], and therefore can enhance the understanding of the molecular mechanisms underlying the pathophysiology of diseases. Several previous studies have constructed regulatory networks to unravel potential mechanisms for complex diseases by using integrative approaches. For example, Jang et al. [15] used the chromatin interactome and protein interactome for combinatorial regulatory variants to find driving genes in breast and liver cancers. Hsiao et al. [16] performed a network analysis on the gene level and the gene set level. However, these studies didn’t consider the prior information of GWASs signals. Castro et al. [17] created a TF-driven gene regulatory network for breast cancer by combining GWASs variants and eQTLs. Although these studies have proved the effectiveness of integrative network-based models for identification of new driver genes or biological pathways, no studies have constructed a full-scale network by using molecular interactions across multiple biological scales to provide systemic insight into breast cancer.
Therefore, in this study, we developed a framework named integrative multi-omics network-based approach (IMNA) to capture genetic-driven regulatory networks and predict key regulatory genes for breast cancer. We combined the interactions and functional relationship data from multiple biological scales, including GWASs, eQTLs, epigenomic elements, transcriptome, protein interactome and chromatin long-range interactions. We also developed an abnormal gene expression score (AGES) signature based on the gene expression deviation of the top ranked-order candidate genes and found that the AGES signature was correlated with genomic variants and clinical properties in breast cancer patients. Moreover, we identified RNASEH2A as a novel breast cancer-associated gene. Our method will benefit further investigation of molecular pathogenic mechanisms in diseases.
2. Method
2.1. Multi-omics datasets used in this study
The GWAS datasets for breast cancer came from the Breast Cancer Association Consortium (BCAC). The study design, samples characteristic, genotyping and quality control within the datasets have been described previously [18], [19], [20]. Briefly, the BCAC comprised 52,675 cases and 49,436 controls from 52 studies, including 41 European ancestry, 9 Asian ancestry and 2 African-American ancestry studies [18], [19]. The samples were genotyped using a custom Illumina Infinium iSelect array (iCOGS) comprising 211,155 SNPs and imputed to 2.5 million SNPs by IMPUTE2 [21] with the 1000 Genomes Project March 2012 release as the reference.
The multiple functional genomic data included cis-eQTLs, cis-mQTLs, chromosome long-range interactions and epigenomic regulatory annotations in relevant tissues/cells. The cis-eQTLs data in breast tissues came from the Genotype-Tissue Expression (GTEx) [22]and the Cancer Genome Atlas (TCGA) [23] database. Cis-mQTLs dataset was obtained from Pancan-meQTL (http://bioinfo.life.hust.edu.cn/Pancan-meQTL/) [24]. Chromatin interactions were extracted from 4D genome (http://4dgenome.research.chop.edu/) using the disease-related cell lines (MCF7 and GM12878) [25]. The Assay for Transposase-Accessible Chromatin followed by sequencing (ATAC-seq) and 15-state chromatin states were used for epigenomic regulatory annotation. ATAC-seq datasets from 5 cell types (HCC1806, MDA-MB-231, MCF-7, ZR-75-1, T47D) were downloaded from Cistrome Data Browser [26] (http://cistrome.org/db) (Supplementary Table 1). 15-state chromatin states of 3 cell types (breast vHMEC, breast myoepithelial cells and HMEC mammary epithelial cell) were downloaded from the Roadmap Epigenomics project. We merged “Enhancer”, “Genic enhancer” and “Bivalent enhancer” into one type of regulatory element to simplify the interpretation of regulatory elements.
We collected 6 expression-based prognostic and predictive gene sets including MammaPrint, PAM50, OncotypeDX, Wang-76, Zhang-15, and Cell cycle from previous publications (Supplementary Table 2, Fig. 1c) [27], [28], [29], [30], [31], [32]. These gene sets are independent of anatomical markers and could reveal molecular characteristics of breast cancer and contribute to breast cancer prognosis, diagnosis, and therapeutics [33], [34].
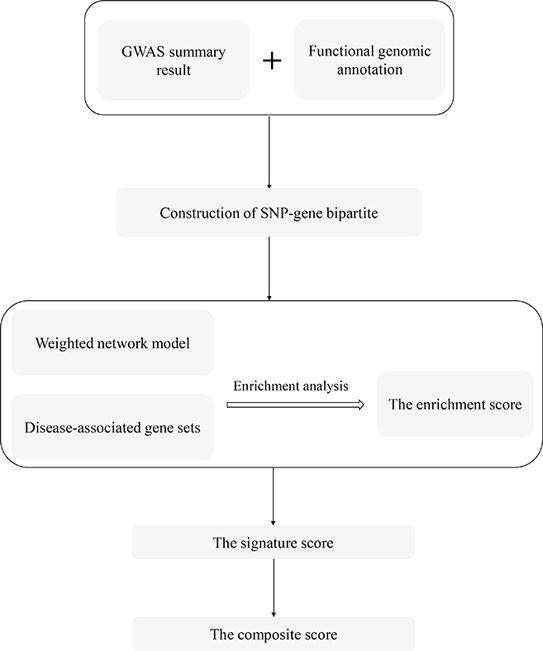
Fig. 1.

An overview of the integrative genomics network-based approach. a) Extraction of SNPs from GWASs and annotation SNPs by multiple tissue-specific functional omics datasets. b) Construction of SNP-gene bipartite with SNP-gene mapping pairs and calculation of degree centrality for each gene node based on the network topology. c) Construction of functional interaction network. Gene nodes in the networks are extracted from the bipartite. Key genes in weighted molecular networks are captured by using enrichment analysis. d) Identification of key genes in the network. Scores of each gene from multiple biological networks are combined to get composite scores. The importance of genes is evaluated by the order of their composite scores.
Gene expression profiles for breast cancer were derived from the GEO (the Gene Expression Omnibus) database, including GSE37751 (n = 108), GSE3744 (n = 47), GSE86374 (n = 159), GSE21422 (n = 19), GSE70947 (n = 296), GSE29044 (n = 109) and GSE14999 (n = 129). We selected these datasets by searching the GEO database using keywords as “breast cancer”, organisms as Homo sapiens, study type as “Expression profiling by array”. Datasets containing breast tumors and adjacent normal breast tissue samples with total sample size more than 100 and the number of controls more than 30 at a single platform were included. Datasets for one or several specific molecular subtypes were not included in our analysis. With the above process, GSE37751, GSE86374, GSE70947, GSE29044 and GSE14999 were chosen. We also included GSE27562 which contained human blood samples. GSE3744 and GSE21422 were used to investigate gene expression profiles between early-stage tumor samples and late-stage tumors by Zhang et al. [35], we also included these two datasets in our analyses. In addition to the above-mentioned microarray data, RNA-seq data of breast tumor samples and controls (GSE115577 and TCGA BRCA dataset) were also included.
The genomic variants, mRNA expression, clinical information for TCGA Breast Invasive Carcinoma (TCGA-BRCA) dataset were downloaded from cBioPortal [37]. Samples with missing clinical information and male samples were removed before analyses.
2.2. Construction of SNP-gene bipartite network
2.2.1. Construction of SNP-gene mapping pairs
To capture disease-associated SNP-gene mapping pairs, we extracted SNPs with their GWASs P-values < 5 × 10−5 from white populations. We excluded SNPs located in the extended MHC region (chr6: 25–35 Mb). For the SNPs located in the gene-body regions, we annotated these SNPs and corresponding genes as SNP-gene mapping pairs. For the other SNPs, their target genes were assigned through multiple aspects, including cis-eQTLs, cis-mQTLs, promoter-enhancer interactions, and regulatory elements annotation. The significant threshold for cis-eQTL was set as P-value < 0.05 after multiple-testing corrections by Benjamini and Hochberg (BH) procedure. The collection of statistically significant cis-meQTLs pairs for breast cancer has been included in the analysis. For chromatin interactions, chromatin states and ATAC-seq peaks were firstly used to get the enhancer region information and annotate the interacting regions. Then we collected the interaction pairs for which there were SNPs located in enhancer in one locus that was paired to a gene promoter in the other locus. All of the above results were merged together to obtain the final SNP-gene mapping pairs. To investigate the function of the target genes, we used clusterProfiler R package [38] to perform gene sets enrichment analysis in KEGG gene sets.
2.2.2. Construction of SNP-gene bipartite network
We constructed a bipartite network based on the SNP-gene mapping pairs as edges connecting SNP and gene nodes. To analyze the structure of the bipartite, we calculated the degree centrality of each SNP and gene node by using NetworkX package Version 2.1 (http://networkx.github.io/) in python. In the bipartite, the degree centrality for a node v is defined in Eq. 1, where the sets U are SNP nodes with n nodes and the sets V are gene nodes with m nodes, deg(v) is the degree of a node v. The degree of the node v is the total number of edges linking to in the opposite node set.
| (1) |
| (1) |
The degree centralities for all genes in the bipartite network are normalized to 1–2 (Eq. (2)).
| (2) |
where is the max value of degree centrality in the gene set V and is the minimum degree centrality.
2.3. Construction of functional interaction network based on SNP-gene bipartite network
We constructed two types of functional interaction networks based on PPI and GIANT network [14]. A total of 11,255,250 known protein-protein interactions (PPIs) for human were derived from the STRING database [39]. To reduce the complexity of network structure and improve calculation speed, we set the threshold of interaction as confidence score over 0.4, which is the default threshold of the STRING database. The confidence score of interactions was used as weights of edges in the PPI network. The GIANT networks identify tissue-specific interactions by using a Bayesian framework to integrate heterogeneous public data [14]. For each tissue, only edges with evidence supporting a tissue-specific functional interaction were included in the network (https://hb.flatironinstitute.org/). A total of 10,857,540 interactions from the mammary gland tissue were used in our analysis. Similarly, the confidence scores of the tissue-specific functional interaction were also used as weights of edges in the GIANT networks. The SNP-gene bipartite provided topological and GWAS signal-driven information on SNP-gene directly regulatory relationships. We extracted subnetworks from the PPI and GIANT networks that only contain genes in the bipartite and their directly connected genes.
2.4. Identification of key drivers
We evaluated the regulatory relationships between genes by considering three types of regulatory networks. The information from networks was incorporated to improve the coverage of functional association between genes. We performed key driver analysis (KDA) to identify key driver genes [40], [41], [42]. The above mentioned 6 expression-based prognostic and predictive gene sets (MammaPrint, PAM50, OncotypeDX, Wang-76, Zhang-15, and Cell cycle) were firstly collected. For each gene, we extracted 1-layer neighborhoods directly connected with it in the PPI or GIANT network as the subnetwork. Fisher’s exact test was used to test whether genes in this subnetwork were enriched in these 6 gene sets. The background included all genes in the 6 gene sets. The enrichment P-values were -log10 transformed and normalized to a scale of 0 to 1 using the min–max normalization method. For each gene, the normalized value was set as the enrichment score. For a given network, each gene has 6 enrichment scores in the current study for breast cancer.
To incorporate with the GWAS signals (i.e., the SNP-gene bipartite network), we defined a signature score for each gene as shown in Eq. (3):
| (3) |
where is the signature score for gene g in a given network (PPI or GIANT); is the enrichment score for gene g in the gene set i; n is the total number of gene sets (here equals to 6 for breast cancer). is the normalized degree centralities for gene g in the SNP-gene bipartite network. If a gene did not exist in the bipartite network, its gene weight was set to 1. Again, the signature scores were normalized to a scale of 0 to 1.
Lastly, we combined the signature scores for each gene from different networks (e.g., PPI or GIANT). For a gene, the average of signature scores from different networks was combined and normalized to get a final score as Eq. (4):
| (4) |
where is the composite score of gene g, N is the number of networks. In this study, N equaled to 2 (refers to the PPI and GIANT network). The composite score across networks was indicated as the numerical value of genes (0–1). Composite scores were ranked according to values and provided a criterion for determining the significance of genes in biological mechanisms for a given disease.
2.5. Gene expression profiling and heat maps
Limma package (Version 3.30.13) [43], [44] in R was used to detect differentially expressed genes between tumor and normal samples. P-values were adjusted for multiple-testing by the BH procedure. For plotting heat maps, hierarchical clustering was performed in R to cluster samples on the gene expression profiles by centroid.
2.6. Survival analysis and Kaplan-Meier plots
The survival analysis for utilization of multiple gene expression signatures was performed using the TCGA-BRCA dataset. We developed an abnormal gene expression score (AGES) for each sample i as Eq. (5):
| (5) |
where represents the counts of genes whose expression in sample i was above the third quartile or below the first quartile of the expression values in all samples. μ is the mean value, and σ is the standard deviation. Here we only included genes with the top 20 composite scores in AGES. The individuals were divided into two groups of high (above the third quartile) and low (below the first quartile) according to the AGES. Univariate survival analyses between two groups were calculated by using Kaplan-Meier curves and the log-rank test in R. The P-values were determined by using the log-rank test, and the significance threshold was set as P-value < 0.05.
3. Result
3.1. Identification of disease-associated gene nodes in SNP-gene bipartite network
We integrated GWASs results with multiple functional omics data, including cis-eQTLs, cis-mQTLs, chromatin interactions, chromatin states and ATAC-seq, to construct SNP-gene pairs (Fig. 1a). After annotation, we obtained 7,500 SNP-gene mapping pairs, including 6,647 SNPs and 274 protein-coding genes. To investigate the functional mapping structure between disease-associated SNPs and genes, we constructed the mapping pairs as a bipartite network. About 0.037% of SNPs were connected with at least one gene (6,647 in total 18,134,195 SNPs). Meanwhile, 1.35% of protein-coding genes were mapped to at least one SNP (274 in total 20,345 genes).
In order to find the contribution of the GWAS signal-driven genes to biological mechanisms, we performed pathway enrichment analysis for these genes by using canonical pathways from KEGG (Figure Supplementary Fig. 1). The top-ranked pathways included cancer-related pathways, such as gastric cancer, thyroid cancer, acute myeloid leukemia, pancreatic cancer, and breast cancer. We also found several signaling pathways, such as MAPK signaling pathway, TGF-beta signaling pathway and WNT signaling pathway, as well as cellular processes pathways, like cell cycle pathway and homologous recombination.
We incorporated all SNP-gene mapping pairs into a bipartite network with two types of nodes as SNPs and genes, and edges for SNP-gene functional mapping pairs (Fig. 1b). To investigate the relevance between the bipartite topology structure and regulatory function of nodes, we calculated the degree centrality for each gene and SNP in the bipartite network (Supplementary Tables 3 and 4). Genes with higher degree centrality values connected with more nodes in SNP sets. Likewise, SNPs with high degree centrality values connected with numerous genes. These results suggested that degree centrality coincided with the genetic variant-disease association and reflected potential genetic perturbation to genes in the mechanism of disease.
3.2. Subnetworks and key genes of biological tissue-Specific networks
To reveal key genes based on disease-associated information and further investigate regulatory mechanisms of those genes in diseases, we constructed two types of tissue-specific gene interaction networks for those genes in the bipartite network and performed KDA on these networks (Fig. 1c). For each network, we identified key genes of which the neighbor-genes were enriched in the gene expression-based signatures. The full list of KDA results for genes in the two types of networks are in Supplementary Table 5. We calculated a composite score for each gene based on the PPI and GIANT networks to provide a quantitative evidence to evaluate the importance of regulatory function (Fig. 1d). The full list of composite scores are provided in Supplementary Table 5 and the top 20 genes are summarized in Table 1.
Table 1.
Top 20 key genes in breast cancer based on composite scores by considering criteria from 3 types of network.
| Key gene | Gene name | ScorePPI | ScoreGIANT | Composite score |
|---|---|---|---|---|
| CCND1 | Cyclin D1 | 1.000 | 0.579 | 1.000 |
| CDKN2A | Cyclin dependent kinase inhibitor 2A | 0.735 | 0.768 | 0.952 |
| MYC | MYC proto-oncogene, bhlh transcription factor | 0.728 | 0.708 | 0.910 |
| ESR1 | Estrogen receptor 1 | 0.760 | 0.430 | 0.754 |
| XRCC5 | X-ray repair cross complementing 5 | 0.184 | 1.000 | 0.750 |
| CHEK2 | Checkpoint kinase 2 | 0.606 | 0.534 | 0.722 |
| XRCC6 | X-ray repair cross complementing 6 | 0.352 | 0.697 | 0.664 |
| EXO1 | Exonuclease 1 | 0.399 | 0.648 | 0.663 |
| RNASEH2A | Ribonuclease H2 subunit A | 0.346 | 0.644 | 0.627 |
| BRCA2 | BRCA2 DNA repair associated | 0.286 | 0.632 | 0.581 |
| ADSL | Adenylosuccinate lyase | 0.000 | 0.847 | 0.536 |
| PPP2R1B | Protein phosphatase 2 scaffold subunit Abeta | 0.380 | 0.344 | 0.459 |
| JUND | Jund proto-oncogene, AP-1 transcription factor subunit | 0.277 | 0.437 | 0.452 |
| PES1 | Pescadillo ribosomal biogenesis factor 1 | 0.113 | 0.589 | 0.445 |
| PSAT1 | Phosphoserine aminotransferase 1 | 0.064 | 0.625 | 0.436 |
| ADCY3 | Adenylate cyclase 3 | 0.134 | 0.551 | 0.434 |
| SEPT9 | Septin 9 | 0.041 | 0.633 | 0.426 |
| STAT3 | Signal transducer and activator of transcription 3 | 0.389 | 0.277 | 0.422 |
| HDGF | Heparin binding growth factor | 0.000 | 0.658 | 0.417 |
| EP300 | E1A binding protein p300 | 0.431 | 0.218 | 0.411 |
Footnotes: PPI: protein–protein interaction.
To assure the robustness of our method, we performed IMNA by using the subsets of gene expression-based signatures as follows: For a total of 6 gene sets, one gene set was removed at each time and the remaining 5 gene sets were used to perform analysis. Jaccard index was used to evaluate the similarity of the top 20 genes between different conditions. We found the relatively high similarity (Jaccard index >0.48) between the pairwise comparison of all conditions (Figure Supplementary Fig. 2a). We also detected the same trend (Jaccard index >0.42) for the similarity of the top 100 genes (Figure Supplementary Fig. 2b).
3.3. Identification of the effects of top key genes for breast cancer
We further examined the expression levels and genomic variations of the top 20 ranked-order genes to investigate the performance of these genes in the pathogenesis of breast cancer. Compared with normal samples, these 20 genes showed significantly abnormal expression levels in breast tumor samples, as well as peripheral blood mononuclear cells (PBMCs) from breast cancer patients (adjust P-value < 0.05 in at least 3 datasets, Fig. 2a, Supplementary Table 6). The expression levels of these 20 genes were progressively changed during disease progression (Fig. 2b). The range of genetic alteration frequencies (including copy-number alterations (CNA) and mutation frequency) of these 20 genes were between 0.9% and 21% in breast cancer (Fig. 2c). Among these 20 genes, 9 genes have been identified as cancer driver genes in breast cancer and/or multiple other tumors according to the COSMIC database (http://cancer.sanger.ac.uk/cosmic), including CDKN2A, CCND1, MYC, ESR1, CHEK2, BRCA2, STAT3, SEPT9 and EP300. Moreover, in the top 20 genes, CCND1, MYC, ESR1 and BRCA2 were found in the KEGG breast cancer pathway. In addition to the breast cancer pathway, some of the top 20 genes were found in other breast cancer-related pathways. For example, 2 genes are in the MAPK signaling pathway (MYC and JUND), 3 in the PI3K-Akt pathway (CDKN2A, CCND1 and CHEK2), and 3 in the p53 pathway (MYC, CCND1 and PPP2R1B).
Fig. 2.

The top 20 rank-ordered genes are correlated with breast cancer, disease pathogenesis and genetic variants. The top 20 genes are differentially expressed between breast cancer samples and normal samples. The top 20 gene expression levels in a) various datasets. b) breast ductal carcinoma in situ (DCIS) and invasive ductal carcinomas (IDCs) (GSE21422). Samples were clustered by gene expression levels of the 20 genes by centroid. c) Genetic alternation frequencies of the top 20 genes in TCGA-BRCA patient samples.
Furthermore, to investigate the overall impact of the 20 ranked-order genes expression levels on breast cancer outcomes, we calculated the AGES to represent the abnormal expression levels of the 20 genes in samples and evaluated the relevance of the AGES values to tumorigenesis and clinical survival. We found that the AGES value was significantly positively correlated with CNA fraction (Pearson correlation, P-value = 1.40 × 10−15, r = 0.25, Fig. 3a) in the TCGA-BRCA dataset. The AGES was associated with several tumor clinical characteristics by investigating the clinical information in the TCGA-BRCA dataset. More breast cancer tumors with ER-negative and triple-negative showed significantly higher AGES values (Chi-squared test, P-value < 0.05, Fig. 3b-c). We also evaluated the relevance between the AGES values and breast patient survival outcomes. Categorizing patients in the first tercile into the low group and the third tercile into the high group separately, the low group had better significant disease-free survival and distant metastasis-free survival outcomes compared to the high group (P < 0.05, Fig. 3d-f). These results indicate that the AGES signature may contribute to breast cancer clinical properties, and might be an effective marker of patient survival.
Fig. 3.

The relevance of the AGES values to tumorigenesis, clinical characteristics and clinical survival. a) AGES values are significantly positively correlated with linear copy-number alterations (CNA). b,c) The AGES values are associated with tumor clinical characteristics. Significant P-values are determined by Chi-squared test (P < 0.01). b) ER-negative. c) Triple-negative. d,e,f) Kaplan-Meier survival curves showed that the AGES are predictive of survival outcomes for breast cancer. d) disease-free survival in TCGA-BRCA dataset. e) distant metastasis-free survival in GSE11121. f) distant metastasis-free survival in GSE20685.
3.4. Identification of novel breast cancer-associated genes
To verify the associations between the top 20 genes and breast cancer, we used various databases to assess functions in breast cancer through text mining of published articles and integrating disease databases (Table 2), including PolySearch2 (http://polysearch.cs.ualberta.ca/polysearch), COREMINE (https://www.coremine.com/medical/) and MalaCards (https://www.malacards.org/). The top 20 genes have been linked to breast cancer or neoplasm by using at least one of the tools. We noticed that RNASEH2A (ribonuclease H2 subunit A) was not previously verified as breast cancer-related gene. We further evaluated whether its expression level was related to tumorigenesis and clinical survival. We found that RNASEH2A expression levels were significantly up-regulated in breast cancer samples compared to normal tissues (adjusted P-value < 0.05, Fig. 4a). Univariate survival analyses revealed that low RNASEH2A mRNA expression was significantly associated with better overall and relapse-free survival (P = 1.3 × 10−5 and P < 1.0 × 10−16, Fig. 4b-c) in KM-plotter [45]. Co-expression analysis was performed for RNASEH2A with the most well-evaluated cell proliferation markers, including MKI67, PCNA, MCM2, MCM3, MCM4, MCM5, MCM6 and MCM7) [46], by using the breast cancer samples in TCGA (n = 1100). We observed that these genes were positively co-expressed with each other, and RNASEH2A had positive correlations with the other 8 genes (rho ≥ 0.3, P-value < 0.01) (Figure Supplementary Fig. 3).
Table 2.
In Silico Validation for the top 20 ranked genes for breast cancer.
| Gene | PolySearch2 (Z Score) | COREMINE (Statistical significance) | MalaCards (Score) |
|---|---|---|---|
| CCND1 | 49.43 | 5.12 × 10−5 | 86.94 |
| CDKN2A | 4.59 | 7.97 × 10−4 | 33.58 |
| MYC | 26.82 | 1.59 × 10−4 | 72.97 |
| ESR1 | 30.11 | 3.78 × 10−6 | 1165.88 |
| XRCC5 | 13.23 | 1.51 × 10−2 | – |
| CHEK2 | 14.01 | 1.30 × 10−4 | 1461.53 |
| XRCC6 | – | 1.43 × 10−2 | – |
| EXO1 | 1.23 | 0.135 | – |
| RNASEH2A | 4.71 | 0.502 | – |
| BRCA2 | 11.95 | 3.39 × 10−6 | 1500.88 |
| ADSL | 2.69 | 0.350 | – |
| PPP2R1B | 5.42 | 2.44 × 10−2 | – |
| JUND | – | 7.16 × 10−2 | – |
| PES1 | 2.39 | 1.83 × 10−2 | – |
| PSAT1 | 1.45 | 1.14 × 10−2 | – |
| ADCY3 | 8.19 | 0.335 | – |
| SEPT9 | – | 0.193 | – |
| STAT3 | 20.75 | 3.05 × 10−4 | 42.45 |
| HDGF | – | 0.232 | – |
| EP300 | 1.82 | 1.72 × 10−3 | 50.25 |
Footnotes: - indicates that the value is not available.
Fig. 4.

The expression level of RNASEH2A related to patient outcome in breast cancer. a) Box plots show increased expression of RNASEH2A in breast tumor samples in 7 independent studies. b, c) Kaplan-Meier survival analyses show the differences in overall survival (OS) and relapse-free survival (RFS) between breast cancer patients with high or low RNASEH2A mRNA levels. P-values are calculated by using the log-rank analysis.
3.5. Application of the method to ovarian cancer
To evaluate the reliability of IMNA, we applied our method to ovarian cancer. The GWAS summary dataset was derived from the Ovarian Cancer Association Consortium (OCAC), which comprised 18,174 cases with epithelial ovarian cancer and 26,134 controls of European ancestry from 43 studies [47], [48]. The multiple functional data included cis-eQTLs, chromosome long-range interactions and epigenomic regulatory annotations, since mQTLs data was not available yet. After the similar procedures as breast cancer, we obtained 5,386 SNP-gene mapping pairs, including 4,419 SNPs and 149 protein-coding gene, and constructed the SNP-gene bipartite network. KEGG pathway enrichment analysis was perfomred for investigating the GWAS signal-driven genes to biological mechanisms (Figure Supplementary Fig. 4 Supplementary Fig. 4a).
We collected 6 expression-based ovarian cancer-related gene sets from the published studies [49], [50], [51], [52], [53], [54]. To construct regulatory networks, 11,255,250 PPI interactions were derived from STRING and 6,4594,988 edges from ovary tissue of GIANT were used. In the weighted regulatory networks, we performed KDA to identify key genes. The full list of gene composite scores was listed in Supplementary Table 7. CDK6 and MLLT10 have been identified as cancer driver genes according to the COSMIC database. By manually checking in the PubMed, 7 of the top 20 genes were reported to be relevant to ovarian cancer, including MAPT [55], BECN1 [56], PRC1 [57], CBX1 [58], CDK6 [59], WWOX [60], and PNPO [61]. We performed KEGG pathway enrichment analysis for the top 100 genes (Figure Supplementary Fig. 4b), and found that the top-ranked pathways included the cell cycle pathway, several cancer-related pathways (renal cell carcinoma, proteoglycans in cancer and small cell lung cancer), and important signaling pathways (PI3K-Akt signaling pathway, VEGF signaling pathway and chemokine signaling pathway). These results indicated that our method can also apply to other types of complex diseases.
4. Discussion
Although GWASs have identified thousands of diseases susceptibility variants, the causal genes and their molecular functional mechanisms are largely unknown [62]. Network-based approaches are helpful to understand the mechanisms of complex diseases and capture genetic variants perturbation in the gene regulatory network [8], [63]. We applied IMNA by combining tissue-specific regulatory networks from diverse biological scales, including GWAS signals, cis-eQTLs, cis-meQTLs, long-range interactions and epigenomic regulatory annotations, multi-gene predictive and prognostic signatures and regulatory networks to identify key genes supported by genetic and biological processes. The concentration of functional disease-associated SNPs on genes reflects the importance of gene regulatory roles in diseases. Predictive and prognostic signatures provide intrinsic molecular characteristics of breast cancer based on gene expression analysis [64]. The identified key genes derived from multiple tissue-specific regulatory networks can regulate other disease-associated genes and influence disease susceptibility. We used the AGES signature to present the correlations between the expression levels of the top 20 genes and several tumor characteristics, which demonstrates the utility of the AGES signature for genetic variants, tumor properties and patient survival. We also applied this method for ovarian cancer to demonstrate the effectiveness of this pipeline on other types of diseases.
This approach used integrative multi-omics methods that benefited the research of complex diseases. First, we derived functional data to reveal the mapping between SNPs and genes in different respects (cis-eQTLs, cis-mQTLs, long-range interactions, chromatin states and ATAC-seq) from diseases-related tissues or cell types. Second, knowledge-driven signatures revealed molecular characteristics of breast cancer from the gene expression level. Intrinsic subtyping, prognostic and predictive signatures of breast cancer based on gene expression profiling have been found for correlation with molecular subtypes, including proliferation, prognosis, and therapeutic response [33], [34], [64], [65]. Third, since biological molecules interact within and across multiple biological levels, we constructed disease networks from genomic, transcriptomic and proteomics scales based on data-driven gene sets and identified the key genes [8], [11]. The network models could provide a relatively complete understanding of biological interactions [12], [13], [33], [66]. Therefore, our integrative network-based approach unveiled the biological mechanism of diseases from different scales. Gene expression profiles and survival analyses verified the ability of key genes for prognosis in breast cancer.
Besides the known breast cancer genes, our study also identified a potential new regulator, RNASEH2A, for breast cancer. RNASEH2A encodes the main catalytic subunit of ribonuclease H2 (RNase H2), which mediates the removal of lagging-strand Okazaki fragment RNA primers from the DNA:RNA duplex during DNA replication and degrades the RNA of RNA:DNA hybrids [67]. RNASEH2A is frequently overexpressed in a variety of cancers, including breast, bladder, brain, prostate, seminomas, and leukemia [68]. RNASEH2A could promote DNA replication and cancer cell proliferation, and may regulate signaling pathways responsible for cell proliferation and apoptosis [68], [69], [70].
Although we have discussed the advantages of IMNA, there are still limitations in our work. First, as a GWASs-based method, other potential important genes without genetic support could be missed in our method. Second, this method constructed models by using genetics and molecular data from static biological systems. Due to the lack of dynamic biological data in disease processes, we could not present the dynamics of network to investigate the disease development currently. Third, our method incorporates breast cancer-related signatures information as prior knowledge to construct network. The results might be influenced by selections of prior knowledge inputs compared with other methods which ignored pathway information. However, disease-related gene sets are more stable in different datasets/platforms compared to individual genes or variants [8], [71]. The information we captured might have better generalization capability. Finally, we used molecular interactions from multiple diverse datasets in different biological scales. Since different molecular subtypes for many datasets (e.g., eQTL) are not available currently, we only applied the model for overall breast cancer. If datasets for different molecular subtypes are available in future, the method can be used to characterize key regulator genes for different molecular subtypes.
In conclusion, we utilized IMNA to interpret the GWAS signals of breast cancer and highlight key genes in the diseases-associated regulatory networks. Our findings provide a global perspective to understand the molecular underpinnings in pathogenesis of breast cancer, and point out candidate therapeutic targets. IMNA can also apply to other complex diseases to unveil underlying mechanisms and help for investigating therapeutic targets.
URLs. IMNA, an integrative genomics network-based approach, https://github.com/xjtugenetics/IMNA.
CRediT authorship contribution statement
Yi-Xiao Chen: Conceptualization, Methodology, Software, Data curation, Writing - original draft, Writing - review & editing. Yu Rong: Data curation, Validation. Feng Jiang: Investigation, Resources. Jia-Bin Chen: Software, Data curation. Yuan-Yuan Duan: Data curation. Shan-Shan Dong: Formal analysis, Writing - original draft, Writing - review & editing. Dong-Li Zhu: Resources. Hao Chen: Visualization. Tie-Lin Yang: Resources. Zhijun Dai: Resources. Yan Guo: Supervision, Writing - review & editing, Methodology.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
The authors thank the TCGA and Broad Institute for maintaining critical public databases and services. The normalized expression matrixes we used were obtained through GDAC (Genome Data Analysis Center, http://gdac.broadinstitute.org/, doi:10.7908/C11G0KM9).
Funding
This study is supported by National Natural Science Foundation of China (81872490 and 31871264); Shaanxi Provincial Key Research and Development Project (2019ZDLSF01-09); Innovative Talent Promotion Plan of Shaanxi Province for Young Sci-Tech New Star (2018KJXX-010); Natural Science Foundation of Zhejiang Province of China (LGF18C060002); China Postdoctoral Science Foundation (2018 M643619); and the Fundamental Research Funds for the Central Universities.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.10.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary Fig. 1.

KEGG pathway enrichment analysis for breast cancer GWAS signal-driven genes.
Supplementary Fig. 2.

The similarity between the pairwise comparisons of the robustness tests. a) Top 20 genes. c) Top 100 genes.
Supplementary Fig. 3.

Co-expression between RNASEH2A and 8 cell proliferation markers.
Supplementary Fig. 4.

KEGG pathway enrichment analysis for ovarian cancer identified genes. a) GWAS signal-driven genes in the bipartite network. b) Top 100 genes.
References
- 1.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017;101(1):5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marigorta U.M., Rodriguez J.A., Gibson G., Navarro A. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet. 2018;34(7):504–517. doi: 10.1016/j.tig.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-a Cancer J Clinicians. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 5.Michailidou K., Beesley J., Lindstrom S., Canisius S., Dennis J. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015;47(4):373–380. doi: 10.1038/ng.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michailidou K., Lindstrom S., Dennis J., Beesley J., Hui S. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551(7678):92–94. doi: 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Y.V., Hu Y.J. Integrative Analysis of Multi-omics Data for Discovery and Functional Studies of Complex Human Diseases. Adv Genet. 2016;93:147–190. doi: 10.1016/bs.adgen.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee I., Blom U.M., Wang P.I., Shim J.E., Marcotte E.M. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011;21(7):1109–1121. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karczewski K.J., Snyder M.P. Integrative omics for health and disease. Nat Rev Genet. 2018;19(5):299–310. doi: 10.1038/nrg.2018.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B., Gaiteri C., Bodea L.G., Wang Z., McElwee J. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer's Disease. Cell. 2013;153(3):707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Civelek M., Lusis A.J. Systems genetics approaches to understand complex traits. Nat Rev Genet. 2014;15(1):34–48. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makinen V.P., Civelek M., Meng Q.Y., Zhang B., Zhu J. Integrative Genomics Reveals Novel Molecular Pathways and Gene Networks for Coronary Artery Disease. PLoS Genet. 2014;10(7) doi: 10.1371/journal.pgen.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shu L., Chan K.H.K., Zhang G.L., Huan T.X., Kurt Z. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet. 2017;13(9) doi: 10.1371/journal.pgen.1007040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greene C.S., Krishnan A., Wong A.K., Ricciotti E., Zelaya R.A. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015;47(6):569–576. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang K., Kim K., Cho A., Lee I., Choi J.K. Network perturbation by recurrent regulatory variants in cancer. PLoS Comput Biol. 2017;13(3) doi: 10.1371/journal.pcbi.1005449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsiao T.H., Chiu Y.C., Hsu P.Y., Lu T.P., Lai L.C. Differential network analysis reveals the genome-wide landscape of estrogen receptor modulation in hormonal cancers. Sci Rep. 2016;6 doi: 10.1038/srep23035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castro M.A.A., de Santiago I., Campbell T.M., Vaughn C., Hickey T.E. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat Genet. 2016;48(1) doi: 10.1038/ng.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michailidou K., Hall P., Gonzalez-Neira A., Ghoussaini M., Dennis J. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michailidou K., Beesley J., Lindstrom S., Canisius S., Dennis J. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015;47:373. doi: 10.1038/ng.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bojesen S.E., Pooley K.A., Johnatty S.E., Beesley J., Michailidou K. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45(4) doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howie B., Fuchsberger C., Stephens M., Marchini J., Abecasis G.R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong J., Mei S.F., Liu C.J., Xiang Y., Ye Y.Q. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 2018;46(D1):D971–D976. doi: 10.1093/nar/gkx861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong J., Wan H., Mei S., Ruan H., Zhang Z. Pancan-meQTL: a database to systematically evaluate the effects of genetic variants on methylation in human cancer. Nucleic Acids Res. 2019;47(D1):D1066–D1072. doi: 10.1093/nar/gky814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teng L., He B., Wang J.H., Tan K. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics. 2015;31(15):2560–2564. doi: 10.1093/bioinformatics/btv158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng R.B., Wan C.X., Mei S.L., Qin Q., Wu Q. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47(D1):D729–D735. doi: 10.1093/nar/gky1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.L, J, van't, Veer,, H, Y, Dai, M J van de Vijver, Y D D He, A A M Hart, et al., Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 28.Parker J.S., Mullins M., Cheang M.C.U., Leung S., Voduc D. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paik S., Shak S., Tang G., Kim C., Baker J. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y., Klijn J.G., Zhang Y., Sieuwerts A.M., Look M.P. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365(9460):671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 31.Zhang F., Kaufman H.L., Deng Y.P., Drabier R. Recursive SVM biomarker selection for early detection of breast cancer in peripheral blood. BMC Med Genomics. 2013;6 doi: 10.1186/1755-8794-6-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J.G., Campen A., Huang S.G., Peng S.B. X Ye, et al., Identification of a gene signature in cell cycle pathway for breast cancer prognosis using gene expression profiling data. BMC Med Genomics. 2008;1 doi: 10.1186/1755-8794-1-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taherian-Fard A., Srihari S., Ragan M.A. Breast cancer classification: linking molecular mechanisms to disease prognosis. Briefings Bioinf. 2015;16(3):461–474. doi: 10.1093/bib/bbu020. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z., Zhang X.H., Zhang S., Dai X.F. An integrative view on breast cancer signature panels. Expert Review of Molecular Diagnostics. 2019;19(8):715–724. doi: 10.1080/14737159.2019.1642751. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W., Mao J.H., Zhu W., Jain A.K., Liu K. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nat Commun. 2016;7:12619. doi: 10.1038/ncomms12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269) doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang I.M., Zhang B., Yang X., Zhu J., Stepaniants S. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol Syst Biol. 2012;8 doi: 10.1038/msb.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang X., Zhang B., Molony C., Chudin E., Hao K. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010;20(8):1020–1036. doi: 10.1101/gr.103341.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu J., Zhang B., Smith E.N., Drees B., Brem R.B. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet. 2008;40(7):854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritchie M.E., Phipson B., Wu D., Hu Y.F. C W Law, et al., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7) doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phipson B., Lee S., Majewski I.J., Alexander W.S., Smyth G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann Appl Stat. 2016;10(2):946–963. doi: 10.1214/16-AOAS920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gyorffy B., Lanczky A., Eklund A.C., Denkert C., Budczies J. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 46.Jurikova M., Danihel L., Polak S., Varga I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016;118(5):544–552. doi: 10.1016/j.acthis.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Pharoah P.D.P., Tsai Y.-Y., Ramus S.J., Phelan C.M., Goode E.L. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45:362. doi: 10.1038/ng.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bojesen S.E., Pooley K.A., Johnatty S.E., Beesley J., Michailidou K. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verhaak R.G., Tamayo P., Yang J.Y., Hubbard D., Zhang H. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest. 2013;123(1):517–525. doi: 10.1172/JCI65833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonome T., Levine D.A., Shih J., Randonovich M., Pise-Masison C.A. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68(13):5478–5486. doi: 10.1158/0008-5472.CAN-07-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kernagis D.N., Hall A.H., Datto M.B. Genes with bimodal expression are robust diagnostic targets that define distinct subtypes of epithelial ovarian cancer with different overall survival. J Mol Diagn. 2012;14(3):214–222. doi: 10.1016/j.jmoldx.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 52.Leong H.S., Galletta L., Etemadmoghadam D., George J. S Australian Ovarian Cancer, et al., Efficient molecular subtype classification of high-grade serous ovarian cancer. J Pathol. 2015;236(3) doi: 10.1002/path.4536. [DOI] [PubMed] [Google Scholar]
- 53.Yoshihara K., Tsunoda T., Shigemizu D., Fujiwara H., Hatae M. High-Risk Ovarian Cancer Based on 126-Gene Expression Signature Is Uniquely Characterized by Downregulation of Antigen Presentation Pathway. Clin Cancer Res. 2012;18(5):1374–1385. doi: 10.1158/1078-0432.CCR-11-2725. [DOI] [PubMed] [Google Scholar]
- 54.Denkert C., Budczies J., Darb-Esfahani S., Gyorffy B., Sehouli J. A prognostic gene expression index in ovarian cancer - validation across different independent data sets. J Pathol. 2009;218(2):273–280. doi: 10.1002/path.2547. [DOI] [PubMed] [Google Scholar]
- 55.Yang Y., Wu L., Shu X., Lu Y., Shu X.O. Genetic Data from Nearly 63,000 Women of European Descent Predicts DNA Methylation Biomarkers and Epithelial Ovarian Cancer Risk. Cancer Res. 2019;79(3):505–517. doi: 10.1158/0008-5472.can-18-2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai M., Hu Z., Liu J., Gao J., Liu C. Beclin 1 expression in ovarian tissues and its effects on ovarian cancer prognosis. Int J Mol Sci. 2014;15(4):5292–5303. doi: 10.3390/ijms15045292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bu H., Li Y., Jin C., Yu H., Wang X. Overexpression of PRC1 indicates a poor prognosis in ovarian cancer. Int J Oncol. 2020;56(3):685–696. doi: 10.3892/ijo.2020.4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y., Pan S., Song Y., Pan C., Chen C. The Prognostic Value of the Chromobox Family in Human Ovarian Cancer. J Cancer. 2020;11(17):5198–5209. doi: 10.7150/jca.44475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dall'Acqua A., Sonego M., Pellizzari I., Pellarin I., Canzonieri V. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol Med. 2017;9(10) doi: 10.15252/emmm.201607012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu Y., Yan Y., Xu Y., Yang H., Fang L. Expression and clinical significance of WWOX, Elf5, Snail1 and EMT related factors in epithelial ovarian cancer. Oncol Lett. 2020;19(2):1281–1290. doi: 10.3892/ol.2019.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang L., Zhou D., Guan W., Ren W., Sun W. Pyridoxine 5'-phosphate oxidase is a novel therapeutic target and regulated by the TGF-β signalling pathway in epithelial ovarian cancer. Cell Death Dis. 2017;8(12):3214. doi: 10.1038/s41419-017-0050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muñoz M., Pong-Wong R., Canela-Xandri O., Rawlik K., Haley C.S. Evaluating the contribution of genetic and familial shared environment to common disease using the UK Biobank. Nat Genet. 2016;48(9):980–983. doi: 10.1038/ng.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sinn P., Aulmann S., Wirtz R., Schott S., Marme F. Multigene Assays for Classification, Prognosis, and Prediction in Breast Cancer: a Critical Review on the Background and Clinical Utility. Geburtshilfe Frauenheilkd. 2013;73(9):932–940. doi: 10.1055/s-0033-1350831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tobin N.P., Lundberg A., Lindstrom L.S., Harrell J.C., Foukakis T. PAM50 Provides Prognostic Information When Applied to the Lymph Node Metastases of Advanced Breast Cancer Patients. Clin Cancer Res. 2017;23(23):7225–7231. doi: 10.1158/1078-0432.CCR-17-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao Y.Q., Chen J., Freudenberg J.M., Meng Q.Y., Rajpal D.K. Network-Based Identification and Prioritization of Key Regulators of Coronary Artery Disease Loci. Arterioscler Thromb Vasc Biol. 2016;36(5):928–941. doi: 10.1161/ATVBAHA.115.306725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sparks J.L., Chon H., Cerritelli S.M., Kunkel T.A., Johansson E. RNase H2-initiated ribonucleotide excision repair. Mol Cell. 2012;47(6):980–986. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng S., Cao Z. Is the role of human RNase H2 restricted to its enzyme activity? Prog Biophys Mol Biol. 2016;121(1):66–73. doi: 10.1016/j.pbiomolbio.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 69.Yang C.A., Huang H.Y., Yen J.C., Chang J.G. Prognostic Value of RNASEH2A-, CDK1-, and CD151-Related Pathway Gene Profiling for Kidney Cancers. Int J Mol Sci. 2018 doi: 10.3390/ijms19061586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dai B., Zhang P., Zhang Y., Pan C., Meng G. RNaseH2A is involved in human gliomagenesis through the regulation of cell proliferation and apoptosis. Oncol Rep. 2016;36(1):173–180. doi: 10.3892/or.2016.4802. [DOI] [PubMed] [Google Scholar]
- 71.Mooney M.A., Nigg J.T., McWeeney S.K., Wilmot B. Functional and genomic context in pathway analysis of GWAS data. Trends Genet. 2014;30(9):390–400. doi: 10.1016/j.tig.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.