

Summary
Psoriasis is characterized by excessive growth and aberrant differentiation of epidermal keratinocytes due to persistent inflammation. However, the underlying mechanism that triggers immune activation in psoriasis is not clear. In this study, we explored excessive DNA as a potential trigger of psoriasis using cultured human keratinocytes and psoriatic skin tissues. We demonstrated that human genomic DNA fragments induced tumour necrosis factor (TNF)‐α expression, hyperproliferation and over‐expression of heparin‐binding epidermal‐like growth factor (HB‐EGF) and transforming growth factor (TGF)‐α, accompanied by defective expression of keratins 1 and 10 in cultured normal human epidermal keratinocytes, which have a similar phenotype to that of keratinocytes in psoriatic skin lesions. In psoriatic lesions, we found high levels of double‐stranded (ds)DNA fragments, accompanying keratinocytes expressing Ki‐67, HB‐EGF and TNF‐α. In addition, we showed that 1,25‐dihydroxyvitamin D3 inhibited genomic DNA fragment‐induced TNFA and interleukin‐1β (IFNB) expression in human keratinocytes, and an intact function of cathelicidin anti‐microbial peptide (CAMP) was required for this effect. These results suggest that excessive dsDNA fragments probably act as a risk factor for immune activation in psoriasis, and the active form of vitamin D can prevent genomic DNA‐mediated skin inflammation via CAMP.
Keywords: active vitamin D, genomic DNA, inflammation, keratinocyte, psoriasis
Cytosolic dsDNA fragments upregulated inflammatory cytokines (TNF‐α, IFN‐β) and growth factors (HB‐EGF, TGF‐α) in cultured human primary keratinocytes, accompanied by increased proliferation and impaired differentiation. Aberrant dsDNA fragments were found in psoriatic skin lesions, but not in unaffected areas, in association with keratinocytes overexpressing Ki‐67, HB‐EGF and TNF‐α. Active vitamin D suppressed dsDNA fragment‐inducible TNF‐α and IFN‐β in a cathelicidin antimicrobial peptide‐dependent manner in human primary keratinocytes. These results suggest that excessive dsDNA are potential triggers of skin inflammation, and vitamin D creams can exert therapeutic effects on psoriasis by suppressing dsDNA fragment‐inducible skin inflammation via cathelicidin antimicrobial peptide.

Introduction
Psoriasis is a chronic skin disease characterized by excessive growth and aberrant differentiation of epidermal keratinocytes [1]. Normally, keratinocytes that reside in the stratum basale of the epidermis proliferate until they permanently withdraw from the cell cycle to undergo differentiation. Differentiating keratinocytes migrate towards the upper layers of the epidermis, where they gradually lose their cytoplasmic organelles and nuclei, eventually morphing into nuclei‐free corneocytes upon arriving at the stratum corneum [2]. Psoriatic keratinocytes proliferate abnormally to initiate/fulfil differentiation, thus arriving at the skin surface with a retained nucleus, a phenomenon termed ‘parakeratosis’ [3].
Psoriatic lesions are associated with over‐expression of inflammatory mediators, such as tumour necrosis factor α (TNF‐α), interleukin‐1β (IL‐1β) and type I interferons (IFNs) [4, 5]. Immunosuppressive agents and monoclonal antibodies targeting inflammatory mediators are extensively used to treat psoriasis [1, 6, 7, 8], supporting the notion that psoriasis is primarily an immune disorder; however, the trigger for such immune activation remains unclear.
We previously showed that cytosolic exposure to fragments of self‐DNA activates both innate and acquired immune responses [9, 10, 11, 12]. In addition, impaired clearance of self‐DNA due to defects in DNases has been implicated in several autoimmune diseases [13, 14]. It has been suggested that extra‐nuclear genomic DNA is a danger signal that contributes to the breakdown of self‐tolerance, ultimately leading to an autoimmune response [15]. Moreover, it was found that in psoriatic skin, some epidermal anti‐microbial peptides [such as the cathelicidin anti‐microbial peptide (CAMP) LL37] are able to form a complex with self‐DNA and self‐RNA that are endocytosed by plasmacytoid dendritic cells (pDCs), leading to activation of Toll‐like receptor (TLR)‐mediated inflammatory cascades [16, 17]. From this viewpoint, defectively degraded self‐DNA in parakeratotic keratinocytes, once it ‘leaks’ out of the nucleus, readily poses a threat to skin immune homeostasis.
In this study, to explore the mechanism underlying the pathogenesis of psoriasis, we investigated the effect of self‐DNA on cell proliferation, cytokine production and cell differentiation in cultured normal human keratinocytes and skin tissue sections from individuals with psoriasis. We also examined the effect of the active form of vitamin D, 1,25‐dihydroxyvitamin D3 (VD3), on dsDNA‐mediated skin inflammation.
Materials and methods
Cell culture and reagents used for treatments
Normal human epidermal keratinocytes (NHEKs; Kurabo, Osaka, Japan) were originally cultured in KG2, i.e. HuMedia‐KB2 basal medium supplemented with insulin, bovine pituitary extract, epidermal growth factor, hydrocortisone and amphotericin B (Kurabo). Once 50–60% confluence was reached, KG2 was replaced with HuMedia‐KB2 basal medium and then cultured overnight for further experiments. Other reagents used were VD3 (Santa Cruz Biotechnology, Dallas, TX, USA), LL‐37 anti‐microbial peptide (AnaSpec, Fremont, CA, USA) and anti‐TNF‐α monoclonal antibody (R&D Systems, Minneapolis, MN, USA).
Preparation and transfection of human genomic DNA fragments
Genomic DNA was prepared from NHEKs using QIAamp DNA mini kits (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. The DNA was digested by EcoRI and PstI and then purified using MinElute reaction cleanup kits (Qiagen). To prepare ssDNA, genomic DNA fragments were boiled at 95°C for 5 min and then immediately chilled on ice for 5 min. DNA fragments were transfected into cells maintained in KB2 medium in 24‐well plates at 0·5 μg/well, using FuGENE HD transfection reagent (Roche Diagnostics, Basel, Switzerland).
Construction and transfection of CAMP‐knock‐out CRISPR/Cas9 vectors
Three guide RNAs (1, 2 and 3) targeting CAMP (ID: NM_004345.4) were designed using the CRISPR design tools (http://crispr.dbcls.jp), as listed in Supporting information, Table S1. Each guide RNA was inserted into BbsI‐digested backbone vectors (Addgene, Watertown, MA, USA; plasmid 62988) [18], according to the manufacturer’s protocol, to create a recombinant vector. The vectors were transfected into cells in 24‐well plates containing KB2 medium at 1 μg/well using FuGENE HD transfection reagent (Roche Diagnostics). After 6 h of transfection, the KB2 medium was replaced with fresh KG2 medium, and 48 h later VD3 treatment and DNA transfection were performed. CAMP knock‐out was confirmed by sequencing of the target region following polymerase chain reaction (PCR) amplification of mRNA and subcloning into a plasmid.
Total RNA extraction and real‐time PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen) and then reverse‐transcribed into cDNA using the high‐capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA), as described previously [11, 19]. Real‐time PCR was performed using fast SYBR green Master Mix (Applied Biosystems) and the StepOnePlus real‐time PCR system (Applied Biosystems), as described previously [11, 19]. The PCR primers used are listed in Supporting information, Table S2. The relative mRNA expression levels were normalized to the corresponding ACTB expression level.
Flow cytometry
Cells were fixed and stained using the bromodeoxyuridine (BrdU) flow kit (Becton Dickinson, Franklin Lakes, NJ, USA), according to the manufacturer’s instructions. Flow cytometric analyses of BrdU‐ or 7‐amino‐actinomycin D‐labelled cells were performed and analysed using the fluorescence activated cell sorter (FACS)Calibur flow cytometer (Becton Dickinson).
Enzyme‐linked immunosorbent assay (ELISA)
Culture supernatants were collected 18 and 24 h after transfection, and the level of TNF‐α was measured using the human TNF‐α Quantikine ELISA kit (R&D Systems), according to the manufacturer’s instructions.
Western blot analysis
Cellular proteins were prepared and analysed as described previously [19]. The membranes were incubated with rabbit anti‐keratin‐1 (Covance, Princeton, NJ, USA; 1 : 1,000), rabbit anti‐keratin‐10 (Covance; 1 = 1000) or goat anti‐β‐actin (Santa Cruz Biotechnology; 1 : 2000) as the primary antibodies. Then, the membranes were incubated with donkey anti‐rabbit immunoglobulin (IgG)–biotin conjugates (GE Healthcare, Little Chalfont, UK; 1 : 20 000) or donkey anti‐goat IgG–biotin conjugates (Millipore, Billerica, MA, USA; 1 : 20 000), followed by incubation with streptavidin horseradish peroxidase (GE Healthcare; 1 : 20 000). Specific bands were visualized using ECL Plus reagent (GE Healthcare) and captured using the C‐DiGit blot scanner (LI‐COR, Lincoln, NE, USA).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and immunofluorescence staining
Formalin‐fixed, paraffin‐embedded skin tissue sections were deparaffinized and rehydrated. Antigen retrieval was performed in 10 mM citrate buffer, pH 6 (Dako, Tokyo, Japan) by applying 300 W microwave irradiation for 5 min. TUNEL staining was performed using the in‐situ cell death detection kit, POD (Roche Diagnostics), according to the manufacturer’s instructions. The tissue sections were also incubated with mouse anti‐Ki‐67 (Dako), goat anti‐heparin‐binding epidermal‐like growth factor (HB‐EGF) (R&D Systems; 1 : 100) or mouse anti‐TNF‐α (R&D Systems; 1 : 50) antibodies at 4°C overnight. After rinsing with phosphate‐buffered saline with Tween‐20 (PBST), the tissues were incubated with donkey anti‐mouse IgG–biotin conjugates (GE Healthcare; 1 : 1000) or donkey anti‐goat IgG–biotin conjugates (Millipore; 1 : 1000) for 1 h, rinsed with PBST, and then incubated with streptavidin horseradish peroxidase (GE Healthcare; 1 : 1000) for 1 h. For fluorescent signal conversion, the TSA cyanine 3 system (PerkinElmer, Waltham, MA, USA) was used according to the manufacturer’s instructions. The nuclei were counterstained with Hoechst 33258 (Life Technologies, Waltham, MA, USA). Images were captured using the FV10i‐LIV Laser Scanning Microscope (Olympus, Tokyo, Japan).
Statistical analysis
All experiments were performed in triplicate and repeated at least three times. All results are expressed as the mean ± standard deviation (s.d.). Statistical analysis was performed with spss software (SPSS standard version 17.0; SPSS Inc., Chicago, IL, USA). Significant difference was determined by ordinary one‐way analysis of variance (ANOVA) followed by Dunnet’s post‐hoc test. P < 0·05 was considered significant.
Ethics
This study was approved by the institutional ethics boards of Teikyo University (no. 16‐008), essentially following the ethical guidelines of the Declaration of Helsinki. Written informed consent was obtained from all participants. All samples were anonymized before use.
Results
Self double‐stranded DNA induces TNF‐α production in normal human epidermal keratinocytes, accompanied by increased cell growth and impaired differentiation
TNF‐α is a well‐known immune mediator over‐expressed in psoriatic lesions [6, 20]. We first examined TNF‐α production in NHEKs after transfection of the cells with fragmentized human genomic DNA as either double‐stranded (ds)‐ or single‐stranded (ss) molecules. Stimulation with the dsDNA, but not ssDNA, fragments significantly induced TNFA mRNA expression at 12 h post‐transfection (Fig. 1a). In accordance, secretion of TNF‐α in the culture medium was increased at 18–24 h post‐transfection in dsDNA fragment‐stimulated cells (Fig. 1b), confirming that human keratinocytes produce TNF‐α upon cytosolic exposure to self‐dsDNA.
Fig. 1.
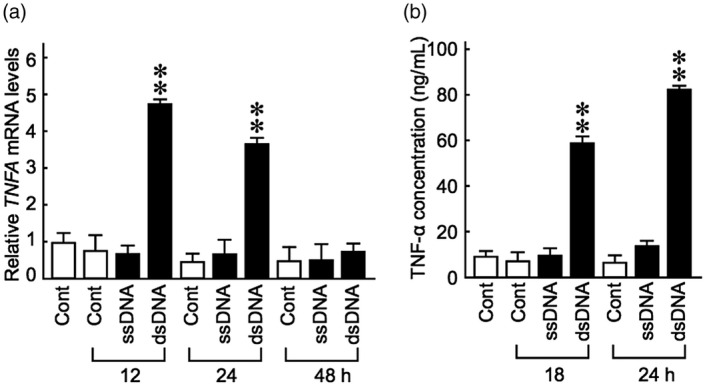
Fragments of genomic dsDNA induce tumour necrosis factor (TNF)‐α production in keratinocytes. Normal human epidermal keratinocytes maintained in KB2 basal medium were transfected with fragments of human genomic dsDNA or ssDNA for the indicated time‐periods. (a) Relative TNFA mRNA levels were evaluated by real‐time polymerase chain reaction (PCR). (b) TNF‐α protein levels in the culture medium were measured by enzyme‐linked immunosorbent assay (ELISA). Data are presented as means ± standard deviation (s.d.) relative to the non‐transfected controls. **P < 0·001, compared with the unstimulated control group.
Flow cytometric analysis showed that dsDNA, but not ssDNA, significantly increased the proportion of cells in the S + G2/M phase of the cell cycle (Fig. 2a), indicating that dsDNA promotes growth of keratinocytes. HB‐EGF and transforming growth factor‐α (TGF‐α) are prominent mitogenic factors for epidermal keratinocytes, and their over‐expression is often detected in hyperproliferative skin diseases, including psoriasis [21, 22, 23, 24, 25]. We found that both HBEGF and TGFA mRNA expression was significantly increased in dsDNA‐stimulated keratinocytes (Fig. 2b). However, when a neutralizing anti‐human TNF‐α monoclonal antibody was added to the culture medium, the induction of HBEGF and TGFA by dsDNA was significantly suppressed (Fig. 2c), indicating that the over‐expression of HB‐EGF and TGF‐α in keratinocytes is largely dependent upon secreted TNF‐α induced by dsDNA.
Fig. 2.

Fragments of genomic dsDNA enhance keratinocyte proliferation, which is correlated with up‐regulation of HBEGF and TGFA mRNA expression. (a) Normal human epidermal keratinocytes maintained in KB2 medium were transfected with human genomic dsDNA or ssDNA fragments for 48 h, after which the cell cycle distribution was analysed by flow cytometry. The proportions of cells in the S + G2/M phases of the cell cycle are indicated. (b) HBEGF and TGFA mRNA expression in keratinocytes transfected with dsDNA or ssDNA fragments for the indicated time‐periods was evaluated by real‐time polymerase chain reaction (PCR). **P < 0·001, compared with the unstimulated control group. (c) Keratinocytes maintained in KB2 basal medium were transfected with dsDNA fragments for 24 h. An anti‐human tumour necrosis factor (TNF)‐α monoclonal antibody or control immunoglobulin (Ig)G was added to the medium at a final concentration of 200 ng/ml at 4 h after transfection, and the HBEGF and TGFA mRNA levels were evaluated by real‐time PCR at 24 h after transfection. Data are presented as means ± standard deviation (s.d.) relative to the control. *P < 0·05; **P < 0·001, compared with the dsDNA‐stimulated IgG‐treated group.
Keratinocyte hyperproliferation has been associated with delayed or impaired differentiation [26, 27, 28]. As hallmarks of differentiated keratinocytes, keratins 1 (KRT1) and 10 (KRT10) are specifically expressed in the spinous and granular layers of the epidermis [29]. The expression of KRT1 and KRT10 was spontaneously up‐regulated in a time‐dependent manner in keratinocytes cultured in KB2 medium (Fig. 3a, open bars). Up‐regulation of these two genes was significantly impaired by dsDNA, but not ssDNA (Fig. 3a, closed bars). In accordance, dsDNA fragments completely abolished the increase in KRT1 and KRT10 protein expression induced by exposure to KB2 medium in keratinocytes (Fig. 3b). These results suggest that genomic dsDNA fragments perturb the normal differentiation process of keratinocytes while simultaneously enhancing cellular proliferation.
Fig. 3.
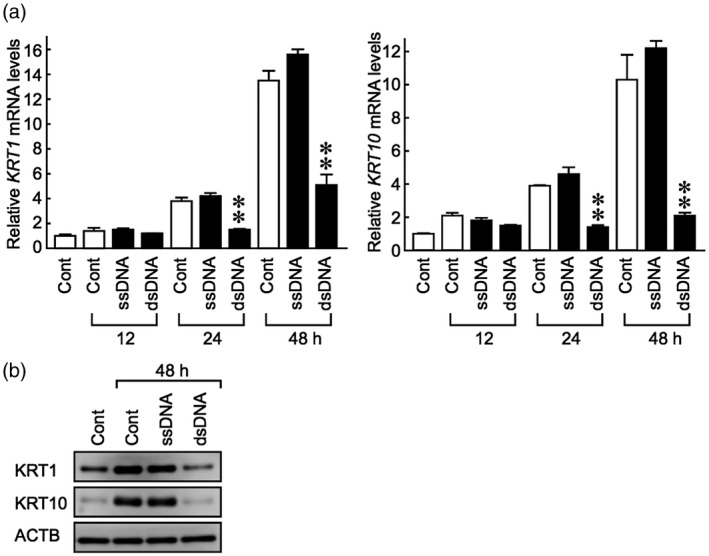
Fragments of genomic dsDNA inhibit keratins 1 and 10 expression in keratinocytes. Normal human epidermal keratinocytes were cultured to 50–60% confluence in KG2 growth medium, which was then replaced with KB2 basal medium for overnight incubation. Cells were then transfected with fragments of human genomic dsDNA or ssDNA for the indicated time‐periods. (a) The mRNA levels of KRT1 and KRT10 were evaluated by real‐time polymerase chain reaction (PCR). (b) The protein levels of KRT1, KRT10 and ACTB (β‐actin) were evaluated by Western blot analysis. Data are presented as means ± standard deviation (s.d.) relative to the original levels. **P < 0·001, compared with the unstimulated control group.
Aberrant levels of dsDNA fragments are associated with expression of Ki‐67, HB‐EGF and TNF‐α in the keratinocytes of psoriatic lesions
To confirm if dsDNA fragments exert similar actions in individuals with psoriasis as those observed in cell culture experiments, we used serial tissue sections obtained from skin biopsies from individuals with psoriasis. A high level of TUNEL‐positive dsDNA fragments was found in psoriatic lesions with severe parakeratosis, but not in non‐involved areas within the same tissue section (Fig. 4 and Supporting information, Fig. S1, TUNEL: green fluorescence). Ki‐67‐positive proliferating keratinocytes were found mainly in the low to middle layers of the epidermis beneath cells harbouring TUNEL‐positive dsDNA fragments (Fig. 4 and Supporting information, Fig. S1, Ki‐67: red fluorescence). Keratinocytes and a few lymphocytes adjacent to the dsDNA fragments also strongly expressed HB‐EGF compared with the non‐involved areas (Fig. 4 and Supporting information, Fig. S1, HB‐EGF: red fluorescence). Moreover, keratinocytes expressing TNF‐α were detected in the epidermal areas associated with dsDNA fragments (Fig. S2, TNF‐α: red fluorescence). Although the cause and effect have yet to be clarified these in‐vivo observations, in line with in‐vitro results, demonstrate that a high level of abnormal dsDNA fragments is associated with keratinocytes over‐expressing Ki‐67, HB‐EGF and TNF‐α in psoriatic lesions.
Fig. 4.
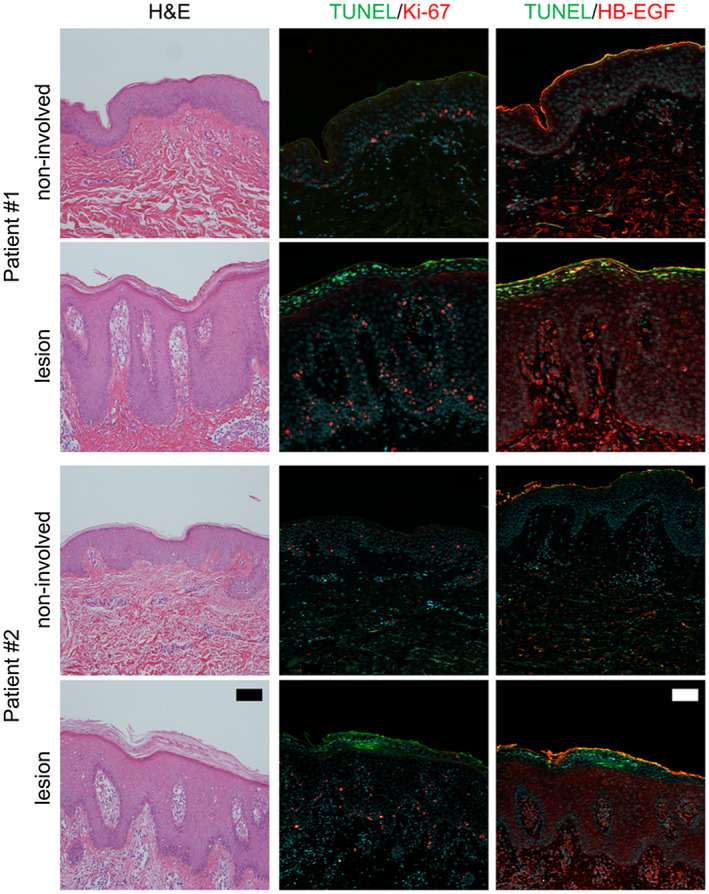
Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL)‐positive dsDNA fragments are present in psoriatic lesions and are associated with keratinocytes expressing Ki‐67 and heparin‐binding epidermal‐like growth factor (HB‐EGF). Skin tissue sections from individuals with psoriasis were subjected to haematoxylin and eosin (H&E) staining or TUNEL staining in combination with immunofluorescence staining of Ki‐67 or HB‐EGF. Representative TUNEL staining (green) in combination with Ki‐67 or HB‐EGF immunofluorescence staining (red) in psoriatic lesions and non‐involved areas within the same tissue sections. Bars = 100 μm.
Vitamin D treatment abolishes dsDNA‐induced inflammatory cytokines in normal human epidermal keratinocytes in a CAMP‐dependent manner
Vitamin D creams are widely prescribed to treat psoriasis; however, the underlying mechanism of their therapeutic effects is not fully elucidated. CAMP is an endogenous anti‐microbial peptide involved in mammalian innate immune responses [30], and its expression in skin lesions is particularly induced by active vitamin D analogues [31, 32]. To determine whether vitamin D regulates dsDNA fragment‐induced inflammation by modulating CAMP expression, keratinocytes cultured in KB2 basal medium were treated with VD3, the active form of vitamin D, followed by stimulation with genomic dsDNA fragments. Real‐time PCR analysis revealed that 0·01 mM and 0·1 mM of VD3 inhibited TNFA expression, comparable with that in non‐stimulated cells (Fig. 5a). IFNB mRNA, which is abnormally expressed in psoriatic lesions [33], was strongly induced by dsDNA and significantly suppressed by VD3 in cultured keratinocytes (Fig. 5a). In contrast, CAMP gene expression was not affected by the dsDNA fragments but was strongly induced by VD3 in a dose‐dependent manner (Fig. 5b). These results confirmed that VD3 suppresses skin inflammatory responses in association with increased CAMP expression [31, 32].
Fig. 5.
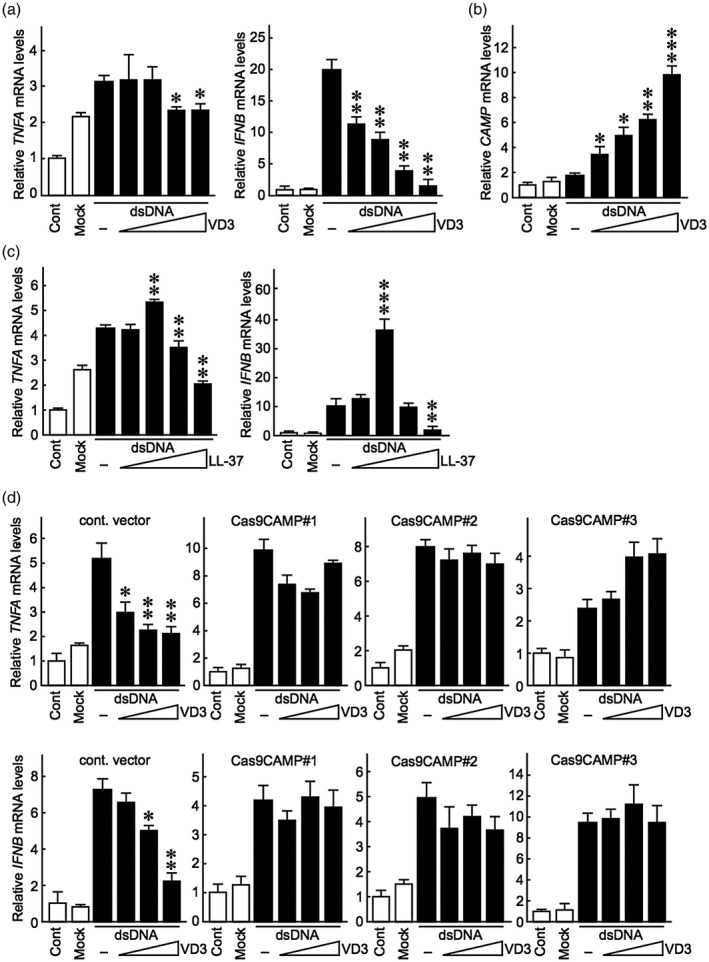
Active vitamin D suppresses dsDNA‐induced TNFA and IFNB expression in a cathelicidin anti‐microbial peptide (CAMP)/LL‐37‐dependent manner. (a,b) Normal human epidermal keratinocytes were maintained in KG2 growth medium, which was replaced with KB2 basal medium containing VD3 (0·1 nM, 1·0 nM, 0·01 mM, 0·1 mM) 24 h before transfection of dsDNA fragments. The medium was then replaced with KB2 medium containing VD3 4 h after transfection. TNFA, IFNB and CAMP mRNA levels were evaluated by real‐time polymerase chain reaction (PCR) 12 h after dsDNA fragment stimulation. (c) Genomic dsDNA fragments were transfected into cells in the presence of LL‐37 (2·5, 5, 10 or 20 μg/ml). TNFA and IFNB mRNA levels were evaluated by real‐time PCR 12 h after dsDNA fragment stimulation. (d) Cells maintained in KG2 medium were transfected with an empty Cas9 vector or a recombinant Cas9 vector carrying guide RNAs targeting cathelicidin anti‐microbial peptide (CAMP) (Cas9CAMP1, 2 or 3) for 48 h, followed by VD3 treatment and dsDNA transfection, as described in the Materials and methods. TNFA and IFNB mRNA levels were evaluated by real‐time PCR 12 h after dsDNA stimulation. Data are presented as means ± standard deviation (s.d.) relative to the control. Statistical analysis was performed against dsDNA fragment‐stimulated cells without VD3 or LL‐37 treatment. *P < 0·05; **P < 0·001; ***P < 0·0001, compared with the dsDNA‐stimulated VD3‐untreated group.
LL‐37 is the active 37‐amino‐acid derivative of CAMP that modifies host immune responses [34]. We evaluated whether LL‐37 exerts the same inhibitory effect of VD3 on dsDNA fragment‐induced cytokine expression. We found that lower concentrations of LL‐37 augmented the level of dsDNA fragments to induce TNFA and IFNB expression, whereas higher concentrations suppressed dsDNA fragment‐induced TNFA and IFNB expression levels (Fig. 5c). To further clarify whether VD3 action on dsDNA fragment‐induced skin inflammation depends on CAMP/LL‐37, we generated CAMP‐deficient keratinocytes using the CRISPR/Cas9 genome‐editing system. We found that the ability of VD3 to suppress dsDNA fragment‐induced TNFA and IFNB expression was abolished in CAMP‐deficient cells (Fig. 5d). Taken together, these results suggest that VD3 exerts an anti‐inflammatory effect on self‐dsDNA‐inducible skin inflammation in a CAMP/LL‐37‐dependent manner.
Discussion
It has been demonstrated previously that cytosolic dsDNA activates innate immune responses and elicits the production of immune mediators [9, 11]. These dsDNA actions are not restricted to immune cells, but also occur in a variety of non‐immune cells, including epithelial cells [11, 12]. In the present study, we demonstrated that genomic DNA fragments induced a psoriasis‐like phenotype in cultured normal human keratinocytes, characterized by over‐expression of inflammatory cytokines (TNF‐α, IFN‐β) and growth factors (HB‐EGF, TGF‐α), and these effects were accompanied by increased cellular proliferation and impaired differentiation. In line with these in‐vitro observations, we found that the presence of aberrant dsDNA fragments was associated with keratinocytes over‐expressing Ki‐67, HB‐EGF and TNF‐α in psoriatic skin lesions, but not unaffected skin areas.
An increased proportion of dsDNA fragments in parakeratotic regions, as demonstrated in the present study, supports the intriguing possibility that parakeratosis might itself be responsible for the leakage of dsDNA fragments. Conceivably, degraded DNA fragments may escape from parakeratotic cells, especially in the presence of impaired barrier function [35, 36]. HB‐EGF and TGF‐α are expressed at low levels in normal epidermis and are over‐expressed in epidermal hyperplasia, indicating their essential role in keratinocyte hyperproliferation [21, 22, 23, 24, 37, 38]. Our results suggest that keratinocyte proliferation induced by dsDNA fragments is probably mediated by TNF‐α, which up‐regulates the above‐mentioned growth factors. Other cytokines, such as IFNs and IL‐1β, might also be responsible for TGFA over‐expression [39, 40, 41], as dsDNA‐induced TGFA expression was not completely inhibited by neutralization of TNF‐α. The inflammatory cytokine profiles probably vary in different disease phases of psoriasis. It was recently reported that IL‐1β over‐expression was predominant in early‐phase psoriasis plague, while TNF‐α, IL‐12, IL‐17, IL‐23 over‐expression was predominant in late‐phase psoriasis plague [42].
Consistent with the in‐vitro results, abundant dsDNA fragments were found in the upper epidermal layers of psoriatic, but not unaffected, skin areas, above keratinocytes over‐expressing Ki‐67, HB‐EGF and TNF‐α. Taken together, these results support the hypothesis that excessive self‐DNA fragments induce hyperproliferation and impair differentiation by activating inflammatory responses in keratinocytes. Thus, an over‐abundance of genomic DNA fragments resulting from defective keratinocyte denucleation may create a vicious cycle ultimately leading to the development of psoriasis (Fig. 6). Additionally, dsDNA‐induced proinflammatory mediators may also recruit lymphocytes to the involved site, together driving skin autoimmunity to the verge of a break‐out.
Fig. 6.
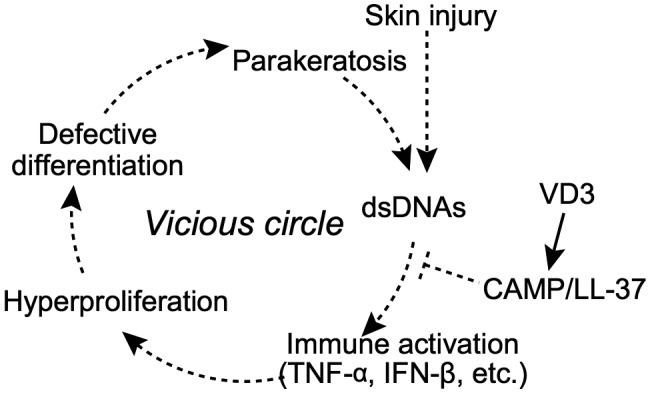
A model of self‐dsDNA fragments as a potential factor aggravating psoriasis. Fragments of self‐dsDNA putatively derived from skin injury promote the growth and inhibit the differentiation of human epidermal keratinocytes by activating an inflammatory cascade characterized by over‐expression of tumour necrosis factor (TNF)‐α and interferon (IFN)‐β. As a result, keratinocytes develop a psoriasis‐like phenotype that exacerbates the progression of parakeratosis associated with excessive genomic DNA fragments. Upon skin irritation/injury, DNA fragments that leak out of parakeratotic keratinocytes further exacerbate inflammation in the lesion, thus forming a vicious cycle resulting in psoriasis. Moreover, active vitamin D analogues may exert a protective effect on the DNA fragments‐mediated skin inflammatory cascade via cathelicidin anti‐microbial peptide (CAMP)/LL‐37.
CAMP has a seemingly tangled role in psoriasis. On one hand, LL‐37 is over‐expressed in inflamed skin, and can form a complex with self‐DNA to activate pCD [16]. On the other hand, CAMP might be a central factor in psoriasis treatment with vitamin D analogues. Topical treatment with vitamin D analogues decreases proinflammatory cytokine levels in psoriatic lesions as well as strongly increasing CAMP expression [32]. An anti‐inflammatory effect of LL‐37 on the absent in melanoma 2 (AIM2) inflammasome pathway probably contributes to this therapeutic effect [43]. The active cathelicidin peptide LL‐37 forms an amphipathic, cationic α‐helix in aqueous solutions that enables its incorporation into lipid bilayers [44] and subsequent binding to nucleic acids, guiding them to their intracellular pathways and destinations [34]. In our experiments, LL‐37 differentially modulated dsDNA‐induced TNFA and IFNB at different concentrations (Fig. 5c). In contrast, VD3 suppressed dsDNA fragment‐induced TNFA and IFNB expression in a dose‐dependent manner. We speculate that VD3 primarily increases the intracellular accumulation of endogenous LL‐37 to somehow shield cytosolic dsDNA, whereas exogenously administrated LL‐37 primarily promotes the uptake of extracellular dsDNA into cells. The overall effect of CAMP on skin inflammation may depend upon where LL‐37 binds self‐DNA and a joint action of various intracellular DNA‐sensing pathways, such as TLR‐9 signalling and AIM2 inflammasome pathways. Elucidation of the role of self‐dsDNA fragments in psoriasis will not only encourage new therapeutic strategies that target excess dsDNA or the components of cytosolic dsDNA signalling pathways, but also broaden our understanding of the current therapies for psoriasis.
In summary, the present results suggest that fragments of genomic dsDNA present in the epidermis are potential triggers of skin inflammation during the development of psoriasis. Furthermore, we showed that VD3 treatment exerts a protective effect and suppresses dsDNA fragment‐induced inflammatory cytokines via CAMP/LL‐37.
Disclosure
All authors have no conflicts of interest to disclose.
Author contributions
All authors approved the final version to be published. K. S. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study conception and design: Y. L., T. H., A. K., N. I., T. K. and K. S. Acquisition of data: Y. L., T. H., Y. I., S. S. and T. K. Analysis and interpretation of data: Y. L., T. H., Y. I. S. S. and K. S. Manuscript drafting and revising: Y. L., T. H., A. K. and K. S.
Supporting information
Fig. S1. TUNEL‐positive dsDNA fragments in psoriatic lesions in association with keratinocytes expressing Ki‐67 and HB‐EGF. Skin tissue sections of psoriatic lesions and non‐involved areas were subjected to hematoxylin and eosin or TUNEL staining together with immunostaining of Ki‐67 or HB‐EGF. Bar = 100 μm. The merged images are presented in Fig. 4.
Fig. S2. TUNEL‐positive dsDNA fragments are present in psoriatic lesions and are associated with keratinocytes expressing Ki‐67, HB‐EGF and TNF‐α. Skin tissue sections of psoriatic lesions were subjected to hematoxylin and eosin (H&E) staining or TUNEL staining (green) in combination with immunofluorescence staining of Ki‐67, HB‐EGF or TNF‐α (red). Bars = 100 μm.
Table S1. 534 Guide RNA sequences used in the CRISP/Cas9 system for CAMP knockout.
Table S2. 537 Primers used for PCR analysis.
Acknowledgements
This study was supported by The LEO Foundation (grant LF16038 to K. S.) and Sasakawa Scientific Research Grant from the Japan Science Society (#28‐403 to Y. L.).
Contributor Information
Y. Luo, Email: yuqianluo31@foxmail.com, Email: koichis0923@med.teikyo-u.ac.jp.
K. Suzuki, Email: yuqianluo31@foxmail.com, Email: koichis0923@med.teikyo-u.ac.jp.
References
- 1. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509. [DOI] [PubMed] [Google Scholar]
- 2. Lippens S, Denecker G, Ovaere P et al Death penalty for keratinocytes: apoptosis versus cornification. Cell Death Diff 2005; 12(Suppl 2):1497–508. [DOI] [PubMed] [Google Scholar]
- 3. Johannesson A, Hammar H. Corneocyte morphology and formation rate in lichen planus and experimental parakeratosis in subjects with and without psoriasis. Acta Derm Venereol 1982; 62:463–70. [PubMed] [Google Scholar]
- 4. Baliwag J, Barnes DH, Johnston A. Cytokines in psoriasis. Cytokine 2015; 73:342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nestle FO, Conrad C, Tun‐Kyi A et al Plasmacytoid predendritic cells initiate psoriasis through interferon‐alpha production. J Exp Med 2005; 202:135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature 2007; 445:866–73. [DOI] [PubMed] [Google Scholar]
- 7. Hueber W, Patel DD, Dryja T et al Effects of AIN457, a fully human antibody to interleukin‐17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med 2010; 2:52ra72. [DOI] [PubMed] [Google Scholar]
- 8. Prieto‐Perez R, Cabaleiro T, Dauden E et al Genetics of psoriasis and pharmacogenetics of biological drugs. Autoimmune Dis 2013; 2013:613086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ishii KJ, Coban C, Kato H et al A Toll‐like receptor‐independent antiviral response induced by double‐stranded B‐form DNA. Nat Immunol 2006; 7:40–8. [DOI] [PubMed] [Google Scholar]
- 10. Ishii KJ, Suzuki K, Coban C et al Genomic DNA released by dying cells induces the maturation of APCs. J Immunol 2001; 167:2602–7. [DOI] [PubMed] [Google Scholar]
- 11. Kawashima A, Tanigawa K, Akama T et al Fragments of genomic DNA released by injured cells activate innate immunity and suppress endocrine function in the thyroid. Endocrinology 2011; 152:1702–12. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki K, Mori A, Ishii KJ et al Activation of target‐tissue immune‐recognition molecules by double‐stranded polynucleotides. Proc Natl Acad Sci USA 1999; 96:2285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Napirei M, Karsunky H, Zevnik B et al Features of systemic lupus erythematosus in Dnase1‐deficient mice. Nat Genet 2000; 25:177–81. [DOI] [PubMed] [Google Scholar]
- 14. Yasutomo K, Horiuchi T, Kagami S et al Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet 2001; 28:313–4. [DOI] [PubMed] [Google Scholar]
- 15. Kawashima A, Tanigawa K, Akama T et al Innate immune activation and thyroid autoimmunity. J Clin Endocrinol Metab 2011; 96:3661–71. [DOI] [PubMed] [Google Scholar]
- 16. Lande R, Gregorio J, Facchinetti V et al Plasmacytoid dendritic cells sense self‐DNA coupled with antimicrobial peptide. Nature 2007; 449:564–9. [DOI] [PubMed] [Google Scholar]
- 17. Ganguly D, Chamilos G, Lande R et al Self‐RNA‐antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 2009; 206:1983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ran FA, Hsu PD, Wright J et al Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 2013; 8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishido Y, Yamazaki K, Kammori M et al Thyroglobulin suppresses thyroid‐specific gene expression in cultures of normal but not neoplastic human thyroid follicular cells. J Clin Endocrinol Metab 2014; 99:E694–702. [DOI] [PubMed] [Google Scholar]
- 20. Winterfield L, Menter A. Psoriasis and its treatment with infliximab‐mediated tumor necrosis factor alpha blockade. Dermatol Clin 2004; 22:437–47, ix. [DOI] [PubMed] [Google Scholar]
- 21. Elder JT, Fisher GJ, Lindquist PB et al Overexpression of transforming growth factor alpha in psoriatic epidermis. Science 1989; 243:811–4. [DOI] [PubMed] [Google Scholar]
- 22. Finzi E, Harkins R, Horn T. TGF‐alpha is widely expressed in differentiated as well as hyperproliferative skin epithelium. J Invest Dermatol 1991; 96:328–32. [DOI] [PubMed] [Google Scholar]
- 23. Mascia F, Mariani V, Girolomoni G et al Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol 2003; 163:303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Romanowska M, al Yacoub N, Seidel H, et al PPARdelta enhances keratinocyte proliferation in psoriasis and induces heparin‐binding EGF‐like growth factor. J Invest Dermatol 2008; 128:110–24. [DOI] [PubMed] [Google Scholar]
- 25. Yoshida A, Kanno H, Watabe D et al The role of heparin‐binding EGF‐like growth factor and amphiregulin in the epidermal proliferation of psoriasis in cooperation with TNFalpha. Arch Dermatol Res 2008; 300:37–45. [DOI] [PubMed] [Google Scholar]
- 26. Richardson RJ, Dixon J, Malhotra S et al Irf6 is a key determinant of the keratinocyte proliferation‐differentiation switch. Nat Genet 2006; 38:1329–34. [DOI] [PubMed] [Google Scholar]
- 27. Lopez RG, Garcia‐Silva S, Moore SJ et al C/EBPalpha and beta couple interfollicular keratinocyte proliferation arrest to commitment and terminal differentiation. Nat Cell Biol 2009; 11:1181–90. [DOI] [PubMed] [Google Scholar]
- 28. Hara T, Miyazaki M, Hakuno F et al PKCeta promotes a proliferation to differentiation switch in keratinocytes via upregulation of p27Kip1 mRNA through suppression of JNK/c‐Jun signaling under stress conditions. Cell Death Dis 2011; 2:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Poumay Y, Pittelkow MR. Cell density and culture factors regulate keratinocyte commitment to differentiation and expression of suprabasal K1/K10 keratins. J Invest Dermatol 1995; 104:271–6. [DOI] [PubMed] [Google Scholar]
- 30. Morizane S, Gallo RL. Antimicrobial peptides in the pathogenesis of psoriasis. J Dermatol 2012; 39:225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vahavihu K, Ala‐Houhala M, Peric M et al Narrowband ultraviolet B treatment improves vitamin D balance and alters antimicrobial peptide expression in skin lesions of psoriasis and atopic dermatitis. Br J Dermatol 2010; 163:321–8. [DOI] [PubMed] [Google Scholar]
- 32. Peric M, Koglin S, Dombrowski Y et al Vitamin D analogs differentially control antimicrobial peptide/‘alarmin’ expression in psoriasis. PLOS ONE 2009; 4:e6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Fits L, van der Wel LI, Laman JD et al In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon‐alpha sensitivity is unaltered. J Invest Dermatol 2004; 122:51–60. [DOI] [PubMed] [Google Scholar]
- 34. Lande R, Gregorio J, Facchinetti V et al Plasmacytoid dendritic cells sense self‐DNA coupled with antimicrobial peptide. Nature 2007; 449:564–9. [DOI] [PubMed] [Google Scholar]
- 35. Roberson ED, Bowcock AM. Psoriasis genetics: breaking the barrier. Trends Genet 2010; 26:415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wolf R, Orion E, Ruocco E et al Abnormal epidermal barrier in the pathogenesis of psoriasis. Clin Dermatol 2012; 30:323–8. [DOI] [PubMed] [Google Scholar]
- 37. Coffey RJ Jr, Derynck R, Wilcox JN et al Production and auto‐induction of transforming growth factor‐alpha in human keratinocytes. Nature 1987; 328:817–20. [DOI] [PubMed] [Google Scholar]
- 38. Piepkorn M, Pittelkow MR, Cook PW. Autocrine regulation of keratinocytes: the emerging role of heparin‐binding, epidermal growth factor‐related growth factors. J Invest Dermatol 1998; 111:715–21. [DOI] [PubMed] [Google Scholar]
- 39. Smith PC, Guerrero J, Tobar N et al Tumor necrosis factor‐alpha‐stimulated membrane type 1‐matrix metalloproteinase production is modulated by epidermal growth factor receptor signaling in human gingival fibroblasts. J Periodont Res 2009; 44:73–80. [DOI] [PubMed] [Google Scholar]
- 40. Valyi‐Nagy I, Jensen PJ, Albelda SM et al Cytokine‐induced expression of transforming growth factor‐alpha and the epidermal growth factor receptor in neonatal skin explants. J Invest Dermatol 1992; 99:350–6. [DOI] [PubMed] [Google Scholar]
- 41. Yoshizumi M, Kourembanas S, Temizer DH et al Tumor necrosis factor increases transcription of the heparin‐binding epidermal growth factor‐like growth factor gene in vascular endothelial cells. J Biol Chem 1992; 267:9467–9. [PubMed] [Google Scholar]
- 42. Fanoni D, Venegoni L, Vergani B et al Evidence for a role of autoinflammation in early‐phase psoriasis. Clin Exp Immunol 2019; 198:283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dombrowski Y, Schauber J. Cathelicidin LL‐37: a defense molecule with a potential role in psoriasis pathogenesis. Exp Dermatol 2012; 21:327–30. [DOI] [PubMed] [Google Scholar]
- 44. Braff MH, Gallo RL. Antimicrobial peptides: an essential component of the skin defensive barrier. Curr Top Microbiol Immunol 2006; 306:91–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. TUNEL‐positive dsDNA fragments in psoriatic lesions in association with keratinocytes expressing Ki‐67 and HB‐EGF. Skin tissue sections of psoriatic lesions and non‐involved areas were subjected to hematoxylin and eosin or TUNEL staining together with immunostaining of Ki‐67 or HB‐EGF. Bar = 100 μm. The merged images are presented in Fig. 4.
Fig. S2. TUNEL‐positive dsDNA fragments are present in psoriatic lesions and are associated with keratinocytes expressing Ki‐67, HB‐EGF and TNF‐α. Skin tissue sections of psoriatic lesions were subjected to hematoxylin and eosin (H&E) staining or TUNEL staining (green) in combination with immunofluorescence staining of Ki‐67, HB‐EGF or TNF‐α (red). Bars = 100 μm.
Table S1. 534 Guide RNA sequences used in the CRISP/Cas9 system for CAMP knockout.
Table S2. 537 Primers used for PCR analysis.