

Abstract
The mitochondrial DNA (mtDNA) depletion syndrome comprises a clinically heterogeneous group of diseases characterized by reductions of the mtDNA abundance, without associated point mutations or rearrangements. We have developed the first in vitro model to study of mtDNA depletion due to reduced mitochondrial thymidine kinase 2 gene (TK2) expression in order to understand the molecular mechanisms involved in mtDNA depletion syndrome due to TK2 mutations. Small interfering RNA targeting TK2 mRNA was used to decrease TK2 expression in Ost TK1− cells, a cell line devoid of endogenous thymidine kinase 1 (TK1). Stable TK2-deficient cell lines showed a reduction of TK2 levels close to 80%. In quiescent conditions, TK2-deficient cells showed severe mtDNA depletion, also close to 80% the control levels. However, TK2-deficient clones showed increased cytochrome c oxidase activity, higher cytochrome c oxidase subunit I transcript levels and higher subunit II protein expression respect to control cells. No alterations of the deoxynucleotide pools were found, whereas a reduction in the expression of genes involved in nucleoside/nucleotide homeostasis (human equilibrative nucleoside transporter 1, thymidine phosphorylase) and mtDNA maintenance (DNA-polymerase γ, mitochondrial transcription factor A) was observed. Our findings highlight the importance of cellular compensatory mechanisms that enhance the expression of respiratory components to ensure respiratory activity despite profound depletion in mtDNA levels.
Keywords: Mitochondrial DNA, siRNA, Thymidine kinase 2, Mitochondrial depletion syndrome, Respiratory chain
1. Introduction
The mitochondrial DNA (mtDNA) depletion syndrome (MDS) comprises a clinically heterogeneous group of diseases characterized by a reduction of the mtDNA copy number without point mutations or rearrangements [1]. mtDNA depletion is transmitted as an autosomal recessive trait and may affect either a specific tissue (most commonly muscle or liver) or multiple organs, including heart, brain and kidney. Myopathic MDS has been associated with mutations in thymidine kinase 2 (TK2) [2] and RRM2B genes [3]. TK2 encodes the mitochondrial thymidine kinase and RRM2B encodes the cytosolic p53-inducible ribonucleotide reductase small subunit (P53R2). The encephalomyopathic form of MDS has been associated with mutations in the gene encoding the β-subunit of the adenosine diphosphate-forming succinyl coenzyme A synthase ligase (SUCLA2) [4]. Hepatocerebral MDS has been linked with mutations in six genes: DGUOK, the gene encoding the mitochondrial deoxyguanosine kinase (dGK) [5]; POLG for the mtDNA polymerase γ [6,7]; MPV17 a small mitochondrial membrane protein of unknown function [8]; PEO1 for the mitochondrial helicase/primase Twinkle [9]; and SUCLG1 for the alpha subunit of succinyl-CoA synthase [10].
Unlike nuclear DNA (nDNA), synthesis of mtDNA is not cell cycle-regulated [11]; hence a constant supply of deoxyribonucleoside triphosphates (dNTPs) is crucial for the maintenance of mitochondrial integrity. TK2 and dGK are involved in the salvage pathways of mitochondrial pyrimidine and purine deoxynucleotides, respectively, which constitute the major source of mtDNA precursors in post-mitotic tissues, while p53R2 is also active in resting cells and contributes to de novo dNTP synthesis [12].
It is still unclear why the phenotypes associated with TK2 and DGUOK mutations are tissue-specific. For a better understanding of the pathogenesis of TK2 mutations, two TK2-deficient mouse models have been generated to assess pathomechanisms of this disease [13,14]. Akman et al. generated a mouse harboring an H126N Tk2 mutation corresponding to the pathogenic human H121N TK2 mutation. Homozygous Tk2 knockin mutant mice manifested a severe and rapid progressive encephalomyelopathy with severe TK2 deficiency, imbalances of mitochondrial dNTP pools, and mtDNA depletion in multiple tissues [13]. Zhou et al. generated a Tk2 knockout mice with several organs affected, including skeletal muscle, heart, liver, spleen, brain, and brown adipose tissue indicating that complete abrogation of Tk2 without any residual enzymatic activity produces a more severe phenotype in mice compared to human patients [14]. It is unclear whether different phenotypes observed in patients with different TK2 mutations, from severe defects causing early childhood mortality to survival into adulthood, correspond to distinct extents of TK2 activity impairment and/or mtDNA depletion.
Although animal models are very useful tools to assess mechanisms of pathogenesis and potential therapies, in vitro cellular models are essential to establish the molecular mechanisms linking mtDNA depletion with altered cellular respiratory activity and overall cellular function. Therefore, we developed the first cellular system of reduced mtDNA levels due to impaired TK2 expression in order to understand the molecular mechanisms involved in this form of the MDS. Small interfering RNA (siRNA) targeting TK2 was used to decrease TK2 expression in Ost TK1− cells, a cell line devoid of endogenous TK1. We generated stable TK2-deficient cell lines and we achieved severe mtDNA depletion in cells as a consequence of impaired TK2 expression. We studied the effects of down-regulation of TK2 and subsequent mtDNA depletion on the expression of respiratory chain components and activity, as well as on the expression of genes involved in the maintenance of mtDNA and nucleoside/nucleotide metabolism.
2. Materials and methods
2.1. Cell line and cell growth
The established human tumor cell line Ost TK1− was kindly provided by Dr. Vera Bianchi (Department of Biology, University of Padova, Italy). Cells were grown in DMEM with 4.5 g/l glucose, 10% dialyzed fetal bovine serum (dFBS), 1% l-glutamine, 1% non-essential amino acids, 100 U/ml antibiotic/antimycotic, 50 μg/ml uridine. To obtain quiescent cells, we transferred confluent cells to 0.1% dFBS medium to enhance quiescence. Flow cytometry was used in order to determine cell-cycle distribution (FACS Calibur. BD Biosciences. Franklin Lakes, NJ, USA).
2.2. RNAi experiments
Ost TK1− cells were stably transfected using Lipofectamine (Invitrogen, Paisley, UK) with the pSilencer 4.1-CMV hygro expression vector (Ambion, Austin, TX, USA) containing the validated TK2 double-stranded oligonucleotide (5′-GGU CUA UGG CUC AUG UCU G-3′). Cells were also transfected with the same vector containing an irrelevant sequence to be used as an RNAi control. Positive clones were selected in the presence of hygromycin (450 μg/ml). To maintain new generated cell lines, hygromycin was added permanently to the culture medium at the same concentration. Selected clones were screened for down-regulation of TK2 mRNA and protein by real-time RT-PCR and immunoblotting, respectively.
2.3. mRNA expression analyses
Total RNA was isolated using a single-column commercial kit (RNeasy Mini Kit, Qiagen, Hilden, Germany) and quantified spectrophotometrically (NanoDrop, Thermo Scientific, Waltham, MA, USA). RNA (0.5 μg) was transcribed into cDNA using MultiScribe reverse transcriptase and random-hexamer primers (TaqMan Reverse Transcription Reagents; Applied Biosystems, Foster City, CA, USA). For quantitative mRNA expression analyses, TaqMan reverse transcriptase-polymerase chain reaction (RT-PCR) was performed on the ABI PRISM 7500HT sequence detection system (Applied Biosystems). Reactions were performed in a final volume of 25 μl using TaqMan Universal PCR Master Mix, No-AmpErase UNG reagent, and the following specific primer pair/probe sets (TaqMan Gene Expression Assays; Applied Biosystems): TK2, Hs00177950_m1; DCK, Hs00176127_m1; DGUOK, Hs00361549_m1; NME4, Hs00359037_m1; NME6, Hs00195083_ m1; TYMS, Hs00426586_m1; TFB1M, Hs00274971_m1; TFB2M, Hs00254651_m1; SLC29A1, Hs00191940_m1); POLG, Hs001-60298_m1; TFAM, Hs00273372_s1; TYMP, Hs00157317_m1; MT-COI, Hs02596864_g1; RRM1, Hs00168784_ m1; RRM2, Hs00357247_g1; RRM2B, Hs00153085_m1; and PPIA, Hs99999904_m1, as reference control.
2.4. Assessment of relative mtDNA content
Total DNA was isolated from cultured cells using a phenol/chloroform extraction method and quantified spectrophotometrically (NanoDrop, Thermo Scientific). mtDNA abundance was determined by quantitative real-time PCR assessment of the 12S rRNA gene (labeled with FAM fluorochrome and the forward primer, 5′-CCA CGG GAA ACA GCA GTG ATT-3′ and reverse primer, 5′CTA TTG ACT TGG GTT AAT CGT GTG A-3′) relative to nuclear DNA, determined by quantification of the nuclear single copy gene RNaseP using the PDARs RNaseP kit and a VIC labeled probe (Applied Biosystems).
2.5. Immunodetection
Protein extracts were prepared by adding protease inhibitor cocktail (Sigma–Aldrich, St. Louis, MO, USA) to the lysis buffer (0.1 M Tris–HCl, pH 7.6, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100), and quantified by Lowry colorimetric assay (Bio-Rad, Hercules, CA, USA). Cell lysates containing 20–30 μg of protein were resolved by SDS-polyacrylamide gel electrophoresis on 15% gels and transferred to PVDF membranes (Westran S., Whatman International Ltd., Kent, UK). Membranes were incubated with blocking buffer (5% BSA/0.01% Tween-20/PBS). Polyclonal TK2 anti-serum [15] and mouse monoclonal anti-cytochrome c oxidase subunit II (Molecular Probes, Invitrogen) were used at 1:3000 and 1:1000 dilution, respectively, in blocking buffer. Primary antibodies were immunodetected using polyclonal anti-mouse or anti-rabbit secondary horseradish peroxidase-conjugated antibodies (Dako, Glostrup, Denmark). Enhanced chemiluminescence reagents were used to analyze the immunoreactive signals (BD Bioscience). Blots were re-probed with mouse monoclonal antibody against voltage dependent anion channel (VDAC) (Calbiochem, Merck KGaA, Darmstadt, Germany) to normalize the amount of mitochondrial protein loaded. Immunoreactive signals were analyzed densitometrically using Quantity One v4.0.3 software (Bio-Rad).
2.6. Mitochondrial respiratory chain activity
Cells where homogenized with SETH buffer (250 mM Sucrose, 2 mM EDTA, 10 mM Tris–HCl, 50 U/ml Heparin, pH 7.6) and sonicated. Complex IV (cytochrome c oxidase) activity was measured using reduced cytochrome c as a substrate and cytochrome c oxidation was monitored at 550 nm [16]. Complex I + III activity was determined as the velocity of reduction of cytochrome c by NADH cytochrome c reductase in the presence of NADH and rotenone. Changes in absorbance at 550 nm were measured with and without rotenone [17]. Citrate synthase activity was determined as already reported [18] and used as a marker of mitochondrial mass. Protein content was determined using the Bradford assay (Bio-Rad).
2.7. Measurement of mitochondrial and cytosolic dNTPs
This analysis was performed following the enzymatic assay described by Pontarin et al. [19]. Briefly, mitochondrial and cytosolic fractions where separated by differential centrifugation and the assay was done using the klenow fragment of the DNA polymerase, the appropriate oligonucleotides as template, and radio-labeled nucleotides as substrate for the reaction.
3. Results
3.1. Silencing of TK2 mRNA in Ost TK1− cells caused reduction in TK2 protein levels
Two TK2-interfered clones (TK2-29 and TK2-33) and two transfection control clones (C-5 and C-11) were selected based on TK2 mRNA expression levels relative to parental untransfected Ost TK1− cells (Fig. 1 A). A strong reduction of TK2 transcript was observed in TK2-29 and TK2-33 cells (70–80% reduction). We assessed whether the abundance of TK2 protein corresponded to mRNA levels observed in these clones and results demonstrated a dramatic reduction of TK2 protein levels in the two interfered clones relative to controls (Fig. 1 B).
Fig. 1.
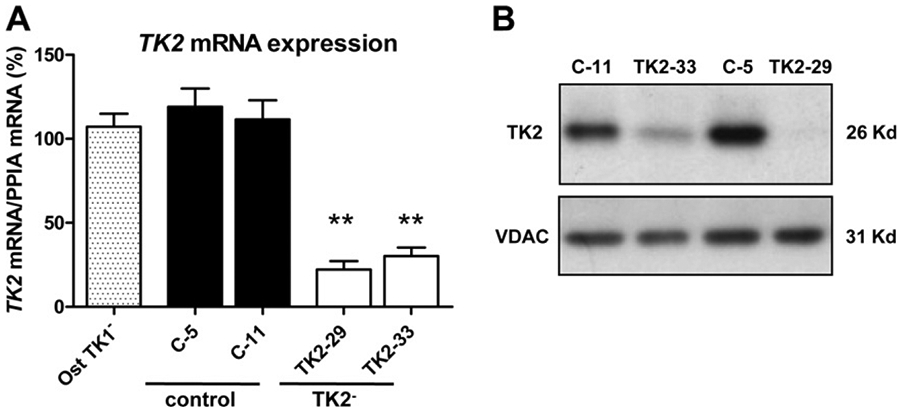
Analysis of TK2 expression in TK2-interfered Ost TK1− cells using quantitative real-time PCR and immunoblotting. (A) TK2 relative mRNA expression in Ost TK1− parental cell line (Ost TK1−), 2 control cell lines (C-5, C-11) and 2 TK2-interfered cell lines (TK2-29, TK2-33). Bars are means ± SEM of 4–5 independent cultures and are expressed as percentage values. **P < 0.001. (B) Representative Western blot showing the reduction of TK2 protein in TK2-deficient clones. VDAC immunoblot signal showing similar mitochondrial protein load.
3.2. TK2. down-regulation caused mtDNA depletion in Ost TK1− cells
We subsequently studied the amount of mtDNA in TK2− and control cells. Although Ost TK1− cells had lost contact inhibition, flow cytometry analyses revealed that less than 47% of confluent cells in 10% dFBS were in G1/G0, while the majority of confluent cells cultured for 10 days in 0.1% dFBS were arrested (87%) and these were considered optimal conditions of quiescent cell status for our study. mtDNA levels were measured in TK2− and control cells and representative results of C-5 and TK2-33 cells are depicted in Fig. 2. mtDNA levels in TK2-33 cells were similar to C-5 cells when cultured in 10% dFBS at confluence, 5 days and 10 days post-confluence. Significant depletion of mtDNA was observed (around 50%) at days 15 and 20 (Fig. 2 A). These results indicate that mtDNA depletion takes place in TK2-deficient Ost TK1− cells even at high concentration of dFBS. However, when cultured in 0.1% dFBS, a more rapid and marked mtDNA reduction was achieved in quiescent TK2-33 cells (Fig. 2 B). Similar results were found in C-11 and TK2-29 cell clones (data not shown).
Fig. 2.
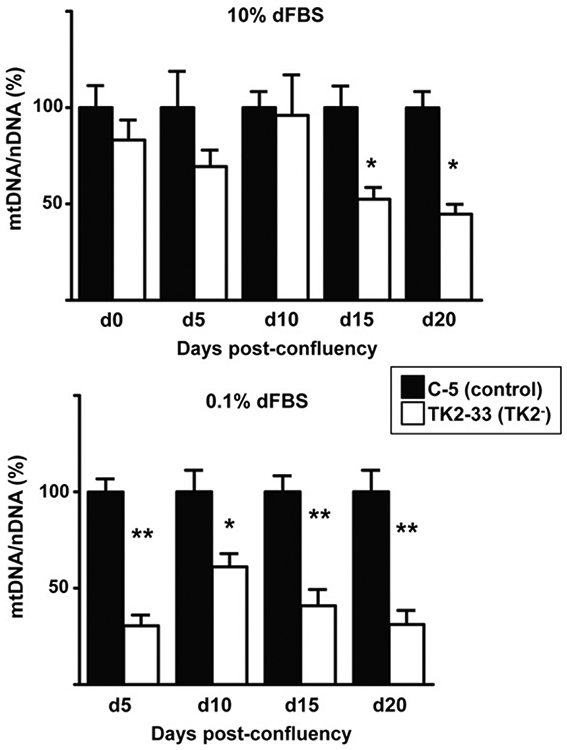
Mitochondrial DNA levels in TK2-interfered cells. Control (C-5) and TK2-interfered (TK2-33) cell clones were grown in 10% dFBS (top) until reaching confluence (d0) and then either shifted to 0.1% dFBS (bottom) or maintained in 10% dFBS (top). Cells in both conditions were harvested at days 5 (d5), 10 (d10), 15 (d15) and 20 (d20) after confluence. Bars are means ± SEM data from 4 to 5 independent cultures and are expressed as percentage values. *P < 0.05; **P < 0.001.
3.3. Analysis of mitochondrial respiratory chain in TK2-deficient Ost TK1− cells
To further characterize TK2-deficient Ost TK1− cells, we measured activities of respiratory chain complexes as well as levels of the mtDNA-encoded transcript MT-COI and cytochrome c oxidase (COX) subunit II protein. Fig. 3A summarizes the observed COX activities in TK2-deficient and control cells at confluence and 10 days after culture in the presence of 0.1% or 10% dFBS. Upon reaching confluence, both TK2-29 and TK2-33 cells showed COX activities higher than control cells, although the increase was more marked in the TK2-33 cell clone. COX activity increased in quiescent mtDNA-depleted TK2-deficient cells maintained in 0.1% dFBS for 10 days, but was very similar in cells cultured in 10% dFBS for the same period of time. No changes were observed in complex I + III respiratory activity (data not shown). To get insight on how COX activity is increased in quiescent TK2-deficient cells despite mtDNA depletion, we measured the expression of the mtDNA-encoded transcript MT-COI. TK2-29 and TK2-33 showed higher levels of MT-COI mRNA than control cells 10 days after confluence in 0.1% dFBS (Fig. 3 B). Similarly, these cell clones showed higher levels of the mtDNA-encoded COXII protein respect to controls, especially when cultured in 0.1% dFBS (Fig. 3 C).
Fig. 3.
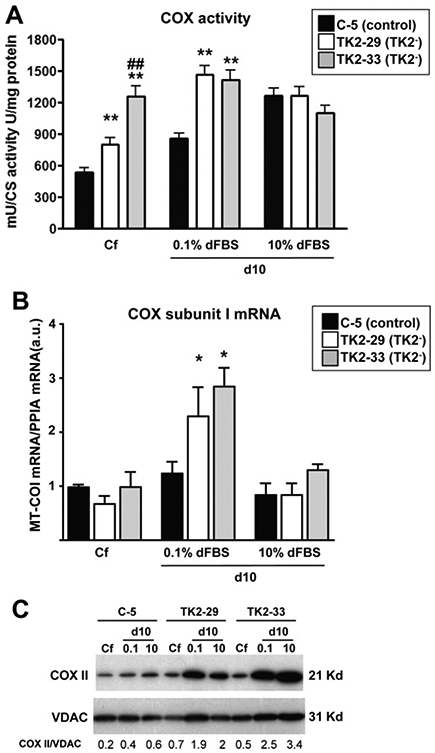
Activity and expression of cytochrome c oxidase components in TK2-interfered cells. (A) cytochrome c oxidase (COX) activity in control and TK2-deficient cells at confluence (Cf) and 10 days post-confluence in 0.1% and 10% dFBS. Data are expressed as milliunits of COX activity per units of citrate synthase (CS) activity per milligram of total protein. (B) cytochrome c oxidase subunit I mRNA levels in control and TK2-deficient cells at distinct culture conditions. Data are expressed as arbitrary units. Bars are means ± SEM from 5 to 6 independent cultures. *P < 0.05, **P < 0.001; TK2− versus control; ##P < 0.001; TK2-29 versus TK2-33. (C) Representative immunoblot of COXII protein content in TK2-deficient and control cells. VDAC indicates mitochondrial protein leading control, and numbers indicate COII/VDAC ratios after densitometric quantification.
3.4. TK2-deficient and control cells showed no differences in mitochondrial and cytosolic dNTP pools
Steady-state levels of mitochondrial and cytosolic dNTP pools were determined in quiescent (10 days post-confluence, 0.1% dFBS) C-5 and TK2-33 cells. No differences were found in the levels of the four dNTPs between TK2-deficient and control cells, either in the mitochondrial or in the cytosolic compartment (Fig. S1).
3.5. Expression of genes involved in mtDNA transcription and replication in TK2-deficient cells
We studied how TK2 deficiency could be affecting the expression of genes encoding proteins related to mtDNA transcription and replication machinery, as well as other enzymes involved in maintaining the homeostasis of the mitochondrial dNTP pool. No significant changes were found in transcript levels for deoxycytidine (DCK) and DGUOK, nucleoside diphosphate kinases nm23-H4 and nm23-H6, thymidylate synthetase (TYMS), ribonucleotide reductase subunits R1 (RRM1), R2 (RRM2), and P53R2 (RRM2B), and mitochondrial transcription factors TFB1M and TFB2M in TK2-deficient versus control cells. However, we found a significant decrease in the expression of SLC29A1 (ENT1) and POLG genes in quiescent TK2-33 cells compared to controls (Fig. 4, top). TFAM and thymidine phosphorylase gene TYMP were also decreased in TK2-33 cells, even at confluence stage (Fig. 4, bottom).
Fig. 4.
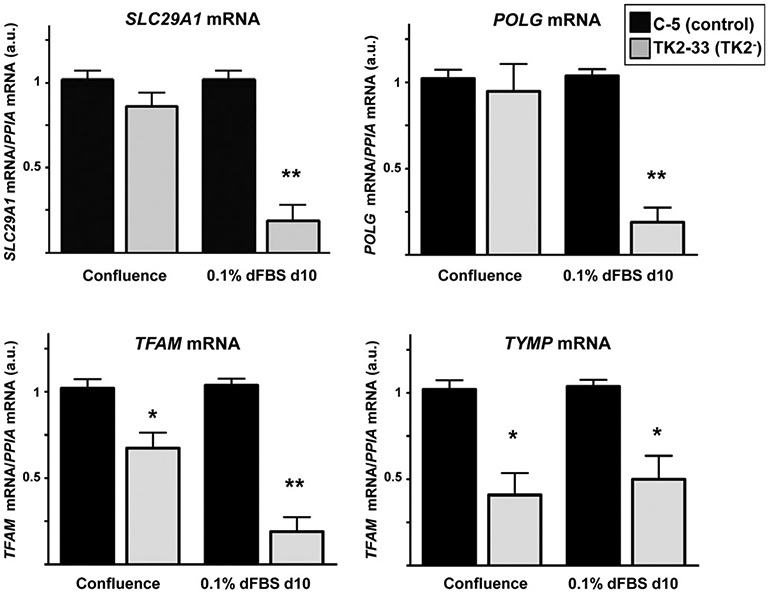
Expression of SLC29A1, POLG, TFAM, and TYMP transcripts in TK2-interfered cells. Bars are means ± SEM from 4 to 5 independent cell cultures. Data are expressed as fold-change versus controls (arbitrary units) for each culture condition. *P < 0.05; **P < 0.001.
4. Discussion
In this study, we used RNA interference to down-regulate the expression of TK2 in Ost TK1-deficient cells, thus generating stable OstTK1−/TK2− cell lines that represent the first in vitro model of mtDNA depletion due to TK2 deficiency. We have characterized these cell lines by analyzing the activities of respiratory chain complexes, the mitochondrial and cytosolic dNTP pools, and how TK2-deficiency alters genes involved in the maintenance and replication of mtDNA. Quiescent TK2-deficient clones showed mtDNA depletion, increased COX activity and increased expression of the catalytic subunits I and II of COX, no alterations of the dNTP pools, and marked reductions in SLC29A1, POLG, TFAM, and TYMP transcript levels. Because cultured fibroblasts derived from patients harboring TK2 mutations do not show mtDNA depletion [15,20,21] our cellular model may represent a relevant system in vitro in order to analyze pathophysiological events taking place in patients.
mtDNA depletion is expected to lead to impaired synthesis of respiratory chain complexes [1]. However, in our TK2-deficient cells, we observed an increase not only in COX activity, but also in the expression of COXI transcript and COXII protein. These results are similar to what we observed in muscle from a patient with severe mtDNA depletion due to TK2 mutations but normal activities of the respiratory chain subunits [22], that we attributed to compensatory mechanisms that may retard progression of the disease. The present data highlight the importance of mtDNA expression regulatory processes to enhance mtDNA-encoded protein synthesis in conditions of lowered mtDNA amounts as a mechanism to ensure cellular function.
The absence of imbalanced nucleotide pools in TK2-deficient cells exhibiting mtDNA depletion may be viewed as a surprising finding but, in fact, it is consistent with previous observations. In non-proliferating cells, an important function of the R1/p53R2 ribonucleotide reductase is to provide deoxynucleotides for mtDNA replication [12], and the small dTTP pool depends on both R1/p53R2 and TK2 activities [23]. In a study exploring the role of R1/p53R2 and TK2 in nucleoside metabolism, treatment with hydroxyurea, a known ribonucleotide reductase inhibitor, evidenced a rapid drop of the cellular dTTP pool in TK2- cells whereas, surprisingly, TK2 down-regulation alone did not affect the kinetics of dNTP changes [23]. In our TK2-deficient cells lines, expression levels of genes for ribonucleotide reductase subunits R1, R2, and p53R2 were unaltered respect to controls. It is possible that changes in dNTP pools and induction of mtDNA depletion were early events in confluent TK2-deficient cells making that cytosolic and mitochondrial dNTPs re-equilibrate to baseline levels when we performed the measures, whereas synthesis of the mtDNA could not recover.
In TK2-deficient cells we observed marked reductions in SLC29A1, POLG, TFAM, and TYMP transcript levels. SLC29A1 gene product ENT1 is an equilibrative nucleoside transporter ubiquitously expressed and functionally present in mitochondria [24,25]. ENT1 abundance in cultured human cancer cells appears to be coordinated with cell cycle; levels of the transporter approximately double between G1 and G2-M phases [26]. In our study, SLC29A1 was down-regulated in quiescent TK2-deficient clones, but not in control cells at the same culture conditions (0.1% dFBS), suggesting that other factors rather than cell cycle status affect expression of this transporter. It has been also suggested that transporter synthesis and/or cell surface expression are regulated in response to cellular deoxynucleotide levels via an unknown mechanism [26]. Whether ENT1 plays a central role preventing imbalance of dNTP pools in TK2-deficient clones remains to be established. We have recently described that differential expression patterns of ENT1 in fibroblasts derived from patients harboring TK2 or DGUOK mutations contribute to the mtDNA depletion phenotype observed in dGK-deficient, but not in TK2-deficient fibroblasts [15]. Furthermore, we have also shown that down-regulation of ENT1, but not TK1, could induce mtDNA depletion in quiescent TK2-deficient fibroblasts, suggesting an important role for this transporter in patients with TK2-deficiency [15].
In our mtDNA-depleted cells, decreases in TFAM and POLG transcripts are probably secondary responses to the reduced requirements for these two factors. In addition to its essential function in transcription and replication, TFAM also serves a role in packaging mtDNA [27]. Thus, TFAM is considered one of the primary factors controlling the mtDNA copy number in mammalian cells [28]. On the other hand, to the light of the present results, neither TFAM nor TFB1M or TFB2M appear as likely candidates for mediating, via compensatory up-regulation, the enhanced expression of mtDNA-encoded transcripts observed in the mtDNA-depleted cells.
Thymidine phosphorylase (TP) degrades thymidine in the cytoplasm, thus limiting the availability of thymidine for being phosphorylated by TK2 in the mitochondria. We observed a decrease in the expression of TP in both confluent and quiescent TK2− cells. Consequently, dTMP synthesized by R1/P53R2 in the cytoplasm [23] would be degraded by the appropriate cytosolic nucleotidase to form thymidine, which would not be degraded to thymine due to the lack of TP. Down-regulation of ENT1 may be preventing the entry of the accumulated cytosolic thymidine into mitochondria inducing the mtDNA depletion observed in these cells. It is worth noting that down-regulation of TP gene expression takes place is TK2-deficient cells before mtDNA levels are reduced, thus indicating that the reduction in TK2 in mitochondria elicits cellular perturbations capable of altering nuclear gene expression before the appearance of mtDNA depletion.
In summary, we have shown that impairment of TK2 expression leads to profound mtDNA depletion but fully preserved or even enhanced mitochondrial respiratory activity. This is associated with a pattern of secondary events including altered expression of several genes involved in mtDNA maintenance and nucleoside/nucleotide metabolism. Thus, it appears that, in addition to malfunction of TK2, other events would be necessary to induce mtDNA depletion-associated impairment in respiratory activity and cellular function in TK2-deficient patients. The cellular mechanisms capable of compensating severe mtDNA depletion, evidenced in the present study, constitute potential targets for therapeutic strategies to alleviate disease symptoms in patients with MDS, and, specifically in TK2 deficiency-associated encephalomyopathies.
Supplementary Material
Acknowledgments
We thank Ramon Martí, Isabel Huber and Marçal Pastor-Anglada for critical review of the manuscript, Vera Bianchi for Ost TK1- cells, and David Lligé for technical support. This work was supported by Fondo de Investigación Sanitaria (FIS 04/0415); CIBER Obesidad y Nutrición, Spain. Fundismun Foundation, and Muscular Dystrophy Association, USA.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbrc.2011.03.018.
References
- [1].Moraes CT, Shanske S, Tritschler HJ, et al. , MtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases, Am. J. Hum. Genet 48 (1991) 492–501. [PMC free article] [PubMed] [Google Scholar]
- [2].Saada A, Shaag A, Mandel H, et al. , Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy, Nat. Genet 29 (2001) 342–344. [DOI] [PubMed] [Google Scholar]
- [3].Bourdon A, Minai L, Serre V, et al. , Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion, Nat. Genet 39 (2007) 776–780. [DOI] [PubMed] [Google Scholar]
- [4].Elpeleg O, Miller C, Hershkovitz E, et al. , Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion, Am. J. Hum. Genet 76 (2005) 1081–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mandel H, Szargel R, Labay V, et al. , The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA, Nat. Genet 29 (2001) 337–341. [DOI] [PubMed] [Google Scholar]
- [6].Ferrari G, Lamantea E, Donati A, et al. , Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gammaA, Brain 128 (2005) 723–731. [DOI] [PubMed] [Google Scholar]
- [7].Naviaux RK, Nguyen KV, POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion, Ann. Neurol 55 (2004) 706–712. [DOI] [PubMed] [Google Scholar]
- [8].Spinazzola A, Viscomi C, Fernandez-Vizarra E, et al. , MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion, Nat. Genet 38 (2006) 570–575. [DOI] [PubMed] [Google Scholar]
- [9].Hakonen AH, Isohanni P, Paetau A, et al. , Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion, Brain 130 (2007) 3032–3040. [DOI] [PubMed] [Google Scholar]
- [10].Ostergaard E, Christensen E, Kristensen E, et al. , Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion, Am. J. Hum. Genet 81 (2007) 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bogenhagen D, Clayton DA, Thymidylate nucleotide supply for mitochondrial DNA synthesis in mouse L-cells. Effect of 5-fluorodeoxyuridine and methotrexate in thymidine kinase plus and thymidine kinase minus cells, J. Biol. Chem 251 (1976) 2938–2944. [PubMed] [Google Scholar]
- [12].Pontarin G, Ferraro P, Hakansson P, et al. , P53R2-dependent ribonucleotide reduction provides deoxyribonucleotides in quiescent human fibroblasts in the absence of induced DNA damage, J. Biol. Chem 282 (2007) 16820–16828. [DOI] [PubMed] [Google Scholar]
- [13].Akman HO, Dorado B, Lopez LC, et al. , Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance, Hum. Mol. Genet (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhou X, Solaroli N, Bjerke M, et al. , Progressive loss of mitochondrial DNA in thymidine kinase 2-deficient mice, Hum. Mol. Genet 17 (2008) 2329–2335. [DOI] [PubMed] [Google Scholar]
- [15].Villarroya J, de Bolos C, Meseguer A, et al. , Altered gene transcription profiles in fibroblasts harboring either TK2 or DGUOK mutations indicate compensatory mechanisms, Exp. Cell Res 315 (2009) 1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rustin P, Chretien D, Bourgeron T, et al. , Biochemical and molecular investigations in respiratory chain deficiencies, Clin. Chim. Acta 228 (1994) 35–51. [DOI] [PubMed] [Google Scholar]
- [17].Sottocasa GL, Kuylenstierna B, Ernster L, et al. , An electron-transport system associated with the outer membrane of liver mitochondria. A biochemical and morphological study, J. Cell Biol 32 (1967) 415–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Srere P, [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)], Methods Enzymol. 13 (1969) 3–11. [Google Scholar]
- [19].Pontarin G, Gallinaro L, Ferraro P, et al. , Origins of mitochondrial thymidine triphosphate: dynamic relations to cytosolic pools, Proc. Natl. Acad. Sci. USA 100 (2003) 12159–12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mariotti C, Uziel G, Carrara F, et al. , Early-onset encephalomyopathy associated with tissue-specific mitochondrial DNA depletion: a morphological, biochemical and molecular-genetic study, J. Neurol. 242 (1995) 547–556. [DOI] [PubMed] [Google Scholar]
- [21].Saada A, Ben-Shalom E, Zyslin R, et al. , Mitochondrial deoxyribonucleoside triphosphate pools in thymidine kinase 2 deficiency, Biochem. Biophys. Res. Commun 310 (2003) 963–966. [DOI] [PubMed] [Google Scholar]
- [22].Vila MR, Villarroya J, Garcia-Arumi E, et al. , Selective muscle fiber loss and molecular compensation in mitochondrial myopathy due to TK2 deficiency, J. Neurol. Sci 267 (2008) 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rampazzo C, Fabris S, Franzolin E, et al. , Mitochondrial thymidine kinase and the enzymatic network regulating thymidine triphosphate pools in cultured human cells, J. Biol. Chem 282 (2007) 34758–34769. [DOI] [PubMed] [Google Scholar]
- [24].Lai Y, Tse CM, Unadkat JD, Mitochondrial expression of the human equilibrative nucleoside transporter 1 (hENT1) results in enhanced mitochondrial toxicity of antiviral drugs, J. Biol. Chem 279 (2004) 4490–4497. [DOI] [PubMed] [Google Scholar]
- [25].Lee EW, Lai Y, Zhang H, et al. , Identification of the mitochondrial targeting signal of the human equilibrative nucleoside transporter 1 (hENT1): implications for interspecies differences in mitochondrial toxicity of fialuridine, J. Biol. Chem 281 (2006) 16700–16706. [DOI] [PubMed] [Google Scholar]
- [26].Pressacco J, Wiley JS, Jamieson GP, et al. , Modulation of the equilibrative nucleoside transporter by inhibitors of DNA synthesis, Br. J. Cancer 72 (1995) 939–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alam TI, Kanki T, Muta T, et al. , Human mitochondrial DNA is packaged with TFAM, Nucleic Acids Res. 31 (2003) 1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Larsson NG, Wang J, Wilhelmsson H, et al. , Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice, Nat. Genet 18 (1998) 231–236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.