

Abstract
Powered by the energy of ATP binding and hydrolysis, protease-containing ABC transporters (PCATs) export amphipathic and hydrophilic bacteriocin and quorum-sensing proteins across the membrane hydrophobic barrier. The cargo proteins have N-terminal leader peptides that are cleaved off by the cysteine protease domain, referred to as the C39 domain, or referred to as the peptidase (PEP) domain. The sequence and structural determinants of the interaction between PCATs and cargo proteins are poorly understood, yet this interaction is a central aspect of the transport mechanism. Here, we demonstrate the ATP-dependent, equilibrium binding of the cargo protein to the transmembrane domain (TMD) of a PCAT subsequent to the removal of the leader peptide by the PEP domain. Binding of the cargo protein to PCAT1 variants devoid of the PEP domain is detected through changes in the spectroscopic properties of fluorescent or spin label. Moreover, we find similar energetics of binding regardless of the presence of the leader peptide, suggesting that although the PEP domain serves for recognition and orientation, interaction with the TMD is the main contributor to the affinity. These findings are in direct contradiction with a recent study claiming that the TMD does not interact with the cargo protein; rather acting as a “Teflon-like” conduit across the bilayer (Kieuvongngam, V., Olinares, P. D. B., Palillo, A., Oldham, M. L., Chait, B. T., and Chen, J. (2020) Structural basis of substrate recognition by a polypeptide processing and secretion transporter. eLife 9, e51492). A distinctive feature of the transport model emerging from our data invokes a stable complex between PCATs and their cargo proteins following processing of the leader peptide and prior to ATP-dependent alternating access that translocates the cargo protein to the extracellular side.
Keywords: ABC transporters, protease-containing ABC transporter (PCAT1), membrane export, fluorescence, EPR spectroscopy, spin labeling, ABC transporter, electron paramagnetic resonance (EPR), transporter, ATP
Secretion of proteins across membranes play a central role in a number of critical biochemical and physiological processes. Multiple export mechanisms have evolved to address the challenge of transferring amphipathic and hydrophilic proteins of a wide range of sizes across this hydrophobic barrier. Gram-negative bacteria, for instance, have secretion systems in the inner and outer membranes consisting of proteins that transport, chaperone, or interact with cargo proteins possessing a Sec-independent signal sequence (1–3). For Gram-positive bacteria, a secretion system consisting exclusively of an ABC transporter serves the dual role of exporting bacteriocin and quorum-sensing peptides and conferring resistance to its peptide cargo (4–7). A subclass of these transporters consists of a core canonical ABC exporter covalently linked to a peptidase domain and referred to as peptidase-containing ABC transporters (PCATs). The peptidase module (also referred to as C39 domains or PEP domain) is a cysteine protease that cleaves off the signal sequence peptide at the N terminus of the cargo protein prior to export.
Crystal structures of protein exporting ABC transporters from multiple organisms (8–10), including PCAT1 from thermophilic Clostridium thermocellum (8), reveal a core homodimeric ABC transporter with an architecture similar to that of solute ABC exporters. Each protomer consists of a transmembrane domain (TMD), a PEP domain, and nucleotide-binding domain (NBD), the latter containing the conserved sequence motifs for ATP binding and hydrolysis. The apo conformation of PCAT1 profiles two NBDs in a loosely packed dimer primed for ATP binding and subsequent hydrolysis (11). The two TMDs cradle a large α-helical barrel of approximately 440 Å2 area lined with charged residues and spanning the length of the lipid bilayer. The two cysteine protease domains are positioned at opposite ends of the core transporter at the entryway of the TM barrel, making relatively weak contacts with TMs 3, 4, and 6 and the NBDs. A structure of an ATPγS-bound PCAT1 mutant, impaired for ATP hydrolysis by replacement of the catalytic glutamate 648, provided a glimpse of structural changes induced by ATP binding. In this structure, the NBDs form the closed dimer considered the catalytic intermediate for ABC hydrolysis. Rearrangements of the TMD involve the movement of TMs 3 and 6 toward the center, thus rupturing the limited interface with the PEP domains. The absence of a visible electron density for this domain was interpreted as evidence of its undocking and flexible attachment to the rest of the transporter.
The cargo protein of PCAT1, hereafter referred to as Sub, was identified as a 90-residue protein containing an N-terminal signal sequence (leader peptide) and encoded in the same operon as PCAT1 (8). PCAT1 specifically cleaves the Sub at a double glycine leader sequence, resulting in the removal of a 2500-Da fragment. Site-directed mutagenesis of the PEP domain confirmed that cleavage occurs at its active site and involves critical cysteine and histidine residues. Direct structural evidence that the cargo protein specifically interacts with the PEP domain was advanced by recent cryo-EM structure of protease-deficient PCAT1 (12), which revealed two cargo protein molecules bound to the PEP domain but only one with density extending to the TMD. To evaluate the determinants of Sub specificity to PCAT1, site-directed mutagenesis of the PEP domain and the leader peptide of Sub was carried out, and binding was evaluated by pulldown assays (12). Mutations of conserved residues led to reduction in the intensity of Sub bands resolved by SDS gel electrophoresis. The authors interpreted these assays, which can be confounding particularly for low affinity interactions, along with the diffuse and amorphous density of Sub in the cryo-EM structure of the complex, to imply that substrate specificity is conferred only through the leader peptide. Contrasting their findings with recent structures of substrate-bound ABC exporter P-glycoprotein (Pgp) (13), they advanced the model that, unlike other ABC exporters, the TMD of PCAT1 does not interact directly with its cargo protein.
To test this model and address the aspect of transport that involves recognition and binding of the cargo protein, we investigated the direct interactions and environment of Sub in multiple biochemical intermediates of WT and engineered PCAT1 variants. A unique native cysteine of Sub (Cys31) was either bimane or spin-labeled (14, 15), and its spectroscopic properties in the presence of PCAT1 were investigated by fluorescence and EPR (16). Together, the data provide evidence of nucleotide-dependent contacts between Sub and the core TMD subsequent to the removal of the leader peptide by the PEP domain in direct contradiction to the conclusions of Kieuvongngam et al. (12). Our model entails specific interactions that involve segments beyond the N-terminal helical leader peptide, which gets clipped by the PEP domain. Thus, PCATs must have evolved specific sequence determinants for binding of their cognate cargo proteins.
Results and discussion
Biochemical characterization of PCAT1 variants and the cargo protein (Sub)
To enable spectroscopic studies, we generated a number of variants of PCAT1 where the PEP domain was deactivated or deleted. In addition, to avoid the possibility of disulfide bond formation with Sub, the variants were either devoid of cysteine residues in the TMD or in the TMD and PEP domain (see “Experimental procedures”). A variant referred to as PCAT1-CL has all the cysteines, including at the active site of the PEP domain, replaced and thus is expected to bind but not process Sub. PCAT1-CT is devoid of cysteines except for PEP domain, which includes the catalytic cysteine of the C39 domain (i.e. PCAT-CL + C39). Finally, a cysteine-less transporter core lacking the PEP domain and hereafter referred to as PCAT1-CC (where “CC” stands for cysteine-less core) was constructed to investigate the specific interactions of Sub with the TMD. Sub was expressed into inclusion bodies (see “Experimental procedures”), purified by nickel-affinity chromatography, using a histidine tag preceding the leader sequence, followed by size-exclusion chromatography (SEC) as described under “Experimental procedures.”
All PCAT1 variants were expressed and purified into micelles of n-dodecyl β-d-maltoside (β-DDM) and appeared homogenous by SEC (data not shown). The β-DDM–solubilized PCAT1 variants displayed robust ATP hydrolysis with a Vmax of 70 ng/mg/min (Fig. 1 and Table 1) similar to that previously reported (9). Only PCAT1-CC had a higher Vmax (112 ng/mg/min) than constructs with an intact PEP domain. Although Vmax for the variants was not dependent on the presence of Sub, a measurable change in Km was observed (Fig. 1 and Table 1). However, the Sub-induced changes in kinetic parameters of ATP turnover were smaller than typically observed for ABC efflux transporters in the presence of their substrates (17).
Figure 1.

ATP turnover by PCAT1-WT and its variants. The kinetic parameters are relatively unaffected by the binding of Sub. However, the core transporter PCAT-CC, which is devoid of the PEP domain, has a significantly different Vmax (Table 1). Each data point is the average of three independent measurements. The error bars represent the standard deviations.
Table 1.
Kinetic parameters of ATP turnover by PCAT1 variants
| PCAT | Vmax | Km |
|---|---|---|
| μmol/min/mg | mm | |
| WT | 0.06 ± 0.002 | 0.15 ± 0.03 |
| WT + Sub | 0.06 ± 0.001 | 0.08 ± 0.01 |
| CC | 0.12 ± 0.002 | 0.21 ± 0.02 |
| CC + Sub | 0.14 ± 0.004 | 0.26 ± 0.04 |
| CT | 0.07 ± 0.002 | 0.19 ± 0.02 |
| CT + Sub | 0.09 ± 0.002 | 0.13 ± 0.02 |
| CL | 0.07 ± 0.002 | 0.13 ± 0.03 |
| CL + Sub | 0.06 ± 0.001 | 0.11 ± 0.02 |
The cargo protein binds specifically to the core transporter of PCAT1
To directly monitor the interaction between Sub and PCAT1, a bimane probe was attached at the native cysteine 31 of Sub (see “Experimental procedures”). Bimane emission fluorescence is exquisitely sensitive to the dielectric constant of its environment (14, 15), allowing direct investigation of Sub binding to PCAT1. As a control, bimane-labeled Sub in detergent micelle solution showed an emission λmax characteristic of an aqueous environment, suggesting that the bimane probe remains in an aqueous environment regardless of whether Sub directly partitions into the micelles (Fig. 2, black trace). In contrast, addition of detergent-solubilized PCAT1 variants resulted in a substantial blue shift in λmax of the bimane-labeled Sub and an increase in the quantum yield consistent with the transfer of Sub into a hydrophobic environment (14, 15) (Fig. 2). This change in fluorescence characteristics demonstrate direct interactions between PCAT1 and Sub. Remarkably, this blue shift was observed for Sub binding to full-length PCAT1, as well as the core transporter PCAT1-CC, demonstrating that the interaction with Sub persists after the removal of the leader peptide and that the presence of the PEP domain is not necessary for Sub interaction with PCAT1. We observed interesting variations in the final emission intensity of the bimane, which were pronounced for PCAT1-CL, suggesting a more hydrophobic environment for bound Sub relative to its environments in the other PCAT1 variants. Interpretation of the structural origin of these changes requires more labeling sites across the cargo protein.
Figure 2.

Fluorescence emissions scans of bimane-labeled Sub in β-DDM micelles in the absence (black trace) and presence of PCAT1 variants (color traces). Binding of Sub to PCAT1 variants result in a blue shift in the maximum emission wavelength (red trace) that is reversed by ATPγS/Mg2+ (violet trace) except for PCAT1-CL, which cannot cleave the leader peptide. ADP/Mg2+ binding has no effect, suggesting that Sub remains bound to PCAT1. Excess ATP/ Mg2+, which mimics turnover conditions, also reset the fluorescence except for PCAT1-CL, which cannot process the leader peptide.
SDS-PAGE analysis of Sub following incubation with the full-length PCAT1-WT shows that the leader sequence is cleaved regardless of whether Sub is labeled (Fig. 3A). Similarly, the other PCAT1 variant with a functional PEP domain, PCAT1-CT, which also forms a stable complex with Sub (Fig. 2), cleaved off the leader peptide of the latter (Fig. 3B). In addition to revealing direct stable interactions between PCAT1 transporter core and Sub, these results also demonstrate that substitution of the cysteines, except at the active site of the peptidase domain, does not compromise processing of the leader peptide by PCAT1.
Figure 3.

SDS-PAGE analysis showing that PCAT1-WT and PCAT1-CT process the leader peptide of bimane-labeled Sub. SDS-PAGE analysis demonstrating that PCAT1-WT (A) and PCAT-CT (B) process the leader peptide of bimane-labeled Sub.
Binding of Sub to the TMD of PCAT1 is modulated by ATP binding
PCAT1 is expected to undergo conformational changes coupled to the ATP hydrolysis cycle. Specifically, the crystal structure determined in the presence of ATPγS revealed altered accessibility of the transmembrane chamber (8). In addition, it has been shown that ATP binding reduces protease activity in the PEP domain (8).
To determine how ATP binding and hydrolysis affect the interactions of PCAT1 with its cargo protein, we measured the fluorescence of bimane-labeled Sub incubated with PCAT1 variants upon addition of the nonhydrolyzable analog of ATP (ATPγS), of ADP and under turnover conditions of excess ATP, all in the presence of Mg2+ (Fig. 2). ATPγS reversed the changes in quantum yield and in λ max induced by binding to PCAT1, suggesting that under these conditions Sub is not bound to PCAT1-WT or PCAT1-CC. Considering the reduced accessibility of the transmembrane barrel, evident in the crystal structure of PCAT1 bound to ATPγS, we conclude that ATP binding disrupts the interactions of the TMD with Sub, resulting in the exclusion of the latter from the transporter core. In contrast, ADP binding had marginal effects on the bimane fluorescence of bound Sub. Under turnover conditions in which ATP is in large excess relative to ADP (Fig. 2, blue trace), the trend was similar to ATPγS for PCAT1-WT and PCAT1-CC. A notable difference in the pattern of fluorescence changes was observed for PCAT-CL, in which the active cysteine of the C39 domain is replaced. For this variant, the presence of ATP did not reverse the increase in fluorescence intensity, suggesting that Sub remains associated with the transporter presumably because of the lack of processing by the PEP domain.
To complement the fluorescence analysis, Sub was spin-labeled using methanethiosulfonate spin label MTSSL (18) (see “Experimental procedures”) at the same cysteine 31 residue used for bimane labeling. Because the disulfide linkage of the spin label to the cysteine is reversible and prone to reduction by sulfhydryls, CW-EPR analysis of Sub was carried out only in the presence of PCAT1-CL and PCAT1-CC, which are devoid of cysteines. A substantial change in the EPR line shape, which reflects the rotational mobility of the spin label, was observed in the presence of detergent-solubilized PCAT1 (Fig. 4, magenta trace). Whereas the EPR line shape of Sub in the presence of detergent micelles reports predominately isotropic fast motion of the spin label with a correlation time faster than 1 ns, binding to PCAT1 leads to a line shape characteristic of restricted motion that is slower than 10 ns (18) (Fig. 4, black arrows). Sub is released upon binding of ATPγS to PCAT1-CC as evidenced by a resetting of the EPR line shape to that characteristic of the fast motion observed in detergent micelles in the absence of PCAT1. This result not only demonstrates direct interactions of Sub with the transporter TMD but also suggests that bound Sub is in a buried environment where spin label mobility is restricted by neighboring side- and main-chain atoms.
Figure 4.

EPR spectra of spin-labeled Sub in the presence of PCAT1 variants: PCAT1-CL and PCAT1-CC. Binding results in a broadening of the EPR line shape, indicating restricted motion of the spin label indicated by the black arrow. The broadening is reversed by binding of ATPγS/Mg2+ to PCAT1-CC but not PCAT1-CL, which cannot process the leader peptide.
In contrast, ATPγS failed to reset the EPR line shape for PCAT1-CL, in agreement with the observation from fluorescence analysis. Therefore, if the leader peptide is not cleaved off by the PEP domain, Sub remains bound to the transporter. Together, the EPR and fluorescence data indicate that whereas the leader peptide binds and interacts with the PEP domain, Sub makes contacts with the hydrophobic transporter core of PCAT1. Release of the Sub is triggered by ATP binding only after the proteolytic processing of the leader segment.
Subaffinity to PCAT1 is predominantly due to interactions with the TMD
We took advantage of the change in bimane fluorescence upon binding to determine the apparent affinity of Sub to PCAT1 variants. As shown in Fig. 5, the addition of increasing concentrations of PCAT1 variants led to progressive change in the normalized bimane emission intensity, resulting in binding isotherms that can be fit to a single binding site yielding the apparent KD of the interaction. Although the apparent KD was in a similar range for the variants, small differences reflect the respective contributions of the TMD and PEP domain to the affinity of PCAT1 to Sub. The binding affinity of Sub to PCAT1-CC, which is devoid of the PEP domain, and to PCAT1-CT, which cleaves off the leader peptide, reflects the interaction of the TMD with Sub in a cysteine-less background (ΔGo ≈ 8.5 kcal/mol). The increase in affinity in the PCAT-CL background is a measure of the contribution of the interactions between the PEP domain and the Sub (ΔGo ≈ 9 kcal/mol). These values indicate that the contacts of Sub with TMD provide the main energetic contribution to the affinity of Sub to PCAT1.
Figure 5.
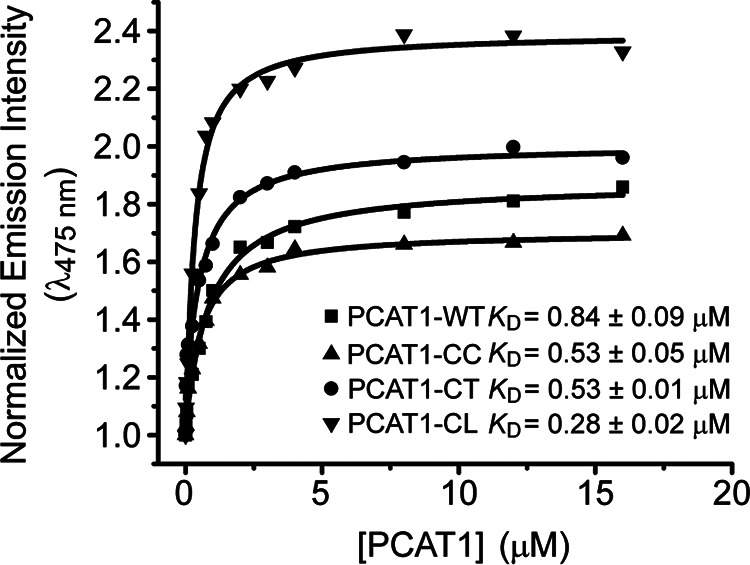
Binding isotherms of bimane-labeled Sub to PCAT1 variants at 37 °C. The fluorescence emission intensity at 475 nm was plotted versus the concentration of PCAT1 variants. Each curve represents the average of two different repeats. The respective KD along with the fit errors were determined by nonlinear square fits to a single binding site.
Unexpectedly, PCAT1-WT has the lowest affinity, although its interaction with Sub should only reflect the contacts with the TMD similar to the PCAT-CC or PCAT1-CL. It is possible that the substitution of the cysteines in the PEP domain is responsible for this small change in affinity.
Conclusion
To our knowledge, the results presented in this paper are the first reported evidence of direct interactions between the TMD of a PCAT and its cargo protein. This interaction is ATP-dependent, and its apparent affinity, below one micromolar range, is not substantially reduced following the processing of the leader peptide or in the absence of the PEP domain. The conclusions of Kieuvongngam et al. (12) that the TMD of PCATs is a Teflon-like conduit for the cargo protein was based on pulldown assays and the diffuse density of Sub in the cryo-EM structure with PCAT1. We contend that the latter reflects the relative moderate affinity reported here and implies that the energetics of the interactions is not sufficient to freeze out the conformational entropy of Sub, which consequently adopts multiple poses in the TMD. Finally, we note that the apparent KD values between Sub and PCAT1 are similar to those of drugs interacting with polyspecific ABC exporters such as Pgp. The structures of Pgp in complex with Taxol6 (13) suggested multiple binding modes not unlike Sub binding to PCAT1 implied by the cryo-EM structure. Overall, our data suggest that the cargo protein forms a stable complex with the transmembrane domain subsequent to the processing of the leader peptide.
The results presented here can be formulated into a minimalist, Sub-centric view of the transport cycle by PCAT1 (Fig. 6). The cycle is expected to be initiated by binding of Sub to apo- or ADP-bound intermediates of PCAT1, which are transiently sampled under typical cellular ATP concentrations (Fig. 6, step 1). Subsequent to Sub binding, the signal peptide is cleaved by the PEP domain (Fig. 6, step 2) prior to ATP binding, whereas the cargo protein is stably bound in the TMD chamber. ATP binding induces a conformational change that closes the chamber, thereby excluding the Sub (Fig. 6, step 3) most likely to the extracellular side through a yet to be identified exit channel. ATP hydrolysis resets the transporter to an inward-facing conformation ready for another round of transport.
Figure 6.
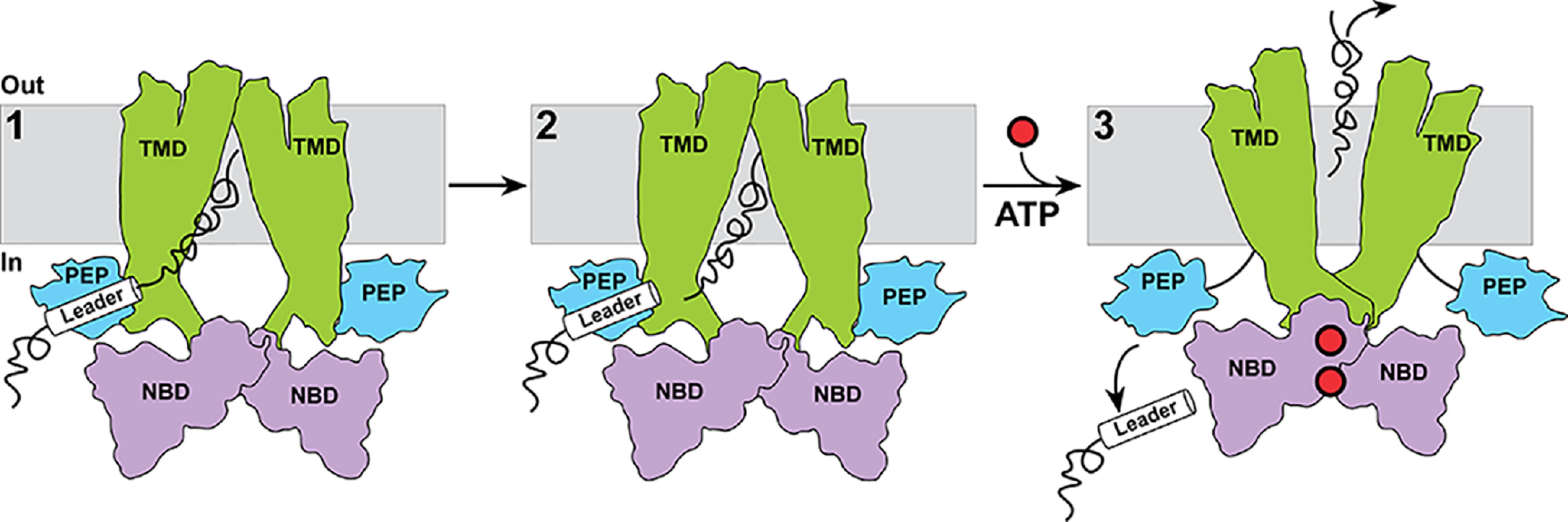
A cargo-protein centric model of transport by PCAT1. Binding of and recognition by the PEP domain of the leader peptide (step 1) enables interactions of the cargo proteins with the TMD subsequent to cleavage of the leader peptide (step 2). ATP binding drives conformational changes in the TMD leading to occlusion of the cargo protein binding chamber (step 3). An outward-facing conformation as depicted is entirely speculative as no such conformation has been observed.
Experimental procedures
Cloning, expression, and purification of PCAT1
The gene from the PCAT1 of C. thermocellum was codon-optimized and synthesized with an N-terminal His10 tag by GenScript for Escherichia coli expression. In addition to the WT transporter, we generated a number of variants. PCAT1 cysteine-less core, or PCAT1-CC, is a core transporter lacking the C39 domain and in which the native cysteines were replaced on the basis of sequence alignment as follows: C171A/C581A/C687A/C713L. Similarly, we constructed full-length cysteine-less transporter named PCAT1-CL in which all the native cysteines were replaced as follows: C12S/C21S/C25S/C129F/C171A/C581A/C687A/C713L. A full-length transporter variant was constructed by mutating cysteines only in the PCAT1 core (as in PCAT1-CC), but the cysteines on the C39 domain remained intact as WT PCAT1 and named as PCAT1-CT. Point mutations were created by site-directed mutagenesis and subsequently confirmed by sequencing (Genewiz).
PCAT1-containing plasmids were transformed into E. coli C43 (DE3) competent cells and a single colony was used to inoculate 1 liter of minimal medium A supplemented with ampicillin as described previously (19). The cells were grown at 37 °C until an absorbance (A600 nm) of 0.6–0.8 was reached, and protein expression was induced at 16 °C by the addition of 1 mm isopropyl β-d-thiogalactopyranoside and allowed to grow for 18 h before harvesting the cells. Briefly, cells were resuspended in 30 ml lysis buffer containing 50 mm Tris-HCl, pH 7.0, 500 mm NaCl, 10% (v/v) glycerol, 1 mm EDTA and lysed by 5–7 passes through a C3 homogenizer (Avestin) and centrifuged at 9000 × g for 15 min. The supernatant was collected, supplemented with 2.5 mm DTT (DTT) where appropriate and centrifuged at 200,000 × g for 1 h to pellet the membrane. PCAT1 membranes were resuspended and solubilized in lysis buffer containing 1% β-DDM and 1.0 mm DTT (DTT) by constant stirring for 2 h in an ice bath and the insoluble material was removed by centrifugation at 193,357 × g for 30 min at 4 °C. The supernatant was incubated Ni-NTA resin for 2 h at 4 °C. The membrane-resin mixture was equilibrated in Ni-NTA buffer containing 50 mm Tris-HCl, pH 7.0, 500 mm NaCl, 10% (v/v) glycerol, 0.05% β-DDM (w/v). The Ni-NTA resin was washed with 5 column volumes of Ni buffer containing 50 mm imidazole and PCAT1 was eluted with Ni Buffer containing 300 mm imidazole. Protein fractions were concentrated using Amicon Ultra-100 kDa centrifugal filters (Millipore) and further purified by gel-filtration chromatography (Superdex 200 column GE) in a buffer containing 20 mm Tris-HCl pH 7.0, 150 mm NaCl, 10% (v/v) glycerol, 0.05% β-DDM (w/v) and supplemented with 1 mm DTT if needed. Purified PCAT1 was concentrated using Amicon Ultra-100 kDa centrifugal filters (Millipore) and the final concentration was estimated by absorbance at 280 nm with a predicted extinction coefficient of 53,750 M−1 cm−1 for full-length PCAT1 and 36,330 M−1 cm−1 for PCAT1-core. PCAT protein solutions were stored on ice until further use.
Cloning, expression, purification, and labeling of cargo protein (Sub)
The substrate (cargo protein) of PCAT1 referred to as Sub, previously (8) identified as a 90-residue protein containing an N-terminal signal sequence and encoded in the same operon as PCAT1, was codon-optimized and synthesized for expression in E. coli by cloning into PET19b vector similarly to PCAT1 with an N-terminal 10 × His tag.
Sub was expressed in E. coli BL21 (DE3) Gold competent cells. Cells were grown in 1 liter Terrific Broth supplemented with ampicillin at 37 °C until OD600 reached to 1.8–2.0, induced with 1 mm isopropyl β-d-thiogalactopyranoside and allowed to grow for 4 h more until the cells were harvested by centrifugation at 6,000 × g for 15 min. The cell pellet was resuspended in buffer solution containing 50 mm Tris, pH 7.5, 150 mm NaCl, 5 mm EDTA with protease inhibitors. The cells were lysed by sonication and centrifuged at 17,000 × g for 15 min to pellet unbroken cell debris and inclusion bodies of Sub. The cell pellets were washed in 25 ml of wash buffer containing 50 mm Tris, pH 7.5, 150 mm NaCl, 0.5 mm EDTA, 0.5 mm DTT, 1% (w/v) Triton X-100. The suspension was centrifuged at 17,000 × g for 15 min, and the cell pellet wash was repeated with the same buffer without Triton. Pellet containing the inclusion bodies was solubilized by resuspension in the same wash buffer (2.5 ml) supplemented with 10 mm DTT and 6 m urea under constant stirring at room temperature until dissolved. After centrifugation at 17,000 × g for 15 min, the solution was rapidly diluted using buffer containing 20 mm NaPO4, 50 mm NaCl, pH 7.5, 0.01% β-DDM to initiate refolding. The sample was further centrifuged to remove insoluble material and nickel-affinity chromatography was performed to isolate His10-tagged Sub.
Sub was spin-labeled through two additions of 10-fold molar excess of the MTSSL spin label for 1 h at room temperature (first addition) followed by overnight (second addition) storage on ice. For bimane labeling, purified sub was incubated with 10-fold molar excess of monobromobimane (mBBr; Toronto Research Chemicals) for overnight incubation on ice. Unreacted labels were removed by SEC on a Superdex 75 column (GE Healthcare Life Sciences) using a buffer containing 20 mm NaPO4, 50 mm NaCl, pH 7.5, 0.01% β-DDM. Peak fractions were combined, and spin-labeled proteins were concentrated using an Amicon Ultra filter concentrator (3-kDa molecular-weight cutoff) for binding studies. The labeling efficiency for mBBr labeled sub was calculated from the absorption spectrum measured using a UV-visible dual-beam spectrophotometer using the following extinction coefficients: ε280 = 8940 liter cm−1 mol−1 for Sub and ε380 = 5000 liter cm−1 mol−1 for mBBr. The contribution from mBBr at 280 nm was subtracted prior to calculating Sub concentrations. The labeling efficiency for spin-labeled Sub was calculated by comparing the total protein in molar concentration from the absorption spectrum at 280 nm and from the spin concentration using CW-EPR.
ATPase assay
The ATPase activity of PCAT1 mutants was determined by a calorimetric phosphate assay as previously described (20). For the ATPase assay, purified PCAT1 in mixed micelles of ∼4.5 μm were incubated for 20 min at 37 °C with increasing concentrations of ATP (three repeats for each assay). The reaction was stopped by adding 1 volume of 10% SDS, and the color was developed using 2 volumes of 1:1 solution of ammonium molybdate (2% in 1 m HCl) and ascorbic acid (12% in 1 m HCl). The absorbance at 850 nm was measured on a BioTek Synergy H4 microplate reader. The amount of phosphate released was determined by comparing with Pi standards.
CW-EPR spectroscopy
EPR samples were prepared in a total volume of 25 μl containing 100 μm of PCAT1, 10 μm Sub, 10 mm nucleotides, 10 mm MgSO4 which was loaded into a glass capillary. Spectra were recorded on a Bruker EMX spectrometer (X-band, 9.5 GHz) with a microwave power of 10 milliwatt and a modulation amplitude of 1.6 Gauss. PCAT1 and the Sub reaction mix were allowed to incubate for 15 min at room temperature before collection of EPR spectra.
Fluorescence spectroscopy
Fluorescence measurements were carried out at 37 °C using a PTI fluorometer with the excitation set to 380 nm. Binding isotherms were determined by incubating samples containing 0.1 μm of bimane-labeled Sub with varying concentrations of PCAT1 at 37 °C for 5 min. The resulting fluorescence emission spectra were recorded over a 425–550-nm range. The slit width was 1.0 mm, and the integration time was 1.0 s.
To obtain binding isotherms, fluorescence emission spectra were measured in the presence of a constant concentration of bimane-labeled Sub and increasing concentrations of PCAT1 variants. The intensity at 475 nm was then plotted as a function of PCAT1 concentration. The resulting binding curves were fit to a single binding mode using Levenberg−Marquart nonlinear least squares method in Origin 7.5 (OriginLab Corporation, Northampton, MA) software. The number of binding sites was set to 1, and the KD was allowed to float.
To determine the effects of nucleotides on binding, the reaction mixture contained 16 μm PCAT1, 0.1 μm bimane-labeled Sub, 100 mm NaCl, 50 mm Tris, 10% glycerol, 2 mm MgSO4, 10 mm nucleotides (ADP/ATPγS), and 0.05% β-DDM. The spectrofluorometer was set similarly to the binding experiment (as described above), and the samples were incubated at 37 °C for 5 min.
Data availability
The data are available upon request from the corresponding author (Hassane.mchaourab@vanderbilt.edu).
Acknowledgments
We thank Dr. Kevin Jagessar for help with the final figures and Dr. Reza Dastvan for helpful discussions.
Author contributions—S. R. and H. S. M. data curation; S. R. formal analysis; S. R. investigation; S. R. and H. S. M. writing-review and editing; H. S. M. conceptualization; H. S. M. supervision; H. S. M. funding acquisition; H. S. M. methodology; H. S. M. writing-original draft; H. S. M. project administration.
Funding and additional information—This work was supported by Grant R01-GM128087 from the NIGMS, National institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PCAT
- protease-containing ABC transporter
- TMD
- transmembrane domain
- NBD
- nucleotide-binding domain
- SEC
- size-exclusion chromatography
- β-DDM
- n-dodecyl β-d-maltoside
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- MTSSL
- (1-oxyl-2,2,5,5-tetramethylpyrrolinyl-3-methyl)-methanethiosulfonate
- CW
- continuous wave
- Pgp
- P-glycoprotein
- Ni-NTA
- nickel–nitrilotriacetic acid
- mBBr
- monobromobimane
- PEP
- peptidase domain
References
- 1. Kanonenberg K., Schwarz C. K., and Schmitt L. (2013) Type I secretion systems: a story of appendices. Res. Microbiol. 164, 596–604 10.1016/j.resmic.2013.03.011 [DOI] [PubMed] [Google Scholar]
- 2. Lenders M. H., Reimann S., Smits S. H., and Schmitt L. (2013) Molecular insights into type I secretion systems. Biol. Chem. 394, 1371–1384 10.1515/hsz-2013-0171 [DOI] [PubMed] [Google Scholar]
- 3. Thomas S., Holland I. B., and Schmitt L. (2014) The type 1 secretion pathway: the hemolysin system and beyond. Biochim. Biophys. Acta 1843, 1629–1641 10.1016/j.bbamcr.2013.09.017 [DOI] [PubMed] [Google Scholar]
- 4. Gebhard S. (2012) ABC transporters of antimicrobial peptides in Firmicutes bacteria: phylogeny, function and regulation. Mol. Microbiol. 86, 1295–1317 10.1111/mmi.12078 [DOI] [PubMed] [Google Scholar]
- 5. Gebhard S., and Mascher T. (2011) Antimicrobial peptide sensing and detoxification modules: unravelling the regulatory circuitry of Staphylococcus aureus. Mol. Microbiol. 81, 581–587 10.1111/j.1365-2958.2011.07747.x [DOI] [PubMed] [Google Scholar]
- 6. Havarstein L. S., Diep D. B., and Nes I. F. (1995) A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol. Microbiol. 16, 229–240 10.1111/j.1365-2958.1995.tb02295.x [DOI] [PubMed] [Google Scholar]
- 7. Hiron A., Falord M., Valle J., Débarbouillé M., and Msadek T. (2011) Bacitracin and nisin resistance in Staphylococcus aureus: a novel pathway involving the BraS/BraR two-component system (SA2417/SA2418) and both the BraD/BraE and VraD/VraE ABC transporters. Mol. Microbiol. 81, 602–622 10.1111/j.1365-2958.2011.07735.x [DOI] [PubMed] [Google Scholar]
- 8. Lin D. Y., Huang S., and Chen J. (2015) Crystal structures of a polypeptide processing and secretion transporter. Nature 523, 425–430 10.1038/nature14623 [DOI] [PubMed] [Google Scholar]
- 9. Morgan J. L., Acheson J. F., and Zimmer J. (2017) Structure of a type-1 secretion system ABC transporter. Structure 25, 522–529 10.1016/j.str.2017.01.010 [DOI] [PubMed] [Google Scholar]
- 10. Choudhury H. G., Tong Z., Mathavan I., Li Y., Iwata S., Zirah S., Rebuffat S., van Veen H. W., and Beis K. (2014) Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. U.S.A. 111, 9145–9150 10.1073/pnas.1320506111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oldham M. L., and Chen J. (2011) Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 15152–15156 10.1073/pnas.1108858108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kieuvongngam V., Olinares P. D. B., Palillo A., Oldham M. L., Chait B. T., and Chen J. (2020) Structural basis of substrate recognition by a polypeptide processing and secretion transporter. eLife 9, e51492 10.7554/eLife.51492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alam A., Kowal J., Broude E., Roninson I., and Locher K. P. (2019) Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 363, 753–756 10.1126/science.aav7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mansoor S. E., McHaourab H. S., Farrens D. L., Mansoor S. E., McHaourab H. S., and Farrens D. L. (1999) Determination of protein secondary structure and solvent accessibility using site-directed fluorescence labeling. Studies of T4 lysozyme using the fluorescent probe monobromobimane. Biochemistry 38, 16383–16393 10.1021/bi991331v [DOI] [PubMed] [Google Scholar]
- 15. Mansoor S. E., McHaourab H. S., Farrens D. L., Mansoor S. E., McHaourab H. S., and Farrens D. L. (2002) Mapping proximity within proteins using fluorescence spectroscopy: a study of T4 lysozyme showing that tryptophan residues quench bimane fluorescence. Biochemistry 41, 2475–2484 10.1021/bi011198i [DOI] [PubMed] [Google Scholar]
- 16. Hubbell W. L., McHaourab H. S., Altenbach C., Lietzow M. A., Hubbell W. L., McHaourab H. S., Altenbach C., and Lietzow M. A. (1996) Watching proteins move using site-directed spin labeling. Structure 4, 779–783 10.1016/S0969-2126(96)00085-8 [DOI] [PubMed] [Google Scholar]
- 17. Dastvan R., Mishra S., Peskova Y. B., Nakamoto R. K., and McHaourab H. S. (2019) Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 364, 689–692 10.1126/science.aav9406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mchaourab H. S., Lietzow M. A., Hideg K., and Hubbell W. L. (1996) Motion of spin-labeled side chains in T4 lysozyme: correlation with protein structure and dynamics. Biochemistry 35, 7692–7704 10.1021/bi960482k [DOI] [PubMed] [Google Scholar]
- 19. Amadi S. T., Koteiche H. A., Mishra S., and McHaourab H. S. (2010) Structure, dynamics, and substrate-induced conformational changes of the multidrug transporter EmrE in liposomes. J. Biol. Chem. 285, 26710–26718 10.1074/jbc.M110.132621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smriti, Zou P., and Mchaourab H. S. (2009) Mapping daunorubicin-binding sites in the ATP-binding cassette transporter MsbA using site-specific quenching by spin labels. J. Biol. Chem. 284, 13904–13913 10.1074/jbc.M900837200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data are available upon request from the corresponding author (Hassane.mchaourab@vanderbilt.edu).