

Abstract
Schisandra chinensis, which has a high development value, has long been used as medicine. Its mature fruits (called Wuweizi in Chinese) have long been used in the famous traditional Chinese medicine (TCM) recorded in the “Chinese Pharmacopoeia.” Chloroplasts (CP) are the highly conserved primitive organelles in plants, which can serve as the foundation for plant classification and identification. This study introduced the structures of the CP genomes of three Schisandraceae species and analyzed their phylogenetic relationships. Comparative analyses on the three complete chloroplast genomes can provide us with useful knowledge to identify the three plants. In this study, approximately 5 g fresh leaves were harvested for chloroplast DNA isolation according to the improved extraction method. A total of three chloroplast DNAs were extracted. Afterwards, the chloroplast genomes were reconstructed using denovo combined with reference-guided assemblies. General characteristics of the chloroplast genome and genome comparison with three Schisandraceae species was analyzed by corresponding software. The total sizes of complete chloroplast genomes of S. chinensis, S. sphenanthera, and Kadsura coccinea were 146875 bp, 146842 bp, and 145399 bp, respectively. Altogether, 124 genes were annotated, including 82 protein-coding genes, 34 tRNAs, and 8 rRNAs of all 3 species. In SSR analysis, only S. chinensis was annotated to hexanucleotides. Moreover, comparative analysis of chloroplast Schisandraceae genome sequences revealed that the gene order and gene content were slightly different among Schisandraceae species. Finally, phylogenetic trees were reconstructed, based on the genome-wide SNPs of 38 species. The method can be used to identify and differentially analyze Schisandraceae plants and offer useful information for phylogenetics as well as further studies on traditional medicinal plants.
1. Introduction
Wuweizi (Schisandra chinensis), first recorded in “Shennong's Herbal Classic of Materia Medica,” has long been used in traditional Chinese medicine (TCM) as a top grade medicine; specifically, it has been utilized as a tonic medicine for about 2000 years [1]. In many Asian countries like Japan and South Korea, Schisandra is also listed as a pharmacopoeia variety [2, 3]. Wuweizi (Schisandra chinensis), Nanwuweizi (Schisandra sphenanthera), and Heilaohu (Kadsura coccinea) have displayed medicinal value. Nanwuweizi (Schisandra sphenanthera) is the same as Wuweizi in historical medicine; both of them were listed separately in the 2000 China Pharmacopoeia. Wuweizi is often used as a diarrhea antispasmodic agent in clinical practice, and it has astringent solidifying, Qi and fluid replenishing, as well as kidney and heart tonifying effects [1]. K. coccinea is known as the “magic longevity fruit,” which can be eaten as a fruit, and its root and root skin can be used for medicinal purposes, as recorded in “Gui Medicine.” Besides, it can promote qi and activate blood circulation, reduce swelling, and relieve pain [4].
Chloroplasts are important organelles for photosynthesis in plants, which have distinct physiological characteristics, such as relatively conservative and single parental inheritance [5, 6]. Chloroplasts can be used for plant species identification, hybridization, and phylogenetic analysis. In 1986, Japanese scientists had first obtained the complete chloroplast genome sequence of plants in tobacco [7]. With the rapid development of high-throughput sequencing in recent years, this emerging technology has been applied to investigation in various fields. For instance, Li et al. had investigated the structural characteristics and genetic evolution of Magnoliaceae plants through chloroplast genome-wide analysis of Magnolia grandiflora in 2013, and had verified the negative correlation of the IR region length in some Magnoliaceae plants with the pseudogene length ψycf1 [8]. In 2013, Yang et al. had carried out chloroplast genome-wide structural analysis to identify and analyze various plants of genus Cymbidium, and examined their genetic relationships [9]. Their results proved that the correlation between species based on organelle gene sequencing was reliable.
Magnoliaceae has always been a research hotspot. So far, the chloroplast genomes of about 20 plant species have been sequenced [10]. Typically, Magnolia is also a controversial group in botany, which is mainly attributed to the classification of botany. In the APG IV system, Schisandraceae includes Schisandra and Illicium. In 2007, Hansen et al. analyzed the chloroplast gene structures of four plants, including medicinal anise [11]. In 2017, Guo et al. examined the chloroplast genes of S. chinensis and determined that the Illicium genus was a sister branch of Schisandra [12]. In 2018, Li and Zheng analyzed the chloroplast genomes of K. coccinea and obtained the evolutionary structure of K. coccinea by means of phylogenetic tree analysis [13]. Modern methods have been utilized to analyze chloroplasts, such as simple sequence repeat (SSR) analysis, IR boundary analysis, and phylogenetic analysis. Of them, SSR is frequently used to identify species and analyze the genetic difference. Previous phylogenetic research focusing on the phylogenetic tree of consensus protein clustering has only taken into consideration the coding region variation, which has certain limitations. Therefore, this paper had been carried out with the aim of analyzing the phylogenetic tree of genome-wide single-nucleotide polymorphisms (SNPs), with the consideration of variations in the coding region and the noncoding region. Notably, our analysis was more comprehensive, and our results were more accurate and reliable.
In the 2015 China Pharmacopoeia, all the three medicinal plants examined in this study are classified into Magnoliaceae; however, in the latest APG IV classification system [14], Schisandraceae is classified into a separate family, which includes Illicium L., Kadsura Kaempf., and Schisandra Michx. Liu and Hu suggested that Schisandraceae be considered as one family, and divided and categorized as the Illicium genus through comparing plant morphology, palynology, and cytology [15–18]. It is believed that the Illicium genus is a model genus, which is also known as Illicium L., as suggested by Zhang. From the perspective of chloroplast gene organization of Magnoliaceae, Schisandraceae, and star anise, this paper had provided the novel foundation for plant classification [19]. There are currently about 80 species of Schisandraceae, including 34 medicinal plants [20, 21]. The commonly used agents in Schisandraceae are found in S. chinensis and S. sphenanthera, and many methods can be used for identification, like chemistry and molecular pharmacy [22–24].
Various plants of Schisandraceae have exhibited high economic and medicinal value. This paper is aimed at analyzing the complete chloroplast genomes of S. chinensis, S. sphenanthera, and K. coccinea, so as to explore the basis for identification and genetic relationships among these three. Notably, S. chinensis and S. sphenanthera have not been accurately distinguished in the history of medication use, and mixed phenomena can be observed. Therefore, exploring the differences at the gene level between these two is not only conducive to identifying these two traditional Chinese medicines, but it also lays the vital genetic foundation for the future cultivation of S. chinensis and S. sphenanthera. This study is aimed at exploring the intrinsic relationship and difference in the chloroplast gene structure of S. chinensis through internal analysis of S. chinensis and S. sphenanthera, as well as K. coccinea. Besides, phylogenetic analysis of Schisandraceae with other families was also carried out to obtain the position of Schisandra during phylogenetic evolution.
2. Materials and Methods
2.1. Plant Materials
The fresh S. chinensis leaves were collected from Dalian, China (E121°52′34.96″, N39°03′43.96″). The fresh leaves of S. sphenanthera were collected from Shangluo, China (E110°02′27.55″, N33°55′34.10″). The fresh leaves of K. coccinea were obtained from the channel Xufeng Chinese Herbal Medicine Cooperative (Huaihua, Hunan Province, China (E110°05′03.05″, N27°28′30.10″). These three species were identified by Dr. Tingguo Kang from the Liaoning University of Traditional Chinese Medicine in Shenyang, China. Permission for using plant fruit samples in biological experiments had been granted. Plant samples were deposited in the herbarium of Liaoning University of Traditional Chinese Medicine and the genomic DNA was stored in the Key Laboratory of Traditional Chinese Medicine in the University (Dalian, China, 116600).
2.2. Chloroplast DNA Extraction and Sequencing
Approximately 5 g fresh leaves were harvested for chloroplast DNA isolation according to an improved extraction method [25]. After DNA isolation, 1 μg purified DNA was fragmented, which was then utilized to construct the short-insert libraries (insert size of 430 bp) in accordance with the manufacturer's protocol (Illumina), followed by sequencing on the Illumina HiSeq 4000 [26].
Raw reads were filtered prior to assembly, so as to remove reads with adaptors, or those with a quality score of <20 (Q < 20), or those containing ≥10% uncalled based (“N” characters) and duplicated sequences. Afterwards, the chloroplast genomes were reconstructed using denovo combined with reference-guided assemblies, and the following three steps were adopted for chloroplast genome assembly [27]. First of all, the filtered reads were assembled into contigs using SOAP denovo 2.04 [28]. Secondly, the assembled contigs were aligned to the reference genomes of two species using BLAST, and then the aligned contigs (≥80% similarity and query coverage) were ordered according to the reference genomes. Thirdly, clean reads were mapped to the assembled draft chloroplast genomes, so as to correct the wrong bases, and later the majority of gaps were filled by means of local assembly.
2.3. Genome Assembly and Annotation
The chloroplast genes were annotated by the online DOGMA tool, and default parameters were used to predict protein-coding genes, and to transfer RNA (trnA) genes as well as ribosome RNA (rRNA) genes. Subsequently, a comprehensive chloroplast genome-wide Blast search was performed among 5 databases [29], namely, the Kyoto Encyclopedia of Genes and Genomes (KEGG), the Clusters of Orthologous Groups (COG), the Nonredundant Protein Database (NR), Swiss-Prot, and Gene Ontology (GO) [30–37]. At the same time, the SSR software MIcroSAtellite (MISA) (http://pgrc.ipk-gatersleben.de/misa/) was employed to identify the SSR sequences, and tandem repeats of 1–6 nucleotides were considered as microsatellites. Moreover, MISA (MIcroSAtellite; http://pgrc.ipk-gatersleben.de/misa) was utilized to detect the genomes of SSR in the chloroplast, and the parameters were set as follows: >11 for mononucleotides, >6 for dinucleotides, >5 for trinucleotides, >5 for tetranucleotides, >5 for pentanucleotides, and >5 for hexanucleotides. In addition, the maximal number of bases interrupting 2 SSRs in a compound microsatellite was set as 100. This paper had focused on the perfect repeat sequences. The mVISTA was used for similarity analysis between these five Magnoliaceae species.
2.4. Chloroplast Genome Mapping
The chloroplast genomes of S. chinensis, S. sphenanthera, and K. coccinea were exported in the GenBank format using the Sequin software, and then mapped based on the annotation results (http://ogdraw.mpimp-golm.mpg.de/index.shtml). Finally, their complete chloroplast genomes were submitted to the NCBI GenBank database.
2.5. Phylogenetic Analysis
The genome sequences of 38 species were utilized to analyze the phylogenetic relationships on the basis of genome-wide SNPs of the 38 species. Additionally, the Maximum Likelihood (ML) phylogenetic tree was constructed using MEGA 6.0.
3. Results and Discussion
3.1. General Characteristics
The chloroplast genes of three medicinal plants were detected (Table 1). Results suggested that the full-length chloroplast genes of S. chinensis, S. sphenanthera, and K. coccinea were 146875 bp, 146842 bp, and 145399 bp, respectively, while the GenBank numbers were SRX4282568, SRX4282569, and SRX4282570, respectively, which were similar to the chloroplast genome sizes of other Schisandraceae plants. These results had indicated the relatively conservative evolution of chloroplast genes in Schisandraceae plants. Moreover, the GC contents of S. chinensis, S. sphenanthera, and K. coccinea were 43.11%, 39.60%, and 39.70%, respectively. The LSC lengths were 96686 bp, 95627 bp, and 94287 bp, separately; the SSC lengths were 18270 bp, 18280 bp, and 18039 bp, respectively; and the IR (IRa, IRb) lengths were 15958 bp, 16466 bp, and 16535 bp, separately. Moreover, it was known from the sequencing results that LSC, SSC, and IRa (IRb) had close lengths in the four regions of chloroplast genes, and the numbers of total genes, encoded genes, rRNA genes, and trnA genes were highly consistent among them. Besides, sex indicated quite close kinship of these three chloroplast genes.
Table 1.
Comparison of three chloroplast gene data.
| Species | S. chinensis | S. sphenanthera | K. coccinea |
|---|---|---|---|
| Gene length (bp) | 146875 | 146842 | 145399 |
| GC (%) | 43.11 | 39.60 | 39.70 |
| LSC (bp) | 96686 | 95627 | 94287 |
| SSC (bp) | 18270 | 18280 | 18039 |
| IRa (bp) | 15958 | 16466 | 16535 |
| IRb (bp) | 15958 | 16466 | 16535 |
| Gene number | 124 | 124 | 124 |
| Protein-coding gene number | 82 | 82 | 82 |
| rRNA gene number | 8 | 8 | 8 |
| trnA gene number | 34 | 34 | 34 |
The annotated genes of S. chinensis, S. sphenanthera, and K. coccinea were generally identical, as presented in Figure 1 (the maps of S. sphenanthera and K. coccinea are shown in Figures S1 and S2), but certain differences were also observed (Table 2). For example, S. sphenanthera had an rps12 sequence in both IRa and IRb, which was not seen in S. chinensis. In addition, the clpP sequences of rps16, petB, and rpl16 in the LSC region of S. chinensis were 218 bp, 641 bp, 410 bp, and 1160 bp in length, which were markedly shorter than the 1079 bp, 1408 bp, 1355 bp, and 1901 bp in S. sphenanthera. Additionally, the rpl16 sequence of S. sphenanthera contained two exons and one exon, which were not seen in S. chinensis; thus, the base species of S. chinensis was identified based on these differences. Compared with S. chinensis, K. coccinea had an additional rps12 sequence in the IRa and IRb regions, as well as a D2 type ndhB in the IRb region. Besides, the lengths of rps16, rpl16, petB, petD, and clpP of S. chinensis were 218 bp, 410 bp, 641 bp, 533 bp, and 1160 bp, respectively; while those of K. coccinea (1085 bp, 1371 bp, 1408 bp, 1254 bp, and 1978 bp) were longer than those of S. chinensis. When comparing S. sphenanthera with K. coccinea, the petD in the LSC region of S. sphenanthera was shorter, the detailed data were the same as those presented above, and the IRb region of K. coccinea was D2 type ndhB. The high gene similarity was ascribed to the species similarity, which also indicated the relatively conservative family evolution of S. chinensis.
Figure 1.
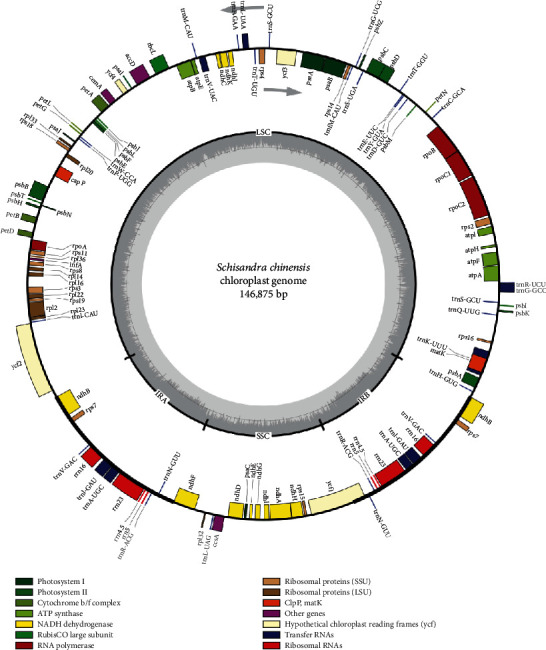
Chloroplast genes map of S. chinensis.
Table 2.
Lists of genomic genes of S. chinensis, S. sphenanthera, and K. coccinea.
| Genes | S. chinensis | S. sphenanthera | K. coccinea |
|---|---|---|---|
| Group of genes | Gene name | Gene name | Gene name |
| Ribosomal RNAs | rRNA23,16d, 5, 4.5 | rRNA23, 16, 5, 4.5 | rRNA23, 16, 5, 4.5 |
| Transfer RNAs | trnH-GUG, trnK-UUUd, trnQ-UUG, trnS-GCU, trnG-GCC, trnG-GCCd, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-UCC, trnM-CAU, trnS-GCU, trnT-UGU, trnL-UAAd, trnF-GAA, trnV-UACd, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnV-GACd, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnV-GAC, trnI-GAUd, trnA-UGCd, trnR-ACG, trnN-GUU, trnL-UAG, trnN-GUU, trnR-ACG, trnA-UGCd, trnI-GAUd, trnV-GAC | trnH-GUG, trnK-UUUd, trnQ-UUG, trnS-GCU, trnG-GCC, trnG-GCCd, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-UCC, trnM-CAU, trnS-GCU, trnT-UGU, trnL-UAAd, trnF-GAA, trnV-UACd, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnV-GACd, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnV-GAC, trnI-GAUd, trnA-UGCd, trnR-ACG, trnN-GUU, trnL-UAG, trnN-GUU, trnR-ACG, trnA-UGC2, trnI-GAUd, trnV-GAC | trnH-GUG, trnK-UUUd, trnQ-UUG, trnS-GCU, trnG-GCC, trnG-GCCd, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-UCC, trnM-CAU, trnS-GCU, trnT-UGU, trnL-UAAd, trnF-GAA, trnV-UACd, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnV-GACd, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnV-GAC, trnI-GAUd, trnA-UGCd, trnR-ACG, trnN-GUU, trnL-UAG, trnN-GUU, trnR-ACG, trnA-UGCd, trnI-GAUd, trnV-GAC |
| Proteins of small ribosomal subunits | rps2, 3, 4, 7d, 8, 11, 12d, 14, 15, 16, 18, 19, | rps2, 3, 4, 7d, 8, 11, 12d, 14, 15, 16, 18, 19, | rps2, 3, 4, 7d, 8, 11, 12d, 14, 15, 16, 18, 19, |
| Proteins of large ribosomal subunits | rpl14, 16, 2, 20, 22, 23, 32, 33, 36 | rpl14, 16, 2, 20, 22, 23, 32, 33, 36 | rpl14, 16, 2, 20, 22, 23, 32, 33, 36 |
| Subunits of RNA polymerase | rpoA, B, C1, C2 | rpoA, B, C1, C2 | rpoA, B, C1, C2 |
| Subunits of NADH dehydrogenase | ndhA, Bd, C, D, E, F, G, H, I, J, K | ndhA, Bd, C, D, E, F, G, H, I, J, K | ndhA, Bd, C, D, E, F, G, H, I, J, K |
| Subunits of photosystem I | psaA, B, C, I, J | psaA, B, C, I, J | psaA, B, C, I, J |
| Subunits of photosystem II | PsbA, B, C, E, D, F, H, I, J, K, L, N, T, Z | PsbA, B, C, E, D, F, H, I, J, K, N, T, Z | PsbA, B, C, E, D, F, H, I, J, K, N, T, Z |
| Large subunit of Rubisco | rbcL | rbcL | rbcL |
| Subunits of cytochrome b/f complex | petA, B, D, G | petA, B, D, G | petA, B, D, G |
| Subunits of ATP synthase | atpA, B, E, F, H, I | atpA, B, E, F, H, I | atpA, B, E, F, H, I |
| Acetyl-CoA carboxylase | accD | accD | accD |
| C-type cytochrome synthesis gene | ccsA | ccsA | ccsA |
| Maturase | matK | matK | matK |
| Protease | clpPd | clpPd | clpPd |
| Envelope membrane protein | cemA | cemA | cemA |
| Conserved hypothetical chloroplast reading frames | ycf1, 2, 3, 4 | ycf1, 2, 3, 4 | ycf1, 2, 3, 4 |
| Translational initiation factor | infA | infA | infA |
d: indicates that there is a double-segment gene.
An intron plays a vital part in regulating gene expression. Recent studies suggest that many introns can increase the expression and timing of foreign genes at specific locations, which can partially determine the plant-specific traits. Therefore, introns can serve as a useful approach to improve the required agronomic traits [38]. There were 12 intron-coding genes in the S. chinensis chloroplast DNA, of which ycf3, rps12, rps12-D2, and clpP contained two introns, whereas the remaining eight genes had only one intron (Table 3). By contrast, there were 14 genes containing introns in the S. sphenanthera chloroplast DNA, among which, ycf3, rps12, rps12-D2, and clpP contained two introns, while the other eight were the same as those of S. chinensis. However, S. sphenanthera had two more exon genes than S. chinensis, which were petD and rpl16; therefore, they could be used to identify north S. chinensis species from the south S. chinensis species. Meanwhile, the number and species of K. coccinea introns were the same as those of S. chinensis. The overall comparison showed that the exons contained in these three medicinal plants were basically the same in size, and the intron size was very small, with only a few bp between them, suggesting the close kinship among these three plants. Investigating the introns among these three plants contributed to improving plant resilience and developing new varieties. In addition, there was a high degree of similarity among the three, which could be used for reference in plant breeding of the latter three.
Table 3.
Characteristics and sizes of intron and exon genes of S. chinensis, S. sphenanthera, and K. coccinea.
| Gene | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) | |
|---|---|---|---|---|---|---|
| S. chinensis | rps16 | 220 | 821 | 39 | ||
| rpoC1 | 1635 | 685 | 454 | |||
| atpF | 411 | 773 | 142 | |||
| ycf3 | 140 | 769 | 220 | 739 | 123 | |
| rps12 | 110 | 27234 | 25 | 537 | 231 | |
| rps12-D2 | 110 | 70401 | 231 | 537 | 25 | |
| clpP | 248 | 559 | 291 | 799 | 70 | |
| PetB | 5 | 762 | 641 | |||
| rpl2 | 442 | 652 | 390 | |||
| ndhA | 517 | 1080 | 561 | |||
| ndhB | 727 | 676 | 777 | |||
| ndhB-D2 | 777 | 676 | 727 | |||
| S. sphenanthera | rps16 | 220 | 820 | 39 | ||
| rpoC1 | 1635 | 685 | 454 | |||
| atpF | 411 | 767 | 142 | |||
| ycf3 | 140 | 769 | 220 | 740 | 123 | |
| rps12 | 113 | 27147 | 25 | 537 | 231 | |
| rps12-D2 | 113 | 70336 | 231 | 537 | 25 | |
| clpP | 248 | 640 | 291 | 802 | 70 | |
| PetB | 5 | 765 | 647 | |||
| PetD | 7 | 691 | 525 | |||
| rpl2 | 442 | 651 | 390 | |||
| rpl16 | 401 | 946 | 8 | |||
| ndhA | 517 | 1080 | 561 | |||
| ndhB | 777 | 676 | 727 | |||
| ndhB-D2 | 727 | 676 | 777 | |||
| K. coccinea | rps16 | 220 | 826 | 39 | ||
| rpoC1 | 1635 | 686 | 454 | |||
| atpF | 411 | 741 | 142 | |||
| ycf3 | 140 | 768 | 220 | 735 | 123 | |
| rps12 | 113 | 27213 | 25 | 537 | 231 | |
| rps12-D2 | 113 | 70281 | 231 | 537 | 25 | |
| clpP | 248 | 566 | 291 | 703 | 70 | |
| PetB | 5 | 762 | 641 | |||
| rpl2 | 442 | 653 | 390 | |||
| ndhA | 517 | 1080 | 561 | |||
| ndhB | 777 | 676 | 727 | |||
| ndhB-D2 | 727 | 676 | 777 |
3.2. Repeat Structure and Simple Sequence Repeat Sequence Analysis
Many repeats are present in gene deserts, although whole-genome sequencing has shown that they can occur in functional regions as well. Repeats of more than 30 bases were considered as the long repeats.
A total of 67, 44, and 44 pairs of repeats were identified in the S. chinensis, S. sphenanthera, and K. coccinea cp genomes (Figure 2(a)). S. chinensis contained 46 forward repeats, 18 palindromic repeats, 2 reverse repeats, and 1 complement repeat. S. sphenanthera and K. coccinea contained 17, 23, 2, and 2 repeats, respectively, with repeat lengths ranging from 30 to 131 bp (Figure 2(b)).
Figure 2.
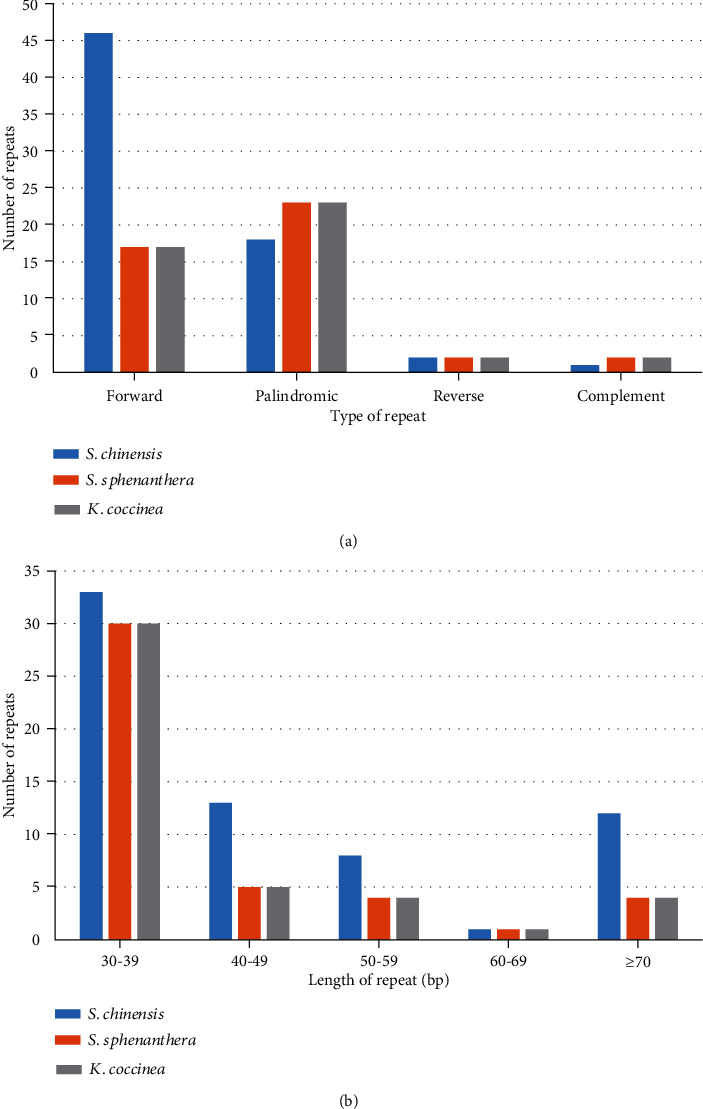
Analysis of repeated sequences in the 3 cp genomes.
A Simple Sequence Repeat (SSR) is a PCR-based highly efficient molecular labeling technique. The SSRs are constantly found to be highly polymorphic, easily visible, stable, and codominant, whereas the structures of chloroplast genomes are simple and relatively conservative. Single parental inheritance, together with other characteristics, has been extensively used in species identification and genetic diversity analysis. Because of the characteristics of neutral markers, the highly variable numbers of repeats, and the relative conservatism of flanking sequences of SSRs, they are widely distributed in the genome of organisms. Microsatellites or SSRs play a major role in polymorphism analysis and in marker-assisted selection [39].
A total of 57 eligible SSR loci were detected in the genomes of three chloroplasts of the S. chinensis family (Table 4, Figure 3). Of them, S. chinensis contained 16 single-nucleotide, 3 dinucleotide, 1 trinucleotide, and 1 hexanucleotide repeat gene sequences. S. sphenanthera contained 18 single-nucleotide and 3 dinucleotide repeat gene sequences. K. coccinea had 13 single-nucleotide, 1 dinucleotide, and 1 trinucleotide repeat gene sequences. Obviously, there was distinct difference between these three plants. Compared with the two Magnoliaceae plants, the three Schisandraceae plants had closer SSR types and abundance.
Table 4.
Comparison of the SSR data of five chloroplast genes.
| Species | S. chinensis | S. sphenanthera | K. coccinea | Magnolia officinalis | Liriodendron chinense |
|---|---|---|---|---|---|
| Mononucleotide | 16 | 18 | 13 | 12 | 31 |
| Dinucleotide | 3 | 3 | 1 | 3 | 3 |
| Trinucleotide | 1 | 0 | 1 | 0 | 1 |
| Hexanucleotide | 1 | 0 | 0 | 0 | 0 |
Figure 3.
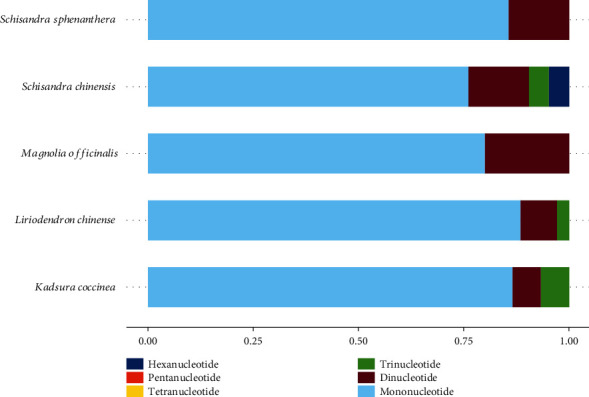
SSR abundance map of 5 species plants.
Gene sequence abundance is inversely related to the length of the repeat gene sequence. Among all the SSRs of these three plants, A/T was the most frequently repeated sequences, followed by AT/AT, and they had accounted for 94.7% of the total sequences. There was a certain base preference regarding the base composition of a single-nucleotide to a trinucleotide repeat sequence, suggesting that the Schisandraceae plant was mainly composed of the A/T-rich repeat sequence. The single nucleotides were identical, and the SSR loci of other Schisandraceae species were searched by the same parameters and their repeat sequence types as well as abundance percentages were compared. Our results suggested that the types and abundance of repeat sequences were conservative between Schisandraceae species, which had laid a certain foundation for further searching for the universal SSR markers between the Schisandraceae species.
3.3. Comparative Analysis of the IR Boundary Regions
The size of chloroplast genomes mainly depends on the contraction and expansion of the IR region [40]. The three medicinal plants of Schisandraceae were internally compared, and the comparison between Schisandraceae and Magnoliaceae families showed that plants within the same family were more similar (Figure 4). S. sphenanthera showed a higher similarity to S. chinensis, the length between LSC and IRa was the same, the gene length between ycf1 and ndhb was 723 bp, and the slightly larger K. coccinea was 732 bp. However, the distance between the ycf1 and ndhB genes of S. chinensis and S. sphenanthera differed greatly from the boundary. The boundary distance between ycf1 and the IR region was also different. K. coccinea was 191 bp, and it was identical to that of S. sphenanthera, while S. chinensis was 693 bp, which was significantly larger than the former two. Such difference could also be detected between LSC and IRb, as well as the distance between the ndhB gene of S. sphenanthera and K. coccinea, with the boundary distances of 531 bp and 540 bp, respectively, which were evidently larger than the 29 bp of S. chinensis. The distance from the trnh-GUG gene of S. chinensis to the IRb region was greater than those of S. sphenanthera and K. coccinea. In summary, the three medicinal plants of Schisandraceae could be distinguished based on the IR region; besides, distance analysis between the IR region and the gene also revealed a closer relationship between S. chinensis and S. sphenanthera. Moreover, the relative analysis of the two Schisandraceae and Magnoliaceae plants showed higher similarity between Magnolia and Liriodendron, and a greater difference between Schisandraceae, which accounted for the correct division of Schisandraceae into one family.
Figure 4.
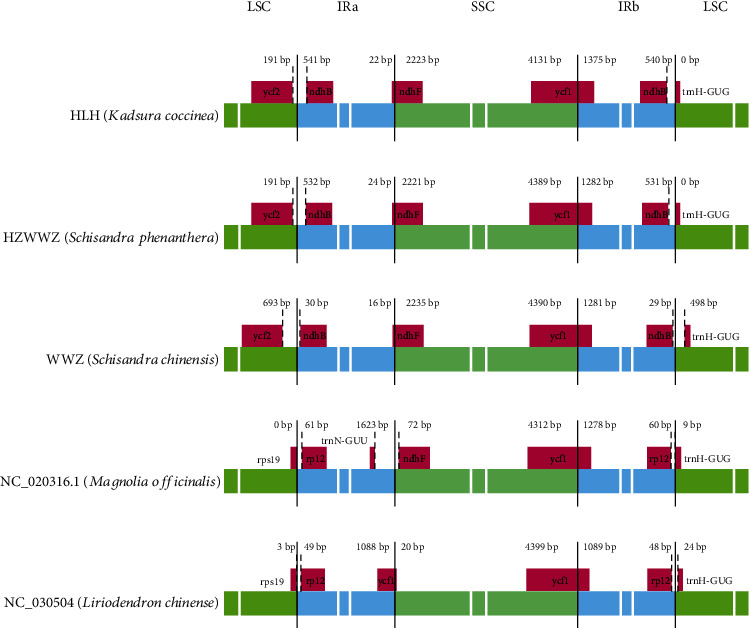
Comparisons of LSC, SSC, and IR boundary regions among three Schisandraceae species and two Magnoliaceae species.
3.4. Phylogenetic Analysis
In this paper, the ML (map) and phylogenetical tree were constructed (Figure 5) based on the genome-wide SNPs of 38 species; 25 of the 36 nodes in the ML tree had ≥90% of the support values, among which, 3 were 100% and only one was <80%. Using the APG IV plant classification system, Schisandraceae was assigned to the Malva sylvestris located in the base group of angiosperms, and consistent results with the latest classification system were obtained. It was observed from Table 4 that Schisandraceae was listed as a separate item. The three Schisandraceae plants had quite close relationships, among which, S. chinensis was closer to S. sphenanthera than K. coccinea, which was consistent with our real-time taxonomy. It was worth mentioning that, in the ML tree, Magnolia officinalis and Liriodendron chinense, together with S. chinensis, were divided into two, which also showed that it was reasonable to modernize S. chinensis from Magnoliaceae and separate it into one family. The Illicium and Schisandraceae had a similar relationship, which was far from the M. officinalis and L. chinense of Magnoliaceae. Hopefully, such data will provide certain help in the subsequent botanical classification of these plants.
Figure 5.
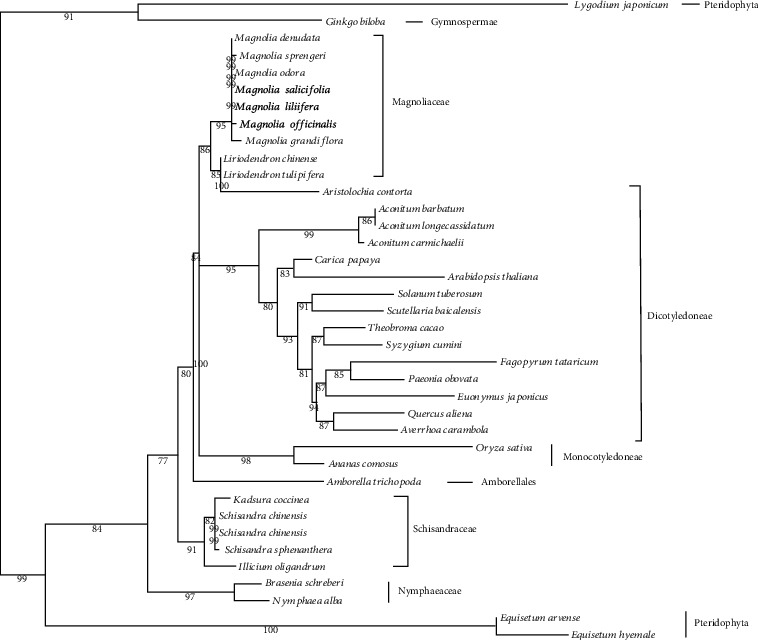
Molecular phylogenetic tree of genome-wide SNPs from 38 species (1000 bootstrap replicates).
The MP tree had indicated the same result as those of the ML tree. Specifically, the three plants and the Illicium genus in Schisandraceae were divided into one, and M. officinalis and L. chinense were also divided into one, indicating that the Schisandraceae and the octagonal fennel were the sister branches that had closer kinship. L. chinense was closer to M. officinalis, which was consistent with our results based on the APG IV classification system, indicating that the chloroplast genomes could accurately identify the genetic relationship between different species.
3.5. Genome Comparison Analysis
Moreover, the genomic structures in the genera of Schisandraceae were compared using the mVISTA software in the Shuffle-LAGAN mode, with the genome-wide S. chinensis chloroplast being used as the reference (Figure 6). The complete genomes of 5 plant species are used for comparison. Genic regions are identified using the DOGMA program, and a comparative map is prepared using mVISTA. The blue block indicates the conserved gene, the sky-blue block stands for trnA and rRNA, and the red block represents the intergenic region. Meanwhile, the white peaks indicate the regions with sequence variation among the five species. Our results suggested that the noncoding region had a higher degree of variation than the coding region, which might be ascribed to the replication correction of the IR region; as a result, genes in the IR region were more conserved than those in the LSC and SSC regions [40]. Comparison of genomes showed that the genes of S. sphenanthera and S. chinensis displayed higher degrees of similarity, followed by K. coccinea, M. officinalis, and L. chinense, respectively. Clearly, the genotypes of three Schisandraceae plants were more similar, and the M. officinalis of Magnoliaceae was more similar to L. chinense. This also provided evidence for the separation of Schisandraceae from Magnoliaceae.
Figure 6.
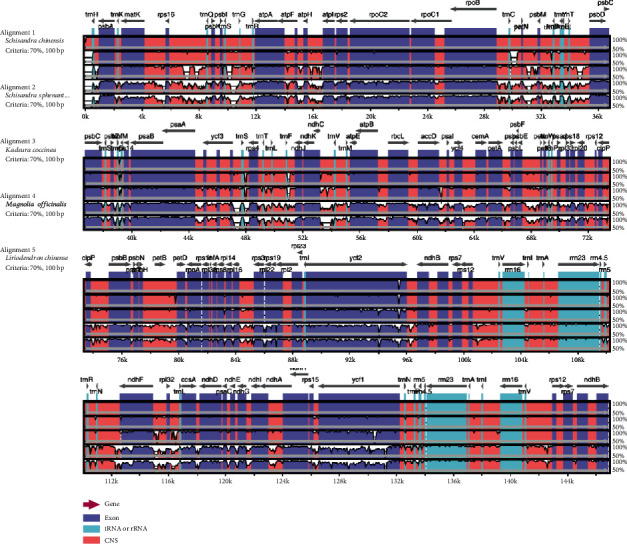
Comparison of chloroplast genomes from five plants species using mVISTA.
4. Conclusions
In this study, the genome-wide chloroplast of three Schisandraceae plants is sequenced, and the genomes are annotated and analyzed. The molecular data identified between these three species are obtained through the comparative analysis of three medicinal plants, and the SSR comparison is the same. Under the analysis conditions, the nucleotide types and numbers of S. chinensis and S. sphenanthera are more similar, while those of K. coccinea are only slightly similar. These three species can be clearly identified through the boundary analysis of the IR region in three Schisandraceae plants. The phylogenetic location of Schisandraceae can be obtained through phylogenetic analysis of three Schisandraceae plants. These results prove that three S. chinensis plants can be identified through genome-wide analysis of chloroplasts, and the phylogenetic relationships among species can also be acquired through chloroplast-to-species evolution and genetic relationship.
Acknowledgments
This research was funded by National Natural Science Foundation of China (General Program, Grant Number 81874338), 2019 Liaoning Provincial Department of Education Scientific Research Project, China (No. L201942), Liaoning Provincial Education Department (Liaoning Higher School Outstanding Young Scholar Growth Plan, Grant Number LJQ2014101), National Key Research and Development in the 13th Five-Year Plan (No. 2017YFC1701205), Major Special Fund for Science and Technology of Inner Mongolia Autonomous Region (No. 2019ZD004), and Natural Science Fund Project of Liaoning Province (No. 2020-MS-224).
Contributor Information
Liang Xu, Email: 861364054@qq.com.
Tingguo Kang, Email: kangtingguo@163.com.
Data Availability
The assembled complete genome sequences of the three species were submitted to NCBI with the accession numbers SRX4282568 (S. chinensis), SRX4282569 (S. sphenanthera), and SRX4282570 (K. coccinea). Users could download the data as a reference for research purposes only.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Supplementary Materials
Figure S1: chloroplast gene map of S. sphenanthera. Figure S2: chloroplast gene map of Kadsura coccinea. Table S3: long repeat sequences in S. chinensis chloroplast genome. Table S4: long repeat sequences in S. sphenanthera chloroplast genome. Table S5: long repeat sequences in K. coccinea chloroplast genome.
References
- 1.Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia. 342nd ed. Beijing: China Medical Science and Technology Press; 2015. [Google Scholar]
- 2.Committee of the Japanese Pharmacopoeia. Japanese Pharmacopoeia. Japan: Bureau Ministry of Health; 2006. Evaluation and Licensing Division Pharmaceuticals and Food Safety; p. p. 9. [Google Scholar]
- 3.Central Pharmaceutical Affairs Council of Korea. Korean Pharmacopoeia. Seoul, Korea: Central Pharmaceutical Affairs Council of Korea; 2002. [Google Scholar]
- 4.Jia M. R., Li X. W. Chinese national medicine history. Beijing: China Medical Science and Technology Press; 2005. [Google Scholar]
- 5.Wicke S., Schneeweiss G. M., Depamphilis C. W., Muller K. F., Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Molecular Biology. 2017;76:273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clegg M. T., Gaut B. S., Learn G. H., Morton B. R. Rates and patterns of chloroplast DNA evolution. Proceedings of the National Academy of Sciences. 1994;91(15):6795–6801. doi: 10.1073/pnas.91.15.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shinozaki K., Ohme M., Tanaka M., et al. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. The EMBO Journal. 1986;5(9):2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X. W., Gao H. H., Wang Y. T., et al. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Science China Life Sciences. 2013;56(2):189–198. doi: 10.1007/s11427-012-4430-8. [DOI] [PubMed] [Google Scholar]
- 9.Yang J. B., Tang M., Li H. T., Zhang Z. R., Li D. Z. Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evolutionary Biology. 2013;13(1):p. 84. doi: 10.1186/1471-2148-13-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu D. L. Research on magnolia. Wuhan Botanical Research. 2001;19:191–198. [Google Scholar]
- 11.Hansen D. R., Dastidar S. G., Cai Z., et al. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae) Molecular Phylogenetics and Evolution. 2007;45(2):547–563. doi: 10.1016/j.ympev.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Guo H. J., Liu J. S., Luo L., et al. Complete chloroplast genome sequences of Schisandra chinensis: genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Science China Life Sciences. 2017;60(11):1286–1290. doi: 10.1007/s11427-017-9098-5. [DOI] [PubMed] [Google Scholar]
- 13.Li B., Zheng Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Scientific Reports. 2018;8(1):9285–9301. doi: 10.1038/s41598-018-27453-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Angiosperm Phylogeny Group, Chase M. W., Christenhusz M. J. M., Fay M. F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society. 2016;181(1):1–20. doi: 10.1111/boj.12385. [DOI] [Google Scholar]
- 15.Liu Y. H. Systematics and evolution of the family Schisandraceae. II. System of Schisandra and evolution of Schisandraceae. Acta Scientiarum Naturalium Universitatis Sunyatseni. 2002;6:67–72. [Google Scholar]
- 16.Liu Y. H. Classification system and evolution trend of Schisandraceae. I. Establishment of Schisandra and classification system of Schisandra in South China. Acta Scientiarum Naturalium Universitatis Sunyatseni. 2002;5:77–82. [Google Scholar]
- 17.Liu Y. H. A new species of Schisandraeaceae in China. Plant Research. 1983;3:148–150. [Google Scholar]
- 18.Zhang H. D. Classification of seed plant system. Journal of Sun Yat-sen University; Natural Science Edition. 1986;1:1–13. [Google Scholar]
- 19.Song W. Z., Xiao P. G. Medicinal plants of Schisandra chinensis and its lignans. Chinese Traditional and Herbal Drugs. 1982;13:40–48. [Google Scholar]
- 20.Lin Q. Medicinal plant resources of Illicium L. Chinese Traditional and Herbal Drugs. 2002;7:81–84. [Google Scholar]
- 21.Wang X. T., Yu J. H., Li W., et al. Characteristics and antioxidant activity of lignans in Schisandra chinensis and Schisandra sphenanthera from different locations. Chemistry & Biodiversity. 2018;15(6, article e1800030) doi: 10.1002/cbdv.201800030. [DOI] [PubMed] [Google Scholar]
- 22.Li X. K., Wang B., Han R. C., et al. Identification of medicinal plant Schisandra chinensis using a potential DNA barcode ITS2. Acta Societatis Botanicorum Poloniae. 2013;82(4):283–288. doi: 10.5586/asbp.2013.032. [DOI] [Google Scholar]
- 23.Chen L., Wu Y. N., Shan H. J., et al. DNA barcoding identification of Schisandrae chinensis fructus and Schisandrae sphenantherae fructus by ITS2 sequences. Chinese Journal of Experimental Traditional Medical Formulae. 2016;1:66–71. [Google Scholar]
- 24.McPherson H., van der Merwe M., Delaney S. K., et al. Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecology. 2013;13(1):p. 8. doi: 10.1186/1472-6785-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borgstrom E., Lundin S., Lundeberg J. Large scale library generation for high throughput sequencing. PLoS One. 2011;6(4, article e19119) doi: 10.1371/journal.pone.0019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cronn R., Liston A., Parks M., Gernandt D. S., Shen R. K., Mockler T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Research. 2008;36(19, article e122) doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo R., Liu B., Xie Y., et al. SOAPdenovo2: an empirically improved memory-efficient short-read denovo assembler. Giga Science. 2012;1(1):p. 18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyman S. K., Jansen R. K., Boore J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 29.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 30.Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Research. 2004;32:277–280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanehisa M., Goto S., Hattori M., et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Research. 2006;34:354–357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanehisa M. A database for post-genome analysis. Trends Genet. 1997;13:375–376. doi: 10.1016/s0168-9525(97)01223-7. [DOI] [PubMed] [Google Scholar]
- 33.Tatusov R. L., Koonin E. V., Lipman D. J. A genomic perspective on protein families. Science. 1997;278(5338):631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 34.Tatusov R. L., Fedorova N. D., Jackson J. D., et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4(1):p. 41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magrane M., Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011 doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashburner M., Ball C. A., Blake J. A., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiao Y., Jia H. M., Li X. W., et al. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra) BMC Genomics. 2012;13(1):p. 201. doi: 10.1186/1471-2164-13-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravi V., Khurana J. P., Tyagi A. K., Khurana P. An update on chloroplast genomes. Plant Systematics and Evolution. 2008;271(1-2):101–122. doi: 10.1007/s00606-007-0608-0. [DOI] [Google Scholar]
- 39.Kaliswamy P., Vellingiri S., Nathan B., Selvaraj S. Microsatellite analysis in the genome of Acanthaceae: an in silico approach. Pharmacognosy Magazine. 2015;11(41):152–156. doi: 10.4103/0973-1296.149731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khakhlova O., Bock R. Elimination of deleterious mutations in plastid genomes by gene conversion. The Plant Journal. 2006;46(1):85–94. doi: 10.1111/j.1365-313X.2006.02673.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: chloroplast gene map of S. sphenanthera. Figure S2: chloroplast gene map of Kadsura coccinea. Table S3: long repeat sequences in S. chinensis chloroplast genome. Table S4: long repeat sequences in S. sphenanthera chloroplast genome. Table S5: long repeat sequences in K. coccinea chloroplast genome.
Data Availability Statement
The assembled complete genome sequences of the three species were submitted to NCBI with the accession numbers SRX4282568 (S. chinensis), SRX4282569 (S. sphenanthera), and SRX4282570 (K. coccinea). Users could download the data as a reference for research purposes only.