

Abstract
Objective:
Thymidine kinase 2, encoded by the nuclear gene TK2, is required for mitochondrial DNA maintenance. Autosomal recessive TK2 mutations cause depletion and multiple deletions of mtDNA that manifest predominantly as a myopathy usually beginning in childhood and progressing relentlessly. We investigated the safety and efficacy of deoxynucleoside monophosphate and deoxynucleoside therapies.
Methods:
We administered deoxynucleoside monophosphates and deoxynucleoside to 16 TK2-deficient patients under a compassionate use program.
Results:
In 5 patients with early onset and severe disease, survival and motor functions were better than historically untreated patients. In 11 childhood and adult onset patients, clinical measures stabilized or improved. Three of 8 patients who were nonambulatory at baseline gained the ability to walk on therapy; 4 of 5 patients who required enteric nutrition were able to discontinue feeding tube use; and 1 of 9 patients who required mechanical ventilation became able to breathe independently. In motor functional scales, improvements were observed in the 6-minute walk test performance in 7 of 8 subjects, Egen Klassifikation in 2 of 3, and North Star Ambulatory Assessment in all 5 tested. Baseline elevated serum growth differentiation factor 15 levels decreased with treatment in all 7 patients tested. A side effect observed in 8 of the 16 patients was dose-dependent diarrhea, which did not require withdrawal of treatment. Among 12 other TK2 patients treated with deoxynucleoside, 2 adults developed elevated liver enzymes that normalized following discontinuation of therapy.
Interpretation:
This open-label study indicates favorable side effect profiles and clinical efficacy of deoxynucleoside monophosphate and deoxynucleoside therapies for TK2 deficiency.
Encoded by the nuclear gene TK2, thymidine kinase 2 (TK2) is the first enzyme in the deoxypyrimidine salvage pathway within mitochondria. TK2 phosphorylates the nucleosides deoxycytidine (dC) and deoxythymidine (dT) to generate deoxycytidine monophosphate (dCMP) and deoxythymidine monophosphate (dTMP). These pyrimidine deoxynucleoside monophosphates are subsequently converted to deoxynucleoside triphosphates required for mitochondrial DNA (mtDNA) replication and maintenance.1,2 Autosomal recessive TK2 mutations cause mtDNA depletion, multiple deletions, or both.3–5
Although the phenotypic spectrum of TK2 deficiency includes late onset cases of mild chronic progressive external ophthalmoplegia,3,6–8 the most frequent clinical presentations are infantile onset and childhood onset progressive limb and bulbar myopathy with restrictive lung disease.3,4,9–13 The infantile onset form manifests rapidly progressive myopathy weakness with motor regression and respiratory insufficiency occasionally accompanied by central nervous system involvement and virtually uniform early fatality with postonset survival of 1 year.4,5,10 The majority of patients with early onset before age 2 years also show severe and rapid progression. The less severe forms begin in childhood through adulthood and exhibit slower progression; however, bulbar, proximal limb, and respiratory weakness can be severe, causing inability to walk or breathe independently.3,4,6,12
We reported that, in the H126N knockin mouse model of TK2 deficiency, oral administration of the TK2 products dCMP and dTMP act as molecular bypass therapy and prolong median life span by 2- to 3-fold.14,15 However, we subsequently observed that after administration, dCMP and dTMP are rapidly catabolized to the nucleosides dC and dT, suggesting that nucleosides, rather than nucleotides, are the major active therapeutic agents. We demonstrated that dC and dT treatment of Tk2-deficient mice delayed disease onset, prolonged life span, and restored mtDNA copy number.16 In this situation, deoxynucleosides function as a substrate enhancement therapy.
Based on our promising preclinical results for this often-lethal disease, we obtained approval to initiate compassionate use of oral dC + dT, dTMP+dCMP, or both sequentially in TK2-deficient patients. Here, we report the outcomes of 16 TK2-deficient patients who received these pharmacological treatments.
Patients and Methods
Study Design
The objective of the study was to evaluate safety and efficacy of oral dC + dT, dCMP+dTMP, or both for TK2 deficiency on a compassionate use basis. In 10 academic medical centers from 5 countries (Spain, USA, Chile, Guatemala, and Italy), treatments were initiated under compassionate use (expanded access as defined by the US Food and Drug Administration [FDA]17) protocols. Compounds were obtained under investigator-sponsored Investigational New Drugs in the United States and under compassionate-use exemptions in other countries. All patients or legal guardians signed informed consent forms for treatment. The FDA, Spanish Drug Agency (Spanish Agency of Medicines and Health Products), and Italian Medicines Agency as well as the Columbia University Medical Center Institutional Review Board and the local pharmacy committees of each center approved the compassionate use treatment. We also obtained written informed consent for publication of the patients’ images and videos.
Patients
We analyzed outcomes of 16 genetically confirmed TK2-deficient patients (P1–P16) who had received treatment for at least 1 year prior to September 1, 2017. Twelve cases were from Spain and 1 from Italy, and 3 were followed in the USA (1 each from the USA, Chile, and Guatemala).
Treatment
In 6 patients, treatment was initiated with oral dCMP+dTMP at 1-to-1 (weight:weight) ratios until 2015, when cell and mouse studies indicated that deoxynucleotides were precursors for deoxynucleosides16,18 (mean duration of treatment with nucleotides = 21.4 months, range = 11–47 months) and therapy was converted to oral dC + dT at 1-to-1 (weight: weight) ratios in all but 1 patient (P11), who opted not to change to nucleosides. The duration of nucleoside treatment varied from 6 to 36 months, with an average of 15.5 months as of August 31, 2017. All 16 patients are continuing treatment.
Doses administered to patients were based on the dosages used in preclinical studies of H126N Tk2 mutant mice.15,16 Doses were titrated up to 400mg/kg/day depending on tolerance. Doses differed and were adjusted based upon frequency of stools, which was observed to increase in proportion to the dose. All patients are currently maintained on doses between 300 and 400mg/kg/day of each nucleoside or nucleotide.
Outcome Measures
Survival Analysis.
Survival was assessed in all patients.
Motor Assessment.
All patients underwent periodic motor assessments using at least 1 of the following scales: 6-minute walk test (6MWT),19 North Star Ambulatory Assessment (NSAA; evaluates motor goals with a score range of 0–34 as values of minimum and maximum motor skills, respectively and expressed in linearized data logit transformed 0–100 scores),20,21 and Egen Klassifikation (EK; evaluates functional capacity in nonambulatory patients with a score range of 30–0 as values of minimum and maximum functional capacity, respectively).22
Respiratory Evaluation.
We measured forced vital capacity (FVC) and maximal inspiratory pressure (MIP) in an upright position in compliant patients. In individuals on mechanical ventilation (MV), daily ventilatory support requirements were recorded.
Nutritional Status.
We collected data on the evolution of weight (percentile) or body mass index (BMI) data. Underweight is defined as BMI < 18.5 or weight percentile <10. Requirements for nasogastric tube and percutaneous endoscopic gastrostomy (PEG) were recorded.
Creatinine Kinase.
Creatine kinase (CK) was measured in serum, before and after treatment.
Serum Growth Differentiation Factor 15.
Serum growth differentiation factor 15 (GDF-15), a proposed biomarker of mitochondrial myopathy, was measured before and during treatment as described.23
Safety Assessments
Serial blood tests and electrocardiograms were performed to assess baseline hematological, renal, hepatic, and cardiac functions as well as possible toxicity of the treatment. All Common Terminology Criteria for Adverse Events 4.03 grade 2 or higher adverse events or new clinical events were recorded.
Statistical Analyses
Kaplan–Meier survival analysis (SAS 9.4 for analysis and R 3.5.1 for replication and graphical output) was used to determine the survival rate of a historical control group of 44 early onset severe myopathy patients (onset before 24 months and rapid progression) from the literature.5,6,9–13,24–30 For other outcomes, 95% confidence intervals (CIs) and associated 2-sided p values were obtained for the mean values of each outcome at every visit and for the change between visits. We evaluated the change in the outcome measures after 6 months of treatment and after ≥12 months of treatment, relative to baseline. The value after ≥12 months was the mean of all values in that period.
Role of the Funding Sources
The sponsors of this study did not contribute to the study design; collection, analysis, and interpretation of data; writing of this paper; and decision to submit this work for publication.
Results
Patients
The phenotype of the 16 TK2-deficient patients (P1–P16) spanned the clinical spectrum of the disease (Tables 1 and 2).4,5,10,12
TABLE 1.
Baseline Characteristics of the Treated TK2-Deficient Patients
| Patient | Age, yr/Sex | Country of Origin | Age at Onset | TK2 Mutations | Muscle mtDNA Depletion (relative to normal) | Muscle mtDNA Multiple Deletions | Weight, BMI or P | CK, U/l |
|---|---|---|---|---|---|---|---|---|
| 1 | 6/M | Spain | 15 mo | p.His121Asn; p.Arg192Lys | 13% | No | <1 P | 284 |
| 2 | 6/M | Spain | 17 mo | p.Tyr208Cys; p.Arg130Trp | 15% | No | 3 P | 148 |
| 3 | 3/F | Spain | 10 mo | p.Thr108Met; p.Pro227Serfs*9 | 30% | No | BMI = 14.3 | 179 |
| 4 | 7/M | USA | 12 mo | p.Lys50Ilefs*99; p.Thr108Met | 11% | No | <3 P | NA |
| 5 | 4/F | Spain | 23 mo | p.Lys202del; p.Asp177Tyr | 25% | No | <3 P | 1,183 |
| 6 | 30/M | Spain | 24 mo | Homozygous p.Thr108Met | 45% | Yes | BMI = 26.9 | 757 |
| 7 | 8/F | Spain | 6 mo | Homozygous p.Thr108Met | 18% | No | <3 P | 1,009 |
| 8 | 6/M | Spain | 18 mo | Homozygous p.Thr108Met | NA | NA | P50 | 393 |
| 9 | 12/M | Spain | 30 mo | Homozygous p.His121Asn | 50% | Yes | <3 P | 538 |
| 10 | 8/F | Chile | 11 mo | p.Thr108Met; p.His121Asn | NA | NA | BMI = 11.8 | 179 |
| 11 | 13/M | Italy | 24 mo | p.Arg183Gln; p.Lys202del | 10% | NA | NA | NA |
| 12 | 28/M | Guatemala | 18 mo | Homozygous p.His121Asn | NA | No | NA | 200–5,500 |
| 13 | 32/F | Spain | 12 yr | Homozygous p.Thr108Met | 17% | Yes | BMI = 13.8 | 2,500 |
| 14 | 33/F | Spain | 16 yr | Homozygous p.Thr108Met | 39% | Yes | BMI = 17.7 | 303 |
| 15 | 60/M | Spain | 50 yr | Homozygous p.Lys202del | NA | Yes | BMI = 27.6 | 400 |
| 16 | 61/F | Spain | 30 yr | Homozygous p.Lys202del | 60% | Yes | BMI = 27.2 | 294 |
Patients 1–5 had early onset severe myopathy patients; patients 6–16 had slower progression.
BMI = body mass index; CK = creatine kinase; F = female; M = male; NA = not available; P = percentile.
TABLE 2.
Baseline Functional Characteristics and Therapeutic Regimen of the Treated TK2-Deficient Patients
| Treatment (until August 2017) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Independent Gait | Ventilatory Support | dTMP+dCMP | dT+dC | ||||||||
| Patient | Yes/No | Age When Lost | Weight, BMI or P | Age at Initiation | h/day | Type of Feeding (oral or enteral) | Age at Treatment Onset | Duration | Daily Dose, Duration mg/kg | Duration | Daily Dose, Duration mg/kg |
| 1 | No | 16 mo | <1 P | 16 mo | 14 | Oral | 31 mo | 11 mo | 400 | 27 mo | 400 |
| 2 | No | 22 mo | 3 P | 29 mo | 11 | NGT | 30 mo | 11 mo | 400 | 27 mo | 400 |
| 3 | No | Never | BMI = 14.3 | 12 mo | 24 | NGT | 16 mo | – | – | 25 mo | 400 |
| 4 | No | Never | <3 P | 19 mo | 24 | NGT | 19 mo | 47 mo | 200 | 21 mo | 400 |
| 5 | No | 28 mo | <3 P | – | – | Oral | 28 mo | – | – | 20 mo | 400 |
| 6 | No | 21 yr | BMI = 26.9 | 21 yr | 8 | Oral | – | – | 16 mo | 400 | |
| 7 | Yes | – | <3 P | – | – | Oral | 6 yr | – | – | 24 mo | 400 |
| 8 | Yes | – | 50 P | – | – | Oral | 4 yr | – | – | 16 mo | 400 |
| 9 | No | 10 yr | <3 P | 7 yr | NA | NGT | 10 yr | – | – | 13 mo | 400 |
| 10 | Yes | – | BMI = 11.8 | – | – | Oral | 4 yr | 27 mo | 200 | 20 mo | 400 |
| 11 | No | – | NA | – | NA | Oral | 8 yr | 65 mo | 300 | – | – |
| 12 | Yes | – | NA | 15 yr | 24 | Oral | 26 yr | 11 mo | – | 23 mo | 350 |
| 13 | Yes | – | BMI = 13.8 | – | – | Oral | 30 yr | – | – | 26 mo | 300 |
| 14 | Yes | – | BMI = 17.7 | 28 yr | 8 | PEG | 32 yr | – | – | 15 mo | 400 |
| 15 | Yes | – | BMI = 27.6 | 50 yr | 8 | Oral | 59 yr | – | – | 20 mo | 400 |
| 16 | Yes | – | BMI = 27.2 | – | – | Oral | 60 yr | – | – | 16 mo | 400 |
Patients 1–5 had early onset severe myopathy patients; patients 6–16 had slower progression.
BMI = body mass index; NA = not available; NGT = nasogastric tube; P = percentile; PEG = percutaneous gastrostomy.
Five patients (P1–P5) had early onset severe myopathy defined by (1) onset before 24 months; and (2) inability to walk, use of MV, or both within 1 year of onset. Four of these 5 patients required mechanical ventilation and enteric feeding. Two of these patients were never able to walk, and the other 3 had lost the ability to walk prior to treatment initiation. All were underweight before the treatment. They did not manifest encephalopathy or systemic involvement other than myopathy. These individuals were compared to a matched historical control group of 44 reported TK2 patients who fulfilled the same criteria.4–6,9–13,24–30
The other 11 patients showed slower progression. Three (27.3%) lost the ability to walk during the course of the disease, whereas 8 (72.7%) were still walking at treatment onset. All had dysphagia; P12 and P14 required enteric feeding. The majority of the patients (60%) had marked weight loss. Of the 11 patients, 5 (45.5%) required MV for an average of 11 hours per day. In the 4 late onset cases (P13–P16; onset ≥12 years old), respiratory symptoms overshadowed limb weakness, and 2 individuals (P14 and P15) required MV at night with only minimal limb weakness.
Survival and Function of Early Onset Severe Myopathy Subjects
Compared to a published cohort,4 our treated early onset severe myopathy patients differed significantly in postonset survival (p = 0.0078; Fig 1). Only 27.3% of historical controls survived at least 2 years after onset (95% CI = 0.17–0.45),4 compared to all 5 treated patients (range = 2.1–6.3 years, mean ± standard deviation = 3.93 ± 1.66). All 5 treated early onset patients achieved clinically meaningful improvements in motor functions (Supplementary Videos 1–5).
FIGURE 1:
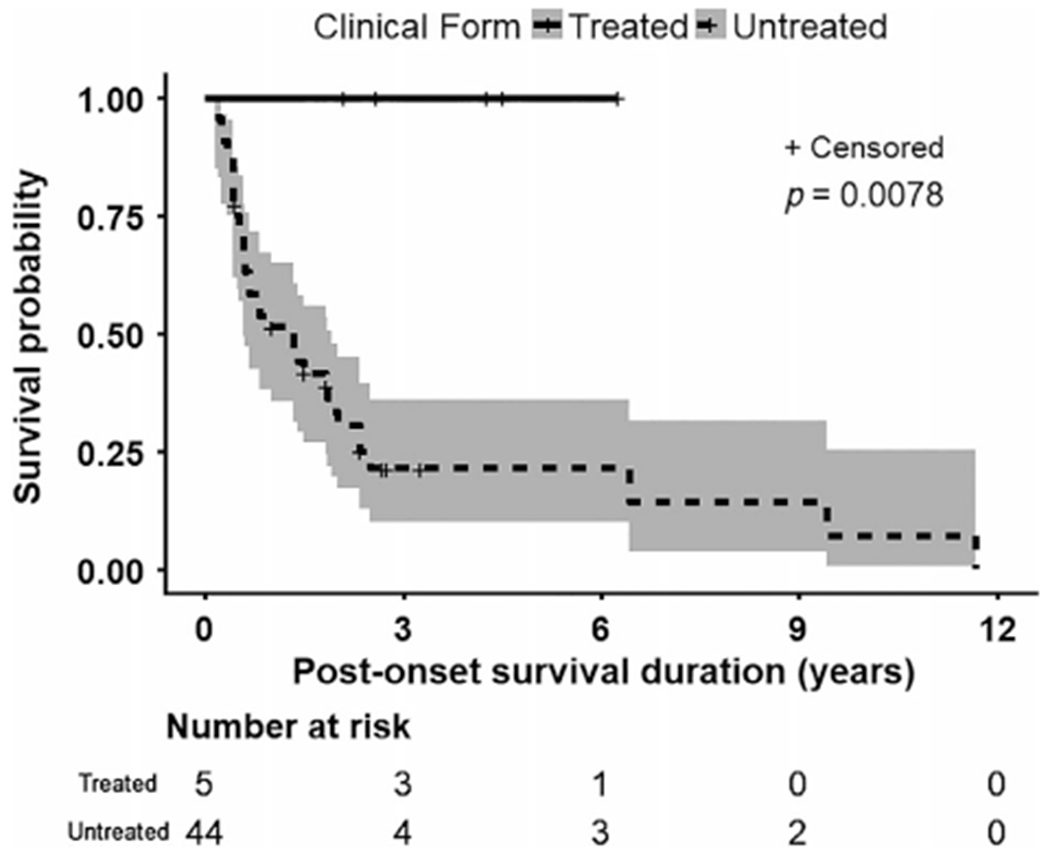
Kaplan–Meier survival curve of the 5 TK2-deficient patients with early onset severe myopathy (onset before 24 months and rapid progression defined by never acquiring ability to walk or loss of ability to walk, to breathe independently, or both within 1 year of onset) showed 100% survival for at least 2 years after treatment (range = 2.1–6.3 years; 3.93 ± 1.66 years, mean ± standard deviation). In contrast, the untreated historical control group with TK2-deficient early onset severe myopathy revealed that only 27.3% of patients survived at least 2 years after onset.4–6,9–13,24–30 Shading indicates upper and lower 95% confidence interval. Comparison of treated versus untreated early onset severe myopathy patients demonstrated a significant difference in postonset survival (p = 0.0078).
Motor Evaluation
6-Minute Walk Test.
6MWT was performed in the 8 patients who were able to ambulate prior to starting treatment and in 1 patient (P11) who regained the ability to walk after 1 year of treatment (Table 3). The distance walked improved in all but 1 (7/8 [87.5%]; P7 lacked baseline 6MWT). Six patients, who were on prolonged treatment (18–36 months), showed protracted improvement (Fig 2). The mean increase was 56m (95% CI = -21.7 to 113.7) after 6 months of treatment and 88.5m (95% CI = −5.47 to 171.5) at last follow-up, which ranged from 12 to 36 months of treatment; the mean increases appear to be clinically meaningful based upon estimates in Duchenne muscular dystrophy (DMD) indicating that mean minimal clinically important differences are 28.5 to 31.7m.19 The subgroup of patients with low baseline 6MWT performance (<300m, range = 0–175m) displayed the greatest improvements; after 6 months of treatment, mean increase over baseline was 146m (95% CI = 133.3–158.7), and after 12 to 36 months, the average increase was 171.9m (95% CI = 84.5–259.2). In contrast, patients with high baseline 6MWT distances (≥300m, range = 386–530m) showed stable or slightly improved performance on therapy. Two patients with early onset severe myopathy (P3 and P5; Table 4, Supplementary Videos 3 and 5) and 1 childhood onset patient (P11) who had lost the ability to walk prior to treatment gained independent ambulation on treatment.
TABLE 3.
Assessments of Ambulation and Respiratory Functions at Baseline and after Treatment of TK2-Deficient Patients with Slower Progression
| Assessment | After 6 Months of Treatment | After 12-36 Months of Treatment |
|---|---|---|
| 6MWT overall | ||
| n | 6 | 7 |
| Mean before treatment, m | 351.5 (188.7 to 514.3) | 304.4 (127.3 to 481.6) |
| Mean at visit, m | 407.5 (304.1 to 510.9) | 392.9 (283.2 to 502.6) |
| Change, m | 56 (−21.70 to 113.7) | 88.46 (5.471 to 171.5) |
| 6MWT < 300m | ||
| n | 2 | 2 |
| Mean before treatment, m | 164 (24.23 to 303.8) | 87.5 (−1024 to 1199) |
| Mean at visit, m | 310 (182.9 to 437.1) | 259.4 (−756.1 to 1284) |
| Change, m | 146 (133.3 to 158.7) | 171.9 (84.5 to 259.2) |
| 6MWT > 300m | ||
| n | 4 | 5 |
| Mean before treatment, m | 445.3 (334.1 to 556.4) | 419.2 (314.9 to 523.5) |
| Mean at visit, m | 456.3 (326.8 to 585.7) | 446.3 (355.3 to 537.3) |
| Change, m | 11 (−40.23 to 62.23) | 27.1 (−32.8 to 87) |
| NSAA | ||
| n | 5 | 4 |
| Mean before treatment | 19.6 (12.2 to 27) | 21.5 (13.9 to 29.1) |
| Mean at visit | 21.2 (13.7 to 28.7) | 27.5 (22.4 to 32.6) |
| Change | 1.6 (−0.6 to 3.8) | 6 (−2.1 to 14.1) |
| FVC | ||
| n | 8 | 6 |
| Mean before treatment, % | 48.7 (25.4 to 72.0) | 46.9 (22.8 to 71.0) |
| Mean at visit, % | 55.9 (27.4 to 84.4) | 50.2 (23.8 to 76.7) |
| Change, % | 7.2 (−3.2 to 17.5) | 3.3 (−0.4 to 7.0) |
| MIP | ||
| n | 4 | 5 |
| Mean before treatment, % | 29.2 (−0.3 to 58.3) | 27.6 (7.1 to 48.1) |
| Mean at visit, % | 34.9 ± (27.0 to 42.7) | 33.6 (18.9 to 48.5) |
| Change, % | 5.6 (−18.3 to 29.5) | 6.1 (−4.8 to 16.9) |
Mean and change in score were estimated by 95% confidence interval.
6MWT = 6-minute walk test; FVC = forced vital capacity; MIP = maximal inspiratory pressure; NSAA = North Star Ambulatory Assessment (evaluates motor goals with a score range of 0–34 as values of minimum and maximum motor skills, respectively).
FIGURE 2:
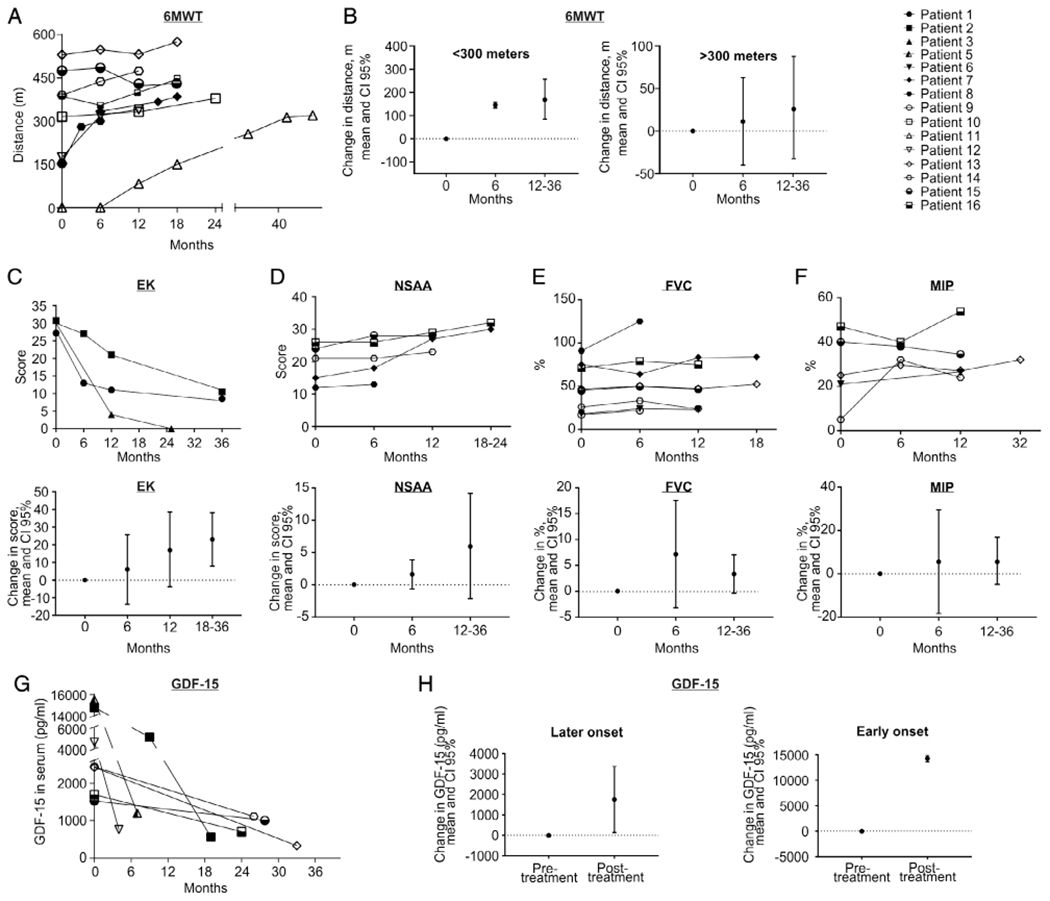
Changes from baseline in different outcome measures. (A) The graph shows individual values of distance walked in the 6-minute walk test (6MWT) at baseline and during the time on treatment, showing continuous improvements in 8 of 9 patients who were able to be evaluated by this test at any point in the treatment (note that P11 was nonambulant at baseline but regained independent gait after 12 months of treatment). (B) The group of patients with low performance (<300m) at baseline in the 6MWT showed more pronounced improvement after ≥12 months of treatment (mean 171.9m, 95% confidence interval [CI] = 84.5–259.2) than patients with higher basal performance (>300m, mean = 27.1m, 95% CI = −32.8 to 87). (C) Individual scores in the Egen Klassifikation (EK) at baseline and after treatment show improvement in every visit compared to the previous one in the 3 early onset severe myopathy patients evaluated by EK. The mean change in the score after 6 months of treatment and after 12 to 36 months, above baseline, showed progressive improvement. (D–F) North Star Ambulatory Assessment (NSAA) scores (D) showed improvement in all the patients evaluated in the group with slower progression, with a trend to improvement in forced vital capacity (FVC; E), and maximal inspiratory pressure (MIP; F) showed mild improvement or stabilization with slight fluctuations between visits, as reflected in the individual graphics. Both FVC and MIP showed a trend to mild improvement, with stabilization of the values during the treatment. Change in score (absolute values) in EK, NSAA, FVC, and MIP were estimated by 95% CI at every period of treatment relative to baseline. There were no statistical differences in any of the outcome measures. Serum levels of growth differentiation factor 15 (GDF-15) in individual subjects (G) and aggregated in later onset and early onset groups (H).
TABLE 4.
Weight, Oral Intake Status, Ventilator Use, and Ability to Walk at Baseline and With Treatment in the TK2-Deficient Patients
| Patients with Early Onset Severe Myopathy | ||||
|---|---|---|---|---|
| Characteristic | Before Treatment | After 6 Months of Treatment | After 12 Months of Treatment | After 18–36 Months of Treatment |
| Underweight, n (%) | 5 (100%) | 3 (60%) | 3 (60%) | 1 (20%) |
| Enteric feeding, n (%) | 3 (60%) | 3 (60%) | 3 (60%) | 1 (20%) |
| MV, n (%) | 4 (80%) | 4 (80%) | 4 (80%) | 3 (60%) |
| Ambulatory, n (%) | 0 | 1 (20%), P5 | 1 (20%), P5 | 2 (40%), P3 + P5 |
| EK mean (95% CI) | 29.3 (26.5–32.2), n = 3 | 20 (−68.9 to 108.9), n = 2 | 12 (−9.2 to 33.2, n = 3) | 6.3 (−7.8 to 20.46, n = 3) |
| Change in EK relative to BT baseline (95% CI) | 6 (−13.7 to 25.7) | 17.3 (−3.8 to 38.5) | 23 (7.9–38.1) | |
| Patients with Later Onset Slower Progressive Myopathy | ||||
| Characteristic | Before Treatment | After 6 Months of Treatment | After 12–36 Months of Treatment | |
| Underweight, n (%) | 6 (60%) | 4 (40%) | 3 (30%) | |
| Enteric feeding, n (%) | 2 (20%) | 0 (0%) | 0 (0%) | |
| MV, n (%) | 5 (45.5%) | 5 (45.5%) | 5 (45.5%) | |
| Ambulatory, n (%) | 8 (72.7%) | 8 (72.7%) | 9 (81.8 %) | |
CI = confidence interval; EK = Egen Klassifikation (evaluates functional capacity in nonambulatory patients with a score range of 30–0 as values of minimum and maximum functional capacity, respectively); MV = mechanical ventilation; P = Patient; BT = before treatment.
There were no statistically significant differences in the motor evaluation outcomes.
Egen Klassifikation.
In the early onset severe myopathy group, EK score was available in 3 of 5 patients, revealing average improvements of 6 points (95% CI = −13.7 to 25.7) after 6 months of treatment, 17.3 points (95% CI = −3.8 to 38.5) after 12 months, and 23 points (95% CI = 7.9–38.1) after 18 to 36 months (see Fig 2C, Table 4). Among patients with disease onset at >2 years old, we obtained the EK score at baseline and after 6 months of treatment in 2 of 3 nonambulant patients, who showed improvements of 3 and 17 points. These changes are not statistically significant; however, they are clinically meaningful based on an estimated 2.39-point change corresponding to functional and global state changes in patient with spinal muscular atrophy and DMD.31
North Star Ambulatory Assessment.
In the sole early onset severe myopathy patient who was able to perform the NSAA, mean score improved by 16 points after 12 months of treatment (see Table 3). In the group of 11 patients with later onset, 5 patients were assessed by the NSAA. Their scores improved at every assessment relative to baseline, with mean improvements of 1.6 (95% CI = −0.6 to 3.8) after 6 months and 6 points (95% CI = −2.1 to 14.1) after 12 to 36 months of treatment (see Fig 2D). This improvement is not statistically significant. The mean NSAA score change is clinically meaningful after 12 to 36 months using a logit transformed scale of 0 to 100, which demonstrates an 11.5-point increase (baseline mean = 60 points and post-treatment = 74.5 points), with estimated minimal important differences of 6.9 to 8.8 points in patients with DMD.21
Respiratory Evaluations
Mechanical Ventilation.
Nine patients required MV at baseline (see Tables 2 and 4). One (P3) was weaned off MV within 18 months of treatment initiation. The other patients who required nocturnal MV remained stable or improved partially after 12 months of treatment. None of the patients increased their time on MV, no patients initiated MV while on therapy, and none had respiratory complications including pneumonias.
FVC and MIP.
Of 8 patients whose respiratory function could be evaluated, 7 showed low baseline FVC (<80% of predicted value), with restrictive lung disease patterns (see Fig 2E). After 6 months of treatment, FVC revealed an average increase of 7.2% (95% CI = −3.2 to 17.5). After 12 to 36 months of treatment, FVC showed a slight decline, but overall mean was 3.3% (95% CI = −0.4 to 7.0) higher than baseline (see Table 3). MIP measurements in 5 adult patients showed a mean increase of 5.6% (95% CI = −18.3 to 29.5) after 6 months of treatment that persisted after 12 to 36 months (see Fig 2F, Table 3). None of these relationships is statistically significant.
Nutrition/Dysphagia
In the early onset severe TK2-deficient patients, BMI and weight percentile increased progressively with prolonged treatment, eventually reaching normal values in 4 of 5 patients (80%; see Table 4 and Supplementary Table 1). Of the 3 patients in this group requiring enteric feeding at baseline, 2 (P1 and P3) were able to permanently discontinue enteric feeding after 18 and 36 months oftreatment.
In the later onset group of 11 patients with slower progression, we obtained body weights in 10. Of six who were underweight at baseline, 5 (83%) gained weight and 4 (67%) normalized after 6 to 36 months of treatment (Supplementary Table 2). Both patients (P9 and P14) who required enteric feeding prior to treatment became able to feed exclusively by mouth after 6 months oftherapy.
Creatine Kinase
In 4 of 16 (25%) patients, baseline CK levels were 5- to 10-fold above normal (see Table 2); in all 4, levels normalized after 6 months of treatment (Supplementary Tables 1 and 2). The remaining 12 cases showed normal or moderately altered CK levels (2–3-fold above normal) without changes on treatment.
Growth Differentiation Factor 15
Baseline serum GDF-15 levels were elevated above the upper limit of normal (550pg/ml) in all 7 patients tested (see Fig 2G, 2H, and Table 5). The highest levels were in P2 and P5 with the early onset severe presentation, followed by P12 with childhood onset and late onset patients with more moderate increases. In all cases, levels of GDF-15 declined between 4 and 33 months of treatment. Normal values were reached in 3 of 7 patients. In the remaining 4 patients, GDF-15 levels decreased between 1.5- and 12-fold.
TABLE 5.
GDF-15 Levels at Pretreatment Baseline and on Deoxynucleoside Treatment
| GDF-15 Levels on Treatment, pg/ml | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Baseline GDF-15, pg/ml | 4 mo | 7 mo | 8 mo | 9 mo | 17 mo | 19 mo | 24 mo | 26 mo | 28 mo | 33 mo |
| P1 | – | – | – | 2,011 | – | 255 | – | – | – | – | – |
| P2 | 14,756 | – | – | – | 5,093 | – | 564 | – | – | – | – |
| P5 | 15,490 | – | 1,200 | – | – | – | – | – | – | – | – |
| P12 | 4,608 | 762 | – | – | – | – | – | – | – | – | – |
| P13 | 2,422 | – | – | – | – | – | – | – | – | – | 330 |
| P14 | 2,439 | – | – | – | – | – | – | – | 1,110 | – | – |
| P15 | 1,529 | – | – | – | – | – | – | – | – | 1,003 | – |
| P16 | 1,695 | – | – | – | – | – | – | 704 | – | – | – |
GDF-15 = growth differentiation factor 15.
Adverse Effects
No treatment-related serious adverse effects were observed in any patient. Safety blood tests and electrocardiogram were normal in all cases. The only drug-related adverse effect observed in 8 patients was diarrhea, which was dose-dependent, transient in most cases, and did not prompt suspension of treatment in any patients. Doses or administration schedules were modified to eliminate diarrhea. Diarrhea prevented 2 patients (P12 and P13) from reaching the recommended dose of 400mg/kg/day. One patient experienced mild and transient abdominal pain. Prior to treatment, P14 had elevated transaminases (approximately 10-fold above upper limit of normal) that were attributed to TK2 deficiency and normalized after 1 year of treatment with dC + dT.
In addition to the 16 patients reported here, 12 TK2-deficient patients initiated dT + dC (each at up to 400mg/kg/day; Supplementary Table 3). Two adults (P10, 58 years old; P11, 63 years old) after 3 to 4 months of treatment developed increased transaminases (alanine aminotransferase 8–13-fold and aspartate aminotransferase 3–6-fold above normal) and y-glutamyl transferase (GGT; 2.1–3.7-fold above normal) with normal bilirubin and alkaline phosphatase levels. In both cases, 3 months after discontinuing therapy, transaminases returned to normal. P10 had a prior episode of spontaneous transient elevated transaminases several years before starting therapy. Twenty days after restarting dT + dC (each at 200mg/kg/day), P11 had recurrent elevated transaminases (3.0–4.1-fold above normal), which again returned to normal after stopping treatment.
Discussion
We have administered pyrimidine deoxynucleoside and deoxynucleotides as novel pharmacological therapies in 16 patients with mitochondrial myopathy due to TK2 deficiency. Although we initially used dTMP and dCMP, after identification of dT and dC as the active agents in vitro and in vivo,16,18 we administered dC and dT to all but 1 patient (P11), who continues dTMP and dCMP treatment. The therapies exerted striking effects on survival in the early onset severe myopathy patients through amelioration of muscle weakness, which enabled reductions or discontinuation of mechanical ventilation and gastrostomy feeding as well as gaining ability to walk in the majority of these severe cases. Oral deoxynucleosides and deoxynucleotides produced no major side effects during this long-term treatment. The beneficial effects of the therapy were verified by functional tests, including 6MWT, EK, and NSAA, with mean changes that appear to be clinically meaningful. In addition, serum levels of GDF-15, a sensitive diagnostic biomarker for mitochondrial myopathy,23,32,33 were highly elevated at baseline and markedly declined in all 7 patients tested. Thus, GDF-15 assessment provides objective biomarker data supporting therapeutic response to deoxynucleosides and deoxynucleotides.
Our data indicate that nucleoside treatment can (1) reverse early onset tetraplegia and enable termination of mechanical ventilation and PEG (P3; Supplementary Video 3), (2) halt early onset disease progression and improve muscle weakness (P1-P5; Supplementary Videos 1–5), (3) produce considerable functional improvements in childhood onset patients (P9, P11), and (4) stabilize weakness in late onset patients after an initial mild improvement (P14, P16). Four treated patients were weaned off invasive respiratory support (P3), gastrostomy feeding (P1, P3, P9, and P14), or both (P3), and 3 gained independent ambulation on treatment (P3, P5, and P11). Furthermore, the 6MWT showed improvements in treated patients with low baseline performance (164m, 95% CI = 24.3–303.8), with clear increases in the distance walked (mean increase 171.9m, 95% CI = 84.5–259.2) at last followup (12–36 months of treatment).
In contrast to the early onset patients, in 4 late onset patients, therapy produced smaller beneficial effects, with stabilization or mild improvements in motor and respiratory functions. Stabilization of respiratory function is an important clinical outcome, because it may reduce morbidity and mortality in late onset patients. Nevertheless, 2 adult patients who started dT + dC therapy after the initial cohort developed elevated transaminases and GGT, which normalized after discontinuing treatment; these findings raise the possibility of hepatic toxicity of the therapy in some older adults. Our observations require confirmation in a larger number of patients with long-term follow-up.
A cytokine member of the transforming growth factor β family, GDF-15, is induced and secreted by muscle cells in response to mitochondrial damage,23 and has been identified as a biomarker for mitochondrial diseases via an unbiased global gene expression screening of muscle from patients with TK2 deficiency.32 Since then, elevated GDF-15 levels have been confirmed to have high sensitivity (67.8–97.9%) and specificity (87.7–96.2%) for mitochondrial diseases, indicating potential utility as a first-line diagnostic test for these diverse disorders.33–35 Although it has been proposed as a potential biomarker for therapeutic efficacy in mitochondrial diseases,36 this is the first report to link GDF-15 levels with clinical improvements in a clinical trial for a mitochondrial disorder. Our data demonstrate that in patients with TK2 deficiency, baseline circulating GDF-15 levels were elevated before deoxynucleoside therapy and decreased significantly with treatment in patients. Furthermore, these results indicate that GDF-15 may be a valuable biomarker in future clinical trials for mitochondrial diseases.
In conclusion, treatment with oral dT + dC, the substrates of TK2, in 15 TK2-deficient patients and dTMP +dCMP in 1 patient provided clinically notable benefit, especially in the infantile and childhood onset forms of the disease. The improvement or stabilization of respiratory function in the late onset patients suggests that this subgroup may also benefit from nucleoside supplementation, although longer longitudinal studies are needed to establish this point. Further studies are ongoing to support potential regulatory approvals.
Supplementary Material
Acknowledgment
This work was supported in part by grants from the Spanish Carlos III Health Institute (PMP15/00025 for C.P., F.Ma., and R.M.; PI16/00579 and CP09/00011 for C.J.-M.), Muscular Dystrophy Association (577391), Arturo Estopinan TK2 Research Fund, Generalitat de Catalunya PERIS program (SLT002/16/00370 for J.T-T.), and European Regional Development Fund.
We thank the patients and their families for collaborating with this study. We are especially grateful to patients J.J.M.P. and A.E. and their families for their exceptional contributions to multiple aspects of this research.
Footnotes
Additional supporting information can be found in the online version of this article.
Potential Conflicts of Interest
C.G., R.M., and M.H. are paid consultants to Modis Therapeutics. R.M. has equity in Modis Therapeutics. These relationships are de minimus for the United Kingdom Medical Research Council (C.G.), Vall d’Hebron Research Institute (R.M.), and Columbia University Medical Center (M.H.). Columbia University has submitted a patent, which has been licensed to Modis Therapeutics; this relationship is monitored by an unconflicted external academic researcher. The other authors declare no conflicts of interest.
References
- 1.Berk AJ, Clayton DA. A genetically distinct thymidine kinase in mammalian mitochondria. Exclusive labeling of mitochondrial deoxyribonucleic acid. J Biol Chem 1973;248:2722–2729. [PubMed] [Google Scholar]
- 2.Wang L, Munch-Petersen B, Herrstrom Sjoberg A, et al. Human thymidine kinase 2: molecular cloning and characterisation of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett 1999;443:170–174. [DOI] [PubMed] [Google Scholar]
- 3.Behin A, Jardel C, Claeys KG, et al. Adult cases of mitochondrial DNA depletion due to TK2 defect: an expanding spectrum. Neurology 2012;78:644–648. [DOI] [PubMed] [Google Scholar]
- 4.Garone C, Taylor RW, Nascimento A, et al. Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet 2018;55: 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saada A, Shaag A, Mandel H, et al. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 2001; 29:342–344. [DOI] [PubMed] [Google Scholar]
- 6.Alston CL, Schaefer AM, Raman P, et al. Late-onset respiratory failure due to TK2 mutations causing multiple mtDNA deletions. Neurology 2013;81:2051–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camara Y, Carreno-Gago L, Martin MA, et al. Severe TK2 enzyme activity deficiency in patients with mild forms of myopathy. Neurology 2015;84:2286–2288. [DOI] [PubMed] [Google Scholar]
- 8.Tyynismaa H, Sun R, Ahola-Erkkila S, et al. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum Mol Genet 2012; 21:66–75. [DOI] [PubMed] [Google Scholar]
- 9.Blakely E, He L, Gardner JL, et al. Novel mutations in the TK2 gene associated with fatal mitochondrial DNA depletion myopathy. Neuromuscul Disord 2008;18:557–560. [DOI] [PubMed] [Google Scholar]
- 10.Chanprasert S, Wang J, Weng SW, et al. Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2) gene. Mol Genet Metab 2013;110:153–161. [DOI] [PubMed] [Google Scholar]
- 11.Mancuso M, Filosto M, Bonilla E, et al. Mitochondrial myopathy of childhood associated with mitochondrial DNA depletion and a homozygous mutation (T77M) in the TK2 gene. Arch Neurol 2003; 60:1007–1009. [DOI] [PubMed] [Google Scholar]
- 12.Oskoui M, Davidzon G, Pascual J, et al. Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch Neurol 2006;63:1122–1126. [DOI] [PubMed] [Google Scholar]
- 13.Paradas C, Gutierrez Rios P, Rivas E, et al. TK2 mutation presenting as indolent myopathy. Neurology 2013;80:504–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akman HO, Dorado B, Lopez LC, et al. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum Mol Genet 2008; 17:2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garone C, Garcia-Diaz B, Emmanuele V, et al. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol Med 2014;6:1016–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, et al. Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol 2017;81:641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guidance for industry: expanded access to investigational drugs for treatment use—questions and answers. Silver Spring, MD: US Food and Drug Administration, 2017. [Google Scholar]
- 18.Camara Y, Gonzalez-Vioque E, Scarpelli M, et al. Administration of deoxyribonucleosides or inhibition of their catabolism as a pharmacological approach for mitochondrial DNA depletion syndrome. Hum Mol Genet 2014;23:2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve 2013;48:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott E, Eagle M, Mayhew A, et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int 2012;17:101–109. [DOI] [PubMed] [Google Scholar]
- 21.Mayhew AG, Cano SJ, Scott E, et al. Detecting meaningful change using the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Dev Med Child Neurol 2013;55:1046–1052. [DOI] [PubMed] [Google Scholar]
- 22.Steffensen B, Hyde S, Lyager S, et al. Validity of the EK scale: a functional assessment of non-ambulatory individuals with Duchenne muscular dystrophy or spinal muscular atrophy. Physiother Res Int 2001; 6:119–134. [DOI] [PubMed] [Google Scholar]
- 23.Montero R, Yubero D, Villarroya J, et al. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS One 2016;11:e0148709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrozzo R, Bornstein B, Lucioli S, et al. Mutation analysis in 16 patients with mtDNA depletion. Hum Mutat 2003;21:453–454. [DOI] [PubMed] [Google Scholar]
- 25.Galbiati S, Bordoni A, Papadimitriou D, et al. New mutations in TK2 gene associated with mitochondrial DNA depletion. Pediatr Neurol 2006;34:177–185. [DOI] [PubMed] [Google Scholar]
- 26.Gotz A, Isohanni P, Pihko H, et al. Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome. Brain 2008;131: 2841–2850. [DOI] [PubMed] [Google Scholar]
- 27.Lesko N, Naess K, Wibom R, et al. Two novel mutations in thymidine kinase-2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscul Disord 2010;20:198–203. [DOI] [PubMed] [Google Scholar]
- 28.Mancuso M, Salviati L, Sacconi S, et al. Mitochondrial DNA depletion: mutations in thymidine kinase gene with myopathy and SMA. Neurology 2002;59:1197–1202. [DOI] [PubMed] [Google Scholar]
- 29.Tulinius M, Moslemi AR, Darin N, et al. Novel mutations in the thymidine kinase 2 gene (TK2) associated with fatal mitochondrial myopathy and mitochondrial DNA depletion. Neuromuscul Disord 2005;15: 412–415. [DOI] [PubMed] [Google Scholar]
- 30.Zhang S, Li FY, Bass HN, et al. Application of oligonucleotide array CGH to the simultaneous detection of a deletion in the nuclear TK2 gene and mtDNA depletion. Mol Genet Metab 2010;99:53–57. [DOI] [PubMed] [Google Scholar]
- 31.Fagoaga J, Girabent-Farrés M, Bagur-Calafat C, et al. Evolution of functional capacity, assessed with the Egen Klassifikation scale, in the Spanish population with spinal muscular atrophy or Duchenne muscular dystrophy. A three year longitudinal study. Rev Neurol 2015;61:344–348. [PubMed] [Google Scholar]
- 32.Kalko SG, Paco S, Jou C, et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genomics 2014;15:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yatsuga S, Fujita Y, Ishii A, et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann Neurol 2015;78: 814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis RL, Liang C, Sue CM. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 2016;86:2010–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji X, Zhao L, Ji K, et al. Growth differentiation factor 15 is a novel diagnostic biomarker of mitochondrial diseases. Mol Neurobiol 2017;54:8110–8116. [DOI] [PubMed] [Google Scholar]
- 36.Koga Y, Povalko N, Inoue E, et al. Biomarkers and clinical rating scales for sodium pyruvate therapy in patients with mitochondrial disease. Mitochondrion (in press). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.