

Abstract
Fatty acids and glucose are the main substrates for myocardial energy provision. Under physiologic conditions, there is a distinct and finely tuned balance between the utilization of these substrates. Using the non-ischemic heart as an example, we discuss that upon stress this substrate balance is upset resulting in an over-reliance on either fatty acids or glucose, and that chronic fuel shifts towards a single type of substrate appear to be linked with cardiac dysfunction. These observations suggest that interventions aimed at re-balancing a tilted substrate preference towards an appropriate mix of substrates may result in restoration of cardiac contractile performance. Examples of manipulating cellular substrate uptake as a means to re-balance fuel supply, being associated with mended cardiac function underscore this concept. We also address the molecular mechanisms underlying the apparent need for a fatty acid–glucose fuel balance. We propose that re-balancing cellular fuel supply, in particular with respect to fatty acids and glucose, may be an effective strategy to treat the failing heart.
Keywords: Long-chain fatty acids, Glucose, Energy homeostasis, Substrate balance, Cardiac contractile function, Heart failure, Diabetes
1. Introduction
An uninterrupted availability of chemical energy in the form of energy-rich phosphate bonds (i.e., adenosine triphosphate, ATP) is required for the physiologic function of all cells. This especially applies to energy conversion in the heart. For example, at any given moment the cellular energy reserve of the heart can maintain contractile activity for only 10 s (ATP) or up to 1 min (creatine phosphate) and, therefore, may not be adequate to support prolonged changes in demand [1,2]. As a result, the continuous and unimpeded production of ATP is vital for cardiac function. This requires sufficient availability/delivery of metabolic substrates, as well as sufficient myocardial oxidative capacity.
The tight coupling of ATP production and utilization, as it is especially apparent in the heart, includes the ability of cells to select, for a given environment, the most appropriate substrate for ATP production. The precise requirements for maintaining such flexibility reside upstream from ATP in the network of energy substrate metabolism. The mechanisms underlying metabolic flexibility are not yet fully understood, but are important to decipher as they may provide targets for metabolic intervention aimed at securing sufficient ATP production to sustain organ function both in response to short and long-term external stimuli.
In this paper, focusing on non-ischemic heart disease, we examine available evidence suggesting that optimal cardiac performance is dependent on a balanced utilization of substrates (e.g., fatty acids and glucose). Based on reported findings, we propose that interventions aimed at manipulating cellular substrate uptake as a means to re-balance fuel supply will help restore impaired organ function, and hold promise as a strategy to treat metabolic disease.
2. Myocardial energy metabolism and contractile function
The heart is a metabolic omnivore because it utilizes virtually all substrates to ensure optimal contractile performance [3]. For the healthy heart, these substrates include long-chain fatty acids (hereafter referred to as ‘fatty acids’; average contribution 60%) and carbohydrates (glucose 30% and lactate 10%), with minor contributions from ketone bodies and amino acids. At any specific time, however, the actual contribution of each of these substrates could be quite different, depending on changes in substrate availability governed by specific (patho)physiologic conditions (e.g., changes during the course of a day) [4]. For both fatty acids and glucose, their supply to the heart and eventual myocellular utilization for energy provision is determined at multiple levels [4,5]. The main steps and regulatory mechanisms involved include (i) substrate uptake into cardiomyocytes by facilitated diffusion through specific membrane-associated proteins (CD36 for fatty acids, GLUT1 and GLUT4 for glucose) [6,7], and (ii) mitochondrial oxidation (whereby substrate preference is determined by mechanisms such as the Randle glucose–fatty acid cycle) [8]. The latter indicates that the intracellular availability of substrates is an important parameter governing their use for energy provision. Additional control is exerted by subcellular localization of metabolites; for instance, allosteric inhibition of carnitine palmitoyl-transferase-1 (CPT-1) by malonyl-CoA limits entry of acyl-CoA esters into the mitochondrial matrix, thus attenuating β-oxidation.
Various signaling pathways coordinate energy metabolism and contractile function in the heart [9]. For example, both insulin- and contraction-induced signaling acutely affect the quantitative utilization of fatty acids and glucose. Moreover, myocardial substrate utilization capacity can be chronically modulated through transcriptional events [10].
In heart failure, which is defined as a myocardial derangement causing systolic and/or diastolic ventricular dysfunction, substrate metabolism is changed towards the utilization of one preferred substrate rather than a mixture of substrates. Although in the hypoxic/ischemic heart the lack of oxygen dictates anaerobic glycolysis as the main pathway for energy provision, in the pressure-overloaded heart and the heart in type 2 diabetes mellitus, which each show a high prevalence [11,12], the specific alterations in substrate preference towards either glucose or fatty acid utilization may be a suitable target for intervention, as will be discussed below.
3. The need for a balance between fatty acid and glucose utilization
Increasing evidence suggests that the heart operates optimally when it uses a mixture of energy providing substrates, especially with respect to fatty acids and glucose [3]. If the balance of substrates is tilted, either towards a predominant use of fatty acids (at the expense of glucose) or a predominant use of glucose (at the expense of fatty acids), this change in substrate preference is associated with impaired cardiac contractile function (Fig. 1). Furthermore, in case of the preferred utilization of one substrate over another, there is a risk that the heart suffers from fuel toxicity, i.e., lipo- or glucotoxicity, each being conditions that elicit major impairments of cardiac functioning [13,14]. Importantly, the association between a tilted substrate balance and impaired cardiac contractile function appears evident from both perspectives, and regardless of the cause of the disease (metabolic versus non-metabolic). As a result, the notion arises that chronic changes in cardiac performance are linked to a change in the fatty acid over glucose substrate balance. Examples of human cardiac diseases of either metabolic or non-metabolic origin and the associated change in the fatty acid over glucose substrate balance illustrate this concept (Table 1).
Fig. 1.
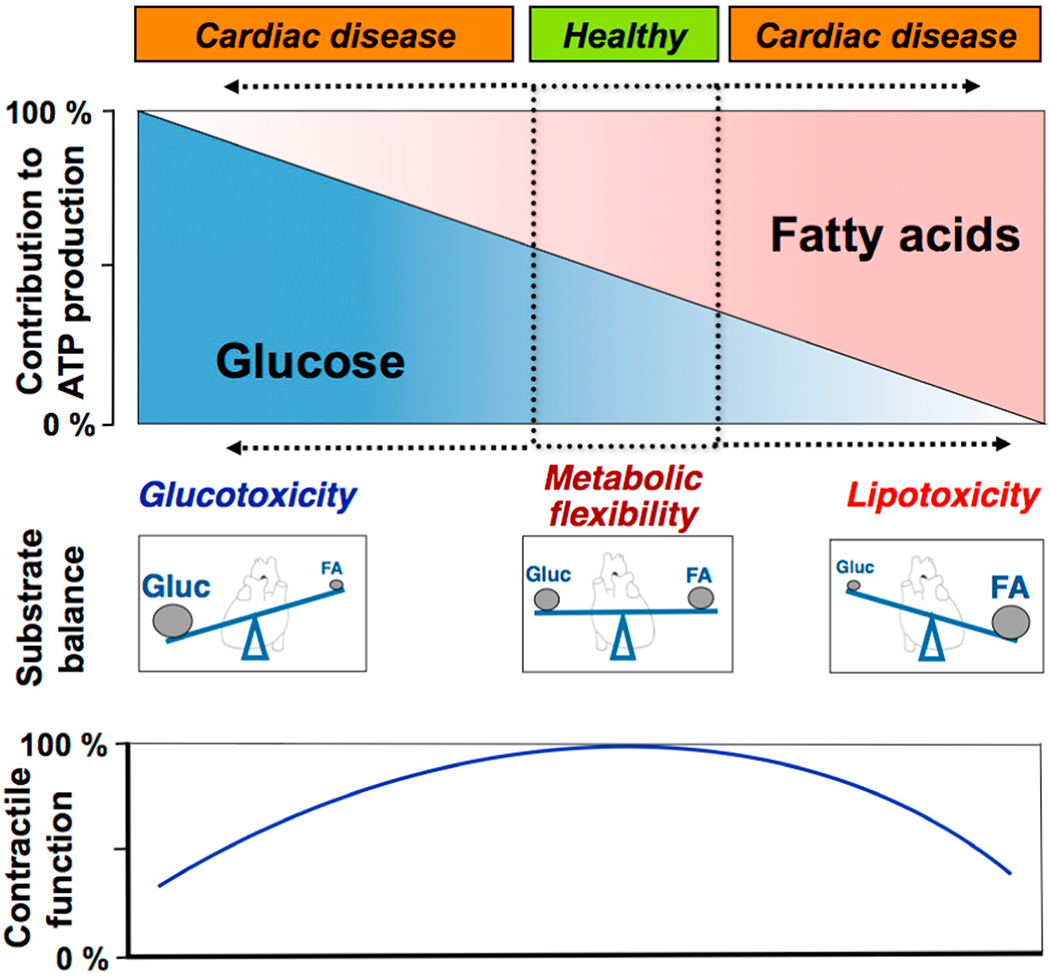
Schematic presentation of the relation between cardiac contractile function and the relative contributions of (long-chain) fatty acids and glucose to overall myocardial ATP production. Minor contributions from other substrates, such as lactate, ketone bodies and amino acids, are not shown. Under healthy conditions, cardiac muscle utilizes a mixture of metabolic substrates, so that the contributions of glucose and of fatty acids are of similar magnitude. However, when this balance of metabolic substrates shifts towards the predominant utilization of a single substrate, i.e., either mostly glucose or mostly fatty acids, this change is accompanied by impaired contractile function. FA, (long-chain) fatty acids; Glue, glucose.
(Adapted from [3]).
Table 1.
Association between a tilted cardiac substrate balance and chronically impaired contractile function.
| • Substrate balance with fatty acids < glucose (predominant glucose utilization) | ||
|---|---|---|
| - Metabolic cause | Inborn error of fatty acid metabolism | [19] |
| (uptake or oxidation) | [69] | |
| - Non-metabolic cause | Pressure-overload hypertrophy | [70] |
| [15] | ||
| [65] | ||
| Atrophy and pulmonary insufficiency (→hypoxia) | [71] | |
| Anemia (→hypoxia) | [72] | |
| Impaired coronary blood flow (→ischemia) | [73] | |
| [74] | ||
| Chronic heart failure | [26] | |
|
| ||
| • Substrate balance with fatty acids > glucose (predominant fatty acid utilization) | ||
| - Metabolic cause | Diabetes type 1, diabetes type 2 | [21] |
| Fatty acid overload (high fat feeding; obesity) | [75] | |
| [14] | ||
| Inborn error of glucose metabolism (uptake or oxidation) | [22] | |
| - Non-metabolic cause | Chronic viral myocarditis (pathogen-induced cardiomyopathy) | [24] |
Examples are taken from the literature (clinical studies and/or reviews). References are indicated in the right column.
Generally, pressure overload-induced cardiac hypertrophy results in a substrate switch towards predominant glucose utilization, and in the chronic setting is associated with worsened cardiac contractile function [15]. Conversely, when the uptake and/or utilization of fatty acids by the heart is restricted, leading to an increased glucose utilization, such condition appears to be associated with impaired contractile function. Examples of the latter are pharmacological blockade of fatty acid oxidation [16,17] and inborn errors of fatty acid oxidation [18–20].
A substrate switch towards predominant fatty acid utilization is seen upon high-fat feeding and obesity, and is well-documented to be accompanied with aberrant control of cardiac metabolism by insulin and contractile dysfunction (diabetic cardiomyopathy) [7,21]. Likewise, an inborn error of glucose metabolism results in a similar substrate switch together with cardiac dysfunction [22]. An example from the other perspective is chronic myocarditis upon pathogen infection that is associated with a predominant utilization of fatty acids [23,24].
Experimental studies in rodents in which metabolic pathways have been genetically modified have provided further evidence for the existence of a close association between marked changes in cardiac substrate preference (fatty acids versus glucose) and cardiac (mal)function [reviewed in [5,25]]. These studies are summarized in Suppl. Fig. 1.
4. Interventions targeting metabolism
The notion that optimal pump function of the heart is dependent on a balanced utilization of fatty acids and glucose for myocardial energy provision suggests that in case of a shifted substrate balance and, hence, impaired contractile function (i) any intervention that will further upset this fuel balance would aggravate cardiac function, while (ii) any intervention that will help rectify the fuel balance may lead to a marked improvement or even full recovery of cardiac contractile performance. Indeed, patients with advanced heart failure (see e.g. [26]) and studies in experimental heart failure models (Table 2) provide convincing evidence for both concepts.
Table 2.
Selected examples of experimental animal intervention studies causing alterations in the cardiac fatty acid–glucose fuel balance.
| Model | Substrate metabolism | Intervention | Metabolism after intervention | Cardiac function | Reference |
|---|---|---|---|---|---|
| • Aggravation of cardiac function | |||||
| CD36 null mice | FAO↓ Gluc↑ | Pressure-overload (TAC) | FAO↓↓ Gluc↑↑ | Worsened function | [28] |
| [27] | |||||
| PGC1α deletion | FAO↓ Gluc↑ | Pressure-overload (TAC) | FAO↓↓ Gluc↑↑ | Worsened function | [76] |
| CPT-1b deletion | FAO↓ Gluc↑ | Pressure-overload (TAC) | FAO↓↓ Gluc↑↑ | Worsened function | [77] |
| • Recovery of cardiac function | |||||
| PPARα overexpr. mice | FAO↑ Gluc↓ | Ablation of CD36 | FAO= Glu= | Function reinstalled | [30] |
| Diabetic db/db mice | FAO↑ Gluc↓ | Pressure-overload (TAC) | FAO= Gluc= | Function maintained | [78] |
| ACC2−/− mice | FAO↑ Gluc↓ | Pressure-overload (TAC) | FAO= Gluc= | Function maintained | [79,80] |
| Post-myocardial infarction rats | FAO↓ Gluc↑ | High-fat diet | FAO= Gluc= | Function maintained | [81] |
During the development of cardiac hypertrophy the heart increases its reliance on glucose to eventually use primarily glucose for ATP production. The subsequent progression from compensated cardiac hypertrophy to heart failure, which is characterized by a further reliance on glucose, is accelerated when cardiac fatty acid utilization is severely hampered by ablation of the sarcolemmal fatty acid transporter CD36 [27]. Similarly, subjecting CD36 null mice (in which the fuel balance already has been shifted towards increased glucose utilization) to mechanical stress (transverse aortic constriction) elicits a marked further impairment of contractile function [28].
Several studies have documented that interventions correcting aberrations of the fatty acid–glucose fuel balance have a beneficial effect on cardiac contractile performance. When mice with a pressure overload-induced cardiac hypertrophy and an associated shifted substrate balance towards increased glucose utilization were subjected to a dietary intervention with high-fat, both substrate balance and contractile function normalized [28]. Similarly, mice with a cardiac specific overexpression of protein kinase-D1, which leads to increased utilization of glucose and cardiac hypertrophy, also could be rescued by feeding a high-fat diet, upon which the substrate balance and contractile function each normalized [29]. These observations suggest that increasing cardiac fatty acid supply and utilization may be beneficial in hypertrophic cardiomyopathy.
Likewise, restoring the substrate balance in a mouse model displaying a dominance of fatty acids over glucose utilization was examined by Yang and co-workers on mice with cardiac-restricted overexpression of nuclear receptor PPARα exhibiting myocardial lipid accumulation and contractile dysfunction [30]. Crossing of these mice with mice deficient in CD36 led to an increase in cardiac glucose uptake and oxidation together with an re-balanced fatty acid-glucose ratio, and a rescue of cardiac function [30].
Taken together, these experimental findings underscore the intimate association between the cardiac substrate balance and cardiac function and suggest that restoring the balance is an effective therapy to improve cardiac contractile performance.
5. Molecular mechanism explaining the mandatory fatty acid–glucose fuel balance
It is not fully understood why a balanced mixture of metabolic substrates, in particular fatty acids and glucose, is a pivotal requirement for optimal cardiac function. The underlying molecular mechanism may involve several possible parameters.
5.1. The need for metabolic flexibility to sustain contraction
Securing metabolic flexibility requires a very rapid alteration of the type of substrate utilized. For instance, the circadian rhythm of changes in substrate delivery and energetic demand imposes the requirement to continuously adapt substrate metabolism in order to maintain optimal ATP production and utilization [31]. According to the ‘futile cycle’ hypothesis of Newsholme, each of the metabolic processes involved should run idle (a certain basal rate) so as to increase instantaneously when needed [32,33]. As a result, rates of both fatty acid and glucose utilization pathways should proceed at least at a certain minimum flux. In the event that cardiac energy provision be maintained from predominantly a single type of metabolic substrate, e.g. glucose, we hypothesize that in the chronic setting this will lead to downregulation of enzymes involved in fatty acid metabolism (e.g., as found in pressure overload-induced hypertrophy [34]), thereby impairing a rapid increase in flux through the fatty acid oxidation pathway, and thus markedly affecting cardiac fuel flexibility.
5.2. Subsidiary metabolic pathways require fatty acids and glucose
Apart from their utilization as major substrates for ATP production, fatty acids and glucose also serve in various other metabolic pathways. This includes the conversion of fatty acids into various lipid species, such as phospholipids, ceramides, diacylglycerols, etc., and the funneling of glucose into the pentose phosphate pathway and the hexosamine biosynthetic pathway (reviewed in [13,35,36]). Of special relevance is the role of fatty acids and glucose in anaplerosis of the Krebs cycle, i.e., the replenishment of the pool of intermediates of this metabolic cycle. Although the role of anaplerosis is well recognized as a major biosynthetic pathway in liver [37], it is only beginning to be appreciated in heart [38,39].
Replenishment of Krebs cycle intermediates is essential because of their continuous efflux from mitochondria for anabolic processes taking place in the cytosol. The flux through the Krebs cycle, which governs the maximal ATP production rate, depends on the presence of balanced amounts of each of the constituting intermediates. Distinction is made between the first span of the Krebs cycle (efflux of citrate and 2-oxoglutarate) and the second span (efflux of succinate and malate) with mitochondrial citrate efflux being quantitatively most important [40]. The major anaplerotic reaction is that of pyruvate carboxylation to produce oxaloacetate, which subsequently reacts with acetyl-CoA to form citrate, thereby directly replacing the loss of citrate. Fatty acids serve a role in the latter process as major provider of acetyl-CoA [38]. However, the regulatory mechanisms underlying the maintenance of the total pool and of individual Krebs cycle intermediates in heart is highly complex and also involves roles for specific amino acids (glutamine, branched-chain amino acids) and odd-chain fatty acids, while citrate released into the cytosol acts as an allosteric inhibitor of phosphofructokinase and as a signaling compound affecting both fatty acid and glucose utilization [41]. A recent study [42] supports a role for the pool of Krebs cycle intermediates in cardiac metabolic flexibility and that this pool is influenced by the fatty acid over glucose fuel balance.
In conclusion, a limited supply of Krebs cycle intermediates in cardiomyocytes will negatively impact on mitochondrial capacity and thus hamper the tuning of energy demand to provision. The fact that both fatty acids and glucose are required for replenishing Krebs cycle intermediates may contribute to the need for their balanced cellular utilization.
5.3. Post-translational protein modification involves both fatty acids and glucose
Most proteins undergo reversible post-translational modifications, often on multiple sites, which may regulate their subcellular localization, stability, activity, and interactions with other proteins [43]. Post-translational protein modifications involving metabolites include phosphorylation, acetylation, acylation (e.g., myristoylation, palmitoylation), and glycosylation (e.g., O-GlcNAcylation) [43]. Because fatty acids and glucose are key metabolic substrates for these modifications, any dysbalance in their availability for these reactions may impact on the proper post-translational modification of selected proteins (especially transcription factors such as PGC-1α and PPARα), thereby potentially affecting cardiac function [44]. For instance, palmitoylation of metabolic transcription factors has been reported to alter differentiation programs in (non-cardiac) cells [45]. It is important to stress that the key enzymes involved in post-translational modifications (e.g., protein acyl transferase, O-GlcNAc transferase) are constitutively active [46], so that changes in post-translational modifications are mostly substrate (i.e., palmitate, glucose) driven. Hence, changes in glucose or lipid-induced post-translational modifications of metabolic transcription factors and their consequences are part of the overall pleiotropy of toxic actions of both substrates.
5.4. Lipotoxicity and glucotoxicity
A predominant ATP production from either fatty acids or glucose renders the heart at risk for so-called lipotoxicity and glucotoxicity, respectively. Lipotoxicity refers to the pathological accumulation of lipid intermediates which may lead to cellular dysfunction [47]. When there is a mismatch between fatty acid uptake and oxidation, the excess fatty acids may be converted into lipid intermediates such as diacylglyceroles and ceramides, which impair cellular function (e.g., through inhibition of insulin signaling and altering intracellular Ca2+ dynamics) and even may cause cellular injury [26,47,48].
During excess glucose availability, glucose 6-phosphate accumulates due to a mismatch with glucose oxidation. This increase in glucose 6-phosphate leads to hexokinase-II disattachment from the mechanistic target of rapamycin (mTOR), mTOR activation, and increased protein synthesis resulting in cardiac hypertrophy [49,50]. Moreover, long-term increases in glucose supply and utilization are associated with a shift to a fetal transcriptional program in adult cardiomyocytes. This fetal switch is often maladaptive whereby contractile force decreases [44]. Also, a main destructive combination of events involved in glucotoxicity is the combination of ROS formation and increased glycation [51].
6. Role of alternative substrates
The omnivoric character of the heart implies that alternative substrates for cardiac energy provision, in particular ketone bodies and amino acids, may also be involved in maintaining a distinct fatty acid–glucose fuel balance or, alternatively, mitigate the consequences of a shifted fuel balance. For instance, should the substrate preference change towards increased fatty acid utilization because of impaired glucose uptake (as in insulin resistance) these alternative substrates may help substitute for the limited intracellular glucose availability thereby preventing excessive fatty acid uptake and lipotoxicity. In the normal heart the contribution of these alternative substrates to total ATP production is limited with ketone bodies and amino acids contributing at most to 10% and 2%, respectively [52]. However, under specific conditions (e.g., prolonged fasting, exercise), or in disease states (e.g., diabetes, heart failure) their contribution to myocardial energy provision may become significant [34,53,54]. Importantly, shifts in ketone body and amino acid metabolism may result in generation of intracellular signaling molecules influencing contractile function.
Ketone bodies are readily oxidized in proportion to their delivery and in priority to fatty acids and glucose [55]. Ketone bodies are not subject to transport regulation and suppress both fatty acid and glucose oxidation via competition of the produced acetyl-CoA for Krebs cycle intermediates [56,57]. This suggests that elevated utilization of ketone bodies does not affect the fatty acid-glucose fuel balance. In addition, ketone bodies may deplete Krebs cycle intermediates (decreased anaplerosis) leading to a decrease in mitochondrial oxidative metabolism [58].
The significance of amino acids for cardiac energy metabolism extends beyond ATP production, and includes protein turnover, anaplerosis to supplement Krebs cycle intermediates, substrate competition, and specific signaling roles. Branched chain amino acids (BCAAs; leucine, isoleucine, valine) serve as an excellent example. The heart has a high demand for amino acids because all cellular proteins are renewed within 30 days [59]. BCAAs influence protein turnover through activation of mTOR complex-1 (mTORC1) which promotes protein synthesis and attenuates autophagy [60]. Additionally, BCAA repress general control nonderepressible 2-kinase (GCN2) thereby relieving inhibition of translation initiation [61]. During heart failure there appears to be an imbalance between BCAA availability (increased circulating levels) and myocardial utilization (due to repression of branched chain 2-oxo acid dehydrogenase), resulting in an accumulation of BCAAs and their 2-oxo acids in the heart (observed in both humans and animal models) [62]. This in turn would be anticipated to promote adverse remodeling, through simultaneous augmentation of protein synthesis and attenuation of autophagy. Indeed, augmentation of BCAA utilization (through pharmacologic activation of branched chain 2-oxo acid dehydrogenase) protects against heart failure progression [62]. Similar to ketone bodies, BCAAs will diminish the oxidation of fatty acids and glucose, not only via competition of the produced acetyl-CoA for Krebs cycle intermediates, but also through inhibition of mitochondrial function [62]. Interestingly, recent studies in skeletal muscle suggest an important role of BCAAs in fatty acid uptake. More specifically, a muscle derived valine metabolite (3-hydroxyisobutyrate, 3-HIB) acts in a paracrine manner, promoting trans-endothelial movement of fatty acids, subsequently increasing muscle cell uptake [63]. Given that 3-HIB is downstream of branched chain 2-oxo acid dehydrogenase, we speculate that decreased 3-HIB levels during heart failure may selectively diminish fatty acid uptake, thereby contributing towards the maladaptive shift towards increased glucose utilization.
Taken together, given the multiple effects of both ketone bodies and certain amino acids on cardiac energy metabolism, it currently remains unclear whether changes in their use by the diseased heart are beneficial or detrimental for cardiac efficiency and functioning [34,52,64].
7. Conclusion and future outlook
The unimpeded flux through the pathways of energy substrate metabolism is vital for the physiologic function of all tissues. In the heart, various metabolic substrates are used for ATP production, of which fatty acids and glucose are quantitatively most important. Data available for the heart indicate that rapid switching between these substrates, elicited by endogenous or exogenous factors such as diet, diurnal rhythm, neurohumoral changes and exercise (i.e., an altered metabolic milieu), are well tolerated and on short-term remain inconsequential for cardiac contractile performance. This virtually instantaneous switching between substrates is referred to as ‘metabolic flexibility’. However, in case such external influences are persisting, the cardiac fuel balance is permanently shifted towards the utilization of a predominant type of substrate, in particular either fatty acids or glucose, leading to an impaired metabolic flexibility and an impairment of contractile function (Fig. 2). The mechanism underlying this phenomenon most likely relates to the notion that the preferential utilization of a single type of substrate causes alterations in subsidiary metabolic pathways, post-translational protein modifications and gene expression, together making the heart more vulnerable for the short-term external influences mentioned above. These findings are expected to apply also to other cell types with an active energy substrate metabolism, such as skeletal muscle and liver. In conclusion, evidence is accumulating that maintaining a distinct and finely tuned fatty acid–glucose fuel balance is crucial for optimal organ performance. The corollary is that rectifying a tilted fuel balance, as occurs in several (e.g., cardiovascular) diseases, holds promise as therapeutic approach.
Fig. 2.
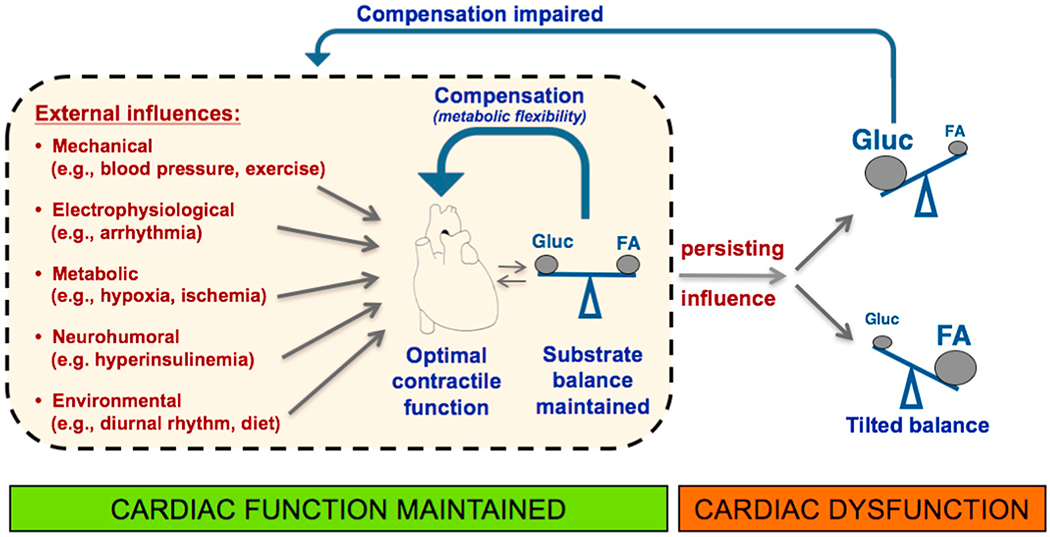
Summary of the association of the myocardial fatty acid–glucose fuel balance and cardiac contractile function. The scheme illustrates the ability of the heart to adapt its substrate preference to short-term external influences so as to secure an adequate ATP production for optimal cardiac contractile function, together referred to as metabolic flexibility. In case of a persisting external influence, however, whereby the balance of metabolic substrates is permanently shifted towards the utilization of a predominant substrate (either glucose or fatty acids), this metabolic flexibility is markedly impaired leading to suboptimal ATP production and impaired contractile function.
FA, (long-chain) fatty acids; Gluc, glucose.
The apparent need for a tuned fatty acid–glucose balance is further illustrated by the observation that a single metabolic perturbation in many cases is tolerated, i.e., cardiac performance is virtually maintained, but that a second perturbation that promotes the shift in balance is no longer tolerated and leads to impaired cardiac function. However, when this second intervention induces a rectification of the fatty acid–glucose fuel balance, this does not affect cardiac function or even elicits recovery of function (Table 2). This phenomenon also explains why dietary fat consumption in some cases worsens cardiac function but in other cases appears protective [65].
It is debated whether in myocardial disease changes in substrate metabolism are cause or consequence of cardiac contractile dysfunction [66]. For the hypertrophied and failing myocardium the notion arises that metabolic changes per se are the (initial) trigger that causes myocardial dysfunction [67]. The insulin resistant and diabetic heart also develops impaired contractile function only after a major shift in substrate metabolism has occurred, i.e., being virtually completely dependent on fatty acids for ATP production [7]. This accumulating evidence that metabolic remodeling precedes structural and functional remodeling of the heart is further underscored by various experimental studies reporting that so-called metabolic modulation, either alone or as adjunct to other medication, is an effective strategy to treat cardiac disease [68]. The present review extends this approach by indicating that metabolic modulation therapy may be effective especially when aimed at restoring the fatty acid–glucose fuel balance. It should be noted that when applying such approach in clinical practice, care must be taken because heart failure patients also will be on a range of medications which could influence cardiac metabolism.
From a clinical perspective, monitoring of the metabolic state of the heart with special reference to the overall utilization of fatty acids versus glucose may be of future interest both for early identification of major changes in substrate preference and for the monitoring of treatment responses, especially when using strategies of metabolic modulation. Non-invasive techniques such as positron emission tomography (PET) or magnetic resonance spectroscopy (MRS) are particularly suitable because they permit the non-invasive serial assessment of the metabolic state of the heart [5]. Much more work may be forthcoming in this direction with the development of metabolic strategies for the treatment of heart failure and the possible application of the re-balancing concept to the function of other organs as well.
Supplementary Material
Acknowledgement
The authors thank Arend Bonen (University of Guelph, Canada) for stimulating discussions during the preparation of this manuscript.
Sources of funding
MN is supported by a VENI Innovational Research Grant from the Netherlands Organisation for Scientific Research (ZonMw-NWO grant nr. 916.14.050). MEY is supported by NIH grants HL106199, HL074259, and HL123574. HT is supported by grants from the NHLBI (HL 61483, HL 123627) and the Cancer Prevention and Research Institute of Texas (CPRIT). PCS is supported by grants from the NHLBI and the Else Kroner-Fresenius Foundation.
Abbreviations:
- ACS
acyl-CoA synthetase
- BCAA
branched-chain amino acids
- CD36
cluster of differentiation 36 (SR-B2)
- CPT1
carnitine palmitoyl-transferase-1
- GLUT
glucose transporter
- PET
positron emission tomography
- PGC1α
PPAR-gamma co-activator-1α
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- TAC
transverse aortic constriction
- mTOR
mechanistic target of rapamycin
Footnotes
Transparency document
The Transparency document associated with this article can be found, in online version.
Declaration of competing interest
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://doi.org/10.1016/j.bbadis.2019.165579.
References
- [1].Taegtmeyer H, Energy metabolism of the heart: from basic concepts to clinical applications, Curr. Probl. Cardiol 19 (1994) 59–113. [DOI] [PubMed] [Google Scholar]
- [2].Balaban RS, Domestication of the cardiac mitochondrion for energy conversion, J. Mol. Cell. Cardiol 46 (2009) 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Glatz JF, Bonen A, Ouwens DM, Luiken JJ, Regulation of sarcolemmal transport of substrates in the healthy and diseased heart, Cardiovasc. Drugs Ther 20 (2006) 471–476. [DOI] [PubMed] [Google Scholar]
- [4].Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC, Myocardial fatty acid metabolism in health and disease, Physiol. Rev 90 (2010) 207–258. [DOI] [PubMed] [Google Scholar]
- [5].Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, Des Rosiers C, Gerszten R, Glatz JF, Griffin JL, Gropler RJ, Holzhuetter HG, Kizer JR, Lewandowski ED, Malloy CR, Neubauer S, Peterson LR, Portman MA, Recchia FA, Van Eyk JE, Wang TJ, American Heart S, Association Council on Basic Cardiovascular, Assessing cardiac metabolism: a scientific statement From the American Heart Association, Circ. Res 118 (2016) 1659–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Joost HG, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H, James DE, Lodish HF, Moley KH, Moley JF, Mueckler M, Rogers S, Schurmann A, Seino S, Thorens B, Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators, Am. J. Physiol. Endocrinol. Metab 282 (2002) E974–E976. [DOI] [PubMed] [Google Scholar]
- [7].Glatz JFC, Luiken J, Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization, J. Lipid Res 59 (2018) 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hue L, Taegtmeyer H, The Randle cycle revisited: a new head for an old hat, Am. J. Physiol. Endocrinol. Metab 297 (2009) E578–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chanda D, Luiken JJ, Glatz JF, Signaling pathways involved in cardiac energy metabolism, FEBS Lett. 590 (2016) 2364–2374. [DOI] [PubMed] [Google Scholar]
- [10].Depre C, Shipley GL, Chen W, Han Q, Doenst T, Moore ML, Stepkowski S, Davies PJ, Taegtmeyer H, Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy, Nat. Med 4 (1998) 1269–1275. [DOI] [PubMed] [Google Scholar]
- [11].Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, ESC guidelines for the diagnosis and treatment of acute and chronic heart failure, Rev Esp Cardiol (Engl Ed) 69 (2016) (2016) 1167. [DOI] [PubMed] [Google Scholar]
- [12].Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, Paulus WJ, Rossignol P, Taegtmeyer H, Bauersachs J, Bayes-Genis A, Brutsaert D, Bugger H, Clarke K, Cosentino F, De Keulenaer G, Dei Cas A, Gonzalez A, Huelsmann M, Iaccarino G, Lunde IG, Lyon AR, Pollesello P, Rena G, Riksen NP, Rosano G, Staels B, van Laake LW, Wanner C, Farmakis D, Filippatos G, Ruschitzka F, Seferovic BP, de Boer RA, Heymans S, Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology, Eur Heart J 39 (2018) 4243–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chess DJ, Stanley WC, Role of diet and fuel overabundance in the development and progression of heart failure, Cardiovasc. Res 79 (2008) 269–278. [DOI] [PubMed] [Google Scholar]
- [14].Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H, Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart, FASEB J. 18 (2004) 1692–1700. [DOI] [PubMed] [Google Scholar]
- [15].Abel ED, Doenst T, Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy, Cardiovasc. Res 90 (2011) 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cabrero A, Merlos M, Laguna JC, Carrera MV, Down-regulation of acyl-CoA oxidase gene expression and increased NF-kappaB activity in etomoxir-induced cardiac hypertrophy, J. Lipid Res 44 (2003) 388–398. [DOI] [PubMed] [Google Scholar]
- [17].Horowitz JD, Chirkov YY, Kennedy JA, Sverdlov AL, Modulation of myocardial metabolism: an emerging therapeutic principle, Curr. Opin. Cardiol 25 (2010) 329–334. [DOI] [PubMed] [Google Scholar]
- [18].Coates PM, Tanaka K, Molecular basis of mitochondrial fatty acid oxidation de-fects, J. Lipid Res 33 (1992) 1099–1110. [PubMed] [Google Scholar]
- [19].Guertl B, Noehammer C, Hoefler G, Metabolic cardiomyopathies, Int. J. Exp. Pathol 81 (2000) 349–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman GI, Willis MS, Coleman RA, Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy, Mol. Cell. Biol 31 (2011) 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lehrke M, Marx N, Diabetes mellitus and heart failure, Am. J. Med 130 (2017) S40–S50. [DOI] [PubMed] [Google Scholar]
- [22].Das AM, Steuerwald U, Illsinger S, Inborn errors of energy metabolism associated with myopathies, J. Biomed. Biotechnol 2010 (2010) 340849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dominguez F, Kuhl U, Pieske B, Garcia-Pavia P, Tschope C, Update on myocarditis and inflammatory cardiomyopathy: reemergence of endomyocardial biopsy, Rev Esp Cardiol (Engl Ed) 69 (2016) 178–187. [DOI] [PubMed] [Google Scholar]
- [24].Ahn J, Kim J, Mechanisms and consequences of inflammatory signaling in the myocardium, Curr. Hypertens. Rep 14 (2012) 510–516. [DOI] [PubMed] [Google Scholar]
- [25].Abdurrachim D, Luiken JJ, Nicolay K, Glatz JF, Prompers JJ, Nabben M, Good and bad consequences of altered fatty acid metabolism in heart failure: evidence from mouse models, Cardiovasc. Res 106 (2015) 194–205. [DOI] [PubMed] [Google Scholar]
- [26].Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC, Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure, Circulation 125 (2012) 2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sung MM, Byrne NJ, Kim TT, Levasseur J, Masson G, Boisvenue JJ, Febbraio M, Dyck JR, Cardiomyocyte-specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure, Am. J. Physiol. Heart Circ. Physiol 312 (2017) H552–H560. [DOI] [PubMed] [Google Scholar]
- [28].Steinbusch LK, Luiken JJ, Vlasblom R, Chabowski A, Hoebers NT, Coumans WA, Vroegrijk IO, Voshol PJ, Ouwens DM, Glatz JF, Diamant M, Absence of fatty acid transporter CD36 protects against Western-type diet-related cardiac dysfunction following pressure overload in mice, Am. J. Physiol. Endocrinol. Metab 301 (2011) E618–E627. [DOI] [PubMed] [Google Scholar]
- [29].Dirkx E, van Eys GJ, Schwenk RW, Steinbusch LK, Hoebers N, Coumans WA, Peters T, Janssen BJ, Brans B, Vogg AT, Neumann D, Glatz JF, Luiken JJ, Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance, J. Mol. Cell. Cardiol 76 (2014) 208–217. [DOI] [PubMed] [Google Scholar]
- [30].Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP, CD36 deficiency rescues lipotoxic cardiomyopathy, Circ. Res 100 (2007) 1208–1217. [DOI] [PubMed] [Google Scholar]
- [31].Young ME, Temporal partitioning of cardiac metabolism by the cardiomyocyte circadian clock, Exp. Physiol 101 (2016) 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Newsholme EA, Arch JR, Brooks B, Surholt B, The role of substrate cycles in metabolic regulation, Biochem. Soc. Trans 11 (1983) 52–56. [DOI] [PubMed] [Google Scholar]
- [33].Qian H, Beard DA, Metabolic futile cycles and their functions: a systems analysis of energy and control, Syst Biol (Stevenage) 153 (2006) 192–200. [DOI] [PubMed] [Google Scholar]
- [34].Wende AR, Brahma MK, McGinnis GR, Young ME, Metabolic origins of heart failure, JACC Basic Transl Sci 2 (2017) 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chatham JC, Marchase RB, The role of protein O-linked beta-N-acetylglucosamine in mediating cardiac stress responses, Biochim. Biophys. Acta 1800 (2010) 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Brahma MK, Pepin ME, Wende AR, My sweetheart is broken: role of glucose in diabetic cardiomyopathy, Diabetes Metab. J 41 (2017) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Owen OE, Kalhan SC, Hanson RW, The key role of anaplerosis and cataplerosis for citric acid cycle function, J. Biol. Chem 277 (2002) 30409–30412. [DOI] [PubMed] [Google Scholar]
- [38].Des Rosiers C, Labarthe F, Lloyd SG, Chatham JC, Cardiac anaplerosis in health and disease: food for thought, Cardiovasc. Res 90 (2011) 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gibala MJ, Young ME, Taegtmeyer H, Anaplerosis of the citric acid cycle: role in energy metabolism of heart and skeletal muscle, Acta Physiol. Scand 168 (2000) 657–665. [DOI] [PubMed] [Google Scholar]
- [40].Randle PJ, England PJ, Denton RM, Control of the tricarboxylate cycle and its interactions with glycolysis during acetate utilization in rat heart, Biochem. J 117 (1970) 677–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Garland PB, Randle PJ, Newsholme EA, Citrate as an intermediary in the inhibition of phosphofructokinase in rat heart muscle by fatty acids, ketone bodies, pyruvate, diabetes, and starvation, Nature 200 (1963) 169–170. [DOI] [PubMed] [Google Scholar]
- [42].Mansor LS, Sousa Fialho MDL, Yea G, Coumans WA, West JA, Kerr M, Carr CA, Luiken J, Glatz JFC, Evans RD, Griffin JL, Tyler DJ, Clarke K, Heather LC, Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation, Cardiovasc. Res 113 (2017) 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Csizmok V, Forman-Kay JD, Complex regulatory mechanisms mediated by the interplay of multiple post-translational modifications, Curr. Opin. Struct. Biol 48 (2017) 58–67. [DOI] [PubMed] [Google Scholar]
- [44].Taegtmeyer H, Sen S, Vela D, Return to the fetal gene program: a suggested metabolic link to gene expression in the heart, Ann. N. Y. Acad. Sci 1188 (2010) 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gunaratnam K, Vidal C, Gimble JM, Duque G, Mechanisms of palmitate-induced lipotoxicity in human osteoblasts, Endocrinology 155 (2014) 108–116. [DOI] [PubMed] [Google Scholar]
- [46].Luiken JJ, Chanda D, Nabben M, Neumann D, Glatz JF, Post-translational modifications of CD36 (SR-B2): implications for regulation of myocellular fatty acid uptake, Biochim. Biophys. Acta 1862 (2016) 2253–2258. [DOI] [PubMed] [Google Scholar]
- [47].Wende AR, Abel ED, Lipotoxicity in the heart, Biochim. Biophys. Acta 1801 (2010) 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, Homma S, George IJ, Takayama H, Naka Y, Goldberg IJ, Schulze PC, Increased de novo ceramide synthesis and accumulation in failing myocardium, JCI Insight 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kundu BK, Zhong M, Sen S, Davogustto G, Keller SR, Taegtmeyer H, Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: review of a hypothesis, Cardiology 130 (2015) 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, Scott B, Carranza S, Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML, Taegtmeyer H, Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart, J. Am. Heart Assoc 2 (2013) e004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Faria A, Persaud SJ, Cardiac oxidative stress in diabetes: mechanisms and therapeutic potential, Pharmacol. Ther 172 (2017) 50–62. [DOI] [PubMed] [Google Scholar]
- [52].Lopaschuk GD, Ussher JR, Evolving concepts of myocardial energy metabolism: more than just fats and carbohydrates, Circ. Res 119 (2016) 1173–1176. [DOI] [PubMed] [Google Scholar]
- [53].Cotter DG, Schugar RC, Crawford PA, Ketone body metabolism and cardiovas-cular disease, Am. J. Physiol. Heart Circ. Physiol 304 (2013) H1060–H1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP, The failing heart relies on ketone bodies as a fuel, Circulation 133 (2016) 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stowe KA, Burgess SC, Merritt M, Sherry AD, Malloy CR, Storage and oxide-tion of long-chain fatty acids in the C57/BL6 mouse heart as measured by NMR spectroscopy, FEBS Lett. 580 (2006) 4282–4287. [DOI] [PubMed] [Google Scholar]
- [56].Tardif A, Julien N, Pelletier A, Thibault G, Srivastava AK, Chiasson JL, Coderre L, Chronic exposure to beta-hydroxybutyrate impairs insulin action in primary cultures of adult cardiomyocytes, Am. J. Physiol. Endocrinol. Metab 281 (2001) E1205–E1212. [DOI] [PubMed] [Google Scholar]
- [57].Hasselbaink DM, Glatz JF, Luiken JJ, Roemen TH, Van der Vusse GJ, Ketone bodies disturb fatty acid handling in isolated cardiomyocytes derived from control and diabetic rats, Biochem. J 371 (2003) 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Russell RR 3rd, Taegtmeyer H, Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate, J. Clin. Invest 87 (1991) 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sud M, Wang X, Austin PC, Lipscombe LL, Newton GE, Tu JV, Vasan RS, Lee DS, Presentation blood glucose and death, hospitalization, and future diabetes risk in patients with acute heart failure syndromes, Eur. Heart J 36 (2015) 924–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yoon MS, The emerging role of branched-chain amino acids in insulin resistance and metabolism, Nutrients 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lu Z, Xu X, Fassett J, Kwak D, Liu X, Hu X, Wang H, Guo H, Xu D, Yan S, McFalls EO, Lu F, Bache RJ, Chen Y, Loss of the eukaryotic initiation factor 2alpha kinase general control nonderepressible 2 protects mice from pressure overload-induced congestive heart failure without affecting ventricular hypertrophy, Hypertension 63 (2014) 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y, Catabolic defect of branched-chain amino acids promotes heart failure, Circulation 133 (2016) 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, Baca LG, Kim E, Ghosh CC, Parikh SM, Jiang A, Chu Q, Forman DE, Lecker SH, Krishnaiah S, Rabinowitz JD, Weljie AM, Baur JA, Kasper DL, Arany Z, A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance, Nat. Med 22 (2016) 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].De Jong KA, Lopaschuk GD, Complex energy metabolic changes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction, Can J Cardiol 33 (2017) 860–871. [DOI] [PubMed] [Google Scholar]
- [65].Stanley WC, Dabkowski ER, Ribeiro RF Jr., O’Connell KA, Dietary fat and heart failure: moving from lipotoxicity to lipoprotection, Circ. Res 110 (2012) 764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Taegtmeyer H, Lubrano G, Rethinking cardiac metabolism: metabolic cycles to refuel and rebuild the failing heart, F1000Prime Rep 6 (2014) 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Peterzan MA, Lygate CA, Neubauer S, Rider OJ, Metabolic remodeling in hypertrophied and failing myocardium: a review, Am. J. Physiol. Heart Circ. Physiol 313 (2017) H597–H616. [DOI] [PubMed] [Google Scholar]
- [68].Noordali H, Loudon BL, Frenneaux MP, Madhani M, Cardiac metabolism - a promising therapeutic target for heart failure, Pharmacol. Ther 182 (2017) 95–114. [DOI] [PubMed] [Google Scholar]
- [69].Exil VJ, Roberts RL, Sims H, McLaughlin JE, Malkin RA, Gardner CD, Ni G, Rottman JN, Strauss AW, Very-long-chain acyl-coenzyme a dehydrogenase deficiency in mice, Circ. Res 93 (2003) 448–455. [DOI] [PubMed] [Google Scholar]
- [70].Yonekura Y, Brill AB, Som P, Yamamoto K, Srivastava SC, Iwai J, Elmaleh DR, Livni E, Strauss HW, Goodman MM, et al. , Regional myocardial substrate uptake in hypertensive rats: a quantitative autoradiographic measurement, Science 227 (1985) 1494–1496. [DOI] [PubMed] [Google Scholar]
- [71].Doenst T, Goodwin GW, Cedars AM, Wang M, Stepkowski S, Taegtmeyer H, Load-induced changes in vivo alter substrate fluxes and insulin responsiveness of rat heart in vitro, Metabolism 50 (2001) 1083–1090. [DOI] [PubMed] [Google Scholar]
- [72].Razeghi P, Young ME, Abbasi S, Taegtmeyer H, Hypoxia in vivo decreases peroxisome proliferator-activated receptor alpha-regulated gene expression in rat heart, Biochem. Biophys. Res. Commun 287 (2001) 5–10. [DOI] [PubMed] [Google Scholar]
- [73].Thomassen A, Nielsen TT, Bagger JP, Henningsen P, Myocardial substrate utilization and amino acid metabolism in chronic coronary artery disease, Z. Kardiol 76 (Suppl. 5) (1987) 19–25. [PubMed] [Google Scholar]
- [74].Neubauer S, Horn M, Naumann A, Tian R, Hu K, Laser M, Friedrich J, Gaudron P, Schnackerz K, Ingwall JS, et al. Impairment of energy metabolism in intact residual myocardium of rat hearts with chronic myocardial infarction, J. Clin. Invest 95 (1995) 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bonen A, Parolin ML, Steinberg GR, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Heigenhauser GJ, Dyck DJ, Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36, FASEB J. 18 (2004) 1144–1146. [DOI] [PubMed] [Google Scholar]
- [76].Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM, Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1 alpha, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 10086–10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, Ding Y, Prasain J, Wood PA, Yang Q, Carnitine palmitoyltransferase-lb deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity, Circulation 126 (2012) 1705–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Abdurrachim D, Nabben M, Hoerr V, Kuhlmann MT, Bovenkamp P, Ciapaite J, Geraets IME, Coumans W, Luiken J, Glatz JFC, Schafers M, Nicolay K, Faber C, Hermann S, Prompers JJ, Diabetic db/db mice do not develop heart failure upon pressure overload: a longitudinal in vivo PET, MRI, and MRS study on cardiac metabolic, structural, and functional adaptations, Cardiovasc. Res 113 (2017) 1148–1160. [DOI] [PubMed] [Google Scholar]
- [79].Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R, Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy, Circ. Res 111 (2012) 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, Wang W, Tian R, Kolwicz SC Jr., Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion, J. Mol. Cell. Cardiol 100 (2016) 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Berthiaume JM, Young ME, Chen X, McElfresh TA, Yu X, Chandler MP, Normalizing the metabolic phenotype after myocardial infarction: impact of sub-chronic high fat feeding, J. Mol. Cell. Cardiol 53 (2012) 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.