

Highlights
-
•
SHC014748M was proved to be more selective for PI3Kδ inhibition relative to other class i PI3K enzymes.
-
•
SHC014748M showed in vitro activity in most of 23 B lymphoma cell lines and primary CLL cells and also inhibited phosphorylation of AKT, targets downstream of PI3Kδ.
-
•
In vivo study revealed that SHC014748M significantly reduced lymphoma cell growth in the treatment group compared with control mice.
-
•
SHC014748M seemed to be a novel promising compound in the treatment of B cell lymphomas and CLL.
Keywords: PI3Kδ, Inhibition, SHC014748M, Preclinical, Lymphoma
Abbreviations: 7-AAD, 7-Aminoactinomycin D; ADR, adverse drug reactions; ANOVA, analysis of variance; BCRs, b-cell antigen receptors; BCA, bicinchoninic acid; CIT, chemoimmunotherapy; FBS, fetal bovine serum; FL, follicular lymphoma; iNHL, indolent non-Hodgkin lymphomas; CLL/SLL, lymphocytic lymphoma; mTOR, mammalian target of rapamycin; NHL, non-Hodgkin lymphomas; PI3Ks, phosphatidylinositol 3-kinase; RIT, radioimmunotherapy; SDS, sodium dodecyl sulfate; STS, staurosporine; IC50, the 50% inhibitory concentration; WM/LPL, Waldenstrom's macroglobulinemia/lymphoplasmacytic lymphoma
Abstract
PI3Kδ (phosphatidylinositol 3-kinase-δ), one of the class I PI3Ks, is found expressed primarily in leukocytes and plays an essential role in B-cell development and function. This provides a rationale for the development of small molecule inhibitors that selectively target p110δ for patients with indolent non-Hodgkin lymphomas. Here in this paper, we comprehensively evaluated the in vitro and in vivo antitumor activity of SHC014748M, an oral selective inhibitor of PI3Kδ under Phase I clinical evaluation. Biochemical and cell-based assays were used to measure compound potency and selectivity in lymphoma cell lines as well as primary chronic lymphocytic leukemia (CLL) cells. Scid mice were subcutaneously inoculated with the SU-DHL-6 cell line. SHC014748M was more selective for PI3Kδ inhibition relative to other class I PI3K enzymes and showed in vitro activity in most of 23 B lymphoma cell lines and primary CLL cells. SHC014748M also inhibited phosphorylation of AKT, targets downstream of PI3Kδ, in both lymphoma cells and primary CLL cells. In vivo study revealed that SHC014748M significantly reduced lymphoma cell growth in the treatment group compared with control mice. CCL4, CCL17, CCL22 and CXCL13 in patient serum decreased sharply after SHC014748M treatment. According to the results, SHC014748M appeared to be a novel promising compound in the treatment of B cell lymphomas and CLL.
Background
Non-Hodgkin lymphomas (NHL) is a heterogeneous group of solid tumors of the lymphatic system, nearly 90% of which arise from B lymphocytes [1], [2], [3]. In 2015, approximately 88,200 people were diagnosed with lymphoma, among which 80%−90% are NHL, while approximately 52,100 died of lymphoma in China [4], [5], [6]. Nearly one third of NHLs are indolent while the others are aggressive [7,8]. Indolent NHL (iNHL) is a set of slow-progressive B-cell malignancies, which mainly include follicular lymphoma (FL), chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), marginal zone lymphoma, and Waldenstrom's macroglobulinemia/lymphoplasmacytic lymphoma [9], [10], [11]. Standard treatments for iNHL include monoclonal antibodies (e.g., anti-CD20) often in combination with chemotherapy (chemoimmunotherapy), radioimmunotherapy, and external-beam radiotherapy [12], [13], [14]. Despite initial response to first-line therapy, iNHL is generally incurable, and along with the development of the disease, iNHL commonly relapses or becomes refractory [15,16]. Thus, there is an unmet need for new drugs, either as monotherapy or in combination with other therapeutic drugs, for patients with relapsed/refractory iNHL.
Phosphatidylinositol 3-kinase (PI3Ks) are enzymes that mediate signals from cell surface receptors with a central role in regulation of cell metabolism, proliferation and survival. The class Ⅰ PI3Ks contain 4 isoforms, named PI3Kα, β, δ, and γ. PI3Kα and β are expressed ubiquitously, while PI3Kγ is preferentially expressed in leukocytes [17], [18], [19]. PI3K δ is expressed mainly in the cells of hematopoietic lineage, where it integrates and transmits signals from surface proteins including B-cell antigen receptors (BCRs) and chemokine receptors (CXCR4 and CXCR5) responsible for cell homing and retention in tissue compartments, via downstream activation of AKT (Protein kinase B). AKT is a central node in signaling cascades that regulate cell survival, proliferation and migration, which is also known to activate the pro-growth mammalian target of rapamycin complex [20], [21], [22]. PI3Kδ inhibition targets proliferation, migration and survival of malignant B-cell leukemia and lymphoma cells, whereas PI3K γ inhibition reduces the differentiation and migration of key cells in the tumor microenvironment, such as CD4+ T cells and M2 tumor-associated macrophages, which sustain leukemia and lymphoma cells in a protective niche [23], [24], [25].
Since 2018, the Food and Drug Administration (FDA) has approved 3 PI3K inhibitors for iNHL treatment, namely Idelalisib, Duvelisib, and Copanlisib. Idelalisib is the first p110δ-selective inhibitor used for relapsed or refractory CLL in combination with rituximab, relapsed, or refractory FL after 2 lines of prior therapies and relapsed or refractory SLL after 2 lines of prior therapies. In a cell-based assay, GS-1101 inhibited PI3Kδ signaling with an EC50 of 8.9 nM. The IC50 was 19 nM in vitro assays using recombinant protein. And In a kinase screen, idelalisib inhibited other Class I PI3K isoforms between 83% and 97.3%. In vitro assays demonstrate that GS-1101 is between 110- and 453-fold more selective for inhibition of PI3Kδ [26]. However, Idelalisib has a black box warning on its product label regarding the risks of fatal and serious toxicities including hepatic toxicity, severe diarrhea, colitis, pneumonitis, infections and intestinal perforation [27,28]. Duvelisib is different from Idelalisib as it targets both p110δ and p110γ. In September 2018, Duvelisib received its first approval for the treatment of adult patients with relapsed or refractory CLL/SLL after at least 2 lines of prior therapies. But it also has a black box warning regarding the risks of fatal and serious toxicities including severe diarrhea, colitis, pneumonitis, infections and cutaneous reaction [29,30]. Copanlisib has predominant inhibition against PI3Kα and PI3Kδ isoforms, which has been proved by FDA for the treatment of patients with relapsed FL after at least 2 prior systemic therapies [31]. Although these treatments approved by FDA showed encouraging efficacy for iNHL, serious adverse drug reactions are difficult to avoid. Other PI3K inhibitors such as Umbralisib, INCB050465, ME-401, RP-6530, ACP-319, Pictilisib, Acalisib, and KA-2237 are still under clinical development [32].
From the studies conducted so far, novel oral drugs with improvements in safety and long-term maintenance are still needed. SHC014748M is a small-molecule inhibitor of PI3K with high selectivity to δ isoform. Here, we present the preclinical characterization of SHC014748M as single-agent in vitro and in vivo tumor models of lymphomas and CLL. This trial is registered with ClinicalTrials.gov, number NCT03588598.
Methods
Compounds
SHC014748M was provided by Sanhome Therapeutics (Nanjing, China). Its molecular formula is C23H21ClN6O4S while its structural formula is given in Figure 1. Idelalisib was purchased from Shining Pharmaceutical Technology Co., Ltd (Shandong, China).
Figure 1.
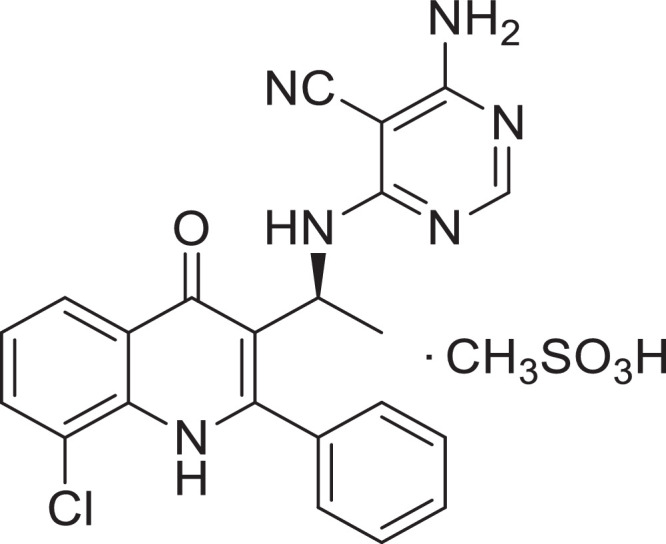
Chemical structural formula of SHC014748M.
B cell lymphoma cell lines
A total of 23 human lymphoma cell lines (from ATCC and he Cell Bank of the Chinese Academy of Sciences) were used 1 (SU-DHL-2) from activated B cell like diffuse large B-cell lymphoma (ABC DLBCL), 8 (DB, Pfeiffer, OCI-LY-19, SU-DHL-4, SU-DHL-6, WSU-DLCL2, TOLEDO, DOHH2) from germinal center B cell DLBCL (GCB DLBCL), 4 (GRANTA-519, MINO,REC-1 and Z-138) from mantle cell lymphoma (MCL), 10 (NAMALWA, CA46, RAJI, DAUDI, ST486, P3HR-1, GA-10, EB2, EB1, Ramos-2G6-4C10) from Burkitt's lymphoma. Cell lines were cultured in RPMI 1640 medium (INVITROGEN, Cat # 61870127) supplemented with 10% fetal bovine serum (GIBCO, Cat # 10099-141), and incubated at 37 °C/5% CO2. All cell lines have been authenticated using STR profiling within the last 3 years and the cells are free of mycoplasma.
Primary CLL cells
Peripheral blood samples were obtained from CLL patients in Jiangsu Province Hospital (The First Affiliated Hospital of Nanjing Medical University). The study was approved by the Institutional Review Board of Jiangsu Province Hospital and was conducted in accordance with Declaration of Helsinki. CLL cells were isolated by B Cell Isolation Kit (Miltenyi Biotec, Cat # 130091151) and were cultured at 37 °C/5% CO2 for 24 hours.
In vitro kinase activity assay
In vitro enzymatic activity was measured using Mobility Shift Assay, Lance Ultra Assay, Lance Screen Assay, Kinases-Glo Assay and ADP-Glo Assay. Kinases were purchased from BPS, Carna, Invitrogen, Millipore and ProQinase. Kinases were seeded in 384-well plates and treated with ten different concentrations of each compound (ranging between 0.05 nM and 10 µM) in duplicate. Staurosporine was used as positive control. Fluorescence polarization were read by Envision 2104 Multilabel (PERKINELMER INC). The 50% inhibitory concentration (IC50) was calculated by fitting the dose-response curve to a 4-parameter log-logistic equation respectively.
Cell proliferation inhibition assay
Human lymphoma cell lines were seeded in 96-well plates and treated with 9 different concentrations of each compound (ranging between 3 nM and 20 µM) in duplicate. Cells were then incubated for 72 hours at 37 °C, 5% CO2. Primary CLL cells were seeded in 96-well plates and treated with 6 different concentrations of each compound (ranging between 100 nM and 100 µM) in duplicate. dimethyl sulphoxide (DMSO) was added to negative control (untreated) cells alone. Wells containing medium only were included on each plate as blanks for absorbance readings. CellTiter-Glo Luminescent Cell Viability Assay (Promega, Cat # G7572,Cat # G7573) was used to measure cell proliferation. IC50 was calculated by fitting the dose-response curve to a 4-parameter log-logistic equation respectively.
Cell apoptotic assay
The percentages of viable and apoptotic lymphoma cell line cells [Annexin V positive/7-Aminoactinomycin D (7-AAD) negative and Annexin V positive/7-AAD positive] were determined by double staining WSU-NHL cells with Annexin V-FITC/7-AAD. Cells were treated with 3 different concentrations of each compound (ranging between 30 nM and 300 nM) in triplicate, then harvested and rinsed with cold phosphate-buffered saline (PBS) once. After centrifugation for 5 minutes, cells were resuspended in 500 µL of 1 × Annexin V binding buffer (Biolegend, Cat # 640934) and then added with 5 µL of PE Annexin V (Biolegend, Cat # B210905) and 5 µL of 7-AAD viability staining buffer (Biolegend, Cat # B219262). After incubation for 15 minutes at room temperature in the dark, the samples were analyzed by flow cytometry.
Cell-based assay
Raji and PC-3 cells were cultured in RPMI Medium 1640 with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 ug/mL streptomycin at 37 °C, 5% CO2. RAW264.7 and C2C12 cells were cultured in DMEM medium with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 ug/mL streptomycin at 37 °C, 5% CO2. PI3Kδ-specific RAJI cells were stimulated with 3 µg/mL F(ab’)2 anti IgM (Jackson ImmunoResearch, Cat # 109006129) for 10 minutes. PI3Kα-specific C2C12 cells were stimulated with 1200 ng/mL insulin like growth factor 1 (IGF-1, Invitrogen, Cat # PHG0078) for 20 minutes. PI3Kβ-specific PC-3 cells were stimulated with 15 µg/mL Lysophosphatidic acid (LPA, SIGMA-ALDRICH, Cat # L7260) for 20 minutes. PI3Kγ-specific RAW 264.7 cells were stimulated with 80 ng/mL C5a (Biotang, Cat # RPR9899) for 5 min. Then cells were treated with ten different concentrations of SHC014748M in triplicate. Phosphorylation of AKT in cells was determined using AlphaLISA technology from PerkinElmer.
AlphaLISA
CLL cells were cultured in 96-well plates with various concentrations of SHC014748M or Idelalisib for 30 min, then stimulated by Anti-Human IgM F (ab’)2 (SIGMA-ALDRICH, Cat # SAB3701394) for 10 minutes. pAKT was measured by AlphaLISA pAKT Kit (PERKINELMER INC, Cat # ALSU-PAKT-B500) and AlphaLISA AKT1 Kit (PERKINELMER INC, Cat # ALSU-TAKT1-A500).
In vivo experiments
Animal experiments were performed under the Guide of animal care and use (NCR 2011) and the Chinese National Standard (GB149252010) without blinded conduct. Scid (CB17 SCID) mice (Ling Chang Biotechnology Co., Ltd, Shanghai, China) were subcutaneously inoculated with the SU-DHL-6 cell line (1 × 107 cells in 0.2 mL of RPMI 1640, 1:1 matrigel). When tumors were palpable, mice were randomly(based on tumor size) assigned into the SHC014748M treatment groups receiving 50 mg/kg,100 mg/kg, or 200 mg/kg once daily, Idelalisib treatment groups receiving 100 mg/kg or 200 mg/kg once daily and the control group receiving vehicle alone once daily(n = 9 in each group to achieve the power of test). Caliper measurements of the longest perpendicular tumor diameters were performed every 2 days to estimate the tumor volume [tumor volume (mm3) =D × d2/2]. Treatments were started when tumor volumes reached approximately 150 mm3. Tumor size was measured using a digital caliper from the first day of treatment until day of first death. Compounds was dissolved in 1% carboxyl methylcellulose/0.5% Tween 80 and stored at 4 °C for in vivo study.
Western blotting analysis
Cells were solubilized in hot sodium dodecyl sulfate lysis buffer and sonicated for 15 seconds. The protein contents were determined using the bicinchoninic acid protein assay. Lysates were fractionated by 8% sodium dodecyl sulfate -PAGE. Membranes were incubated with primary antibodies overnight, followed by the appropriate horseradish peroxidase-conjugated antimouse or antirabbit secondary antibodies for 1 hour. After that, members were scanned by Odyssey imaging system (LI-COR Biosciences). Equal loading of samples was confirmed by probing for Actin. The following antibodies were used PI3Kinase p110δ (D1Q7R) Rabbit mAb (CST, Cat # 34050); The TM beta Actin Antibody Mouse mAb (Genscript, Cat # A00702100); IRDye 800CW Goat anti-Rabbit IgG (H + L) (Li-COR, Cat # 92632211); IRDye 800CW Goat anti-Mouse IgG (H + L) (Li-COR, Cat # 92632210).
Measurement of chemokine concentrations using ELISA
The effects of SHC014748M on CCL4, CCL17, CCL22, and CXCL13 secretion was measured in blood sample of patients receiving 150 mg QD or 200 mg QD. Coated antibody solution was added to each well in duplicate. After 12 hours, the supernatant of each well was harvested and assayed for CCL3, CCL4, and CXCL13 protein by quantitative ELISA according to the manufacturer's instructions (R&D Systems, CAS # P161174).
Pharmacokinetic assessments
Blood samples for measurement of SHC014748M and Idelalisib concentrations were collected at the following time points: prior to and at 10, 30, 60, 120, 360, 600, and 1440 minutes after the start of study drug administration on days 1 and 14. Immediately following withdrawal of Heparin sodium -anticoagulated blood. Blood samples were centrifuged at 4500 rpm for 10 minutes at 4 °C and plasma samples were stored at −20–80 °C until analysis. SHC014748M concentrations were determined by a validated liquid chromatography–tandem mass spectrometry (LS-MS/MS) assay. A tandem mass spectrometer was used for the detection of the compounds, and quantification was based on the peak area ratios of compound and its stable isotope labelled internal standard. Concentrations below the limit of quantification were not included in the PK analyses.
Data analysis and statistics
Results are expressed as mean or mean ± standard error of mean (SEM) or mean ± SD. Statistical analysis was performed using GraphPad Prism version 4.00 for Windows. Analysis of variance was used for comparation among groups, Student's paired or unpaired t test was used for comparation between groups. P < 0.05 was considered statistically significant.
Results
SHC014748M is a highly selective PI3Kδ inhibitor
To determine the selective inhibition of SHC014748M, evaluation against a panel of 50 different kinases which are closely related to BCR signal were conducted. Among 50 kinases, SHC014748M inhibited the activity of all 4 Class I PI3K isoforms. IC50 of PI3Kδ,PI3Kα,PI3Kβ, and PI3Kγ are respectively 0.77, 236, 96, and 101 nM. SHC014748M was 125- to 306-fold more selective for PI3Kδ inhibition compared with other class I PI3K isoforms (Table 1). Inhibition curves of SHC014748M against class I PI3K isoforms were shown in Figure 2A. No significant inhibition was observed against the other 46 diverse protein kinases with 10 µM of SHC014748M (data not shown).
Table 1.
SHC014748M is a potent PI3Kδ inhibitor with highly selectivity against other class I PI3K isoforms.
| Kinase | PI3Kδ | PI3Kα | PI3Kβ | PI3Kγ |
|---|---|---|---|---|
| IC50 (nM) | 0.77 | 236 | 96 | 101 |
| Fold-selectivity | 1x | 306x | 125x | 131x |
Figure 2.

SHC014748M a selective inhibitor of PI3Kδ and downstream AKT signaling, exerted antitumor activity in lymphoma cell lines in vitro. (A) The inhibition curves versus the concentration of SHC014748M against class I PI3K isoforms PI3Kδ, PI3Kα, PI3Kβ, and PI3Kγ were obtained by fitting the data (ranging between 0.05 nM and 10 µM) to a 4-parameter log-logistic equation in duplicate. Data are presented as mean ± SEM. (B) Cell viability was determined by performing CellTiter-Glo Luminescent Cell Viability Assay in lymphoma cell lines after SHC014748M treatment for 72 hours. IC50 were calculated by fitting the dose-response curves (ranging between 3 nM and 20 µM) to a 4-parameter log-logistic equation respectively. (C) Representative stained cells after treatment with DMSO, STS and 30 nM, 300 nM, 3000 nM of SHC014748M for 48 hours were tested using annexin V/7-ADD staining assay and followed by flow cytometric analysis. Q1: cell debris; Q2: late stage apoptotic cells (annexin V+/7-ADD+); Q3: early stage apoptotic cells; (annexin V+/7-ADD-) Q4: live cells (annexin V-/7-ADD-). (D) Qualitative analysis of the percentage ratios of viable, apoptotic, and necrotic cells in SHC014748M and Idelaslisib treated WSU-NHL cells. (E) AKT phosphorylation was determined by AlphaLISA. Plot the Alfa signal read on Envision against the concentration of SHC014748M ranging between 0 and 3000 nM for RAJI cells, and 0 nM and 100 µM for PC-3, RAW 264.7, and C2C12 cells. Data are presented as mean ± SEM. STS, Staurosporine; PI3K, phosphatidylinositol 3-kinase.
SHC014748M exerts antitumor activity in B cell lymphoma cell lines in vitro
The effects of SHC014748M (ranging between 3 and 20 µM) on cell proliferation were evaluated on 23 B cell lymphoma cell lines. IC50 of SHC014748M varies from 27 nM to over 20 µM respectively, showing in vitro activity in most of them with a median IC50 value of 7383 nM. SHC014748M produced more robust inhibition on cell proliferation in GCB DLBCL cell lines with a median IC50 value of 4790 nM, while showed less activity in highly invasive MCL cell lines with a median IC50 value of 12,850 nM (Figure 2B). Idelalisib had weak antiproliferative activity in most of the B cell lymphoma cell lines with IC50 values of over 20 µM. According to the results above, SHC014748M is a highly selective PI3Kδ inhibitor with more potent antitumor activity in lymphoma cells compared to Idelalisib in vitro.
SHC014748M concentration-dependently promotes apoptosis in a B cell lymphoma cell line in vitro
The functional role of PI3Kδ was evaluated by measuring apoptosis in WSU-NHL cell line, exposed concentrations from 30 to 3000 nM. As shown in Figure 2C and D, SHC014748M at the concentration of 30 nM led to a 2.4-fold increase (Control, 4.9%; SHC014748M, 12.0%) in the proportion of apoptotic cells in Annexin V+/7-ADD+ straining, while a 3.7-fold increase (Control, 5.2%; SHC014748M, 19.1%) in annexin V+/7-ADD- staining, indicating that both early stage apoptosis and late stage apoptosis were significantly induced by SHC014748M even at the minimum concentration. We also found that, SHC014748M at the concentration of 30, 300, and 3000 nM resulted in 3.1-, 4.2-, and 5.5-fold increase (Control, 10.1%; 30 nM, 31.1%; 300 nM, 42.1%, 3000 nM, 55.7%) in the total apoptotic cells respectively, showing that SHC014748M could induce apoptosis in B cell lymphoma cells in a concentration-dependent manner. Idelalisib exhibited comparable induction of apoptotic cell death as SHC014748M at the concentration of 300 nM and 3000 nM, but its efficacy at 30 nM was much lower than that of SHC014748M (1.7-fold vs. 3.1-fold).
SHC014748M selectively inhibits phosphorylation of class I PI3K isoform-dependent AKT in vitro
To determine the cellular selectivity of SHC014748M against the Class I PI3K isoforms, inhibition of phosphorylation of AKT was measured. PI3Kα activity was reflected by pAKT in IGF-1 stimulated C2C12 cells, while PI3Kβ was reflected by pAKT in LPA stimulated PC-3 cells, PI3Kγ was reflected by pAKT in C5a stimulated RAW264.7 cells and PI3Kδ was reflected by pAKT in anti-IgM stimulated Raji cells. The results in Figure 2E showed that SHC014748M potently inhibited phosphorylation of AKT, specific downstream of PI3Kδ, on threonine-308 or serine-473 (IC50 = 3.32 nM). On the other hand, no significant inhibition was observed against AKT phosphorylation in other Class I PI3K: IC50 of pAKT in PI3Kα is >30,000 nM (>9036-fold vs PI3Kδ); IC50 of pAKT in PI3Kβ is 453.3 nM (137-fold vs PI3Kδ) and IC50 of pAKT in PI3Kγ is 3040 nM (916-fold vs PI3Kδ). Idelalisib inhibited pAKT of PI3Kδ as potent as SHC014748M (IC50 =5.55 nM), whereas its selectivity was much lower: IC50 of pAKT in PI3Kα is >30,000 nM (>5405-fold vs PI3Kδ); IC50 of pAKT in PI3Kβ was 380.3 nM (69-fold vs PI3Kδ) and IC50 of pAKT in PI3K γ is 1123 nM (202-fold vs PI3Kδ). Thus, in cell-based assays, SHC014748M had 137- to >9036-fold selectivity for PI3Kδ over the other class I PI3K isoforms while Idelalisib had 69- to >5405-fold selectivity.
SHC014748M shows antitumor activity in primary CLL cells in vitro
To confirm the in-vitro antitumor activity of SHC014748M on CLL, the viability of primary leukemia cells derived from CLL patients were determined by Cell Titer-Glo Assay, exposed concentrations from 3.0 nM to 10 µM. SHC014748M showed in vitro efficacy in most of the primary CLL cells, with the IC50 values varied from 850 nM to 37,040 nM. As illustrated in Figure 3A, the IC50 values of SHC014748M in 12 CLL cells were less than 20 µM while in 3 CLL cells were beyond 20 µM. IC50 of Idelalisib varied from 25,650 nM to >200,000 nM respectively, which are much higher than that of SHC014748M (Figure 3A). According to the results above, SHC014748M had more potent antitumor activity in primary CLL cells compared with Idelalisib.
Figure 3.

SHC014748M shows antitumor activity in primary CLL cells in vitro. (A) Cell viability was determined by performing CellTiter-Glo Luminescent Cell Viability Assay in lymphoma cell lines after SHC014748M treatment for 24 hours. IC50 were calculated by fitting the dose-response curves (ranging between 100 nM and 100 µM) to a 4-parameter log-logistic equation respectively. (B) CLL cells from patients, normal B cells from healthy persons and Raji cell line were examined for PI3Kδ expression by Western blot. Normal B cells were isolated from PBMC of 20 healthy persons. β-actin was served as a loading control. (C) The bar diagram represents relative expression of PI3Kδ. The expression of PI3Kδ in normal B cells were regarded as 100%. (D) Anti-IgM was used to activate BCR signal in CLL cells in the presence or absence of SHC014748M or Idelalisib. Cells were lysed and AKT phosphorylation was determined by AlphaLISA. (E) The bar diagram represents the inhibition rate of SHC014748M or Idelalisib at different concentrations. CLL, chronic lymphocytic leukemia.
PI3Kδ is highly expressed in primary CLL cells
Given the importance of PI3Kδ signaling in B-cell development, we sought to determine the protein expression of PI3Kδ in primary CLL cells in patient serum. As the results shown in Figure 3B and C, in CLL samples derived from 6 different patients, high expression of PI3Kδ was found with relatively little variability among patients. In addition to primary CLL cells, we also evaluated the expression profile of PI3Kδ in normal B cells and Raji cell line, a lymphoblast-like derived cell line from Burkitt's lymphoma. As expected, the protein level of PI3Kδ in Raji cell line was as abundant as that in primary CLL cells, whereas much higher than that in normal B cells.
AKT phosphorylation was decreased by SHC014748M in primary CLL cells
To corroborate our in vitro findings in isoform-specific cell-based assay, the pAKT levels in circulating CLL cells with and without SHC014748M treatment was evaluated by AlphaLISA. Not surprisingly, the phosphorylation of AKT was rapidly raised after anti-IgM stimulation and decreased in the presence of SHC014748M or Idelalisib at the concentration ≥100 nM. SHC014748M at the concentration ≥100 nM could efficiently decrease the levels of pAKT to the unstimulated levels, comparing with Idelalisib only at the concentration ≥300 nM (Figure 3D and E).
SHC014748M exhibited in vivo antitumor activity in lymphoma
The in vivo efficacy of SHC014748M was next evaluated by xenograft model in which SCID mice are injected subcutaneously with SU-DHL-6 cells. SHC014748M reduced tumor growth in all treatment groups and showed statistically significant differences in tumor volume between the control group versus SHC014748M treatment groups (50, 100, vs 200 mg/kg: P < 0.05, Figure 4A), so did the Idelalisib treatment groups (100 mg/kg, 200 mg/kg: P < 0.05). Significant differences in tumor weight were also found between the control group versus all SHC014748M treatment groups (50 mg/kg, 100 mg/kg, 200 mg/kg: P < 0.05, Figure 4B) and Idelalisib 100 mg/kg treatment group (P < 0.05), while SHC014748M has more potent antitumor activity than Idelalisib at the dose of 200 mg/kg. At the same time, treatment with either does of SHC014748M did not affect the body weight (data not shown). Taken together, these data demonstrated that SHC014748M significantly inhibits lymphoma cell growth in mice xenograft model in vivo.
Figure 4.

SHC014748M inhibits tumor growth in human NHL xenografts model. (A) Mice injected with 5 × 106 SU-DHL-6 cells were treated orally once a day with control vehicle, SHC014748M and Idelalisib. Mean tumor volume was calculated every 2 days. (B) The bar diagram represents the mean tumor weight. (C) Concentration - time pharmacokinetic profile of SHC014748M and Idelalisib in vivo xenografts model on (C) Day 1 and (D) Day 14. Data are represented as mean ± SEM. * means P < 0.05.
The pharmacokinetic parameters on days 1 and 14 in this SCID mice xenograft model are summarized in Table 2. Concentration-time pharmacokinetic profiles are illustrated in Figure 4C. Cmax for SHC014748M were observed at 1 hour indicating rapid absorption after single dose and multiple doses. Cmax and AUC(0-t) demonstrated only slight increases in exposure above the level of 50 mg for SHC014748M repeated dosing. Accumulation ratio (mean ratio of Day 14/Day 1 AUC) was 1.54 for QD dosing. Mean apparent terminal elimination half-life (t1/2) following 14 days of dosing ranged from 6.5 to 11.7 hours. In addition, as expected, blood concentration of SHC014748M in SCID mice was higher than the effective concentration obtained in-vitro study (Figure 4A).
Table 2.
PK Parameters of SHC014748M and Idelalisib in vivo xenografts model of SCID mice.
| Compound | Dose (mg/kg) | Days | PK Parameters |
|||
|---|---|---|---|---|---|---|
| T1/2 (h) | Tmax (h) | Cmax (nM) | AUC(0-t) (h*ng/mL) | |||
| SHC014748M | 50 | 1 | 5.3 ± 4.4 | 1.7 ± 0.6 | 26,612 ± 9856.1 | 311,514.3 ± 75,938.6 |
| 14 | 2.7 ± 0.7 | 1.3 ± 0.6 | 30,324.6 ± 6027.8 | 348,031.8 ± 39,235.1 | ||
| 100 | 1 | 4.4 ± 2.3 | 2 ± 0 | 27,310.2 ± 4901.2 | 324,080.8 ± 27,532.9 | |
| 14 | 2.6 ± 0.6 | 2 ± 0 | 36,012.6 ± 2520.3 | 441,954.3 ± 24,908 | ||
| 200 | 1 | 4.3 ± 0.1 | 2 ± 0 | 35,777.6 ± 5755.5 | 478,082.1 ± 28,807.5 | |
| 14 | 2.3 ± 0.1 | 1.7 ± 0.6 | 38,625.6 ± 4553.4 | 493,884.9 ± 76,201.9 | ||
| Idelalisib | 100 | 1 | 3.4 ± 2 | 0.7 ± 0.3 | 4174.3 ± 9891.7 | 6933.1 ± 76,212.9 |
| 14 | 1.8 ± 0.6 | 0.4 ± 0.2 | 5049 ± 6049.6 | 5518.4 ± 39,376.8 | ||
| 200 | 1 | 1.9 ± 0.5 | 0.8 ± 0.3 | 11,141.5 ± 2997.2 | 29,943.5 ± 29,943.5 | |
| 14 | 4.3 ± 1.1 | 0.7 ± 0.3 | 8092 ± 930.3 | 22,395.9 ± 4275.7 | ||
SHC014748M inhibits secretion of the chemokines and cytokines in iNHL patients
To confirm our findings in the preclinical study, we evaluated the concentrations of CCL4, CCL17, CCL22 and CXCL13 in iNHL patients before and after treatment with SHC014748M. As illustrated in Figure 5, secretion of CCL4, CCL17, CCL22 and CXCL13 in blood samples from 6 patients (3 from the 150 mg QD group and 3 from 200 mg QD group) were significantly inhibited after the oral administration of SHC014748M. The respective mean CCL4, CCL17, CCL22 and CXCL13 concentrations of patients before taking drugs were 124.4, 315.1, 3799.3, and 734.5 pg/mL, which are comparable with the reference [33]. Durative treatment of patients with SHC014748M once a day for 14 days remarkably abrogated the secretion of CCL4, CCL17, CCL22 and CXCL13, reducing the respective mean values for CCL4, CCL17, CCL22, and CXCL13 to 81.9, 35.9, 971.8, and 184.3 pg/mL. Levels of CCL4, CCL17, CCL22, and CXCL13 remained the same after treatment of SHC014748M once a day for 28 days. SHC014748M showed nearly universal reductions in concentrations of circulating chemokines and cytokines in iNHL patients. The presence or levels of any one or any combination of the chemokine biomarkers CCL2, CCL3, CCL4, CCL5, CXCL13, CCL17, CCL22, and tumour necrosis factor (TNF)-alfa can be used to select a patient with a hematological disorder for treatment [34]. A key spot of this paper is the evaluation of chemokines, such as CCL4, CCL17, and CCL22, that tie together BCR signaling and the lymph node microenvironment [35]. We demonstrated a remarkable inhibition of CCL4, CCL17, and CCL22 production in patients treated with SHC014748M. These data corroborated that CCL4, CCL17, and CCL22 might function as surrogate markers of B cell activation, and hence may also function as markers for response to therapy. Plasma concentrations of circulating CXCL13, a chemokine that attracts B cells to lymph nodes [36], among patients receiving SHC014748M was substantially reduced. Loss of CXCL13 production may be particularly important in explaining the lymphocytosis observed during early treatment with drug.
Figure 5.

Levels of chemokines and cytokines in patients were decreased by SHC014748M treatment in iNHL patients. The bar diagram represents the mean supernatant concentrations of (A) CCL4, (B) CCL17, (C) CCL22, and (D) CXCL13 of 6 patients after treatment of SHC014748M for 14 or 28 days. C0D1, C1D15, and C2D1 represents Cycle 0 Day 1, Cycle 1 Day 15, and Cycle 2 Day 1. Cycle 0 Day 1 was the baseline value, taken before the first dose of SHC014748M.
Discussion
In this paper, we have shown that (1) SHC014748M, a novel PI3Kδ inhibitor, is potently effective and highly selective in both kinase assay and isoform specific cell-based assay; (2) Cell proliferation is inhibited by SHC014748M not only in B cell lymphoma cell lines but also in primary CLL cells; (3) expression of AKT phosphorylation decreases because of SHC014748M treatment in B cell lymphoma cell line as well as primary CLL cells; (4) SHC014748M promotes apoptosis of lymphoma cell line in a concentration-dependent manner; (5) SHC014748M has antilymphoma activity in SU-DHL-6 xenograft model. (6) SHC014748M administration inhibits secretion of the chemokines and cytokines in patients.
Based upon basic medical research and strong preclinical data, intensive efforts have been made to develop PI3K inhibitors for the treatment of iNHL [37,38]. Idelalisib is a potent, orally bioavailable PI3K inhibitor that is specific for the δ isoform, approved by FDA for the treatment of CLL and FL. PI3K inhibitors approved by FDA also include Duvelisib, which inhibit both the PI3Kδ and γ isoforms, and Copanlisib, which is a combined PI3K α/δ inhibitor. Other PI3K inhibitors such as Umbralisib, INCB050465, ME-401, RP-6530, ACP-319, Pictilisib, Acalisib, KA-2237 are still under clinical investigation [39,40]. In this paper, a detailed comparison of SHC014748M and Idelalisib has been carried out, and the results suggested that SHC014748M had efficacy equivalent to or better than that of Idelalisib. Together with the data in the literature, we also compared the nonclinical efficacy of SHC014748M with that of other PI3K inhibitors (Tables 3 and 4). It turns out that SHC014748M tends to be a selective inhibitor of PI3K-δ rather than a generic inhibitor [26], [41].
Table 3.
Kinase inhibition assays of SHC014748M and other PI3K inhibitors.
| Compound | Kinase | PI3Kδ | PI3Kα | PI3Kβ | PI3Kγ |
|---|---|---|---|---|---|
| SHC014748M | IC50 (nM) | 0.77 | 236 | 96 | 101 |
| Fold-selectivity | 1x | 306x | 125x | 131x | |
| Idelalisib | IC50 (nM) | 19 | 8600 | 4500 | 2100 |
| Fold-selectivity | 1X | 453X | 210X | 110X | |
| Duvelisib | IC50 (nM) | 2.5 | 1602 | 85 | 27 |
| Fold-selectivity | 1X | 640X | 34X | 11X | |
| Copanlisib | IC50 (nM) | 0.7 | 0.5 | 3.7 | 6.4 |
| Fold-selectivity | 1X | 0.7X | 5X | 9X |
Table 4.
Antitumor activities of SHC014748M and Copanlisib in vitro.
| Classification | cell line | IC50 (nM) |
||
|---|---|---|---|---|
| SHC014748M | Copanlisib | SHC014748M/ Copanlisib | ||
| ABC DLBCL | Pfeiffer | 4641.98 | 0.849 | 5467.58 |
| OCI-LY19 | 9957.66 | 1994 | 4.99 | |
| Toledo | 3210.91 | 3498 | 0.92 | |
| WSU-DLCL2 | 784.00 | 123.4 | 6.35 | |
The major toxicities of PI3Kδ inhibitors can be divided into 3 aspects. First, there are increased rates of bacterial infection. Second, there appears to be an increased risk for opportunistic infections. Prophylaxis for Pneumocystis jirovecii and monitoring for cytomegalovirus reactivation is now mandatory during the treatment. In addition to the 2 types of infectious toxicities, inflammatory toxicities of presumed autoimmune origin including colitis, hepatitis, and pneumonitis, have also been seen. However, the exact role of PI3Kδ in the adverse reactions remains unclear and efforts should be made to alleviate the adverse reaction [29,41]. SHC014748M was found to have a lethal dose of 200 mg/kg in SD rats, and a maximum tolerated dose of 60 mg/kg. The main side-effects of SHC014748M were gastrointestinal reactions and bone marrow toxicity, and the target organ sites of toxicity included adrenal, bone marrow (sternum and femur), immune organs (mesenteric lymph nodes, spleen, and thymus), liver, and stomach. Pneumonia was a major adverse effect in the Phase I trial of SHC014748M involving iNHL patients. Most of the adverse side effects are thought to be consequences of on-target inhibition in T cells, producing both susceptibility to infections and autoimmune effects, so there may be no reason to think that a highly-specific PI3K-γ inhibitor will fare better [41].
There are 8 mammalian PI3K enzymes, which are grouped into 3 classes [42,43]. The most important in cancer are the 4 class I enzymes, termed PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ [44,45]. Among which p110α and p110β are widely expressed by all cell types while p110γ and p110δ are expressed primarily in hematopoietic cells and have important roles in the functions of both innate and adaptive immune cells [46,47]. Bi et al. have generated p110α- and p110β-deficient mice, both of which die as embryos, after embryonic day (E) 9.5 or shortly after implantation, respectively. Besides, PI3Kα inhibition was found to cause hyperglycemia and hypertension in clinical trial [48,49]. Sasaki et al. showed that T-cell development and function were impaired, whereas B cells were unaffected, in p110γ-deficient mice [50,51]. Considering the information above, we aim to improve the in-vitro selectivity of SHC014748M against other PI3K class I isoforms. The results of kinase assay showed that, SHC014748M was 125- to 306-fold more selective for PI3Kδ inhibition relative to other PI3K class I enzymes. In the cell-based assay, SHC014748M was strongly potent against PI3Kδ and highly selective versus PI3Kα/γ (>900 × ) , with ∼137 × selectivity versus PI3Kβ. This result is in accordance with the result in kinase assay and shows that SHC014748M has achieved our ultimate goal.
NHL is a heterogeneous group of human lymphoid malignancies of primarily B-cell lineage (B-NHL) that appear to correspond to various stages in normal B-cell ontogeny [39]. These lymphomas range from low-grade (indolent) to high-grade (aggressive) histiotypes that correspond in part to various cellular proliferation indices (flow cytometry, cell cycle analysis, tumor growth fraction, labeling index, etc.) of the lymphoma cells [52,53]. Thus, efficacy of drugs differs among NHL subtypes in clinical research. As a result, we evaluated SHC014748M in 23 B cell lymphoma cell lines and primary CLL cells. The results of the experiment showed that SHC014748M produced more robust inhibition on cell proliferation in GCB DLBCL cell lines with a median IC50 value of 4790 nM, while showed less effects in highly aggressive MCL cell lines with a median IC50 value of 12,850 nM. In 15 primary CLL cells, SHC014748M showed in vitro antiproliferation activity in most of them. The results above would help to figure out the potential clinical indications of SHC014748M.
INHL is a group of B cell malignancies which are characterized by slow-progression so that it is difficult to establish an animal model with long disease duration for drug validation. Considering that proliferation of SU-DHL-6 could be remarkably inhibited by SHC014748M in vitro study. SU-DHL-6, an aggressive B cell lymphoma cell line, was used to establish the mouse model of B cell lymphoma. The results of the experiment showed that SHC014748M administration delayed tumor growth at all dosage. However, there were no differences of tumor volume among the 3 doses. Possible reason for this may be that 50 mg/kg may be the saturation dose of SHC014748M in SCID mice. Besides SHC014748M at the dose of 50 mg/kg might achieved the maximum antitumor effect in this aggressive lymphoma model. According to the pharmacokinetic results, exposure of SHC014748M was much higher than Idelalisib at the same dosage. It might be because that SHC014748M was more metabolic stable than Idelalisib in liver microsome of mice (data not shown).
Phase I study has been developed with SHC014748M as a single agent in patients with relapsed or refractory indolent B cell NHL, including CLL, MZL, FL, and Waldenstrom's macroglobulinemia (NCT03588598). In the Dose escalation trial, SHC014748M exhibits an acceptable safety profile in patients treated up to 3 months. Steady-state concentration, a pharmacokinetic parameter which seems to be closely related to efficacy according to the clinical research of Idelalisib [54,55], of SHC014748M in patients at the dose above 150 mg/day are far higher than the concentration which is effective in preclinical in-vitro study.
In conclusion, SHC014748M, a novel selective PI3Kδ inhibitor, presents promising antitumor activity in iNHL.
CRediT authorship contribution statement
Lei Fan: Conceptualization, Supervision, Methodology, Writing - review & editing. Chao Wang: Software, Data curation, Writing - original draft. Liwen Zhao: Data curation, Investigation, Visualization. Zhiqiang Wang: Software, Data curation, Validation. Xian Zhang: Software, Formal analysis, Validation. Xiaorong Liu: Software, Data curation, Validation. Lei Cao: Data curation, Investigation, Visualization. Wei Xu: Project administration, Supervision, Writing - review & editing. Jianyong Li: Project administration, Funding acquisition, Supervision, Writing - review & editing.
Acknowledgments
Acknowledgments
We wish to thank all the patients who took part in this research, their families, and the investigators.
Footnotes
Funding: This work was supported by National Natural Science Foundation of China (81720108002); Jiangsu Provincial Special Program of Medical Science (BE2017751), and National Science and Technology Major Project (2018ZX09734007); Excellent Youth Foundation Project of JiangSu Province (BK20160099); “Liu Da Ren Cai Gao Feng” of JiangSu Province (2015-WSN-050).
Declaration of Competing Interest: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Contributor Information
Lei Fan, Email: fanlei3014@126.com.
Jianyong Li, Email: lijianyonglm@126.com.
References
- 1.Shankland K.R., Armitage J.O., Hancock B.W. Non-Hodgkin lymphoma. Lancet. 2012;380:848–857. doi: 10.1016/S0140-6736(12)60605-9. [DOI] [PubMed] [Google Scholar]
- 2.Chiu B.C., Hou N. Epidemiology and etiology of non-Hodgkin lymphoma. Cancer Treat Res. 2015;165:1–25. doi: 10.1007/978-3-319-13150-4_1. [DOI] [PubMed] [Google Scholar]
- 3.SEER . National Cancer Institute; Bethesda, MD: 2012. SEER Stat Fact Sheets: Non-Hodgkin lymphoma. [Google Scholar]
- 4.Chen W., Zheng R., Baade P.D., Zhang S., Zeng H., Bray F., Jemal A., Yu X.Q., He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 5.Siegel R., Ma J., Zou Z., Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 6.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 7.Gopal A.K., Kahl B.S., de Vos S., Wagner-Johnston N.D., Schuster S.J., Jurczak W.J., Flinn I.W., Flowers C.R., Martin P., Viardot A. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–1018. doi: 10.1056/NEJMoa1314583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowzyk A.A., Ajithkumar T., Behan S., Hodson D.J. Non-Hodgkin lymphoma. BMJ. 2018;362:k3204. doi: 10.1136/bmj.k3204. [DOI] [PubMed] [Google Scholar]
- 9.Lunning M.A., Vose J.M. Management of indolent lymphoma: where are we now and where are we going. Blood Rev. 2012;26:279–288. doi: 10.1016/j.blre.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swerdlow F. ed4th. Lyon: International Agency for Research on Cancer; 2008. WHO Classification of Tumours of Haematopoietic and Lympoid Tissues. [Google Scholar]
- 11.Cheson B.D., Chua N., Mayer J., Dueck G., Trneny M., Bouabdallah K., Fowler N., Delwail V., Press O., Salles G. Overall survival benefit in patients with rituximab-refractory indolent non-Hodgkin lymphoma who received obinutuzumab plus bendamustine induction and obinutuzumab maintenance in the GADOLIN Study. J Clin Oncol. 2018;36:2259–2266. doi: 10.1200/JCO.2017.76.3656. [DOI] [PubMed] [Google Scholar]
- 12.Flinn I.W., Patel M., Oki Y., Horwitz S., Foss F.F., Allen K., Douglas M., Stern H., Sweeney J., Kharidia J. Duvelisib, an oral dual PI3K-δ, γ inhibitor, shows clinical activity in indolent non-Hodgkin lymphoma in a phase 1 study. Am J Hematol. 2018;93:1311–1317. doi: 10.1002/ajh.25228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.National Comprehensive Cancer Network: NCCN clinical practice guidelines in oncology (NCCN Guidelines): B-cell lymphomas (version 1.2017), 2016. Available online: https://www.nccn.org.
- 14.Valla K., Flowers C.R., Koff J.L. Targeting the B cell receptor pathway in non-Hodgkin lymphoma. Expert Opin Investig Drugs. 2018;27:513–522. doi: 10.1080/13543784.2018.1482273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horning S.J. Natural history of and therapy for the indolent non-Hodgkin's lymphomas. Semin Oncol. 1993;20:75–88. [PubMed] [Google Scholar]
- 16.Casulo C., Byrtek M., Dawson K.L., Zhou X., Farber C.M., Flowers C.R., Hainsworth J.D., Maurer M.J., Cerhan J.R., Link B.K. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the National LymphoCare Study. J Clin Oncol. 2015;33:2516–2522. doi: 10.1200/JCO.2014.59.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fung-Leung W.P. Phosphoinositide 3-kinase δ (PI3Kδ) in leukocyte signaling and function. Cell Signal. 2011;23:603–608. doi: 10.1016/j.cellsig.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Bilanges B., Posor Y., Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019;20:515–534. doi: 10.1038/s41580-019-0129-z. [DOI] [PubMed] [Google Scholar]
- 19.Vanhaesebroeck B., Stephens L., Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 20.Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoellenriegel J., Meadows S.A., Sivina M., Wierda W.G., Kantarjian H., Keating M.J., Giese N., O'Brien S., Yu A., Miller L.L. The phosphoinositide 3′-kinase δ inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118:3603–3612. doi: 10.1182/blood-2011-05-352492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanhaesebroeck B., Guillermet-Guibert J., Graupera M., Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 23.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Henau O., Rausch M., Winkler D., Campesato L.F., Liu C., Cymerman D.H., Budhu S., Ghosh A., Pink M., Tchaicha J. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bilanges B., Posor Y., Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019;20:515–534. doi: 10.1038/s41580-019-0129-z. [DOI] [PubMed] [Google Scholar]
- 26.Center for Drug Evaluation and Research: Application Number: 205858Orig1s000. Available online: https://www.fda.gov/drugs.
- 27.ZYDELIG (idelalisib tablets) Gilead Sciences, Inc.; Foster City, CA: 2014. Full prescribing information. Available online: https://www.fda.gov/media/100575/download.
- 28.Smith S.M., Pitcher B.N., Jung S.H., Bartlett N.L., Wagner-Johnston N., Park S.I., Richards K.L., Cashen A.F., Jaslowski A., Smith S.E. Safety and tolerability of idelalisib, lenalidomide, and rituximab in relapsed and refractory lymphoma: the alliance for clinical trials in oncology A051201 and A051202 phase 1 trials. Lancet Haematol. 2017;4:e176–e182. doi: 10.1016/S2352-3026(17)30028-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janku F., Yap T.A., Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–291. doi: 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- 30.Flinn I.W., O'Brien S., Kahl B., Patel M., Oki Y., Foss F.F., Porcu P., Jones J., Burger J.A., Jain N. Duvelisib, a novel oral dual inhibitor of PI3K-δ, γ, is clinically active in advanced hematologic malignancies. Blood. 2018;131:877–887. doi: 10.1182/blood-2017-05-786566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eltantawy A., Vallejos X., Sebea E., Evans K. Copanlisib. An intravenous phosphatidylinositol 3-kinase (PI3K) inhibitor for the treatment of relapsed follicular lymphoma. Ann Pharmacother. 2019;53:954–958. doi: 10.1177/1060028019833992. [DOI] [PubMed] [Google Scholar]
- 32.Rodgers T.D., Reagan P.M. Targeting the B-cell receptor pathway: a review of current and future therapies for non-Hodgkin's lymphoma. Expert Opin Emerg Drugs. 2018;23:111–122. doi: 10.1080/14728214.2018.1479396. [DOI] [PubMed] [Google Scholar]
- 33.Badoux X.C., Keating M.J., Wen S., Lee B.N., Sivina M., Reuben J., Wierda W.G., O'Brien S.M., Faderl S., Kornblau S.M. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood. 2011;118:3489–3498. doi: 10.1182/blood-2011-03-339077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Methods of Treating Hematological Disorders with Quinazolinone Compounds in Selected Subjects. 2011.
- 35.Burger J.A., Quiroga M.P., Hartmann E., Burkle A., Wierda W.G., Keating M.J., Rosenwald A. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113:3050–3058. doi: 10.1182/blood-2008-07-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burkle A., Niedermeier M., Schmitt-Graff A., Wierda W.G., Keating M.J., Burger J.A. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood. 2007;110:3316–3325. doi: 10.1182/blood-2007-05-089409. [DOI] [PubMed] [Google Scholar]
- 37.Fruman D.A., Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lampson B.L., Brown J.R. PI3Kδ-selective and PI3Kα/δ-combinatorial inhibitors in clinical development for B-cell non-Hodgkin lymphoma. Expert Opin Investig Drugs. 2017;26:1267–1279. doi: 10.1080/13543784.2017.1384815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrientos J.C. Idelalisib for the treatment of indolent non-Hodgkin lymphoma: a review of its clinical potential. Onco Targets Ther. 2016;9:2945–2953. doi: 10.2147/OTT.S102573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lien E.C., Dibble C.C., Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017;45:62–71. doi: 10.1016/j.ceb.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esposito A., Viale G., Curigliano G. Safety, tolerability, and management of toxic effects of phosphatidylinositol 3-kinase inhibitor treatment in patients with cancer: a review. JAMA Oncol. 2019 doi: 10.1001/jamaoncol.20190034. [DOI] [PubMed] [Google Scholar]
- 42.Vanhaesebroeck B., Guillermet-Guibert J., Graupera M., Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 43.Hirsch E., Ciraolo E., Franco I., Ghigo A., Martini M. PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene. 2014;33:3083–3090. doi: 10.1038/onc.2013.265. [DOI] [PubMed] [Google Scholar]
- 44.Fritsch R., de Krijger I., Fritsch K., George R., Reason B., Kumar M.S., Diefenbacher M., Stamp G., Downward J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell. 2013;153:1050–1063. doi: 10.1016/j.cell.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samuels Y., Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 46.Rommel C., Camps M., Ji H. PI3Kδ and PI3Kγ: partners in crime in inflammation in rheumatoid arthritis and beyond. Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Echeverria C., Sellers W.R. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 48.Bi L., Okabe I., Bernard D.J., Wynshaw-Boris A., Nussbaum R.L. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem. 1999;274:10963–10968. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- 49.Bi L., Okabe I., Bernard D.J., Nussbaum R.L. Early embryonic lethality in mice deficient in the p110β catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13:169–172. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- 50.Sasaki T., Irie-Sasaki J., Jones R.G., Oliveira-dos-Santos A.J., Stanford W.L., Bolon B., Wakeham A., Itie A., Bouchard D., Kozieradzki I. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 51.Fruman D.A., Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–272. doi: 10.1111/j.1600-065X.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 52.Bryant J., Pham L., Yoshimura L., Tamayo A., Ordonez N., Ford R.J. Development of intermediate-grade (mantle cell) and low-grade (small lymphocytic and marginal zone) human non-Hodgkin's lymphomas xenotransplanted in severe combined immunodeficiency mouse models. Lab Invest. 2000;80:557–573. doi: 10.1038/labinvest.3780061. [DOI] [PubMed] [Google Scholar]
- 53.Cabanillas F., Rivera N., Pardo W.I. Indolent lymphomas that present with clinically aggressive features: a subset of low-grade lymphomas with a behavior inconsistent with the histologic diagnosis. Clin Lymphoma Myeloma Leuk. 2016;16:550–557. doi: 10.1016/j.clml.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 54.Flinn I.W., Kahl B.S., Leonard J.P., Furman R.R., Brown J.R., Byrd J.C., Wagner-Johnston N.D., Coutre S.E., Benson D.M., Peterman S. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-δ, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood. 2014;123:3406–3413. doi: 10.1182/blood-2013-11-538546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown J.R., Byrd J.C., Coutre S.E., Benson D.M., Flinn I.W., Wagner-Johnston N.D., Spurgeon S.E., Kahl B.S., Bello C., Webb H.K. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123:3390–3397. doi: 10.1182/blood-2013-11-535047. [DOI] [PMC free article] [PubMed] [Google Scholar]