

Abstract
Neutrophils are the most abundant leukocytes and usually the first immune cell-type recruited to a site of infection or tissue damage. In asphyxiated neonates, elevated peripheral neutrophil counts are associated with poorer neurological outcomes. Induced neutropenia provides brain protection in animal models of neonatal hypoxic-ischemic (HI) injury, but the anti-neutrophil serum used in past studies heavily cross-reacts with monocytes, thus complicating the interpretation of results. Here we examined neutrophil influx and extravasation, and used a specific anti-Ly6G antibody for induced neutropenia against lipopolysaccharide (LPS)-pretreated HI injury in murine neonates, a model for inflammation-sensitized hypoxic-ischemic encephalopathy (HIE). As early as 6 h after the LPS/HI insult, the mRNAs for neutrophil-recruiting and mitogenic chemokines ascended in the ipsilateral hemisphere, coinciding with immuno-detection of neutrophils. However, neutrophils mainly resided within blood vessels, exhibiting signs for neutrophil extracellular traps (NETs), before 48 h post-LPS/HI. Prophylactic anti-Ly6G treatment blocked the brain infiltration of neutrophils, but not monocytes or lymphocytes, and markedly decreased LPS/HI-induced pro-inflammatory cytokines, matrix metalloproteinase 9 (MMP-9), and brain tissue loss. In contrast, anti-Ly6G treatment at 4 h post-LPS/HI failed to prevent the influx of neutrophils and brain damage. Together, these results suggest important pathological functions for early-arriving neutrophils in inflammation-sensitized HIE.
Keywords: Hypoxic-ischemic encephalopathy, neutrophil extracellular trap, lipopolysaccharide, matrix metalloproteinases, anti-Ly6G
Introduction
Inflammation is a critical contributor to normal development and injury outcomes in the immature brain.1 For example, asphyxiated neonates with intrauterine infection (chorioamnionitis) showed poorer neurological outcomes after therapeutic hypothermia than those without.2,3 Animal research also showed that hypothermia was unable to prevent lipopolysaccharide (LPS)-sensitized neonatal hypoxic-ischemic (HI) injury that involves unique immune responses.4,5 Better understanding of the brain–immune interactions in neonatal HI injury may lead to improved clinical therapies.1,6
Multiple studies have suggested critical functions by neutrophils in adult ischemic stroke.7,8 In experimental adult stroke, neutrophils were first enriched in the leptomeninges from 6 h after ischemia, followed by presence in the cortical Virchow-Robin space from 15 h, and in the brain parenchyma at 24 h.8 Neutrophils have a short half-life of ∼6.5 h in the blood, but a 48-fold reservoir in the bone marrow (120 million compared to <2.5 millions) in a naïve mouse.7 Upon injury, the damaged tissue produces danger signals and chemokines to recruit neutrophils from the blood, where they promote tissue injury or assist wound healing depending on the severity and stage of insult.9 Tissue injury also leads to a rise of granulocyte colony-stimulating factor (G-CSF) to stimulate neutrophil production in the bone marrow.7 Hence, neutrophils are early responders to tissue injury, but they are usually sequestered within blood vessels for a protracted period, where they can injure the endothelial cells by neutrophil extracellular trap (NET)-related mechanisms.8,10–13 Further, neutrophils are the principal source of matrix metalloproteinases (MMP-9) that disrupts the blood–brain barrier (BBB) in adult ischemic stroke.14 Conversely, the ischemic brain tissue secretes IL-1 to promote endothelial transmigration by neutrophils of enhanced neurotoxicity.15,16 In animal models of adult stroke, interference with neutrophil-endothelium adhesion diminishes the infarct size, but this treatment was unsuccessful in clinical trials, suggesting the need for alternative approaches to counter the neutrophil-mediated detrimental effects.7
Less is understood regarding the roles of neutrophils in neonatal cerebrovascular disorders. Yet, an elevated peripheral neutrophil count from 12 to 96 h after birth is associated with poorer neurological outcomes in asphyxiated neonates, despite impaired transendothelial migratory ability by neonatal neutrophils.17,18 In a rodent model of birth asphyxia/hypoxic-ischemic encephalopathy (HIE), the brain-infiltrating neutrophils were detected as early as 4 h post-injury, but they showed a striking lack of extravasation for at least 24 h.19,20 Using polyclonal anti-neutrophil serum (ANS), previous studies showed that prophylactic neutrophil depletion diminished neonatal HI brain injury, whereas ANS treatment after the HI insult was ineffectual.21,22 Although significant, these previous studies share a caveat of cross-reactivity by ANS to monocytes, eosinophils, and to some degree, lymphocytes.19 This is problematic, since neutrophils and monocytes are often considered “partners in crime” for exerting similar or complementary functions in tissue injury.23 Thus, unlike in adult ischemic stroke, the roles of neutrophils in neonatal HI brain injury warrant further investigation.
In the present study, we examined the expression of neutrophil-mitogenic and recruitment chemokines, as well as, the timing of neutrophil influx and extravasation in LPS/HI-injured mouse neonates. We also compared the effects of prophylactic and post-LPS/HI treatment by a neutrophil-specific anti-Ly6G monoclonal antibody (1A8).24,25 Our results suggest pathological functions by early-infiltrating neutrophils in inflammation-sensitized HIE, which may be an important target for therapeutic intervention.
Materials and methods
Animals and LPS/HI surgery
C57BL/6J mice were purchased from the Jackson Laboratory and bred in a specific-pathogen-free facility. The LPS-sensitized HI brain injury model was performed as previously described.5 Briefly, 10-day-old C57BL/6J mouse pups were intraperitoneally injected with 0.3 mg/kg body-weight of lipopolysaccharides (LPS from E. coli O55:B5, Sigma-Aldrich, St. Louis, MO) 4 h before inducing hypoxia and ischemia by the Rice-Vannucci HI procedure.5 Mouse pups were anesthetized by 2% isoflurane mixed with room air when the right common carotid artery was ligated with 5.0 surgical silk suture through a midline neck incision. Mouse pups were allowed to recover with their dams in the ambient atmosphere for 1 h. Then, to induce hypoxia, mouse pups were placed in glass chambers infused with 10% oxygen balanced by 90% nitrogen and submersed in a water bath equilibrated to 37°C for 45 min. Mouse pups were returned after hypoxia to their dams for recovery in ambient atmosphere until further experimental analyses. Mouse pups of both genders were used in this study. All procedures were approved by the University of Virginia School of Medicine, the Institutional Animal Care and Use Committee (IACUC) and conducted according to the National Institutes of Health Guide for Care and Use of Laboratory Animals. Experiments are performed and reported in accordance with the ARRIVE (Animal Research: Reporting in vivo Experiments) guidelines (https://www.nc3rs.org.uk/arrive-guidelines).
Quantitative real-time PCR
Total RNAs were extracted from mouse brains collected at indicated time-points using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions, followed by quantification and quality check using a NanoDrop 2000 (Thermo Fisher Scientific). Two micrograms of total RNA were used to synthesize cDNA using the High Capacity cDNA Synthesis Kit (Applied Biosystems, Waltham, MA). All cDNAs were diluted 4-fold with water before using as a template for real-time PCR. Quantitative real-time PCR was performed using a CFX96 Touch System (Bio-Rad, Hercules, CA) and detected by IQ™ SYBR Green Supermix (Bio-Rad) as described previously.5 The primers used in this study are listed as follows: Il1a Forward, 5ʹ- GCAAC GGGAAGATTCTGAAG-3′, Il1a Reverse, 5′-TGAC AAACTTCTGCCTGACG-3′, Il1b Forward, 5′-CT TTCGACAGTGAGGAGAATGAC-3′, Il1b Reverse, 5′-CAAGACATAGGTAGCTGCCACAG-3′, Il6 Forward, 5′-GGAGAGGAGACTTCACAGAGGAT-3′, Il6 Reverse 5′-AGTGCATCATCGCTGTTCATAC-3ʹ, Tnf Forward, 5′-CCACCACGCTCTTCTGT
CTA-3′, Tnf Reverse, 5′-CTCCTCCACTT
GGTGGTTTG-3′, Cxcl1 Forward, 5′-GCTAAAA GGTGTCCCCAAGTAA-3′, Cxcl1 Reverse, 5′-TAG GACCCTCAAAAGAAATTGTA-3′, Cxcl2 Forward, 5ʹ- CAAGAACATCCAGAGCTTGAGTGT-3′, Cxcl2 Reverse, 5ʹ- TTTTGACCGCCCTTGAGAGT-3′, Ccl2 Forward, 5′-ACCACTATGCAGGTCTCTGTCAC-3′, Ccl2 Reverse, 5′-GCTGCTGGTGATTCTCTTGTAG T-3′, Ccl3 Forward, 5′-TTCTCTGTACCATGACACT CTGC-3′, Ccl3 Reverse, 5′-CGTGGAATCTTCCGG CTGTAG-3′, Csf3 Forward, 5′-GTTGTGTGCCACC TACAAGC-3′, Csf3 Reverse, 5′-CCATCTGCTGCCA GATGGTGGT-3′, β-actin Forward, 5′-GGCACCACA CTTTCTACAATGA-3′, β-actin Reverse, 5ʹ- AGTGGT ACGACCAGAGGCATAC-3′. Transcript quantification was performed as fold changes using the 2−ΔΔCt method compared to reference β-actin gene with the threshold cycle (Ct) determined within the exponential phase of amplification reaction. The mean value in untouched groups was set to 1.
Brain immune cell isolation and flow cytometric analysis
Mice were anesthetized and transcardially perfused with ice-cold PBS. Right cerebral hemispheres were collected, gently homogenized in ice-cold PBS, and passed through 40 µm nylon cell strainers followed by centrifugation. Cell pellet were re-suspended in 30% isotonic Percoll (GE Healthcare, Pittsburgh, PA) solution and overlaid on the top of 70% isotonic Percoll. Percoll density gradients were centrifuged at 500 ×g for 30 min at room temperature. Leukocytes were collected from the 30–70% Percoll interface, washed, and centrifuged at 500 ×g for 10 min at room temperature followed by staining with fluorophore-conjugated antibodies for flow cytometry analysis. Briefly, isolated cells were incubated with anti-mouse CD16/32 antibodies (BD Biosciences, San Jose, CA) for 10 min to reduce nonspecific binding. Cells were further stained with fluorophore-conjugated primary antibodies against mouse CD11b (BD Biosciences), CD45 (BioLegend, San Diego, CA), CD3ε (BD Biosciences), Ly6G (BD Biosciences), Ly6C (Thermo Fisher Scientific, Waltham, MA), MHC class II (BD Biosciences), ICAM1 (BioLegend), and CD206 (BioLegend) for 30 min at 4°C followed by wash and centrifugation. Stained cells were acquired on LSRII (BD Biosciences) equipped with 405, 488, 561, and 640 nm lasers, and the data were analyzed with the FlowJo software (version 10, TreeStar).
Western blotting
Total proteins were extracted from mouse brains with RIPA buffer (Thermo Fisher Scientific) containing with proteinase inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Protein concentrations were determined by protein assay dye reagent concentrate (Bio-Rad) according to the manufacturer’s instructions. Equal amount of proteins was separated by 12% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA). Membranes were probed with antibodies against human/mouse myeloperoxidase (R&D systems, Minneapolis, MN) and GAPDH (Cell Signaling Technologies) followed by appropriate horseradish peroxidase-conjugated secondary antibodies. The immunoreactivity was visualized with an ECL reagent (PerkinElmer, Waltham, MA) and recorded with the Azure C600 imaging system (Azure Biosystems, Dublin, CA). ImageJ software was used to quantify band intensities.
Immunohistochemistry
Mouse brains were collected at indicated time-points by transcardial perfusion of 4% PFA, followed by post-fix overnight at 4°C, cryoprotected in 30% sucrose, frozen, and embedded in the optimum cutting temperature (OCT) compound. The coronal brain sections (20 µm) were permeabilized with 0.2% Triton X-100 in PBS, and blocked with 3% normal goat serum in PBS. Brain sections were further incubated with primary antibodies specific to mouse Histone H3 (citrulline R2+R8+R17, Abcam, Cambridge, MA), myeloperoxidase (Abcam), and Alexa594-conjugated isolectin GS-IB4 (Thermo Fisher Scientific) that were diluted in PBS containing 1% goat serum overnight at 4°C, followed by staining with corresponding fluorophore-conjugated secondary antibodies for 2 h at room temperature. Stained sections were mounted on microscope slides with Fluoro-gel II mounting medium (Electron Microscopy Sciences, Hatfield, PA). All images were collected on an Olympus FV1300 confocal microscope (Olympus, Waltham, MA).
Neutrophil depletion and blood neutrophil assessment
For the kinetic and efficiency of neutrophil depletion, naïve mouse pups were untreated or treated daily with anti-Ly6G antibodies (20 µg/g body-weight, Bio X cell, West Lebanon, NH) or isotype control IgG (Bio X Cell) by intraperitoneally injection in two consecutive days, and the blood was collected at 6, 24, and 48 h after the first dose of antibody. For the prophylactic neutrophil depletion, mouse pups were treated with two doses of anti-Ly6G antibodies (20 µg/g body-weight) or isotype control IgG by intraperitoneally injection one day before and immediately after HI induction. For delayed neutrophil depletion, mouse pups were intraperitoneally injected with one dose of 20 µg/g anti-Ly6G antibodies or isotype control IgG 4 h after the LPS/HI insult. Neutrophil depletion in the blood was assessed by flow cytometry. Briefly, mouse blood (20 µL) was collected from the submandibular vein at 24 or 48-h recovery into microtubes containing EDTA, and incubated with anti-CD16/32 antibodies. Blood samples were further incubated with fluorophore-conjugated antibodies specific to mouse CD45 (BioLegend), CD11b (BD Biosciences), Ly6C (Thermo Fisher Scientific), and Ly6G (BD Biosciences) at room temperature for 30 min followed by red blood cell lysis. Samples were washed, resuspended, and analyzed by LSRII as described above. The abundance of neutrophils (Ly6G+Ly6C−) and monocytes (Ly6G−Ly6C+) in the blood was expressed as percentage in CD45+CD11b+ leukocytes.
MMP zymogram
The MMP activity in mouse brains was determined as previously described.5 Briefly, mice were perfused with ice-cold PBS, and the brains were snap frozen immediately after collection. Total proteins were homogenized and extracted with RIPA buffer (Thermo Fisher Scientific) containing with proteinase inhibitor cocktail (Roche Diagnostics). Equal amount of proteins was incubated at 4°C overnight with 50 µL of gelatin Sepharose 4B (GE HealthCare) to obtain the gelatin-binding fraction. The gelatin-binding fractions were separated in 10% sodium dodecyl sulfate-polyacrylamide gels containing 0.15% porcine skin gelatin (Sigma-Aldrich). The gels were washed twice with renaturing solution (2.5% Triton X-100 in distilled water) for 30 min/wash, and then incubated with the developing buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, and 5 mM CaCl2 at 37°C overnight. Gels were then stained with Coomassie blue dye at room temperature for 20 min followed by de-staining. Recombinant active mouse MMP-2 (R&D systems) and mouse MMP-9 (R&D systems) were used as positive controls. Gels were photographed and presented in inverted images. The area showing MMP activity was quantified and normalized to GAPDH using the NIH Image J software.
Measurement of brain damage
Brain tissue loss was performed seven days after LPS/HI injury as previously described.5 Briefly, mice were deeply anesthetized and perfused with PBS. Mouse brains were collected and fixed in 4% PFA overnight at 4°C followed by serial sectioning into 1-mm thick slices on a Vibratome (Stoelting, Wood Dale, IL). Each section was photographed, and the extent of tissue loss was quantified as the mean area ratio in all sections on the ipsilateral to contralateral hemispheres using ImageJ software.
Statistical analysis
Statistics were performed using Prism (version 7, GraphPad). All values are expressed as mean ± standard deviation (SD). Experimental groups were compared by one-way ANOVA followed by post hoc analysis of Tukey test or unpaired two-tailed Student’s t test. Significance was defined as p < 0.05.
Results
Rapid induction of neutrophil chemoattractant and their influx in neonatal LPS/HI injury
To test whether neonatal LPS/HI injury leads to rapid induction of pro-inflammatory or neutrophil-recruiting chemokines in murine brains, we extracted mRNAs from unchallenged, ipsilateral (right), and contralateral (left) hemisphere at 6-h recovery for RT-qPCR comparison. LPS injection alone failed to increase chemokine expression in both hemispheres (Figure 1(a), n = 4 for LPS samples). At 6-h recovery, HI insult in the absence of LPS-sensitization significantly increased the transcripts of Il1β, Il1α, Tnf, and Ccl3/Mip1α (encoding Macrophage Inflammatory Protein 1α) in the ipsilateral hemispheres compared to contralateral hemisphere and unchallenged mouse brains, and the transcripts of Cxcl2/Gro2 in the ipsilateral hemispheres compared to unchallenged brains (Figure 1(a), n = 7 for HI samples). HI insult also slightly increased the Il6, Cxcl1/Gro1, and Ccl2/Mcp1 (encoding Monocyte Chemoattractant Protein 1) transcripts in the ipsilateral hemispheres. At 6-h post-LPS/HI, the transcript levels for Il1α, Il6, Ccl2/Mcp1, and Ccl3/Mip1α were significantly increased in the ipsilateral hemisphere compared to contralateral hemisphere and unchallenged mouse brains (Figure 1(a), n = 6 for post-LPS/HI samples). The transcripts for Il1β, Tnf, Cxcl1/Gro1, Cxcl2/Gro2, and Csf3/Gcsf (encoding Granulocyte Colony Stimulating Factor) also showed a trend of increase in the ipsilateral hemisphere. Interestingly, LPS/HI significantly increased Csf3 and Ccl2 in the ipsilateral hemispheres compared to HI insult alone, which could lead to rapid recruitment of both neutrophils and monocytes after neonatal LPS/HI brain injury. Hence, we focused our analysis on the combined LPS/HI injury model for the rest of this study.
Figure 1.

LPS/HI triggers rapid inflammatory responses in neonatal mouse brains. (a) RT-qPCR analysis suggested elevated expression of multiple pro-inflammatory and neutrophil/monocyte-recruiting cytokines in the ipsilateral (R) hemisphere at 6 h after HI or LPS/HI injury compared with unchallenged (UN), LPS-injected, or the contralateral (L) hemisphere, although the cytokine induction patterns by HI-versus-LPS/HI were not identical. Of note, LPS/HI injury is far more potent for inducing the Csf3 mRNA. (b, c) Detection of leukocyte infiltration in LPS/HI-injured murine brains by flow cytometry. Representative flow plots (b) and quantification (c) showed increase of CD45hiCD11b+ leukocytes, CD45hiCD11b+Ly6G+ neutrophils, and MHC class II+ myeloid cells in the brain from 6 to 48 h after LPS/HI injury. Each dot on bar graphs represents an individual animal. Data were shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA followed post hoc analysis of Tukey test and unpaired Student’s t test in panels a and c, respectively.
Next, we applied flow cytometry to compare the number of neutrophils and myeloid cells in brain parenchyma at 6, 24, and 48 h after LPS/HI injury. Figure 1(b) shows our gating strategy that distinguishes myeloid cells (CD45hiCD11b+) from the brain resident microglia (CD45medCD11b+) and separates myeloid cells into neutrophils (CD45hiCD11b+Ly6G+) and non-neutrophils (Ly6G− CD11b+MHCII+). This experiment showed more than twofold increase of myeloid cells in brain parenchyma as early as 6 h after LPS/HI insult, but these are mainly MHCII+ non-neutrophils (e.g. dendritic cells, macrophages, and monocytes). In contrast, strong influx of Ly6G+ neutrophils inside the brain parenchyma was mostly detected at 48 h after LPS/HI insult (Figure 1(c), n = 4–7 for each time-point). Of note, these flow results do not exclude the possibility of intravascular stagnation of neutrophils in LPS/HI-injured brains.
Intravascular stasis of neutrophils with extracellular traps in LPS/HI brain injury
At 24 h post-LPS/HI, there was a marked increase of myeloperoxidase (MPO), a granule protein specifically expressed by neutrophils, in the ipsilateral hemisphere compared to the contralateral hemisphere and untouched naïve brains (Figure 2(a), n = 3–5 as indicated). To locate neutrophils in post-LPS/HI brains, we collected mouse brains at 24 or 48 h post-injury, and performed double-labeling with anti-myeloperoxidase (MPO, a neutrophil marker) and anti-citrullinated histone H3 (H3-cit, a marker for NET) antibodies. This analysis showed absence of neutrophils or H3-cit signals in the contralateral cerebral cortex at 24- or 48-h recovery (Figure 2(b), top row). In contrast, many MPO and H3-cit double-labeled neutrophils were detected in the ipsilateral cerebral cortex, predominantly within blood vessels, at 24-h post-injury (second row, arrows showcase anti-MPO/H3-cit double-labeled cells, and third row illustrates MPO+ neutrophils within the isolectin B4+ endothelial vascular wall). Some MPO+ neutrophils were also detected in the perivascular space, similar to the findings in adult stroke.8 By 48 h after LPS/HI injury, anti-MPO/H3-cit double-labeled cells became more evenly detectable inside and outside blood vessels (bottom row, arrows show anti-MPO/H3-cit double-labeling). These data suggest delayed neutrophil extravasation and NET-mediated endothelial events after neonatal HI insult, similar to adult ischemic stroke.8,10–13,19,20
Figure 2.

Detection of neutrophils and neutrophil extracellular traps (NETs) after LPS/HI injury in mouse brains. (a) Western blot and densitometric analysis of neutrophil myeloperoxidase (MPO) expression in the ipsilateral (Right) and contralateral (Left) hemispheres at 24-h post-LPS/HI. Each dot on bar graphs represents an individual animal. UN: untouched naïve mice. Data were shown as mean + SD. *p < 0.05, by unpaired Student’s t test. (b) Shown are anti-myeloperoxidase (MPO, a neutrophil marker) with anti-citrullated histone H3 (H3-cit, a NET marker) or Alexa 594-conjugated isolectin B4 (IB4, blood vessel marker), and the merged images in contralateral and contralateral hemispheres at 24 h and in the ipsilateral hemisphere at 48-h post-LPS/HI. Shown are the same patterns in two independent experiments. Arrows show examples of anti-MPO/H3-cit double-labeled cells. Scale bars: 20 µm.
Prophylactic anti-Ly6G administration markedly attenuates neonatal LPS/HI brain injury
To evaluate the functions of neutrophils, we administered intraperitoneally two doses of 20 µg/g anti-Ly6G antibody versus control IgG to mice—the first dose one day before and the second dose immediately after LPS/HI insult—and compared the effects one or seven days later (Figure 3(a)). In naïve mice, anti-Ly6G antibody was able to decrease blood neutrophil counts at six post-treatment, and daily anti-Ly6G antibody treatment in two consecutive days maintained blood neutrophils at the nadir for 48 h after the first dose without affecting blood monocytes (Figure 3(a)). Flow cytometry analysis showed an increase of Ly6G+ neutrophils, but not Ly6G−Ly6C+ monocytes, in the blood of control IgG-treated animals at 24 h post-LPS/HI (Figure 3(b)). In contrast, prophylactic anti-Ly6G treatment markedly reduced the number of Ly6G+ neutrophils in the blood, leading to an inverse increase of the percentage of Ly6C+ monocytes in CD45+ CD11b+ myeloid cells (Figure 3(b), ***p < 0.001 compared to control IgG-treated mice). Flow cytometry analysis also showed an increase of myeloid cells (CD45hiCD11b+), neutrophils (Ly6C+Ly6G+), monocytes (Ly6C+Ly6G−) at 24 and 48 h after LPS/HI, and T cells (CD45hiCD3+) at 24 h after LPS/HI in the ipsilateral hemisphere of control IgG-treated mice (Figure 3(c) and (d)). Because neutrophils have been reported to possess functional diversity,26 we compared surface expression of pro-inflammatory (ICAM1) and anti-inflammatory (CD206) markers on the brain-infiltrating neutrophils. At 48 h after LPS/HI, about 33% of brain-infiltrating neutrophils expressed ICAM1 (N1-like) and <0.1% of infiltrating neutrophils expressed CD206 (N2-like), while the majority of neutrophils were negative for ICAM1 and CD206 (Figure 3(c), n = 5 for LPS/HI-injured mice). Among them, only neutrophils were significantly blocked by the prophylactic anti-Ly6G treatment (Figure 3(c), *p < 0.05 compared to control IgG-treated mice). These results support the specificity of anti-Ly6G-mediated neutrophil-depletion and reduction of immune infiltrates in LPS/HI-injured brains.
Figure 3.

Prophylactic anti-Ly6G treatment specifically reduces neutrophil infiltration to the brain. (a) The scheme of prophylactic anti-Ly6G therapy of LPS/HI injury in mice and outcome analysis, and the kinetic of depletion efficiency in the blood of naïve mice. (b) Quantification of neutrophils (CD45+CD11b+Ly6C+Ly6G+) and monocytes (Ly6C+Ly6G−) in the blood at 24 and 48 h after the second dose of anti-Ly6G-versus-control IgG treatment. (c) Comparison of the myeloid cells (CD45hiCD11b+), neutrophils (Ly6C+Ly6G+), monocytes (Ly6C+Ly6G−) at 24 and 48 after neonatal LPS/HI injury, plus characterization of the brain-infiltrating neutrophil subtypes (ICAM1+CD206−, ICAM1−CD206+, and double-negative ICAM1−CD206−) at 48-h post-injury. (d) Comparison of T cells (CD45hiCD3+) in the brain parenchyma at 24 after neonatal LPS/HI injury. For (b)–(d), shown on the left are representative flow plots, and on the right, summarized data and statistical analysis. Each dot on the graphs is an individual sample. Data shown are mean ± SD and *p < 0.05, ***p < 0.001 by unpaired Student’s t test.
At 24-h post-LPS/HI, control IgG-treated mice showed increased expression of Il1α, Il1β, Il6, Tnf, Cxcl/Gro1, Cxcl2/Gro2, Ccl2/Mcp1, and Ccl3/Mip1α transcritps (Figure 4(a)). Prophylactic anti-Ly6G treatment decreased Il1α, Il1β, and Tnf (pro-inflammatory cytokines) and Cxcl2/Gro1, Ccl2/Mcp1, and Ccl3/Mip1α (neutrophil/monocyte chemokines), but not Il6 or Cxcl1/Gro1 mRNAs (Figure 4(a)). Zymogram showed that anti-Ly6G treatment consistently reduced the LPS/HI-induced MMP-9 activity at 24-h recovery (Figure 4(b)). Prophylactic anti-Ly6G treatment also significantly reduced the brain tissue loss seven days after LPS/HI (Figure 4(c), n = 11 for each group). In a subset of anti-Ly6G-versus-control antibody-treated mice, we compared the blood neutrophil counts of individual animal at 24-h post-LPS/HI and the extent of brain tissue loss at day 7 post-injury. This analysis showed clear segregation of blood neutrophil counts between anti-Ly6G- and control antibody-treated mice, without linear correlation to the eventual brain atrophy level within each group (Figure 4(d), n = 5 for each group). Together, these results suggest a critical role for neutrophils in neonatal LPS/HI brain injury.
Figure 4.

Prophylactic neutrophil depletion confers protection against neonatal LPS/HI brain injury. (a) Prophylactic anti-Ly6G treatment, as depicted in Figure 3(a), attenuated the brain mRNA levels for multiple pro-inflammatory cytokines and neutrophil/monocyte-chemoattractants at 24 h after LPS/HI injury. (b) Representative image of MMP zymogram in the brains of untouched and anti-Ly6G- or control IgG-treated mice collected at 24-h recovery (Left) and quantification (Right, n = 5 for untouched, n = 11 for control IgG- and n = 4 for anti-Ly6G-treated mice). (c) Representative images of control IgG- or anti-Ly6G-treated mouse brains at seven days recovery (Left) and quantification of brain tissue loss (Right, n = 11 each for control IgG- and anti-Ly6G-reated animals). (d) The distribution of blood neutrophil frequency at 24 h and the extent of brain tissue loss at seven days recovery in anti-Ly6G- or control antibody-treated mice. Data shown are mean ± SD from two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired Student’s t test.
Post-LPS/HI application of anti-Ly6G fails to prevent neutrophil influx and brain damage
To test the therapeutic potential of induced neutropenia against neonatal HI brain injury, we injected 20 µg/g anti-Ly6G-versus-control IgG to mouse neonates at 4 h after LPS/HI to simulate a more clinically relevant scenario. At 24 h after LPS/HI, control IgG-treated mice showed 1.7-fold increase of neutrophils in the blood, while anti-Ly6G-treated mice showed significant reduction of neutrophils and an inverse increase of Ly6G−Ly6C+ monocytes among CD45+CD11b+ leukocytes (Figure 5(a)). Yet, the delayed anti-Ly6G treatment was unable to consistently avert the increase of neutrophils and monocytes in LPS/HI-injured mouse brains at 24 h (Figure 5(b), n = 6 for each group), possibly due to rapid infiltration of neutrophils in damaged tissues.8,9 Accordingly, anti-Ly6G and control IgG-treated mice showed a similar degree of brain atrophy after seven days of recovery (Figure 5(c), n = 11 for control IgG and n = 12 for anti-Ly6G treatment). These results indicate the limits of induced neutropenia as a therapy of infection-sensitized neonatal HI brain injury.
Figure 5.
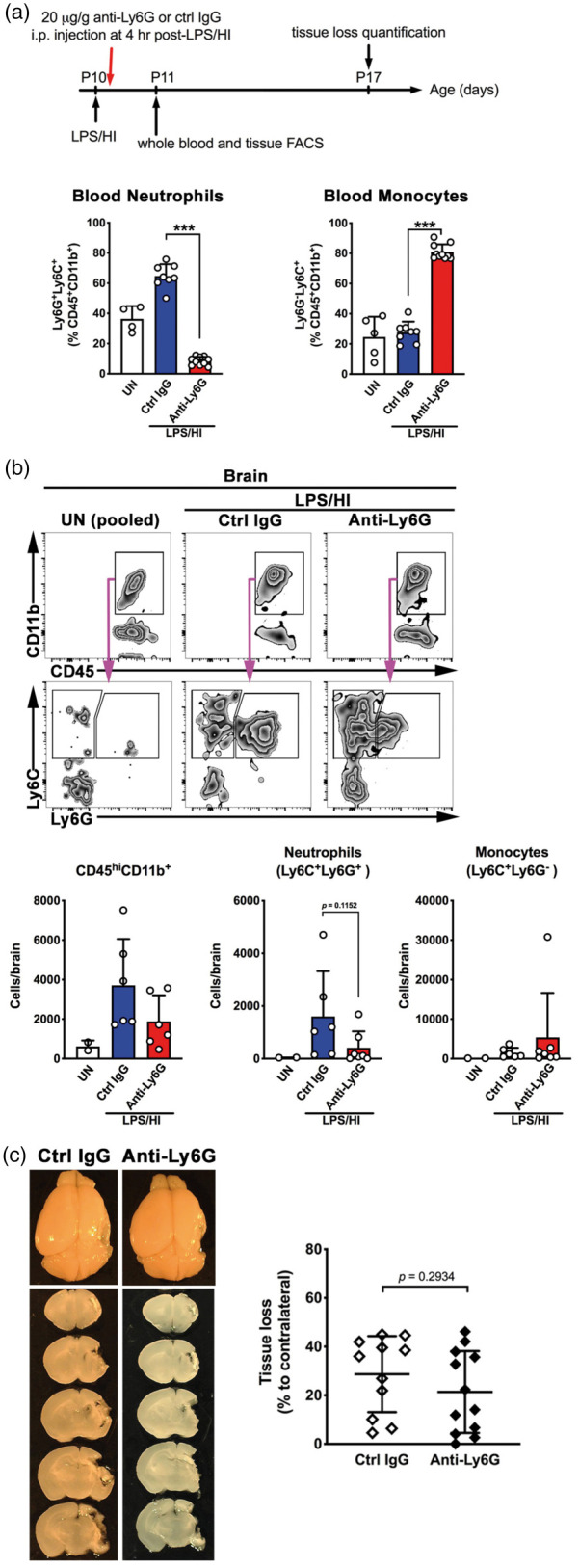
Post-LPS/HI administration of anti-Ly6G antibody failed to prevent brain atrophy and the influx of neutrophils. (a) The scheme of post-LPS/HI anti-Ly6G treatment and outcome analysis in mouse neonates. Comparison of the blood neutrophils (CD45+CD11b+Ly6C+Ly6G+) and monocytes (CD45+CD11b+Ly6C+Ly6G−) at 24 h after LPS/HI in mice post-treated with control IgG (n = 8) or anti-Ly6G (n = 10) antibodies. (b) Representative flow plots and the summarized data of myeloid cells (CD45hiCD11b+), neutrophils (CD45hiCD11b+Ly6C+Ly6G+), and monocytes (Ly6C+Ly6G−) at 24 h recovery in the brains of mice received post-LPS/HI control IgG or anti-Ly6G treatment (n = 6 for each group). (c) (Left) Representative mouse brains and (Right) quantification of brain tissue loss of mice that received post-LPS/HI treatment of control IgG (n = 11) or anti-Ly6G antibodies (n = 12) and collected at seven-day recovery. Each dot on the graphs represents an individual sample. Data shown are mean ± SD and representative of two independent experiments. ***p < 0.001, unpaired Student’s t test.
Discussion
Brain-immune interactions play an important role in birth asphyxia and neonatal HI brain injury, especially in those compounded by intrauterine infection (chorioamnionitis) and carry a greater risk for cerebral palsy.1,3 Histological chorioamnionitis, estimated to occur in 50% of preterm birth and up to 20% of deliveries at term, also reduces the responsiveness to therapeutic hypothermia.2,27 While microglial activation occurs secondary to neural injury in an “sterile inflammation” manner, growing evidence indicates that chorioamnionitis-induced fetal immune responses both intensify and accelerate post-HI neuroinflammation with unique mechanisms.1,5 Determining these immune mechanisms may lead to more successful therapies of neonatal HIE. Thus, we have used the LPS/HI model to assess the roles of neutrophils in infection-sensitized neonatal HI brain injury in this study.
Neutrophils, the most abundant leukocytes in the blood and fortified by a large reservoir in the bone marrow, are commonly the earliest responders to infection and tissue injury by the innate immune responses.6–8 Upon injury, the tissue produces G-CSF to stimulate neutrophil production in the bone marrow and releases danger signals plus chemotactic cytokines to recruit neutrophils from the blood (Figure 6).8,9,28 It is becoming increasingly clear that neutrophils have two critical phases of action—an often protracted intravascular phase and an intra-tissue phase—that may contribute to tissue damage. In the intravascular phase, activated endothelial cells stimulate the NADPH oxidase-mediated oxidative stress inside neutrophils, which in turn promotes degranulation of the protease contents and expulsion of nuclear chromatin, leading to extracellular assembly of NETs to damage the vascular barrier.11–14 Among several granule-stored proteases, MMP-9 has a prominent role in BBB damage, and neutrophils are the principal source for MMP-9 after cerebral ischemia.14 The importance of NETs for barrier damage is suggested by a greater propensity to vascular occlusion in the mice lacking two NET-degrading deoxyribonucleases (DNase1 and DNase1-like 3).10 Besides BBB-disruption, neutrophils may also enter tissue through Mac1 (CD11b/CD18) and intercellular adherence molecule 1 (ICAM1)-mediated endothelial adherence and IL1-stimulated transmigration, which triggers neutrophils to acquire a greater neurotoxic phenotype.15,18 The downstream effectors include neutrophil-mediated phagocytosis plus the release of proteases, reactive oxygen species, and pro-inflammatory cytokines.6 In addition, recent studies showed that some neutrophils also express M2 macrophage markers (e.g. Arginase and CD206) and mediate the resolution of inflammation, a condition called the N2 phenotype.29,30 Blocking neutrophil-endothelium adhesion with anti-Mac1 antibodies markedly reduced the infarct size in experimental cerebral ischemia, but this strategy did not yield benefits in the Acute Stroke Therapy by Inhibition of Neutrophil (ASTIN) clinical trial.31 Although the exact causes for negative results in the ASTIN trial are unclear, one possibility may be undiminished neutrophil toxicity in the intravascular phase.
Figure 6.

Summary of the actions by neutrophils in neonatal HI brain injury and/or stroke. This scheme emphasizes both intravascular and intra-tissue phases of neutrophil functions, as well as, two neutrophil-targeting therapies and their limitations. For induced neutropenia, the challenge is early influx of neutrophils in HI-injured brain tissue. For blocking neutrophil-endothelium adhesion, this approach would not counter neutrophil-mediated endovascular insults through oxidative burst, expulsion of proteases by degranulation, and NET/NETosis effects. The concept of N1 (cytotoxic) and N2 (anti-inflammatory) neutrophils is based on previous studies.26,28,29
Neutrophils from human neonates manifest impaired extravasation from blood vessels, but elevated neutrophil counts in the blood from 12 to 96 h after birth are associated with neurological disability in asphyxiated term neonates.17,18 These findings suggest a critical role for intravascular neutrophils in neonatal HI brain injury. Consistent with this idea, preclinical studies showed rapid brain-influx of neutrophils (as early as 4 h post-HI), but delayed extravasation until 40–48 h after HI.19,20 Previous studies have attempted immune-induced neutropenia on neonatal HI injury, but the employed polyclonal antibodies were either uncharacterized (Accurate Antibodies AIA51140)21,22 or showed strong cross-reactivity to monocytes, eosinophils, and to a lesser degree, lymphocytes (anti-neutrophil serum, ANS).19 These off-target effects make it difficult to distinguish the functions by neutrophils or monocytes, which often have overlapping functions in innate immune responses.23 Nevertheless, prophylactic “neutropenia” appears to be more effective than post-injury “neutrophil depletion,” but the underlying causes were unclear.21
In the present study, we used a Ly6G-specific mAb (1A8) to selectively deplete neutrophils without affecting monocytes (Figure 3(b) and (c) and Figure 5(a) and (b)) and assess the effects in LPS/HI brain injury24,25 We also examined the expression of neutrophil-mitogenic and recruitment cytokines, as well as, the timing and locations of neutrophil infiltrates after LPS/HI injury. Our results indicate that, as early as 6 h post-LPS/HI, the ipsilateral hemisphere already up-regulated the mRNAs for G-CSF (neutrophil-specific growth factor) and mixed neutrophil/monocyte-recruitment chemokines (IL-1α, IL-6, CCL3/MIP1α, and MCP-1), coinciding with the brain parenchyma-influx of myeloid cells, but these were mostly CD11b+MHCII+ non-neutrophils (e.g. dendritic cells, macrophages, and monocytes). In contrast, neutrophils were clustered within blood vessels and showed signs for NETs before 48-h post-LPS/HI (Figure 2(b)). Prophylactic anti-Ly6G treatment blocked neutrophil influx and markedly diminished LPS/HI-induced pro-inflammatory cytokines, MMP-9 activation, and brain tissue loss (Figures 3 and 4). In contrast, anti-Ly6G treatment at 4-h post-LPS/HI appeared too late to prevent the influx of neutrophils and the resultant brain atrophy (Figure 5).
In conclusion, the results from this study suggest important pathological functions for early-arriving neutrophils in infection-sensitized neonatal HI brain injury, and underscore the limits of therapeutic neutropenia presumably due to unfavorable kinetics of neutrophil depletion versus rapid brain influx. Finally, given the protracted intravascular stagnation for neutrophils with NETs after neonatal HI, our findings suggest that the intravascular phase of neutrophil toxicity may be a critical therapeutic target against neonatal HI brain injury.
Acknowledgements
We thank all members of the Kuan laboratory and Center for Brain Immunology and Glia (BIG) for helpful discussion and reagent sharing.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Institute of Health grants (NS084744, NS095064, NS100419, and NS108763 to C-YK).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, or publication of this article.
Authors’ contributions
H-WY and C-YK designed the study and wrote the manuscript. H-WY also performed all experiments.
References
- 1.Hagberg H, Mallard C, Ferriero DM, et al. The role of inflammation in perinatal brain injury. Nat Rev Neurol 2015; 11: 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wintermark P, Boyd T, Gregas MC, et al. Placental pathology in asphyxiated newborns meeting the criteria for therapeutic hypothermia. Am J Obstet Gynecol 2010; 203: 579: e571–579. [DOI] [PubMed] [Google Scholar]
- 3.Wu YW, Escobar GJ, Grether JK, et al. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA 2003; 290: 2677–2684. [DOI] [PubMed] [Google Scholar]
- 4.Osredkar D, Thoresen M, Maes E, et al. Hypothermia is not neuroprotective after infection-sensitized neonatal hypoxic-ischemic brain injury. Resuscitation 2014; 85: 567–572. [DOI] [PubMed] [Google Scholar]
- 5.Yang D, Sun YY, Bhaumik SK, et al. Blocking lymphocyte trafficking with FTY720 prevents inflammation-sensitized hypoxic-ischemic brain injury in newborns. J Neurosci 2014; 34: 16467–16481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B, Concepcion K, Meng X, et al. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog Neurobiol 2017; 159: 50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jickling GC, Liu D, Ander BP, et al. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 2015; 35: 888–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-de-Puig I, Miro-Mur F, Ferrer-Ferrer M, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol 2015; 129: 239–257. [DOI] [PubMed] [Google Scholar]
- 9.McDonald B, Pittman K, Menezes GB, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010; 330: 362–366. [DOI] [PubMed] [Google Scholar]
- 10.Jimenez-Alcazar M, Rangaswamy C, Panda R, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017; 358: 1202–1206. [DOI] [PubMed] [Google Scholar]
- 11.Gupta AK, Joshi MB, Philippova M, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett 2010; 584: 3193–3197. [DOI] [PubMed] [Google Scholar]
- 12.Papayannopoulos V.Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 2018; 18: 134–147. [DOI] [PubMed] [Google Scholar]
- 13.Meegan JE, Yang X, Coleman DC, et al. Neutrophil-mediated vascular barrier injury: role of neutrophil extracellular traps. Microcirculation 2017; 24: e12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang G, Guo Q, Hossain M, et al. Bone marrow-derived cells are the major source of MMP-9 contributing to blood-brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res 2009; 1294: 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen C, Thornton P, Denes A, et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol 2012; 189: 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Bonilla L, Racchumi G, Murphy M, et al. Endothelial CD36 contributes to postischemic brain injury by promoting neutrophil activation via CSF3. J Neurosci 2015; 35: 14783–14793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morkos AA, Hopper AO, Deming DD, et al. Elevated total peripheral leukocyte count may identify risk for neurological disability in asphyxiated term neonates. J Perinatol 2007; 27: 365–370. [DOI] [PubMed] [Google Scholar]
- 18.Anderson DC, Rothlein R, Marlin SD, et al. Impaired transendothelial migration by neonatal neutrophils: abnormalities of Mac-1 (CD11b/CD18)-dependent adherence reactions. Blood 1990; 76: 2613–2621. [PubMed] [Google Scholar]
- 19.Hudome S, Palmer C, Roberts RL, et al. The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr Res 1997; 41: 607–616. [DOI] [PubMed] [Google Scholar]
- 20.Bona E, Andersson AL, Blomgren K, et al. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res 1999; 45: 500–509. [DOI] [PubMed] [Google Scholar]
- 21.Palmer C, Roberts RL, Young PI.Timing of neutrophil depletion influences long-term neuroprotection in neonatal rat hypoxic-ischemic brain injury. Pediatr Res 2004; 55: 549–556. [DOI] [PubMed] [Google Scholar]
- 22.Doycheva DM, Hadley T, Li L, et al. Anti-neutrophil antibody enhances the neuroprotective effects of G-CSF by decreasing number of neutrophils in hypoxic ischemic neonatal rat model. Neurobiol Dis 2014; 69: 192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prame Kumar K, Nicholls AJ, Wong CHY.Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res 2018; 371: 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daley JM, Thomay AA, Connolly MD, et al. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 2008; 83: 64–70. [DOI] [PubMed] [Google Scholar]
- 25.Lee PY, Wang JX, Parisini E, et al. Ly6 family proteins in neutrophil biology. J Leukoc Biol 2013; 94: 585–594. [DOI] [PubMed] [Google Scholar]
- 26.Shaul ME, Levy L, Sun J, et al. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: a transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology 2016; 5: e1232221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conti N, Torricelli M, Voltolini C, et al. Term histologic chorioamnionitis: a heterogeneous condition. Eur J Obstet Gynecol Reprod Biol 2015; 188: 34–38. [DOI] [PubMed] [Google Scholar]
- 28.Castanheira FVS, Kubes P.Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019; 133: 2178–2185. [DOI] [PubMed] [Google Scholar]
- 29.Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009; 16: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuartero MI, Ballesteros I, Moraga A, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke 2013; 44: 3498–3508. [DOI] [PubMed] [Google Scholar]
- 31.Krams M, Lees KR, Hacke W, et al. Acute stroke therapy by inhibition of neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke 2003; 34: 2543–2548. [DOI] [PubMed] [Google Scholar]