

Abstract
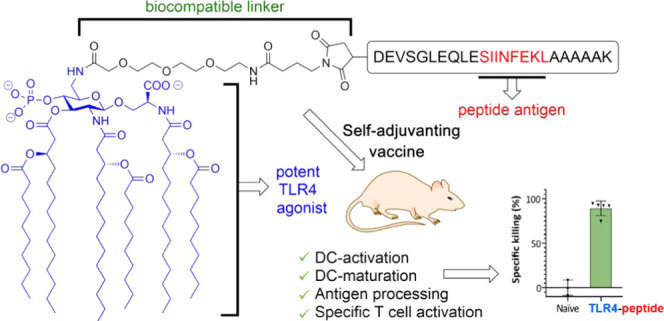
Self-adjuvanting vaccines, wherein an antigenic peptide is covalently bound to an immunostimulating agent, have been shown to be promising tools for immunotherapy. Synthetic Toll-like receptor (TLR) ligands are ideal adjuvants for covalent linking to peptides or proteins. We here introduce a conjugation-ready TLR4 ligand, CRX-527, a potent powerful lipid A analogue, in the generation of novel conjugate-vaccine modalities. Effective chemistry has been developed for the synthesis of the conjugation-ready ligand as well as the connection of it to the peptide antigen. Different linker systems and connection modes to a model peptide were explored, and in vitro evaluation of the conjugates showed them to be powerful immune-activating agents, significantly more effective than the separate components. Mounting the CRX-527 ligand at the N-terminus of the model peptide antigen delivered a vaccine modality that proved to be potent in activation of dendritic cells, in facilitating antigen presentation, and in initiating specific CD8+ T-cell-mediated killing of antigen-loaded target cells in vivo. Synthetic TLR4 ligands thus show great promise in potentiating the conjugate vaccine platform for application in cancer vaccination.
1. Introduction
Immunotherapy has become a powerful strategy to combat cancer. Significant advances have been made in the activation of antitumor T-cell immunity, including the development of immune checkpoint blockade antibodies,1 chimeric antigen receptor T cells (CAR T cells),2 and vaccination strategies, in which the immune system is trained to recognize cancer neoantigens.3,4 To optimally direct an immune reaction against cancer via vaccination, adjuvants are used to activate antigen-presenting cells, such as dendritic cells (DCs) and macrophages. DCs express pathogen recognition receptors (PRRs),5 through which they recognize invading pathogens and initiate an immune response, which eventually leads to the priming of T cells.6 Pathogen-associated molecular patterns (PAMPs) are ligands for these PRRs and can be used as molecular adjuvants. Molecular adjuvants are well-defined single-molecule immunostimulants that act directly on the innate immune system to enhance the adaptive immune response against antigens. Many well-defined PAMPs have been explored over the years, and the most extensively targeted PRR families are the Toll-like receptors (TLRs),7 C-type lectins,8 and nucleotide-binding oligomerization domain (NOD)-like receptors.9,10 To further improve vaccine activity, the antigen and adjuvants have been combined in covalent constructs, delivering “self-adjuvanting” vaccine candidates.11,12 In the immune system, the stimulation of different TLRs can activate distinct signaling cascades and thereby support the generation of polarized types of immune reactions. Hence, targeting of distinct TLRs in vaccination influences the nature of the adaptive immune response induced.13,14 Several TLR agonists11,15,16 have been conjugated to antigenic peptides (often synthetic long peptides, SLPs), including ligands for TLR2,17−22 TLR7,23,24 and TLR9,20,25,26 yielding vaccine modalities with improved activity with respect to their nonconjugated counterparts. Lipid A (Figure 1A), a conserved component of the bacterial cell wall, is one of the most potent immune-stimulating agents known to date, and it activates the innate immune system through binding with TLR4. The high toxicity of lipid A makes it unsuitable for safe use in humans, but monophosphoryl lipid A (MPLA, Figure 1A), a lipid A derivative in which the anomeric phosphate has been removed, has proven its effectiveness as an adjuvant in various approved vaccines.27−29 It has also been used recently in conjugates in which it was covalently attached to a tumor-associated carbohydrate antigen (TACA) or a synthetic bacterial glycan.30−34 The latter conjugate was able to elicit a robust immunoglobulin G (IgG) antibody response in mice, critical for effective antibacterial vaccination.34 MPLA thus represents a very attractive PAMP to be explored in SLP conjugates, targeting cancer epitopes. The physical properties and challenging synthesis of lipid A derivatives, however, limit its accessibility.35−37 Because of its potent immunostimulating activity, many mimics of MPLA have been developed and the class of aminoalkyl glucosamine 4-phosphates (AGPs) has been especially promising.38−41 AGPs have been shown to be efficacious adjuvants and to be clinically safe, resulting in their use in a hepatitis B vaccine.42 CRX-527 (Figure 1A) has been established as one of the most potent AGPs.38
Figure 1.
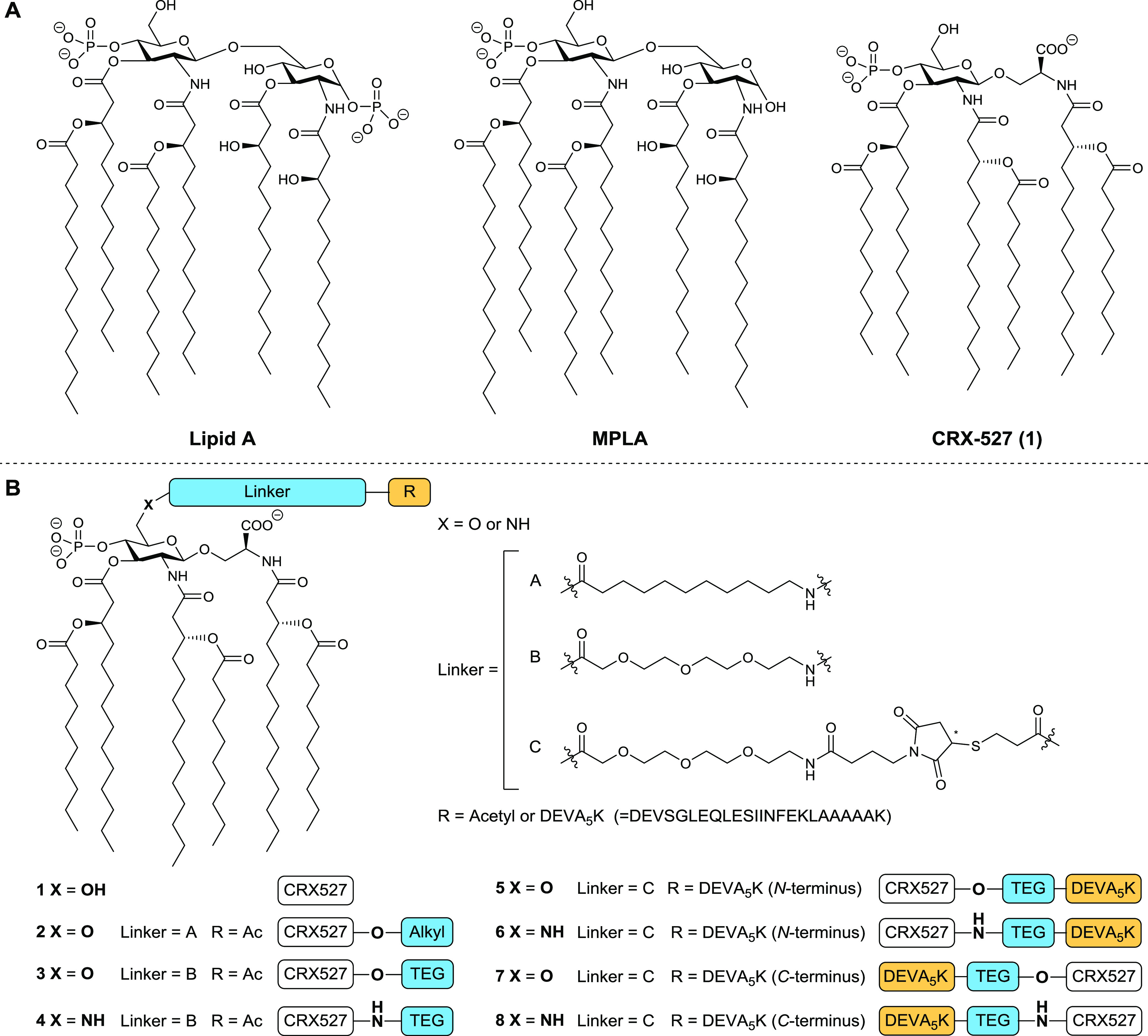
(A) Representative structures of lipid A of Escherichia coli and MPLA of Salmonella enterica serotype minnesota Re 595; Structure of CRX-527 (1). (B) Structures of CRX-527 derivatives 2–4 and CRX-527 conjugates 5–8. The DEVA5K peptide in the conjugates carries the SIINFEKL epitope in its sequence.
We here introduce conjugation-ready derivatives of CRX-527 for application in the development of adjuvant-SLP vaccine conjugates. We have established a robust synthetic route to generate linker-equipped CRX-527 analogues and used these in the assembly of SLP conjugates. The self-adjuvanting SLPs carrying this TLR4 ligand are capable of mobilizing a strong T-cell immune response against the incorporated antigen and are capable of promoting effective and specific killing of target cells expressing the antigen in vivo.
2. Results and Discussion
2.1. Synthesis of Ligands and Conjugates
In the development of the conjugation-ready CRX-527 derivates, we set out to probe both the influence of the nature of the linker and the mode of connectivity of the linker to the TLR4 ligand, CRX-527 (see Figure 1B). In lipopolysaccharides, bacterial O-antigens are connected to a lipid A anchor through the C6 position of the glucosamine-C4-phosphate residue, and from the crystal structure of lipid A in complex to the TLR4–MD2 complex, it is apparent that this position is exposed from the complex.43 The C6 position of the glucosamine-C4-phosphate can thus be used for conjugation purposes. Indeed, previous work on antibacterial MPLA conjugate vaccines has shown that the adjuvant can be modified at this position without compromising adjuvant activity.34 We explored two types of linkers at the C6 position of CRX-527: a hydrophobic alkyl linker (A) and a hydrophilic triethylene glycol (TEG) linker (B). These linkers were connected to CRX-527 through an ester bond34 or via a more stable amide bond. To connect the ligands to the SLPs, the linkers were equipped with a maleimide, to allow for a thiol-ene conjugation to the sulfhydryl functionalized SLP. We used the ovalbumin-derived SLP, DEVSGLEQLESIINFEKLAAAAAK (DEVA5K), as a model antigen.20 Herein, the MHC-I epitope SIINFEKL is embedded in a longer peptide motif to ensure that the peptide will have to undergo proteasomal processing to produce the minimal epitope. The target compounds generated for this study are depicted in Figure 1 and include CRX-527 (1), ester-linkers CRX-527 2 and 3, amide-linker CRX-527 4 as well as the conjugates 5 and 6 having the CRX-527 ligand at the N-terminus of the peptide and conjugates 7 and 8, with the ligand at the C-terminus of the SLP. To obtain CRX-527 derivatives 1–4, building blocks 16a/16b were required, and the assembly of these key intermediates was accomplished as depicted in Figure 2A. Based on the synthesis route to CRX-527 developed by Johnson and co-workers,41 we assembled glucosaminyl serine building block 11 from glucosamine donor 9 and serine 10. Condensation of 9 and 10 under the influence of boron trifluoride etherate proceeded in a completely β-selective manner to give a mixture of the desired product and unreacted donor 9. The mixture could be separated after hydrogenolysis of the benzyl ester, giving acid 11 in 63% yield on a 170 mmol scale. Next, all acetyls were removed, before the benzyl ester was reinstalled using phase-transfer conditions to deliver triol 12. To enable the introduction of the chiral lipid tail, we masked the C4- and C6-hydroxyl groups in 12 with a silylidene ketal. This protecting group strategy proved to be crucial as the use of a C6-O-tert-butyldimethylsilyl (TBDMS) group, as previously reported,41 led to an intractable mixture when the lipid tails were attached. As lipid A analogues bearing fewer lipid tails may have a different immunological response,44 the purity of the ligand is of utmost importance. Next, the Troc-protecting groups were removed from both amine groups, after which an N,N,O-triacetylation event using fatty acid (FA) 14 (Scheme S1) and EDC·MeI and catalytic 4-dimethylaminopyridine (DMAP) (0.03 equiv) delivered compound 15. On a 9.5 mmol scale, this intermediate was obtained in 57% over two steps. The silylidene ketal was removed, and then the primary alcohol was selectively protected with a TBDMS group, and the phosphate triester was installed at the C4-OH. Desilylation provided the key building block 16a on a multigram scale. The alcohol in 16a was transformed into the corresponding primary azide using Mitsunobu conditions delivering 16b.
Figure 2.

(A) Synthesis of building blocks 16a/16b. Reagents and conditions: (a) (i) BF3·OEt2, dichloromethane (DCM), 0 °C to room temperature (rt); (ii) H2, Pd/C, tetrahydrofuran (THF), 63% over two steps. (b) (i) NH4OH, MeOH; (ii) BnBr, tetra-n-butylammonium bromide (TBAB), DCM/NaHCO3 (aq. sat.), 79% over two steps. (c) (tBu)2Si(OTf)2, dimethylformamide (DMF), −40 °C, 94%. (d) (i) Zn dust, AcOH; (ii) 14, EDC·MeI, DMAP, DCM, 57% over two steps. (e) (i) HF·Et3N. THF, 0 °C, 92%; (ii) tert-butyldimethylsilyl chloride (TBDMSCl), pyridine, 88%; (iii) dibenzyl N,N-diisopropylphosphoramidite, tetrazole, DCM, 0°, 1 h; (iv) 3-chloroperbenzoic acid, quant. over two steps; (v) trifluoroacetic acid (TFA), DCM, 84%. (f) PPh3, diethyl azodicarboxylate (DEAD), diphenyl phosphoryl azide (DPPA), THF, 67%. (B) Synthesis of TLR4 ligands 1–4. Reagents and conditions: (g) H2, Pd/C, THF, 1: 89%. (h) (i) 17, EDC·MeI, DMAP, dichloroethane (DCE), 88%; (ii) H2, Pd/C, THF, 2: 56%. (i) (i) 18, EDC·MeI, DMAP, DCE, 74%; (ii) H2, Pd/C, THF, 3: 66%. (j) (i) Zn, NH4Cl, DCM/MeOH/H2O; (ii) 18, EDC·MeI, DMAP, DCE, 40% over two steps; (iii) H2, Pd/C, THF, 4: 61%. (C) Assembly of conjugates 5–8. Reagents and conditions: (k) (i) 19, EDC·MeI, DMAP, DCE, 80%; (ii) H2, Pd/C, THF, 77%; (iii) sulfo-N-succinimidyl 4-maleimidobutyrate sodium salt, Et3N, DCM, 20a: 84%. (l) (i) Zn, NH4Cl, DCM/MeOH/H2O; (ii) 19, EDC·MeI, DMAP, DCE, 56% over two steps; (iii) sulfo-N-succinimidyl 4-maleimidobutyrate sodium salt, Et3N, DCE, 20b: 81%. (m) 21, DMF/CHCl3/H2O, 48 h, 5: 52%, 6: 54%. (n) 22, DMF/CHCl3/H2O, 48 h, 7: 57%, 8: 42%. (D) Liquid chromatography-mass spectrometry (LC-MS) trace of crude and purified C-terminus conjugate 7, and matrix-assisted laser desorption ionization (MALDI) analysis of 7.
With building blocks 16a/16b available in sufficient amounts, attention was directed to the assembly of ligands 1–4, having either an alkyl or a triethylene glycol (TEG) linker (Figure 2B). Debenzylation of 16a using Pd/C gave the original CRX-527 (1). Elongation of 16a with the N-acetylated linkers 17 or 18, under the influence of EDC·MeI and DMAP, furnished the fully protected linker-CRX-527 compounds that were subjected to a hydrogenation reaction to obtain ligands 2 and 3. In contrast to the findings of Guo and co-workers, in their synthesis of linker functionalized MPLA derivatives, where the C6–ester bond was found unstable,34 no hydrolysis of esters 2 and 3 was observed. Ligand 4 was obtained from azide 16b by zinc-mediated reduction, condensation with linker 18, and subsequent hydrogenation.
Next, the synthesis of the CRX-527-peptide conjugates 5–8 was undertaken (Figure 2C). Based on the immunological evaluation of ligands 1–4 (vide infra, Figure 3), the TEG linker was used for the assembly of the peptide–antigen conjugates. First, 16a and azido linker 19 were conjugated under the agency of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). Reduction of the azide and benzyl esters was then followed by the introduction of the maleimide functionality using sulfo-N-succinimidyl 4-maleimidobutyrate to give conjugation-ready CRX-527 20a. The amide congener of this compound was assembled from 16b in an analogous manner. The DEVA5K peptides with a thiol function at the N-terminus (21) or the C-terminus (22) were assembled using a semiautomated solid-phase peptide synthesis protocol and purified to homogeneity by reversed-phase high-performance liquid chromatography (RP-HPLC) (see Supporting Information for full synthetic details). The conjugation of the ligand and the peptide antigen required significant optimization because of the physical properties of the ligand and the peptides. We found that the thiol–maleimide coupling could be achieved by dissolving 21 or 22 in DMF/H2O (4/1 v/v) followed by the addition of a solution of maleimide 20a or 20b in CHCl3. After shaking for 2 days, LC-MS analysis confirmed the full conversion of the maleimide and the conjugates were purified by C18 column chromatography (see the Supporting Information for details). Figure 2D shows the analysis of the conjugation of maleimide 20a with an excess of thiol 22, providing conjugate 7 (Figure 2D, left panel), which was purified by HPLC to provide the pure conjugate (Figure 2D, middle panel). The integrity and purity of the synthesized N-terminus conjugates 5 and 6 and the C-terminus conjugates 7 and 8 were ascertained by LC-MS analysis and MALDI-TOF MS (see Figure 2D, right panel for the MALDI-TOF MS spectrum of 7).
Figure 3.

TLR4 ligands and the cognate conjugates induce IL-12 production and T-cell activation in vitro. (A) The D1 dendritic cell line was stimulated for 24 h with the synthetic compounds 1–4, and the induction of DC maturation was analyzed by measuring IL-12p40 production. MPLA was included as a control for TLR4 stimulation. (B) The activity of the conjugates 5–8 together with their reference ligands 3 and 4 was analyzed by measuring IL-12p40 production. Unconjugated peptide was included as the negative control. (C) Antigen uptake and presentation were measured by incubation of compound-pulsed D1 with the SIINFEKL-specific hybridoma T-cell line B3Z. B3Z activation was determined by colorimetric reaction of the CPRG reporter enzyme and measurement of absorbance.
2.2. In Vitro Activity
Immunological evaluation of TLR4 ligands 1–4 and conjugates 5–8 was performed by first assessing their ability to induce maturation of dendritic cells and to present antigen to T cells in vitro. Binding of lipid A to the TLR4/MD-2 complex triggers the production of inflammatory cytokines and maturation of the DCs. Production of the subunit IL-12p40 of the proinflammatory cytokine IL-12 is a marker of this activation. We first analyzed if the addition of the peptide would affect the interaction with TLR4 and impede the activity of the ligand. To probe activation of the DCs by the CRX-527 ligands and conjugates, murine DCs were stimulated for 24 h with different compounds and the amount of IL-12p40 in the supernatant was measured. First, the effect of the different linkers (compounds 2–4) on the activity of the CRX-527 ligand was evaluated. As can be seen in Figure 3A, stimulation of the DCs with the CRX-527 ligand 1 induces strong IL-12p40 secretion and its activity is higher than the commercially available TLR4 ligand MPLA. The addition of an ester-alkyl linker (2) significantly decreased the ability of the ligand to induce DC activation. However, the ester– or amide–TEG linker functionalized ligands mostly preserve the induction of IL-12p40, as shown for compounds 3 and 4. For these two ligands, the activity was comparable to the unmodified ligand 1 up to 1.56 nM and slightly reduced at the lowest concentrations. However, substantial levels of IL-12p40 could still be detected at these concentrations. Possibly, the hydrophobic nature of the alkyl linker of compound 2 induces a different configuration of the ligand that affects binding to the MD-2/TLR4 pocket, preventing activation of the signaling cascade.43 The DC-activating capacity of compounds 1, 3, and 4 indicates that functionalization at the C6 position with a hydrophilic linker does not inhibit binding of the ligand to the receptor.
Next, the DEVA5K peptide conjugates 5–8 were evaluated for their ability to induce IL-12p40 in DCs (Figure 3B). We analyzed whether conjugation of the peptide via the ester– (compounds 5 and 7) or the amide–TEG linker (compounds 6 and 8) could differently modulate the activity of the conjugates. Moreover, we investigated whether conjugation of the ligand to the N- or C-terminus of the peptide could also influence activity. Interestingly, we found that even if ligands 3 and 4 do not display any differences in activity (Figure 3A,B), their respective peptide conjugates show differential potencies (Figure 3B). Specifically, the ester conjugates 5 and 7 induce high levels of IL-12p40, similar to the ligands 3 and 4, while the amide conjugates 6 and 8 display overall lower levels of IL-12p40 production. No difference in activity was observed between the N- or C-terminal DEVA5K conjugates. The activity of the ester conjugates 5 and 7 titrates faster than the free ligands 3 and 4, as can be observed at the lowest concentrations.
These data show that conjugation of CRX-527 to a long peptide via different linkers results in immunologically active compounds and has therefore potential for vaccination. Importantly, the efficacy of peptide vaccines relies on the ability of DCs to take up the peptide and process it to enable surface presentation of the epitope on MHC molecules. Recognition of the antigen–MHC complex by the T-cell receptor and simultaneous costimulation by mature DCs then result in the initiation of a T-cell response.45 The uptake and processing of conjugates 5–8 were evaluated in an antigen presentation assay using the T-cell hybridoma reporter line B3Z, which is specific for the SIINFEKL epitope contained in the DEVA5K peptide. The B3Z cell line possesses a T-cell receptor, specific for the SIINFEKL epitope, which controls the expression of the β-galactosidase reporter gene. Recognition of the SIINFEKL epitope induces the expression of the enzyme, which can subsequently be detected through a colorimetric reaction caused by the conversion of a substrate. The efficiency in antigen presentation of the conjugates was compared to free peptide 23 and to a mixture of peptide 23 and the CRX-527 ligand 1. Figure 3C shows that incubation of DCs with the ester conjugates 5 and 7 leads to similar levels of T-cell activation as the free peptide and the mixture of the peptide and CRX-527 1. Therefore, conjugation of the peptide to the CRX-527 ligand does not affect its uptake and processing, after both N-terminus and C-terminus conjugation. Notably, incubation with the amide conjugates 6 and 8 resulted in slightly enhanced antigen presentation. In this case, the N-terminus conjugate 6 displayed a higher antigen presentation and consequent T-cell activation than its C-terminal counterpart. It is possible that hydrolysis takes place for the ester conjugates 5 and 7 prior to uptake in the DCs, leading to diminished uptake of the peptide moiety (as compared to their amide counterparts) and resulting in lower antigen presentation. Importantly, this readout system is not influenced by the costimulatory signals provided by mature DCs and only reports whether uptake and processing occur in DCs.
To summarize, in vitro evaluation of the conjugates revealed that the ester conjugates display higher potency in inducing DC maturation, while the amide conjugates are presented more efficiently. Therefore, the combined action of costimulation, induced by the triggering of the TLR4 and antigen presentation to CD8+ T cells was evaluated in an in vivo immunization study.
2.3. In Vivo Activity
Having established that the conjugates maintained the capacity to activate DCs and to induce antigen presentation, the conjugated vaccines were compared for their ability to induce de novo T-cell responses in vivo. Mice were injected intradermally with 5 nmol each of conjugates 5–8, or a mixture of peptide 23 and TLR4 ligand 1, and the presence of SIINFEKL-specific T-cell responses was monitored in blood via SIINFEKL-Kb tetramer staining. Analysis of blood after the first vaccine injection demonstrated the successful induction of SIINFEKL-specific T-cell responses in all groups vaccinated with the CRX-527 conjugates. No significant differences could be distinguished between the groups (Figure S1A). Two weeks after the first injection, mice were boosted with the same formulations and the SIINFEKL-specific responses were measured in blood 7 days later. As shown in Figure 4A,B, the strongest induction of SIINFEKL-specific CD8+ T cells was detected in the group that received the N-terminal ester conjugate 5. Overall, the two N-terminal conjugates 5 and 6 displayed higher T-cell induction than their C-terminal counterpart conjugates 7 and 8. T-cell responses were detectable also in mice vaccinated with a mixture of CRX-527 and peptide; however, these responses displayed a higher spread than all conjugate groups. One day later, the spleens and lymph nodes draining the vaccination site were harvested to analyze the presence and the phenotype of the SIINFEKL-specific responses in these organs. Analysis of T-cell responses in the spleen displayed a similar trend to that observed in blood (Figure S1B). However, in the inguinal lymph nodes (Figure 4C), a higher percentage of SIINFEKL-specific T-cell responses was detected for the N-terminal conjugates 5 and 6. In this organ, mice vaccinated with the C-terminus conjugates 7 and 8 or the mixture display low T-cell responses. Next, we investigated whether the phenotype of the SIINFEKL-specific T cells induced by the N-terminal conjugates 5 and 6 was different compared to the group that was vaccinated with the mixture of TLR4 ligand 1 and peptide 23. The induction of differentiation into memory CD127+/KLRG1low responses is a marker for T-cell quality, which is associated with improved functions and tumor clearance.46 This phenotype was pronounced in the groups vaccinated with the conjugates 5 and 6 and was clearly less present in the group that was vaccinated with the mixture (Figure 4D). Within the T-cell memory population, two further subsets can be distinguished based on the expression of CD62L, a surface protein that, when present, determines homing at lymphoid tissues rather than circulation in the blood vessels and tissues. High expression of CD62L defines central memory T cells, while lower expression of this surface protein determines effector memory T cells. This last subset recirculates in tissues and can exert immediate effector functions upon antigen re-encounter. We observed significantly higher differentiation into effector memory T cells when mice were immunized with the conjugated vaccines 5 and 6 rather than the mixture (Figure 4E). It has been shown that the promotion of differentiation into effector memory T cells rather than short-lived effector cells is dependent on optimal priming conditions, such as the presence of helper T cells46−48 and proper costimulatory signals.49,50 These data indicate that conjugation of the TLR4-adjuvant and peptide represents an effective strategy to achieve an increased effector memory T-cell phenotype, which is an important hallmark for effective vaccination.51−53
Figure 4.

Conjugates induce SIINFEKL-specific T-cell responses with an effector memory phenotype in vivo. (A) Representative plots of SIINFEKL-Kb tetramer-positive T cells in blood of vaccinated mice. (B) Percentage of SIINFEKL-specific T-cell responses measured by tetramer staining in blood on day 21 after booster vaccination. Every dot represents a single animal. (C) On day 22, inguinal lymph nodes were harvested and the presence of SIINFEKL-specific T cells was measured by tetramer staining in all groups. Statistical significance was determined by the Kruskal–Wallis test followed by the multiple comparison rank test and Dunn’s correction, *p < 0.05. (D, E) The phenotype of SIINFEKL-specific T cells was characterized in the higher-responding groups in spleen and lymph nodes by analyzing the expression of the surface markers KLRG1, CD127 (D), and CD62L (E) by flow cytometry. Statistical significance was determined by two-way analysis of variance (ANOVA) followed by multiple comparison and Tukey’s correction, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Finally, we investigated the functionality of the induced T-cell responses upon vaccination with conjugate 6 in an in vivo cytotoxicity assay. Mice were immunized with the N-terminus conjugate 6, and the kinetics of the T-cell response was followed in blood by SIINFEKL-Kb tetramer staining (Figure 5A). After 14 days, a boost was administered, and 1 week later animals were intravenously injected with cells loaded with either SIINFEKL peptide or an irrelevant peptide, to measure the ability of the induced T cells to specifically kill SIINFEKL-loaded cells. Prior to injection, the two groups of target cells were differentially labeled with the fluorescent dye carboxyfluorescein succinimidyl ester (CFSE), to be able to distinguish the two populations during later analysis by flow cytometry. After 18 h, the spleens were harvested and the killing of the two peptide-loaded populations was determined in naïve and vaccinated mice via flow cytometric analysis (Figure 5A,B). As expected, naïve mice displayed similar relative frequencies of the two CFSE-labeled populations. On the contrary, specific killing of the SIINFEKL-loaded target cells was observed in the vaccinated mice. Notably, four out of five mice that were vaccinated with conjugate 6 displayed >90% killing. The killing degree reflected the levels of specific CD8+ T cells present, as detected in the inguinal lymph nodes by tetramer staining (Figure 5C).
Figure 5.
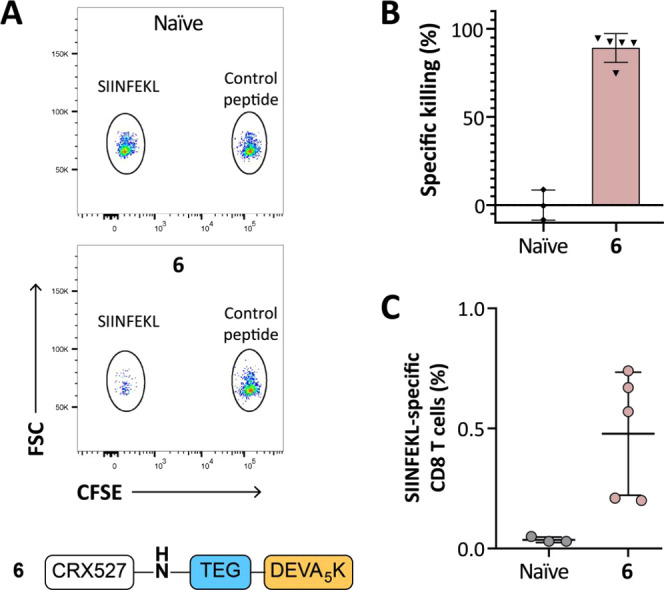
Immunization with CRX-527 conjugate 6 results in efficient specific killing of peptide-loaded target cells. Naïve C57BL/6 mice (n = 5) were injected intradermally with 5 nmol of CRX-527 conjugate. (A) After booster vaccination, mice were injected with differentially CFSE-labeled target cells. Then, 18 h after injection, spleens were analyzed for the presence of the two CFSE-labeled target populations. (B) Calculated specific killing of SIINFEKL-loaded cells. (C) The amount of SIINFEKL-specific T cells was determined in the lymph nodes by tetramer staining.
To conclude, in vivo evaluation of the CRX-527–peptide conjugates shows that conjugates 5–8 are effective in initiating antigen-specific T-cell responses. In particular, the N-terminus conjugates could raise higher responses than their C-terminal counterparts. Phenotypic and functional analyses of these responses revealed that CRX-527 conjugate 6 was capable of raising an adequate protective immune response, effectively killing cells presenting the antigen against which the vaccination was directed at and underscoring the potential of these new conjugates for anticancer immunotherapy.
3. Conclusions
Adjuvant–antigen conjugates are promising agents for cancer immunotherapy. Well-defined molecular adjuvants are essential to stimulate relevant immune subsets and generate the most appropriate type of immunity against distinct tumor types. MPLA is one of the most potent innate immune-stimulating agents, which is currently used as an adjuvant in vaccines, but the application of this TLR4 ligand in adjuvant–antigen constructs is hampered by its challenging synthesis. CRX-527 is a potent MPLA analogue, and we have here disclosed an expeditious synthesis of conjugation-ready derivatives of this immune-stimulating agent and demonstrated the preparation of TLR4-ligand–peptide antigen conjugates for the first time. The assembly of the conjugation-ready ligand critically depended on the protecting group strategy, and the use of a silylidene ketal in the glucosaminyl serine proved crucial for the efficient introduction of lipid tails. The developed route of synthesis is high-yielding and could be executed on a multigram scale to allow the generation of several peptide conjugates. Different linker systems and connection modes were probed to conjugate the TLR4 ligand to a synthetic long peptide antigen. In vitro evaluation of the conjugates showed that the attachment of a lipophilic linker at the C6 of CRX-527 abrogates the activity of the ligand. The use of a hydrophilic glycol-based linker provided conjugates that could induce strong DC maturation and allowed effective antigen processing and presentation. In vivo evaluation of the conjugates demonstrated the efficacy of the vaccine modalities in priming de novo CD8+ T-cell responses. Conjugation of the TLR4 ligand at the N-terminus of the peptide stimulated the best induction of T-cell responses, promoting differentiation into effector memory T-cell responses. Finally, it was shown that the CRX-527 peptide, in which the TLR4 ligand was conjugated to the N-terminus of the SLP through an amide-linked spacer, was a potent inducer of antigen-specific effector CD8+ T-cell responses in vivo. Overall, we have developed a platform to potentiate synthetic peptide vaccines with a potent and well-defined TLR4 ligand, a powerful addition to the toolbox available to generate self-adjuvanting vaccines. CRX-527 conjugates hold great promise for the development of anticancer SLP vaccines, and the availability of the conjugation-ready ligand and chemistry to fuse the ligand to peptide antigens will enable the generation of conjugates bearing other peptide epitopes, such as defined oncoviral antigens or cancer neoantigens. The generated conjugation-ready CRX-527 may also find application in the generation of well-defined antibacterial, viral, or fungal vaccines.
4. Experimental Section
4.1. Materials and Methods
All reagents were of commercial grade and used as received unless stated otherwise. Reaction solvents were of analytical grade and when used under anhydrous conditions stored over flame-dried 3 Å molecular sieves. All moisture- and oxygen-sensitive reactions were performed under an argon atmosphere. Column chromatography was performed on silica gel (Screening Devices BV, 40–63 μm, 60 Å). For thin-layer chromatography (TLC) analysis, precoated silica gel aluminum sheets (Merck, silica gel 60, F254) were used with detection by UV absorption (254/366 nm) where applicable. Compounds were visualized on TLC by UV absorption (245 nm) or by staining with one of the following TLC stain solutions: (NH4)6Mo7O24·H2O (25 g/L), (NH4)4Ce(SO4)4·2H2O (10 g/L), and 10% H2SO4 in H2O; bromocresol (0.4 g/L) in EtOH; KMnO4 (7.5 g/L) and K2CO3 (50 g/L) in H2O. Staining was followed by charring at ∼150 °C. 1H, 13C, and 31P NMR spectra were recorded on a Bruker AV-300 (300/75 MHz) spectrometer, an AV-400 (400/100 MHz) spectrometer, a Bruker AV-500 Ultrashield (500/126 MHz) spectrometer, a Bruker AV-600 (600/151 MHz) spectrometer, or a Bruker AV-850 (850/214 MHz) spectrometer, and all individual signals were assigned using two-dimensional (2D) NMR spectroscopy. Chemical shifts are given in parts per million (ppm) (δ) relative to tetramethylsilane (TMS) (0 ppm) in CDCl3 or via the solvent residual peak. Coupling constants (J) are given in hertz (Hz). LC-MS analyses were done on an Agilent Technologies 1260 Infinity system with a C18 Gemini 3 μm, C18, 110 Å, 50 × 4.6 mm2 column or a Vydac 219TP 5 μm diphenyl, 150 × 4.6 mm2 column with a flow of 1, 0.8, or 0.7 mL/min. Absorbance was measured at 214 and 256 nm, and an Agilent Technologies 6120 Quadrupole mass spectrometer was used as the detector. Peptides, TLR2 ligand, and conjugate were purified with a Gilson GX-281 preparative HPLC with a Gemini-NX 5 μm, C18, 110 Å, 250 × 10.0 mm2 column or a Vydac 219TP 5 μm diphenyl, 250 × 10 mm2 column. Peptide fragments were synthesized with automated solid-phase peptide synthesis on an Applied Biosystems 433A peptide synthesizer. Optical rotations were measured on an Anton Paar modular circular polarimeter MCP 100/150. High-resolution mass spectra (HRMS) were recorded on a Synapt G2-Si or a Q Exactive HF Orbitrap equipped with an electron spray ion source positive mode. Mass analysis of the TLR4 ligands and TLR4-ligand conjugates was performed on an Ultraflextreme MALDI-time-of-flight (TOF) or a 15T MALDI-FT-ICR MS system. Infrared spectra were recorded on a PerkinElmer spectrum 2 Fourier transform infrared (FT-IR). Unprotected lipid A derivatives were dissolved in a mixture of CDCl3/MeOD 5/1 v/v for NMR analysis. DC activation and B3Z assay results were analyzed with GraphPad Prism version 7.00 for Windows, GraphPad Software. Purity of all compounds was >95% as determined by NMR or LC-MS analysis. FA represents fatty acid.
4.1.1. Automated Solid-Phase Synthesis General Experimental Information
The automated solid-phase peptide synthesis was performed on a 250 μmol scale on a Protein Technologies Tribute-UV IR peptide synthesizer applying the Fmoc-based protocol starting from the Tentagel S RAM resin (loading 0.22 mmol/g). The synthesis was continued with Fmoc amino acids specific for each peptide. The following consecutive steps were performed in each cycle for HCTU chemistry on a 250 μmol scale: (1) deprotection of the Fmoc group with 20% piperidine in DMF for 10 min; (2) DMF wash; and (3) coupling of the appropriate amino acid using a fourfold excess; generally, the Fmoc amino acid (1.0 mmol) was dissolved in 0.2 M HCTU in DMF (5 mL), and the resulting solution was transferred to the reaction vessel followed by 2 mL of 1.0 M N,N-diisopropylethylamine (DIPEA) in DMF to initiate the coupling; the reaction vessel was then shaken for 30 min at 50 °C; (4) DMF wash; (5) capping with 10% Ac2O in 0.1 M DIPEA in DMF; (6) DMF wash; and (7) DCM wash. Aliquots of resin of the obtained sequences were checked on an analytical Agilent Technologies 1260 Infinity system with a Gemini 3 μm, C18, 110 Å, 50 × 4.6 mm2 column or a Vydac 219TP 5 μm diphenyl, 150 × 4.6 mm2 column with a 1 mL/min flow. The Fmoc amino acids applied in the synthesis were Fmoc-Ala-OH, Fmoc-Asn(Trt)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Gly-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH, Fmoc-Lys(MMT)-OH, Fmoc-Phe-OH, and Fmoc-Ser(OtBu)-OH Fmoc-Val-OH.
4.1.2. General Procedure for Cleavage from the Resin, Deprotection, and Purification
First, 30 μmol of resin was washed with DMF and DCM and dried after the last synthesis step followed by a treatment for 180 min with 0.6 mL cleavage cocktail of 95% TFA, 2.5% triisopropylsilane (TIS), and 2.5% H2O. The suspension was filtered, the resin was washed with 0.6 mL of the cleavage cocktail, and the combined TFA solutions were added dropwise to cold Et2O and stored at −20 °C overnight. The obtained suspension of the product in Et2O was centrifuged, Et2O was removed, and the precipitant was dissolved in CH3CN/H2O/tBuOH (1/1/1 v/v/v) or dimethyl sulfoxide (DMSO)/CH3CN/H2O/tBuOH (3/1/1/1 v/v/v/v). Purification was performed on a Gilson GX-281 preparative RP-HPLC with a Gemini-NX 5 μm, C18, 110 Å, 250 × 10.0 mm2 column or a Vydac 219TP 5 μm diphenyl, 250 × 10 mm2 column.
4.1.3. General Purification Method for CRX-527-O-Conjugates
A C18 column was washed subsequently with CH3CN, MeOH, DCM/MeOH (1/1 v/v), MeOH, CH3CN, CH3CN/H2O, and H2O. The reaction mixture was added on the column, and the Eppendorf was rinsed with a mixture of CH3CN/tBuOH/Milli-Q H2O (1/1/1 v/v/v, 0.50 mL), which was also added to the C18 column. The column was subsequently flushed with 6 mL of the following solvent systems: H2O, CH3CN/H2O (1/1 v/v), CH3CN, DMSO, and CH3CN/tBuOH/Milli-Q H2O (1/1/1 v/v/v) and collected in Eppendorfs containing 1.0 mL of each solvent system. The column was then flushed with MeOH (6.0 mL), followed by DCM/MeOH (1/1 v/v, 6.0 mL), which were collected in separate flasks, concentrated in vacuo at 35 °C, and lyophilized by dissolving in CH3CN/tBuOH/Milli-Q H2O (1/1/1 v/v/v), yielding the conjugate as a white solid.a
4.1.4. MALDI-TOF Measurements
MALDI-TOF measurements: First, 1 μL of a DMSO solution of the compound was spotted on a 384-MTP target plate (Bruker Daltonics, Bremen, Germany) and air-dried. Subsequently, 1 μL of 2,5-dihydroxybenzoic acid (2,5-DHB; Bruker Daltonics) matrix (20 mg/mL in ACN/water; 50:50 (v/v)) was applied on the plate, and the spots were left to dry prior to MALDI-TOF analysis. An UltrafleXtreme MALDI-TOF (Bruker Daltonics), equipped with Smartbeam-II laser, was used to measure the samples in the reflectron positive ion mode. The MALDI-TOF was calibrated using a peptide calibration standard prior to measurement.
4.2. Synthesis and Characterizations
The syntheses and characterizations for compounds 14, 17, 18, and 18, together with the characterizations of the intermediates, are described in the Supporting Information.
4.2.1. Acetyl 3,4,6-Tri-O-acetyl-2-N-trichloroethoxycarbonyl-α/β-d-glucopyranoside (9)
NaHCO3 (144 g, 1.65 mol, 3.0 equiv) and 2,2,2-trichloroethoxycarbonyl chloride (93 mL, 0.68 mol, 1.2 equiv) were added to a solution of d-glucosamine·HCl (0.12 kg, 0.55 mol, 1.0 equiv) in H2O (1.1 L). The reaction was stirred vigorously at room temperature overnight, after which the resulting white suspension was filtered and the residue was washed with cold H2O. The white solid was coevaporated with toluene (3×) before dissolving in pyridine (0.60 L). The reaction mixture was cooled to 0 °C, and Ac2O (0.30 L, 3.2 mol, 5.8 equiv) was added. The reaction mixture was allowed to warm up to room temperature and stirred overnight. The reaction mixture was cooled to 0 °C, quenched by the addition of H2O, and subsequently diluted with EtOAc. The organic layer was washed several times with 1 M HCl, dried over MgSO4, filtered, and concentrated in vacuo. TLC analysis showed no full conversion; therefore, the oil was dissolved in pyridine (0.60 L) and cooled to 0 °C. Ac2O (0.45 L, 4.8 mol, 8.7 equiv) was added, and after 30 min, the mixture was allowed to warm up to room temperature. After 2.5 h, TLC analysis showed full conversion. The reaction was quenched by the addition of MeOH and concentrated in vacuo. Coevaporation with toluene (3×) gave compound 9 (189 g, 362 mmol, 66%), which was used without further purification. Rf: 0.20 (7/3 pentane/EtOAc); 1H NMR (CDCl3, 400 MHz, HH-correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC)): δ 6.16 (d, 1H, J = 3.8 Hz, H-1), 5.43 (d, 1H, J = 9.5 Hz, NH), 5.27–5.17 (m, 1H, H-3), 5.13 (t, 1H, J = 9.9 Hz, H-4), 4.76 (d, 1H, J = 12.1 Hz, CHH Troc), 4.56 (d, 1H, J = 12.1 Hz, CHH Troc), 4.23–4.11 (m, 2H, H-2, CHH-6), 4.02–3.94 (m, 2H, H-5, CHH-6), 2.13 (s, 3H, CH3 Ac), 2.02 (s, 3H, CH3 Ac), 1.98–1.96 (m, 6H, 2× CH3 Ac); 13C-attached proton test (APT) NMR (CDCl3, 101 MHz, HSQC): δ 171.2, 170.7, 169.2, 168.7 (C=O Ac), 154.1 (C=O Troc), 95.3 (Cq Troc), 90.4 (C-1), 74.6 (CH2 Troc), 70.3 (C-3), 69.6 (C-5), 67.6 (C-4), 61.5 (CH2-6), 53.1 (C-2), 20.9, 20.7, 20.6, 20.5 (CH3 Ac); FT-IR (neat, cm–1): 3329, 2958, 2258, 2126, 1742, 1536, 1432, 1368, 1212, 1172, 1141, 1123, 1095, 1080, 1031, 1012, 952, 910, 820, 728, 681, 648, 599, 568, 526, 475; HRMS: [M + Na]+ calcd for C17H22Cl3NO11Na: 544.0151, found 544.0159.
4.2.2. Benzyl N-Trichloroethoxycarbonyl-l-serinate (10)
l-Serine (49.6 g, 472 mmol, 1.0 equiv) was dissolved in a mixture of CCl4/benzyl alcohol (1/1 v/v, 0.46 L). p-Toluenesulfonic acid (96.6 g, 508 mmol, 1.1 equiv) was added, and the white suspension was heated to 100 °C using a Dean–Stark apparatus. After stirring overnight, a clear solution was obtained, which was cooled down to room temperature before concentrating in vacuo. The residue was dissolved in DCM and washed with sat. aq. NaHCO3 (3×). The organic layer was extracted with 1 M HCl (3×), and the combined aqueous layers were concentrated in vacuo. Coevaporation with toluene yielded the intermediate as a white solid (46.6 g, 201 mmol), which was dissolved in DCM (1.0 L) under an argon atmosphere. Succinimidyl-2,2,2-trichloroethyl carbonate54 (61.5 g, 212 mmol, 1.05 equiv) was added to the reaction mixture, followed by the addition of Et3N (42 mL, 0.30 mol, 1.5 equiv) under a flow of argon. After 1 h, TLC analysis showed complete conversion of the starting material and the reaction mixture was washed with 1 M HCl (1×) and H2O (1×). The aqueous layers were extracted with DCM (1×), and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification by column chromatography (20 → 100% EtOAc in pentane) yielded the title compound (69.4 g, 187 mmol, 40% over two steps). HRMS: [M + H]+ calcd for C13H15O5NCl3: 370.00103, found 370.00105. Analytic data were in agreement with reported data.54
4.2.3. N-Trichloroethoxycarbonyl-O-[3,4,6-tri-O-acetyl-2-N-trichloroethoxycarbonyl-β-d-glucopyranosyl]-l-serine (11)
Compounds 9 (88.9 g, 170 mol, 1.0 equiv) and 10 (69.3 g, 187 mmol, 1.1 equiv) were coevaporated with toluene (2×) under an argon atmosphere and dissolved in DCM (0.28 L). The mixture was cooled to 0 °C, followed by the slow addition of BF3·OEt2 (42 mL, 0.34 mol, 2.0 equiv). The mixture was allowed to warm up to room temperature and stirred for an additional 48 h. The mixture was quenched with Et3N and washed with sat. aq. NaHCO3 (1×). The aqueous layer was extracted with DCM (3×), and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification by column chromatography (20 → 100% EtOAc in pentane) gave an oil (117 g), which was a mixture of unreacted donor 9 and benzyl N-trichloroethoxycarbonyl-O-[3,4,6-tri-O-acetyl-2-N-trichloroethoxycarbonyl-β-d-glucopyranosyl]-l-serinate. After coevaporating with toluene (3×) under an argon atmosphere, the oil was dissolved in THF (1.2 L), followed by the addition of Pd/C (10%, 11.7 g). The black suspension was purged with argon for 15 min, followed by purging with H2(g), and after 15 min, a H2(g)-filled balloon was applied. After stirring at room temperature overnight, the mixture was filtered over a Whatmann filter and concentrated in vacuo. Purification by column chromatography (10 → 100% acetone in pentane) yielded the title compound (79.7 g, 107 mmol, 63% yield over two steps) and unreacted donor 11 (18.5 g, 35.4 mmol). Rf: 0.13 (9/1 DCM/MeOH); [α]D25 +11.3° (c = 0.23, CHCl3); 1H NMR (MeOD, 400 MHz, HH-COSY, HSQC): δ 5.31–5.21 (m, 1H, H-3), 4.98 (t, 1H, J = 9.7 Hz, H-4), 4.86 (d, 1H, J = 5.1 Hz, CHH Troc), 4.83–4.74 (m, 2H, 2× CHH Troc), 4.74–4.68 (m, 2H, H-1, CHH Troc), 4.46–4.38 (m, 1H, CH serine), 4.31 (dd, 1H, J = 12.4, 4.6 Hz, CHH-6), 4.25–4.07 (m, 2H, CHH-6, CHH serine), 3.95 (dd, 1H, J = 10.5, 3.9 Hz, CHH serine), 3.81 (ddd, 1H, J = 10.0, 4.3, 2.3 Hz, H-5), 3.58 (dd, 1H, J = 10.4, 8.6 Hz, H-2), 2.07 (s, 3H, CH3 Ac), 2.00 (s, 3H, CH3 Ac), 1.97 (s, 3H, CH3 Ac); 13C-APT NMR (MeOD, 101 MHz, HSQC): δ 172.5, 172.4, 171.7 (C=O Ac), 171.3 (C=O serine), 156.7, 156.5 (C=O Troc), 101.9 (C-1), 97.1 (Cq Troc), 75.7, 75.4 (CH2 Troc), 73.7 (C-3), 73.0 (C-5), 70.2 (C-4), 70.1 (CH2 serine), 63.1 (CH2-6), 57.1 (C-2), 55.7 (CH serine), 20.6 (CH3 Ac); FT-IR (neat, cm–1): 3340, 2958, 1744, 1532, 1369, 1232, 1170, 1102, 1048, 819, 769, 734, 569; HRMS: [M + Na]+ calcd for C21H26Cl6N2O14Na: 762.9407, found 762.9416.
4.2.4. Benzyl N-Trichloroethoxycarbonyl-O-[2-N-trichloroethoxycarbonyl-β-d-glucopyranosyl]-l-serinate (12)
Compound 11 (79.6 g, 107 mmol, 1.0 equiv) was dissolved in MeOH (1.1 L), and NH4OH (13.4 M, 73.5 mL, 985 mmol, 9.2 equiv) was added. After 2 days of stirring at room temperature, TLC analysis showed complete conversion of the starting material. The reaction mixture was concentrated in vacuo and coevaporated with toluene. The obtained oil was dissolved in a DCM/sat. aq. NaHCO3 mixture (1/1 v/v, 2.6 L), after which tetrabutylammonium bromide (34.9 g, 108 mmol, 1.0 equiv) and benzyl bromide (64 mL, 0.54 mol, 5.0 equiv) were added. The reaction mixture was stirred overnight. The layers were separated, and the aqueous layer was extracted with CHCl3 (2×) and DCM (1×). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification by column chromatography (20 → 100% EtOAc in pentane and then 20% MeOH in EtOAc) afforded compound 12 (44.2 g, 65.5 mmol, 79% yield over two steps). Rf: 0.49 (9/1 DCM/MeOH); [α]D25 −15.2° (c = 0.48, MeOH); 1H NMR (MeOD, 400 MHz, HH-COSY, HSQC): δ 7.41–7.28 (m, 5H, Ar), 5.25–5.12 (m, 2H, CH2 Bn), 4.85 (d, 1H, J = 12.2 Hz, CHH Troc), 4.79–4.69 (m, 3H, CHH Troc, CH2 Troc), 4.49 (t, 1H, J = 4.4 Hz, CH serine), 4.45 (d, 1H, J = 8.2 Hz, H-1), 4.24 (dd, 1H, J = 10.2, 5.2 Hz, CHH serine), 3.93–3.83 (m, 2H, CHH serine, CHH-6), 3.67 (dd, 1H, J = 11.8, 5.5 Hz, CHH-6), 3.45 (dd, 1H, J = 10.2, 8.2 Hz, H-3), 3.41–3.33 (m, 1H, H-2), 3.31–3.21 (m, 2H, H-4, H-5); 13C-APT NMR (MeOD, 101 MHz, HSQC): δ 171.2 (C=O serine), 157.1, 156.5 (C=O Troc), 137.0 (Cq Ar), 129.6, 129.3, 129.1 (Ar), 102.8 (C-1), 96.8 (Cq Troc), 78.0 (C-5), 75.6 (CH2 Troc), 75.5 (C-3), 72.0 (C-4), 69.6 (CH2 serine), 68.2 (CH2 Bn), 62.7 (CH2-6), 58.9 (C-2), 56.2 (CH serine); FT-IR (neat, cm–1): 3423, 2955, 2487, 1729, 1431, 1332, 1293, 1173, 1060, 820, 731, 569; HRMS: [M + Na]+ calcd for C22H26Cl6N2O11Na: 726.9560, found 726.9576.
4.2.5. Benzyl N-Trichloroethoxycarbonyl-O-[4,6-O-di-tert-butylsilylidene-2-N-trichloroethoxycarbonyl-β-d-glucopyranosyl]-l-serinate (13)
A solution of compound 12 (2.01 g, 2.84 mmol, 1.0 equiv) in DMF (14 mL) was cooled to −40 °C. Di-tert-butylsilanediyl-bistriflate (0.92 mL, 3.1 mmol, 1.1 equiv) was added dropwise. After 1 h, the reaction was allowed to warm up to room temperature and stirred overnight. The reaction mixture was quenched by the addition of pyridine (1.6 mL, 19.9 mmol, 7.0 equiv). The mixture was diluted with Et2O, and the organic layer was washed with H2O (1×) and sat. aq. NaHCO3 (3×), dried over Na2SO4, filtered, and concentrated in vacuo. After purification by column chromatography (2 → 3% acetone in DCM), the title compound (2.07 g, 2.44 mmol, 86%) was obtained as a white foam. Rf: 0.60 (1/1 pentane/Et2O); [α]D25 −24.0° (c = 0.86, CHCl3); 1H NMR (CDCl3, 400 MHz, HH-COSY, HSQC): δ 7.38–7.28 (m, 5H, Ar), 6.30 (s, 1H, NH serine), 5.80 (d, 1H, J = 7.7 Hz, NH GlcN), 5.23–5.12 (m, 2H, CH2 Bn), 4.81–4.65 (m, 5H, H-1, 2× CH2 Troc), 4.53 (dt, 1H, J = 7.8, 3.4 Hz, CH serine), 4.25 (dd, 1H, J = 10.3, 3.3 Hz, CHH serine), 4.14 (dd, 1H, J = 10.1, 5.0 Hz, CHH-6), 3.89–3.80 (m, 2H, CHH-6, CHH serine), 3.80–3.72 (m, 1H, H-3), 3.67 (t, 1H, J = 8.9 Hz, H-4), 3.42–3.34 (m, 1H, H-5), 3.34–3.25 (m, 1H, H-2), 3.22 (br, 1H, OH), 1.04 (s, 9H, 3× CH3tBu), 0.97 (s, 9H, 3× CH3tBu); 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 169.3 (C=O serine), 154.6, 154.5 (C=O Troc), 135.1 (Cq Ar), 128.6, 128.5, 128.2 (Ar), 100.8 (C-1), 95.5, 95.4 (Cq Troc), 77.4 (C-4), 74.6 (CH2 Troc), 73.5 (C-3), 70.3 (C-5), 68.9 (CH2 serine), 67.6 (CH2 Bn), 66.0 (CH2-6), 57.4 (H-2), 54.5 (CH serine), 27.4, 27.0 (CH3tBu), 22.6, 19.9 (CqtBu); FT-IR (neat, cm–1): 3340, 2935, 2886, 2860, 1730, 1523, 1473, 1387, 1365, 1336, 1243, 1201, 1160, 1076, 1009, 943, 909, 826, 765, 730, 697, 653, 618, 569, 476; HRMS: [M + Na]+ calcd for C30H42Cl6N2O11SiNa: 867.0581, found 867.0599.
4.2.6. Benzyl N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[4,6-O-di-tert-butylsilylidene-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-β-d-glucopyranosyl]-l-serinate (15)
To a solution of compound 13 (2.55 g, 3.00 mmol, 1.0 equiv) in THF (30 mL) were added activated zinc (4.0 g, 61 mmol, 20 equiv) and AcOH (0.69 mL, 12 mmol, 4.0 equiv) under an argon atmosphere. The suspension was stirred for 25 min, and the mixture was subsequently sonicated for 5 min. The mixture was stirred again for 25 min, followed by sonicating for 5 min. TLC and LC-MS analyses showed complete conversion of the starting material. The suspension was filtered over a Whatmann filter, and the residue was washed with DCM and EtOAc. The combined filtrates were concentrated in vacuo and coevaporated with toluene (3×), and the obtained solid was dissolved in EtOAc. The solution was subsequently washed with 0.1 M HCl (1×), sat. aq. NaHCO3 (1×), and brine (1×). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. A mixture of the obtained yellow oil and acid 14 (5.39 g, 13.5 mmol, 4.5 equiv) was coevaporated with toluene (1×) and dissolved in DCM (30 mL) under an argon atmosphere. EDC·MeI (4.01 g, 13.5 mmol, 4.5 equiv) and DMAP (11 mg, 90 μmol, 0.03 equiv) were added, and the reaction mixture was stirred for 4 h, after which the mixture was concentrated in vacuo. Several purifications by column chromatography (2 → 20% EtOAc in DCM + 0.1% Et3N and 0 → 10% acetone in DCM + 0.1% Et3N) gave compound 15 (3.07 g, 1.87 mmol, 62% over two steps) as a white foam. Rf: 0.58 (95/5 DCM/acetone); [α]D25 −15.4° (c = 0.50, CHCl3); 1H NMR (CDCl3, 500 MHz, HH-COSY, HSQC): δ 7.37–7.27 (m, 5H, Ar), 7.01 (d, 1H, J = 7.8 Hz, NH serine), 6.27 (d, 1H, J = 8.3 Hz, NH GlcN), 5.22–5.05 (m, 5H, 3× CH FA, CH2 Bn), 5.06–4.98 (m, 1H, H-3), 4.72–4.66 (m, 2H, H-1, CH serine), 4.21 (dd, 1H, J = 10.7, 3.0 Hz, CHH serine), 4.14 (dd, 1H, J = 10.2, 5.0 Hz, CHH serine), 3.88–3.77 (m, 3H, H-4, CHH-6, CHH serine), 3.73–3.65 (m, 1H, H-2), 3.44–3.37 (m, 1H, H-5), 2.67–2.20 (m, 12H, 6× CH2 FA), 1.71–1.50 (m, 12H, 6× CH2 FA), 1.40–1.17 (m, 90H, 45× CH2 FA), 1.02 (s, 9H, 3× CH3tBu), 0.94 (s, 9H, 3× CH3tBu), 0.87 (t, 18H, J = 6.7 Hz, 6× CH3 FA); 13C-APT NMR (CDCl3, 126 MHz, HSQC): δ 173.9, 173.8, 173.3, 170.6, 170.3, 170.2 (C=O FA), 169.5 (C=O serine), 135.5 (Cq Ar), 128.6, 128.4, 128.1 (Ar), 101.6 (C-1), 75.1 (C-4), 74.4 (C-3), 71.5, 71.5 (CH FA), 70.8 (C-5), 70.1 (CH FA), 68.8 (CH2 serine), 67.3 (CH2 Bn), 66.3 (CH2-6), 54.7 (C-2), 52.8 (CH serine), 42.2, 41.3, 39.2, 34.7, 34.6, 34.6, 34.5, 34.0, 32.1, 32.0, 32.0, 29.9, 29.8, 29.8, 29.8, 29.7, 29.7, 29.7, 29.6, 29.6, 29.6, 29.5, 29.5, 29.5, 29.4, 29.4, 29.3 (CH2 FA), 27.5, 27.0 (CH3tBu), 25.5, 25.4, 25.2, 25.2, 25.1, 22.8 (CH2 FA), 22.7, 20.0 (CqtBu), 14.2 (CH3 FA); FT-IR (neat, cm–1): 3285, 3068, 2956, 2923, 2854, 1734, 1652, 1540, 1450, 1466, 1378, 1364, 1246, 1173, 1075, 1030, 1011, 837, 827, 769, 723, 696, 652, 581, 463; HRMS: [M + H]+ calcd for C96H173N2O16Si: 1638,2549, found 1638.2493.
4.2.7. Benzyl N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[4-O-bis(benzyloxy)phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-β-d-glucopyranosyl]-l-serinate (16a)
Compound 15 (1.92 g, 1.17 mmol, 1.0 equiv) was dissolved in THF (12 mL) under an argon atmosphere and cooled to 0 °C. HF·Et3N (0.58 mL, 3.6 mmol, 3.0 equiv) was added, and the reaction mixture was stirred for 1.5 h, after which TLC analysis showed complete conversion of the starting material. The reaction was quenched with sat. aq. NaHCO3, diluted with EtOAc, and washed with brine (1×). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. Purification by column chromatography (0 → 4% MeOH in DCM) gave the diol intermediate (1.61 g, 1.08 mmol, 92%). TBDMSCl (290 mg, 1.92 mmol, 1.5 equiv) was added to a solution of the previously obtained diol (1.81 g, 1.21 mmol, 1.0 equiv) in pyridine (8.0 mL). After stirring at room temperature for 3 h, TLC analysis showed complete conversion of the starting material. The reaction mixture was diluted with EtOAc, washed with 1 M HCl (2×) and sat. aq. NaHCO3 (2×), dried over MgSO4, filtered, and concentrated in vacuo. Purification by column chromatography (5 → 20% EtOAc in toluene) afforded the intermediate (1.71 g, 1.06 mmol, 88%), which was coevaporated with toluene (2×) under an argon atmosphere and dissolved in dry DCM (18 mL). Dibenzyl diisopropylaminephosphoramidite (0.70 mL, 1.9 mmol, 1.8 equiv) and tetrazole (186 mg, 2.65 mmol, 2.5 equiv) were added. After stirring for 35 min, the reaction mixture was cooled to 0 °C, followed by the addition of meta-chloroperoxybenzoic acid (m-CPBA) (0.74 g, 3.0 mmol, 2.8 equiv). After 40 min, TLC analysis showed complete conversion into the phosphate. The reaction was diluted with aq. sat. NaHCO3 and extracted with DCM (3×). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by column chromatography (0 → 20% EtOAc in toluene) and several size exclusions (DCM/MeOH: 1/1) gave the phosphate intermediate in quantitative yield (2.00 g). TFA (0.81 mL, 11 mmol, 10 equiv) was added to a solution of the previously obtained phosphate (2.00 g, 1.06 mmol, 1.0 equiv) in DCM (21 mL) at 0 °C. After 20 min, the resulting yellow solution was allowed to warm up to room temperature and stirred for an additional 3 h. TLC analysis showed complete conversion, and the reaction was quenched with aq. sat. NaHCO3 at 0 °C. The reaction mixture was further diluted with H2O and extracted with DCM (3×). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. After purification by column chromatography (10 → 50% EtOAc in toluene), compound 16a (1.57 g, 0.893 mmol, 84%) was obtained as a white foam. Rf: 0.70 (1/1 pentane/EtOAc); [α]D25 +2.2° (c = 0.33, CHCl3); 1H NMR (CDCl3, 400 MHz, HH-COSY, HSQC): δ 7.38–7.24 (m, 15H, Ar), 7.19–7.08 (m, 1H, NH serine), 6.57 (d, 1H, J = 7.7 Hz, NH GlcN), 5.36 (t, 1H, J = 9.7 Hz, H-3), 5.23–5.07 (m, 5H, 3× CH FA, CH2 CO2Bn), 5.05–4.88 (m, 5H, H-1, 2× CH2 dibenzyl phosphate), 4.76–4.67 (m, 1H, CH serine), 4.44 (q, 1H, J = 9.3 Hz, H-4), 4.28–4.18 (m, 1H, CHH serine), 3.93–3.84 (m, 1H, CHH serine), 3.82–3.71 (m, 3H, CH2-6, OH), 3.60–3.51 (m, 1H, H-2), 3.37 (d, 1H, J = 9.7 Hz, H-5), 2.69 (dd, 1H, J = 14.7, 6.1 Hz, CHH FA), 2.54 (dd, 1H, J = 14.7, 5.8 Hz, CHH FA), 2.43–2.23 (m, 8H, 4× CH2 FA), 2.20 (t, 2H, J = 7.5 Hz, CH2 FA), 1.74–1.48 (m, 12H, 6× CH2 FA), 1.48–1.06 (m, 90H, 45× CH2 FA), 0.94–0.80 (m, 18H, 6× CH3 FA); 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 173.5, 173.4, 173.1, 170.6, 170.1, 169.7 (C=O FA), 169.4 (C=O serine), 135.3, 135.2, 135.2, 135.1, 135.1 (Cq Bn), 128.8, 128.7, 128.6, 128.5, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8, 127.8 (Ar), 100.4 (C-1), 74.7, 74.7 (C-5), 73.1, 73.0 (C-4), 72.3, 72.2 (C-3), 71.2, 70.8 (CH FA), 69.9, 69.9 (CH2 dibenzyl phosphate), 69.8 (CH FA), 68.7 (CH2 serine), 67.0 (CH2 CO2Bn), 60.3 (CH2-6), 55.1 (C-2), 52.8 (CH serine), 41.4, 41.0, 39.0, 34.4, 34.3, 34.2, 31.8, 31.8, 29.6, 29.6, 29.6, 29.5, 29.5, 29.5, 29.5, 29.4, 29.4, 29.4, 29.3, 29.3, 29.2, 29.2, 29.2, 29.1, 29.1, 29.1, 25.2, 25.2, 25.0, 24.9, 24.9, 22.6, 22.6 (CH2 FA), 14.0 (CH3 FA); 31P-APT NMR (CDCl3, 162 MHz, heteronuclear multiple bond correlation (HMBC)): δ −0.05; FT-IR (neat, cm–1): 3317, 3066, 2956, 2923, 2853, 1733, 1654, 1640, 1541, 1499, 1466, 1456, 1379, 1238, 1166, 1128, 1106, 1080, 1034, 1016, 914, 736, 696, 602, 531, 498; HRMS: [M + H]+ calcd for C102H170N2O19P: 1758,2130, found 1758.2065.
4.2.8. N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[6-azide-4-O-phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-β-d-glucopyranosyl]-l-serine (16b)
After coevaporating with toluene (2×), compound 16a (82 mg, 47 μmol, 1.0 equiv) was dissolved in THF under an argon atmosphere. PPh3 (48 mg, 0.18 mmol, 3.9 equiv) was added, and the reaction mixture was cooled to −20 °C. DEAD (15 μL, 96 μmol, 2.0 equiv) and DPPA (20.5 μL, 96 μmol, 2.0 equiv) were added subsequently, and the stirring was continued for 1 h, followed by the addition of DEAD (15 μL, 96 μmol, 2.0 equiv) and DPPA (20.5 μL, 96 μmol, 2.0 equiv). After stirring for 1 h at −20 °C, the reaction mixture was slowly warmed up to room temperature overnight. The mixture was concentrated in vacuo. Purification by column chromatography (10 → 30% EtOAc in pentane) afforded the title compound (56 mg, 31 μmol, 66%). Rf: 0.52 (7/3 pentane/EtOAc); 1H NMR (CDCl3, 400 MHz, HH-COSY, HSQC): δ 7.38–7.24 (m, 15H, Ar), 7.02 (d, 1H, J = 8.1 Hz, NH serine), 6.44 (d, 1H, J = 7.4 Hz, NH GlcN), 5.34 (dd, 1H, J = 10.5, 8.9 Hz, H-3), 5.21–5.12 (m, 5H, 3× CH FA, CH2 CO2Bn), 5.03 (d, 1H, J = 8.2 Hz, H-1), 5.00–4.94 (m, 2H, 2× CHH Bn), 4.91 (dd, 2H, J = 7.9, 3.2 Hz, 2× CHH Bn), 4.78–4.70 (m, 1H, CH serine), 4.32–4.21 (m, 2H, H-4, CHH serine), 3.85 (dd, 1H, J = 11.2, 2.8 Hz, CHH serine), 3.55–3.48 (m, 1H, H-5), 3.46–3.37 (m, 2H, H-2, CHH-6), 3.28 (dd, 1H, J = 13.4, 6.2 Hz, CHH-6), 2.67 (dd, 1H, J = 14.9, 6.2 Hz, CHH FA), 2.55–2.44 (m, 2H, 2× CHH FA), 2.41–2.19 (m, 9H, CHH FA, 4× CH2 FA), 1.71–1.43 (m, 12H, 6× CH2 FA), 1.37–1.14 (m, 90H, 45× CH2 FA), 0.93–0.81 (m, 18H, 6× CH3 FA); 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 173.8, 173.7, 171.1, 170.3, 170.0 (C=O FA), 169.5 (C=O serine), 135.5, 135.5, 135.4 (Cq Bn), 128.9, 128.8, 128.8, 128.7, 128.6, 128.4, 128.3, 128.3, 128.2 (Ar), 100.3 (C-1), 74.5, 74.5 (C-5), 74.0, 73.9 (C-4), 72.3, 72.3 (C-3), 71.4, 71.0, 70.3 (CH FA), 70.0, 70.0, 69.9, 69.9 (CH2 dibenzyl phosphate), 69.2 (CH2 serine), 67.3 (CH2 CO2Bn), 55.8 (C-2), 52.8 (CH serine), 50.7 (CH2N3), 41.6, 41.2, 39.8, 34.7, 34.6, 34.6, 34.6, 32.0, 32.0, 29.9, 29.8, 29.8, 29.8, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.5, 29.4, 29.4, 29.4, 29.3, 25.5, 25.4, 25.3, 25.2, 25.2, 25.1, 22.8, 22.8 (CH2 FA), 14.2 (CH3 FA); 31P-APT NMR (CDCl3, 162 MHz, HMBC): δ −1.03; FT-IR (neat, cm–1): 3276, 2992, 2853, 2361, 2102, 1733, 1654, 1543, 1498, 1457, 1274, 1248, 1171, 1108, 1010, 904, 734, 696, 601, 506; HRMS: [M + H]+ calcd for C102H169N5O18P: 1783.22059, found 1783.22059.
4.2.9. N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[4-O-phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-β-d-glucopyranosyl]-l-serine (1)
After coevaporating with toluene (3×) under an argon atmosphere, compound 16a (21.7 mg, 12.3 μmol, 1.0 equiv) was dissolved in THF (1.0 mL), followed by the addition of Pd/C (10%, 21 mg). A H2(g)-filled balloon was applied on the obtained black suspension. After stirring at room temperature for 3 h, the mixture was filtered over a Whatmann filter. The filter was washed with DCM, followed by the addition of Et3N (3.4 μL, 24 μmol, 2.0 equiv). After mixing for 5 min, the clear solution was concentrated in vacuo and purified by size exclusion (DCM/MeOH: 1/1). Lyophilization gave compound 1 (18.2 mg, 12.2 μmol, 99%) as a white solid. 1H NMR (CDCl3, 850 MHz, HH-COSY, HSQC) δ 5.20–5.15 (m, 1H, CH FA), 5.14–5.07 (m, 3H, H-3, 2× CH FA), 4.54–4.49 (m, 2H, H-1, CH serine), 4.19–4.08 (m, 2H, H-4, CHH serine), 3.88 (d, J = 13.1 Hz, 1H, CHH-6), 3.84–3.78 (m, 1H, CHH serine), 3.72–3.66 (m, 2H, H-2, CHH-6), 3.28 (d, J = 9.8 Hz, 1H, H-5), 2.63–2.45 (m, 4H, 2× CH2 FA), 2.40 (dd, J = 14.7, 7.3 Hz, 1H, CHH FA), 2.29 (dd, J = 14.7, 5.7 Hz, 1H, CHH FA), 2.28–2.20 (m, 6H, 3× CH2 FA), 1.60–1.47 (m, 12H, 6× CH2 FA), 1.30–1.15 (m, 90H, 45× CH2 FA), 0.85–0.81 (m, 18H, 6× CH3 FA); 13C NMR (CDCl3, 214 MHz, HSQC) δ 173.8, 173.8, 173.7, 170.8, 170.6, 170.5 (C=O), 100.7 (C-1), 75.3 (C-5), 73.5 (C-3), 71.1, 70.8 (CH FA), 70.3 (C-4), 70.1 (CH FA), 69.2 (CH serine), 59.9 (CH2-6), 54.1 (C-2), 52.6 (CH serine), 41.0, 40.5, 38.8, 34.4, 34.4, 34.2, 34.1, 34.0, 31.8, 31.8, 31.8, 31.8, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.5, 29.4, 29.4, 29.4, 29.4, 29.3, 29.3, 29.3, 29.3, 29.2, 29.2, 29.2, 29.2, 29.1, 29.1, 29.1, 25.2, 25.1, 25.1, 24.9, 24.9, 22.6, 22.5, 22.5 (CH2 FA), 13.9 (CH3 FA); 31P NMR (CDCl3, 202 MHz, HMBC) δ 2.40; HRMS: [M + H]+ calcd for C81H152N2O19P: 1488.0721, found 1488.0725.
4.2.10. N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[4-O-phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-6-O-(11-acetamidoundecanoyl)-β-d-glucopyranosyl]-l-serine (2)
Compound 16a (57.6 mg, 32.8 μmol, 1.0 equiv) and acid 17 (20.8 mg, 85.5 μmol, 2.6 equiv) were coevaporated twice with toluene under an argon atmosphere before being dissolved in dry DCE (0.5 mL). The solution was cooled to 0 °C, followed by the addition of EDC·MeI (20.8 mg, 68.6 μmol, 2.1 equiv) and DMAP (5.3 mg, 43 μmol, 1.3 equiv). The obtained yellow suspension was allowed to warm up to room temperature and was stirred overnight. The white suspension was diluted with aq. sat. NaHCO3 and extracted with DCM (2×). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by column chromatography (10 → 60% EtOAc in pentane) yielded the intermediate (57.4 mg, 28.9 μmol, 88%), of which 26.7 mg (13.5 μmol, 1 equiv) was coevaporated with toluene (2×) under an argon atmosphere and dissolved in THF (1.0 mL). Pd/C (10%, 20.8 mg) was added, and the reaction mixture was stirred for 2.5 h at room temperature under a blanket of H2(g). The black suspension was filtered over a Whatmann filter, and the filter was washed with CHCl3. Et3N (4.0 μL, 28.6 μmol, 2.1 equiv) was added to the combined filtrates and mixed for 5 min, and the solution was concentrated in vacuo. After purification by size exclusion (DCM/MeOH: 1/1) and lyophilization, compound 2 (12.0 mg, 7.00 μmol, 52%) was obtained as a white solid. 1H NMR (CDCl3, 600 MHz, HH-COSY, HSQC): δ 5.21–5.15 (m, 2H, CH FA), 5.15–5.06 (m, 2H, H-3, CH FA), 4.60 (d, 1H, J = 7.0 Hz, H-1), 4.57–4.53 (m, 1H, CH serine), 4.49 (d, 1H, J = 11.0 Hz, CHH-6), 4.22–4.07 (m, 3H, H-4, CHH-6, CHH serine), 3.71–3.62 (m, 3H, H-2, H-5, CHH serine), 3.11 (t, 2H, J = 7.2 Hz, CH2NHAc), 2.62–2.54 (m, 3H, CH2 FA, CHH FA), 2.50 (dd, 1H, J = 14.6, 5.8 Hz, CHH FA), 2.40 (dd, 1H, J = 14.6, 7.3 Hz, CHH FA), 2.34–2.21 (m, 9H, CHH FA, 3× CH2 FA, CH2 linker), 1.90 (s, 3H, CH3 Ac), 1.62–1.48 (m, 14H, 6× CH2 FA, CH2 linker), 1.46–1.41 (m, 2H, CH2 linker), 1.33–1.15 (m, 102H, 45× CH2 FA, 6× CH2 linker), 0.83 (t, 18H, J = 6.9 Hz, 6× CH3 FA); 13C-APT NMR (CDCl3, 151 MHz, HSQC): δ 174.5, 174.2, 174.0, 171.7, 171.0, 170.9, 170.7 (C=O), 100.1 (C-1), 73.5 (C-3), 73.3, 73.3 (C-5), 71.8, 71.7 (C-4), 71.3, 71.2, 70.5 (CH FA), 68.7 (CH2 serine), 63.9 (CH2-6), 54.3 (C-2), 52.6 (CH serine), 41.2, 40.7 (CH2 FA), 39.8 (CH2NHAc), 39.3, 34.7, 34.6, 34.6, 34.5, 34.5, 34.4, 34.2, 32.1, 32.1, 32.1, 32.0, 29.9, 29.9, 29.9, 29.9, 29.8, 29.8, 29.8, 29.7, 29.7, 29.7, 29.6, 29.6, 29.6, 29.5, 29.5, 29.5, 29.4, 29.4, 29.3, 29.2, 27.0, 25.5, 25.4, 25.4, 25.2, 25.2, 25.0, 22.8, 22.8 (CH2 FA, CH2 linker), 22.6 (CH3 Ac), 14.2 (CH3 FA); 31P-APT NMR (CDCl3, 202 MHz, HMBC): δ 0.59; HRMS: [M + H]+ calcd for C94H175N3O21P: 1713.2450, found 1713.2458.
4.2.11. N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[4-O-phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-6-O-(13-acetamido-3-oxo-2,5,8,11-tetraoxatridecyl)-β-d-glucopyranosyl]-l-serine (3)
Compound 16a (49.6 mg, 28.2 μmol, 1.0 equiv) and acid 18 (22.5 mg, 90.3 μmol, 3.2 equiv) were coevaporated twice with toluene under an argon atmosphere before being dissolved in dry DCE (0.43 mL). The solution was cooled to 0 °C, followed by the addition of EDC·MeI (17.7 mg, 59.6 μmol, 2.1 equiv) and DMAP (2.2 mg, 18 μmol, 0.6 equiv). The obtained yellow suspension was allowed to warm up to room temperature and was stirred overnight. The resulting white suspension was diluted with DCM (0.6 mL) and stirred for an additional 4 h. The reaction mixture was subsequently diluted with aq. sat. NaHCO3 and extracted with DCM (2×). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by column chromatography (10 → 30% acetone in DCM) yielded the intermediate (41.3 mg, 20.8 μmol, 74%), of which 19.7 mg (9.90 μmol, 1.0 equiv) was coevaporated with toluene (2×) under an argon atmosphere and dissolved in THF (1.0 mL). Pd/C (10%, 19.8 mg) was added, and the reaction mixture was stirred for 3 h at room temperature under a blanket of H2(g). The black suspension was filtered over a Whatmann filter. The filter was washed with CHCl3, and Et3N (3.0 μL, 22 μmol, 2.2 equiv) was added to the combined filtrates. The solution was mixed for 5 min and concentrated in vacuo. After purification by size exclusion (DCM/MeOH: 1/1) and lyophilization, compound 3 (6.7 mg, 3.9 μmol, 39%) was obtained as a white solid. 1H NMR (CDCl3, 600 MHz, HH-COSY, HSQC): δ 5.22–5.15 (m, 2H, 2× CH FA), 5.15–5.04 (m, 2H, H-3, CH FA), 4.60–4.47 (m, 3H, H-1, CH serine, CHH-6), 4.33–4.12 (m, 5H, H-4, CHH-6, CHH serine, CH2 linker), 3.78–3.71 (m, 3H, H-2, CHH serine, CHH linker), 3.69–3.53 (m, 10H, H-5, 4× CH2 linker, CHH linker), 3.44–3.32 (m, 2H, CH2NHAc), 2.68–2.46 (m, 4H, 2× CH2 FA), 2.41 (dd, 1H, J = 14.5, 7.2 Hz, CHH FA), 2.30 (dd, 1H, J = 14.5, 5.7 Hz, CHH FA), 2.28–2.19 (m, 6H, 3× CH2 FA), 1.95 (s, 3H, CH3 Ac), 1.63–1.46 (m, 12H, 6× CH2 FA), 1.33–1.15 (m, 90H, 45× CH2), 0.83 (t, 18H, J = 7.0 Hz, 6× CH3); 13C-APT NMR (CDCl3, 151 MHz, HSQC): δ 174.1, 174.0, 173.8, 172.6, 171.2, 170.8, 170.7 (C=O), 100.6 (C-1), 73.9 (C-3), 72.7, 72.7 (C-5), 71.3 (CH FA), 71.1 (C-4, CH FA), 70.4 (CH2 linker), 70.3 (CH FA), 70.1, 69.9 (CH2 linker), 69.4 (CH2 serine, CH2 linker), 68.5 (CH2 linker), 62.5 (CH2-6), 53.8 (C-2), 52.4 (CH serine), 41.4, 40.7, 39.3 (CH2 FA), 39.1 (CH2NHAc), 34.7, 34.6, 34.6, 34.5, 34.5, 34.3, 32.1, 32.1, 32.0, 32.0, 29.9, 29.9, 29.9, 29.9, 29.8, 29.8, 29.8, 29.8, 29.7, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.5, 29.5, 29.4, 29.4, 29.3, 25.5, 25.4, 25.3, 25.2, 25.2, 25.2, 22.8, 22.8 (CH2 FA), 22.6 (CH3 Ac), 14.1 (CH3 FA); 31P-APT NMR (CDCl3, 202 MHz, HMBC): δ 1.52; HRMS: [M + H]+ calcd for C91H169N3O24P: 1719.18282, found 1719.18284.
4.2.12. N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[4-O-phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-6-N-(13-acetamido-3-oxo-5,8,11-trioxa-2-azatridecyl)-β-d-glucopyranosyl]-l-serine (4)
Compound 16b (23.6 mg, 13.2 μmol, 1.0 equiv) was dissolved in a mixture of DCM/MeOH/H2O (1,1,0.1 v/v/v, 1.2 mL). Activated zinc powder (9.1 mg, 0.15 mmol, 11.6 equiv) and NH4Cl (7.9 mg, 0.15 mmol, 11.2 equiv) were added, and the reaction mixture was stirred for 6 h. The reaction mixture was subsequently diluted with DCM and washed with aq. sat. NaHCO3 (1×). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The obtained amine (13.2 μmol, 1.0 equiv) and acid 18 (10.6 mg, 42.5 μmol, 3.2 equiv) were coevaporated with toluene (2×) under an argon atmosphere before being dissolved in dry DCE (0.4 mL). The solution was cooled to 0 °C, followed by the addition of EDC·MeI (8.5 mg, 29 μmol, 2.2 equiv) and DMAP (0.7 mg, 6 μmol, 0.4 equiv). The resulting yellow suspension was allowed to warm up to room temperature and was stirred overnight. The obtained white suspension was diluted with aq. sat. NaHCO3 and extracted with DCM (2×). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by column chromatography (10 → 40% acetone in DCM) and size exclusion (DCM/MeOH: 1/1) afforded the intermediate (10.6 mg, 5.33 μmol, 40% over two steps), which was coevaporated with toluene (2×) under an argon atmosphere and dissolved in THF (1.0 mL). Pd/C (10%, 21 mg) was added, and the reaction mixture was stirred for 3.5 h at room temperature under a blanket of H2(g). The black suspension was filtered over a Whatmann filter, and the filter was washed with CHCl3. Et3N (1.5 μL, 10.8 μmol, 2.1 equiv) was added to the combined filtrates. The solution was mixed for 5 min and concentrated in vacuo. After purification by size exclusion (DCM/MeOH: 1/1) and lyophilization, the title compound (5.8 mg, 3.4 μmol, 67%) was obtained as a white solid; 1H NMR (CDCl3, 850 MHz, HH-COSY, HSQC) δ 5.11–5.06 (m, 1H, CH FA), 5.07–5.03 (m, 1H, CH FA), 5.03–4.97 (m, 1H, CH FA), 4.97–4.93 (m, 1H, H-3), 4.45 (s, 1H, CH serine), 4.39 (d, J = 7.6 Hz, 1H, H-1), 4.02 (d, J = 9.9 Hz, 1H, CHH serine), 4.00–3.87 (m, 3H, H-4, CH2 linker), 3.84–3.78 (m, 1H, CHH-6), 3.69 (d, J = 8.3 Hz, 1H, CHH serine), 3.64–3.47 (m, 9H, H-2, 4× CH2 linker), 3.45–3.39 (m, 2H, CH2 linker), 3.36 (s, 1H, H-5), 3.27–3.24 (m, 2H, CH2NHAc), 3.23–3.20 (m, 1H, CHH-6), 2.54–2.49 (m, 1H, CHH FA), 2.49–2.42 (m, 2H, 2× CHH FA), 2.42–2.38 (m, 1H, CHH FA), 2.29 (dd, J = 14.6, 7.2 Hz, 1H, CHH FA), 2.20 (dd, J = 14.6, 5.6 Hz, 1H, CHH FA), 2.18–2.09 (m, 6H, 3× CH2 FA), 1.84 (s, 3H, CH3 Ac), 1.51–1.37 (m, 12H, 6× CH2 FA), 1.21–1.06 (m, 90H, 45× CH2 FA), 0.74 (t, J = 7.2 Hz, 18H, 6× CH3 FA); 13C-APT NMR (CDCl3, 214 MHz, HSQC): δ 173.7, 173.7, 170.9, 170.7 (C=O), 100.6 (C-1), 73.2, 73.2 (C-3/4/5), 71.1, 71.0 (CH FA), 70.9, 70.9 (CH FA), 70.6 (CH2 linker), 70.1, 70.1 (CH FA), 69.9, 69.9, 69.8, 69.7 (CH2 linker), 69.1 (CH2 serine), 53.8 (C-2), 52.6 (CH serine), 41.1, 40.4 (CH2 FA), 39.0 (CH2NHAc), 39.0 (CH2-6), 34.4, 34.4, 34.3, 34.2, 34.1, 31.8, 31.8, 29.6, 29.6, 29.6, 29.5, 29.4, 29.4, 29.4, 29.3, 29.2, 29.2, 29.1, 29.1, 25.2, 25.1, 25.1, 24.9, 22.5 (CH2 FA), 13.9 (CH3 FA); 31P-APT NMR (CDCl3, 202 MHz, HMBC): δ 1.19; HRMS: [M + H]+ calcd for C91H170N4O23P: 1718.19880, found 1718.19982.
4.2.13. N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[6-O-(11-N-(4-maleimidobutanoyl)-11-amino-3,6,9-trioxa-undecanoyl)-4-O-phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-β-d-glucopyranosyl]-l-serine (20a)
A solution of alcohol 16a (0.57 g, 0.32 mmol, 1.0 equiv) and acid S19 (0.19 g, 0.81 mmol, 2.5 equiv) was cooled to 0 °C under an argon atmosphere. EDC·MeI (0.20 g, 0.67 mmol, 2.0 equiv) and DMAP (2.0 mg, 16 μmol, 0.05 equiv) were added. After stirring for 15 min, the reaction mixture was allowed to warm up to room temperature and stirring continued for 3 h. Silica was added, and the suspension was concentrated in vacuo. Purification by column chromatography (20 → 50% EtOAc in toluene + 0.1% Et3N) gave the intermediate (0.51 g, 0.26 mmol, 80%).
After coevaporating with toluene (3×) under an argon atmosphere, the intermediate (0.51 g, 0.26 mmol, 1.0 equiv) was dissolved in dry THF (2.6 mL), followed by the addition of Pd/C (10%, 53 mg). A H2(g)-filled balloon was applied on the obtained black suspension. After stirring at room temperature overnight, the black suspension was filtered over washed silica. The silica was washed with CHCl3, followed by the addition of Et3N (0.14 mL, 1.0 mmol, 4 equiv). After mixing for 10 min, the clear solution was concentrated in vacuo. Lyophilization gave the free amine (0.34 g, 0.20 mmol, 77%) as a white solid. Then, 32.9 mg of the previously obtained amine (19.6 μmol, 1.0 equiv) was dissolved in DCM (1.6 mL), followed by the addition of sulfo-N-succinimidyl 4-maleimidobutyrate sodium salt (9.1 mg, 23.8 μmol, 1.2 equiv) and Et3N (16.4 μL, 0.12 mmol, 6.0 equiv). After stirring for 4 h, DCM (0.8 mL) was added to the obtained white suspension and the mixture was stirred overnight. The reaction mixture was diluted with brine and extracted with DCM (3×). The combined organic layers were concentrated in vacuo. Purification by size exclusion (DCM/MeOH: 1/1) afforded compound 20a (30.3 mg, 16.4 μmol, 84%). 1H NMR (CDCl3, 600 MHz, HH-COSY, HSQC): δ 6.64 (s, 2H, HC=CH), 5.11–5.01 (m, 3H, CH FA), 4.98 (t, 1H, J = 9.0 Hz, H-3), 4.63–4.54 (m, 1H, CHH-6), 4.45–4.32 (m, 2H, H-1, CHH-6), 4.28 (s, 1H, CH serine), 4.20–3.91 (m, 5H, H-4, CH2 serine, CH2 linker), 3.71–3.33 (m, 14H, H-2, H-5, 6× CH2 linker), 3.24–3.17 (m, 2H, CH2 linker), 2.56 (dd, 1H, J = 16.1, 7.0 Hz, CHH FA), 2.51–2.38 (m, 2H, 2× CHH FA), 2.38–2.28 (m, 2H, 2× CHH FA), 2.21 (dd, 1H, J = 14.6, 5.3 Hz, CHH FA), 2.17–2.11 (m, 6H, 3× CH2 FA), 2.08 (t, 2H, J = 7.1 Hz, CH2 linker), 1.82–1.72 (m, 2H, CH2 linker), 1.51–1.37 (m, 12H, 6× CH2 FA), 1.26–1.02 (m, 90H, 45× CH2 FA), 0.73 (t, 18H, J = 7.0 Hz, 6× CH3 FA); 13C-APT NMR (CDCl3, 151 MHz, HSQC): δ 173.8, 173.7, 173.6, 171.3, 171.1, 171.0, 170.9, 170.2 (C=O), 134.2 (C=C), 100.7 (C-1), 73.5 (C-3), 72.9 (C-5), 71.0 (CH FA), 70.9 (C-4), 70.8 (CH FA), 70.6, 70.3, 70.3, 70.2 (CH2 linker), 70.1 (CH FA), 69.9, 69.7, 69.5, 69.5, 69.1, 69.0, 68.3 (CH2 linker), 68.0 (CH2 serine), 62.7 (CH2-6) 53.6 (C-2), 53.3 (CH serine), 41.0, 40.7, 39.1, 39.0, 39.0, 38.7, 37.1, 36.9, 34.4, 34.3, 34.2, 34.2, 34.1, 32.6, 31.8, 31.8, 31.8, 31.8, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.4, 29.4, 29.4, 29.3, 29.3, 29.3, 29.2, 29.2, 29.2, 29.1, 29.1, 29.1, 25.2, 25.2, 25.1, 25.0, 24.9, 24.2, 22.6, 22.5 (CH2 FA, CH2 linker), 13.9 (CH3 FA); 31P-APT NMR (CDCl3, 202 MHz, HMBC): δ 0.82; HRMS: [M + H]+ calcd for C97H174N4O26P: 1842.21484, found 1842.21478.
4.2.14. N-[(R)-3-(Decanoyloxy)tetradecanoyl]-O-[6-N-(11-N-(4-maleimidobutanoyl)-11-amino-3,6,9-trioxa-undecanoyl)-4-O-phosphoryl-2-N-[(R)-3-(decanoyloxy)tetradecanoyl]-3-O-[(R)-3-(decanoyloxy)tetradecanoyl]-β-d-glucopyranosyl]-l-serine (20b)
Azide 16b (76 mg, 43 μmol, 1.0 equiv) was dissolved in a mixture of DCM/MeOH/H2O (1/1/0.1 v/v/v, 3.8 mL). Freshly prepared activated zinc powder (30 mg, 0.47 mmol, 11 equiv) and NH4Cl (23.1 mg, 0.43 mmol, 11 equiv) were added, and the suspension was stirred vigorously for 5.5 h. The reaction mixture was subsequently diluted with DCM and washed with sat. aq. NaHCO3 (1×). The aqueous layer was extracted with DCM (2×). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The obtained amine and acid S19 (27 mg, 0.12 mmol, 2.8 equiv) were dissolved in DCE (1.3 mL) under an argon atmosphere and cooled to 0 °C. EDC·MeI (26 mg, 87 μmol, 2.0 equiv) and DMAP (2.8 mg, 23 μmol, 0.53 equiv) were added, and the reaction mixture was allowed to warm up to room temperature overnight. The white suspension was diluted with DCM and washed with sat. aq. NaHCO3 (1×). The organic layer was dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (30 → 70% EtOAc in pentane + 0.1% Et3N) and size exclusion (DCM/MeOH: 1/1) afforded the intermediate (48 mg, 24 μmol, 56% over two steps), of which 21.4 mg (10.8 μmol, 1.0 equiv) was coevaporated with toluene (2×) under an argon atmosphere and dissolved in THF (1.0 mL). Pd/C (10%, 21.1 mg) was added, and the reaction mixture was stirred for 5.5 h at room temperature under a blanket of H2(g). The black suspension was filtered over a Whatmann filter. The filter was washed with CHCl3, and Et3N (3.0 μL, 22 μmol, 2.0 equiv) was added to the combined filtrates. The solution was mixed for 5 min and concentrated in vacuo. Purification by size exclusion (DCM/MeOH: 1/1) afforded the free amine (15.1 mg, 9.0 μmol, 83%). A solution of the obtained free amine in DCE (22.4 mM, 375 μL, 1.0 equiv) was added to an Eppendorf tube containing sulfo-N-succinimidyl 4-maleimidobutyrate sodium salt (4.1 mg, 10.7 μmol, 1.2 equiv) and Et3N (7.0 μL, 50 μmol, 6.0 equiv). After mixing overnight at 850 rpm, the mixture was diluted with brine and extracted with DCM (3×). The combined organic layers were concentrated in vacuo. Purification by size exclusion (DCM/MeOH: 1/1) afforded the title compound (12.5 mg, 6.8 μmol, 81%). 1H NMR (CDCl3, 850 MHz, HH-COSY, HSQC): δ 6.64 (s, 2H, HC=CH), 5.13–5.00 (m, 3H, 3× CH FA), 4.97–4.89 (m, 1H, H-3), 4.35 (d, 1H, J = 7.7 Hz, H-1), 4.23 (s, 1H, CH serine), 3.99–3.88 (m, 3H, H-4, CHH serine, CH2 linker), 3.76–3.69 (m, 1H, CHH-6), 3.69–3.61 (m, 2H, H-2, CHH serine), 3.61–3.48 (m, 7H, 3× CH2 linker, CHH linker), 3.48–3.39 (m, 4H, 2× CH2 linker), 3.39–3.33 (m, 2H, H-5, CHH linker), 3.26–3.19 (m, 1H, CHH-6), 2.54 (dd, 1H, J = 16.2, 7.2 Hz, CHH FA), 2.49–2.39 (m, 2H, 2× CHH FA), 2.39–2.34 (m, 1H, CHH FA), 2.30 (dd, 1H, J = 14.6, 7.4 Hz, CHH FA), 2.20 (dd, 1H, J = 14.6, 5.4 Hz, CHH FA), 2.18–2.10 (m, 6H, 3× CH2 FA), 2.08 (t, 1H, J = 7.2 Hz, CH2 linker), 1.78–1.73 (m, 2H, CH2 linker), 1.51–1.37 (m, 12H, 6× CH2 FA), 1.26–1.05 (m, 90H, 45× CH2 FA), 0.73 (t, 18H, J = 7.2 Hz, 6× CH3 FA); 13C-APT NMR (CDCl3, 214 MHz, HSQC): δ 173.9, 173.9, 173.8, 173.6, 171.2, 171.1, 170.9, 170.4 (C=O), 134.2 (C=C), 100.9 (C-1), 73.9 (C-5), 73.5 (C-3), 72.0 (C-4), 71.0, 70.8 (CH FA), 70.3 (CH2 linker), 70.1 (CH FA), 70.0, 69.5, 69.3, 69.3 (CH2 serine, CH2 linker), 53.6 (C-2, CH serine), 41.0, 40.7, 39.0, 39.0 (CH2 FA), 38.7 (CH2-6), 34.4, 34.3, 34.2, 32.7, 31.8, 31.8, 31.8, 31.8, 30.7, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.5, 29.5, 29.4, 29.4, 29.3, 29.3, 29.3, 29.2, 29.2, 29.2, 29.1, 29.1, 29.1, 25.2, 25.2, 25.1, 25.0, 25.0, 24.9, 24.3, 22.6, 22.5 (CH2 FA, CH2 linker), 13.9 (CH3 FA); 31P-APT NMR (CDCl3, 202 MHz, HMBC): δ 1.24; HRMS: [M + H]+ calcd for C97H175N5O25P: 1841.23083, found 1841.23006.
4.2.15. 3-Mercaptoproponamide-Asp-Glu-Val-Ser-Gly-Leu-Glu-Gln-Leu-Glu-Ser-Ile-Ile-Asn-Phe-Glu-Lys-Leu-Ala-Ala-Ala-Ala-Ala-Lys-NH2 (21)
Tentagel S Ram resin loaded with H-Asp(OtBu)-Glu(OtBu)-Val-Ser(tBu)-Gly-Leu-Glu(OtBu)-Gln(Trt)-Leu-Glu(OtBu)-Ser(tBu)-Ile-Ile-Asn(Trt)-Phe-Glu(OtBu)-Lys(Boc)-Leu-Ala-Ala-Ala-Ala-Ala-Lys(MMT) on a 70 μmol scale was washed with DMF (5×), followed by the addition of a solution of 3-(tritylthio)propionic acid (52 mg, 150 μmol, 2.1 equiv) and HCTU (58 mg, 140 μmol, 2.0 equiv) in DMF (1.4 mL) and DIPEA (49 μL, 280 μmol, 4.0 equiv). The reaction vessel was shaken overnight at 850 rpm, after which it was washed with DMF (3×) and DCM (3×). The peptide was cleaved from the resin after treatment with TFA/TIS/H2O/1,2-ethanedithiol (94/2.5/2.5/1 v/v/v/v, 2.8 mL) for 3 h, the suspension was filtered, and the residue was washed with the cleavage cocktail (2.8 mL). The product was precipitated with Et2O. After purification by RP-HPLC and lyophilization, compound 21 (14 mg, 5.3 μmol, 8%) was obtained as a white solid. LC-MS: Rt = 5.15 min (C18 Gemini, 10–90% MeCN, 11 min run); electrospray ionization (ESI)-MS (m/z): 1317.7 [M + 2H]2+; HRMS: [M + 2H]2+ calcd for C115H191N29O39S: 1317.17819, found 1317.17784.
4.2.16. Asp-Glu-Val-Ser-Gly-Leu-Glu-Gln-Leu-Glu-Ser-Ile-Ile-Asn-Phe-Glu-Lys-Leu-Ala-Ala-Ala-Ala-Ala-(Nε-(3-mercaptoproponamide))-Lys-NH2 (22)
Tentagel S Ram resin loaded with H-Asp(OtBu)-Glu(OtBu)-Val-Ser(tBu)-Gly-Leu-Glu(OtBu)-Gln(Trt)-Leu-Glu(OtBu)-Ser(tBu)-Ile-Ile-Asn(Trt)-Phe-Glu(OtBu)-Lys(Boc)-Leu-Ala-Ala-Ala-Ala-Ala-Lys(MMT) on a 50 μmol scale was washed with DMF (5×), treated with a mixture of Boc2O (0.11 g, 0.50 mmol, 10 equiv) in 0.1 M DIPEA in DMF (0.5 mL) for 1 h, and washed with DMF (3×) and DCM (3×). The peptide was treated with a continuous flow of a mixture of TFA/TIS/DCM (96/2/2 v/v/v, 15 mL) over 5 min. The resin was washed subsequently with DCM (5×), TFA/TIS/DCM (96/2/2 v/v/v, 2 mL), DCM (5×), 1 M DIPEA in N-methyl-2-pyrrolidone (NMP) (2 mL), DCM (3×), and DMF (3×). A solution of 3-(tritylthio)propionic acid (49 mg, 140 μmol, 2.0 equiv) and HCTU (58 mg, 140 μmol, 2.0 equiv) in DMF (1.4 mL) and DIPEA (49 μL, 280 μmol, 4.0 equiv) was added. The reaction vessel was shaken overnight at 850 rpm, after which it was washed with DMF (3×) and DCM (3×). The peptide was cleaved from the resin after treatment with TFA/TIS/H2O/1,2-ethanedithiol (95/2/2/1 v/v/v/v, 2.8 mL) for 3 h, the suspension was filtered, and the residue was washed with the cleavage cocktail (2.8 mL). The product was precipitated with Et2O. After purification by RP-HPLC and lyophilization, compound 22 (7.9 mg, 3.0 μmol, 6%) was obtained as a white solid. LC-MS: Rt = 5.31 min (C18 Gemini, 10–90% MeCN, 11 min run); ESI-MS (m/z): 1317.2 [M + 2H]2+; HRMS: [M + 2H]2+ calcd for C115H191N29O39S: 1317.17819, found 1317.17916.
4.2.17. N-Terminus 6-O-DEVA5K Conjugate (5)
Thiol-peptide 21 (1.4 mg, 0.53 μmol, 1.5 equiv) was dissolved in a mixture of DMF/Milli-Q H2O (4/1 v/v, 114 μL) in an Eppendorf tube. A solution of compound 20a (5.0 mM, 114 μL, 0.36 μmol, 1.0 equiv) was added, and the mixture was shaken at 850 rpm for 48 h. LC-MS analysis showed complete conversion of the starting material. The reaction mixture was diluted with a mixture of CH3CN/tBuOH/Milli-Q H2O (1/1/1 v/v/v, 0.80 mL) and sonicated for 5 min. After purification using a C18 column, LC-MS analysis showed that the DCM/MeOH (1/1 v/v) flush contained a pure conjugate. After lyophilization, conjugate 5 (0.86 mg, 0.19 μmol, 52%) was obtained as a white solid. LC-MS: Rt = 14.27 min (diphenyl Vydac, 10–90% isopropyl alcohol (IPA), 25 min run, 0.8 mL/min); ESI-MS (m/z): [M + Na + 2H]3+ calcd for C212H364N33O65SPNa: 1500.2, found 1500.3; MALDI-TOF MS (m/z): [M + 2Na – H]+ calcd for C212H362N33O65SPNa2: 4522.406, found 4531.383.
4.2.18. N-Terminus 6-NH-DEVA5K Conjugate (6)
Thiol-peptide 21 (3.65 mg, 1.39 μmol, 1.5 equiv) was dissolved in a mixture of DMF/Milli-Q H2O (4/0.5 v/v, 277 μL) in an Eppendorf tube. A solution of compound 20b (5.0 mM, 182 μL, 0.91 μmol, 1.0 equiv) was added, and the mixture was shaken at 850 rpm for 48 h. LC-MS analysis showed complete conversion of the starting material. The reaction mixture was diluted with a mixture of CH3CN/tBuOH/Milli-Q H2O (1/1/1 v/v/v, 0.59 mL) and sonicated for 5 min. After purification using a C18 column, LC-MS analysis showed that the MeOH and DCM/MeOH (1/1 v/v) flush contained a pure conjugate. After lyophilization, conjugate 6 (2.19 mg, 0.49 μmol, 54%) was obtained as a white solid. LC-MS: Rt = 16.85 min (diphenyl Vydac, 10–90% IPA, 25 min run, 0.7 mL/min); ESI-MS (m/z): [M + Na + 2H]3+ calcd for C212H365N34O64SPNa: 1499.9, found 1499.8; MALDI-TOF MS (m/z): [M + 2Na – H]+ calcd for C212H363N34O64SPNa2: 4521.422, found 4530.340.
4.2.19. C-Terminus 6-O-DEVA5K Conjugate (7)
Thiol-peptide 22 (2.1 mg, 0.80 μmol, 1.5 equiv) was dissolved in a mixture of DMF/Milli-Q H2O (4/0.5 v/v, 161 μL) in an Eppendorf tube. A solution of compound 20a (5.0 mM, 107 μL, 0.53 μmol, 1.0 equiv) was added, and the mixture was shaken at 850 rpm for 48 h. LC-MS analysis showed complete conversion of the starting material. The reaction mixture was diluted with a mixture of CH3CN/tBuOH/Milli-Q H2O (1/1/1 v/v/v, 0.80 mL) and sonicated for 5 min. After purification using a C18 column, LC-MS analysis showed that the MeOH and DCM/MeOH (1/1 v/v) flush contained a pure conjugate. After lyophilization, conjugate 7 (1.37 mg, 0.30 μmol 57%) was obtained as a white solid. LC-MS: Rt = 14.24 min (diphenyl Vydac, 10–90% IPA, 25 min run, 0.8 mL/min); ESI-MS (m/z): [M + 2Na + H]3+ calcd for C212H363N33O65SPNa2: 1507.6, found 1507.6; MALDI-TOF MS (m/z): [M + Na]+ calcd for C212H362N33O65SPNa: 4499.417, found 4497.786.
4.2.20. C-Terminus 6-NH-DEVA5K Conjugate (8)
Thiol-peptide 22 (2.0 mg, 0.76 μmol, 1.5 equiv) was dissolved in a mixture of DMF/Milli-Q H2O (4/0.5 v/v, 152 μL) in an Eppendorf tube. A solution of compound 20b (5.0 mM, 101 μL, 505 nmol, 1.0 equiv) was added, and the mixture was shaken at 850 rpm for 48 h. LC-MS analysis showed complete conversion of the starting material. The reaction mixture was diluted with a mixture of CH3CN/tBuOH/Milli-Q H2O (1/1/1 v/v/v, 0.80 mL) and sonicated for 5 min. After purification using a C18 column, LC-MS analysis showed that the DCM/MeOH (1/1 v/v) flush contained a pure conjugate. After lyophilization, conjugate 8 (0.94 mg, 0.21 μmol, 42%) was obtained as a white solid. LC-MS: Rt = 16.90 min (diphenyl Vydac, 10–90% IPA, 25 min run, 0.7 mL/min); ESI-MS (m/z): [M + 2Na + H]3+ calcd for C212H364N34O64SPNa2: 1507.2, found 1507.4; MALDI-TOF MS (m/z): [M + Na]+ calcd for C212H363N34O64SPNa: 4498.433, found 4508.766.
4.2.21. Asp-Glu-Val-Ser-Gly-Leu-Glu-Gln-Leu-Glu-Ser-Ile-Ile-Asn-Phe-Glu-Lys-Leu-Ala-Ala-Ala-Ala-Ala-Lys-NH2 (23)
Tentagel S Ram resin loaded with H-Asp(OtBu)-Glu(OtBu)-Val-Ser(tBu)-Gly-Leu-Glu(OtBu)-Gln(Trt)-Leu-Glu(OtBu)-Ser(tBu)-Ile-Ile-Asn(Trt)-Phe-Glu(OtBu)-Lys(Boc)-Leu-Ala-Ala-Ala-Ala-Ala-Lys(MMT) on a 30 μmol scale was washed with DCM (5×). The peptide was cleaved from the resin after treatment with TFA/TIS/H2O (95/2/2/1 v/v/v/v) (1.2 mL) for 3 h, the suspension was filtered, and the residue was washed with the cleavage cocktail (1.2 mL). The product was precipitated with Et2O. After purification by RP-HPLC and lyophilization, compound 23 (12 mg, 4.7 μmol, 16%) was obtained as a white solid. LC-MS: Rt = 4.82 min (C18 Gemini, 10–90% MeCN, 11 min run); ESI-MS (m/z): 1273.7 [M + 2H]2+; HRMS: [M + 2H]2+ calcd for C112H187N29O38: 1273.17904, found 1273.17779.
4.3. Immunological Evaluation Procedures
4.3.1. Cell Culture
The D1 cell line is a growth-factor-dependent immature spleen-derived DC cell line from C57BL/6 (H-2b) mice. D1 cells were cultured as described elsewhere.55 The B3Z cell line was cultured in Iscove’s modified Dulbecco’s medium (IMDM) (Lonza, Basel, Switzerland) supplemented with 8% fetal calf serum (FCS) (Greiner, Kremsmünster, Austria), penicillin and streptomycin, glutamine (Gibco, Carlsbad, CA), β-mercaptoethanol (Merck, Kenilworth, NJ), and hygromycin B (AG Scientific Inc, San Diego, CA) to maintain expression of the β-galactosidase reporter gene.
4.3.2. In Vitro DC Maturation Assay
The test compounds were dissolved in DMSO at a concentration of 500 μM and sonicated in a water bath for 15 min. The 50 000 D1 cells were seeded in 96-well round-bottom plates (Corning, Amsterdam, The Netherlands) in 100 μL of an R1-supplemented IMDM medium, and 100 μL of two times concentrated test compounds in the medium was added. After 24 h of incubation at 37 °C, the supernatant was taken from the wells for enzyme-linked immunosorbent assay (ELISA) analysis (BioLegend, San Diego) to measure the amount of produced IL-12p40.
4.3.3. In Vitro Antigen Presentation Assay
The test compounds were dissolved in DMSO at a concentration of 500 μM and sonicated in a water bath for 15 min. The 50 000 D1 cells were seeded in 96-well flat-bottom plates and pulsed for 2 h with 200 μL of the test compounds in a medium at the indicated concentrations. After 2 h, cells were washed once with 200 μL of the fresh medium and 50 000 B3Z was added per well in 200 μL of the medium and incubated with pulsed D1 cells overnight. The following day, TCR activation was detected by measurement of absorbance at 595 nm upon color conversion of chlorophenol red-β-d-galactopyranoside (Calbiochem, Merck, Bullington, MA) by the β-galactosidase enzyme, followed by measurement of absorbance at 595 nm.56,57
4.3.4. Mice
Female C57BL/6 mice were purchased from Charles River Laboratories. Congenic CD45.1+ C57BL/6 mice were bred at the Leiden University Medical Centre animal facility. All animal experiments were in accordance with the Dutch national regulations and received ethical and technical approval by the local Animal Welfare Body.
4.3.5. Immunization of Mice
C57BL/6 female mice (6–8 weeks old) were injected intradermally at the tail base with 5 nmol of each conjugate or an equimolar mix of lipid A and DEVA5K peptide. To prepare the vaccine, the different compounds were dissolved in DMSO at a concentration of 500 μM and sonicated in a water bath for 15 min. The required amounts for vaccination were added to a saline solution, and 30 μL per mouse was injected. Fourteen days later, the animals were boosted with the same vaccine formulations. At different time points during the experiments, 20 μL of blood was collected from the tail vein for detection of SIINFEKL-specific T-cell responses via SIINFEKL-H2-Kb tetramer staining. At the end of the experiments, spleens and lymph nodes were removed and processed in single cell suspensions for ex vivo analysis.
4.3.6. In Vivo Specific Killing
Splenocytes were harvested from CD45.1+ C57BL/6 naïve mice, processed into single cell suspension, and labeled with either 5 or 0.005 μM CFSE for 10 min at 37 °C. Cells that were labeled with 0.005 μM CFSE (CFSE low) were loaded for 1 h at 37 °C with 1 μM SIINFEKL peptide, while cells that were labeled with 5 μM CFSE (CFSE high) were loaded in the same conditions with an irrelevant epitope derived from the E6 protein of human papilloma virus (sequence: RAHYNIVTF). In total, 4 000 000 splenocytes per peptide-loaded group were injected intravenously in vaccinated or naïve mice. One day after transfer, mice were sacrificed and the spleens were harvested and processed into single cell suspensions. Splenocytes were subsequently stained with eFluor450 anti-CD45.1 antibody (eBioscience, San Diego) and analyzed by flow cytometry to detect CD45.1+/CFSE+ target cells. Specific killing was calculated according to the following equation:
 |
Acknowledgments
We thank F. Lefeber and K. B. S. S. Gupta for their help in the NMR measurements and D. A. Johnson for his synthetic advice.
Glossary
Abbreviations
- AcOH
acetic acid
- AGP
aminoalkyl glucosamine 4-phosphates
- CFSE
carboxyfluorescein succinimidyl ester
- DC
dendritic cell
- DCE
dichloroethane
- DCM
dichloromethane
- DEAD
diethyl azodicarboxylate
- DEVA5K
DEVSGLEQLESIINFEKLAAAAAK
- DMAP
4-dimethylaminopyridine
- DPPA
diphenyl phosphoryl azide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HRMS
high-resolution mass spectra
- LC-MS
liquid chromatography-mass spectrometry
- MALDI-TOF
matrix-assisted laser desorption/ionization time of flight
- MHC
histocompatibility complex
- MPLA
monophosphoryl lipid A
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- PAMP
pathogen-associated molecular patterns
- PRR
pathogen recognition receptor
- RP-HPLC
reversed-phase high-performance liquid chromatography
- SLP
synthetic long peptide
- TBDMS
tert-butyldimethylsilyl
- TEG
triethylene glycol
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TIS
triisopropylsilane
- TLC
thin-layer chromatography
- TLR
Toll-like receptor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00851.
Author Contributions
∥ N.R.M.R. and E.T. contributed equally to this work.
The authors declare no competing financial interest.
Footnotes
For the amide conjugates, H2O + 0.1% TFA was used instead of H2O.
Supplementary Material
References
- Wei S. C.; Duffy C. R.; Allison J. P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discovery 2018, 8, 1069–1086. 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- Mchayleh W.; Bedi P.; Sehgal R.; Solh M. Chimeric Antigen Receptor T-Cells: The Future Is Now. J. Clin. Med. 2019, 8, 207 10.3390/jcm8020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher T. N.; Schreiber R. D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Heimburg-Molinaro J.; Lum M.; Vijay G.; Jain M.; Almogren A.; Rittenhouse-Olson K. Cancer Vaccines and Carbohydrate Epitopes. Vaccine 2011, 29, 8802–8826. 10.1016/j.vaccine.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker S. W.; Bonham K. S.; Zanoni I.; Kagan J. C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A.; Medzhitov R. Toll-like Receptor Control of the Adaptive Immune Responses. Nat. Immunol. 2004, 5, 987–995. 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Kawai T.; Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- van Dinther D.; Stolk D. A.; van de Ven R.; van Kooyk Y.; de Gruijl T. D.; den Haan J. M. M. Targeting C-Type Lectin Receptors: A High-Carbohydrate Diet for Dendritic Cells to Improve Cancer Vaccines. J. Leukoc. Biol. 2017, 102, 1017–1034. 10.1189/jlb.5MR0217-059RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaude J.; Kent A.; van Rooijen N.; Blander J. M. Simultaneous Targeting of Toll- and Nod-Like Receptors Induces Effective Tumor-Specific Immune Responses. Sci. Transl. Med. 2012, 4, 120ra16 10.1126/scitranslmed.3002868. [DOI] [PubMed] [Google Scholar]
- Zom G. G.; Willems M. M. J. H. P.; Meeuwenoord N. J.; Reintjens N. R. M.; Tondini E.; Khan S.; Overkleeft H. S.; van der Marel G. A.; Codee J. D. C.; Ossendorp F.; Filippov D. V. Dual Synthetic Peptide Conjugate Vaccine Simultaneously Triggers TLR2 and NOD2 and Activates Human Dendritic Cells. Bioconjugate Chem. 2019, 30, 1150–1161. 10.1021/acs.bioconjchem.9b00087. [DOI] [PubMed] [Google Scholar]
- Zom G. G. P.; Khan S.; Filippov D. V.; Ossendorp F. TLR Ligand–Peptide Conjugate Vaccines: Toward Clinical Application. Adv. Immunol. 2012, 114, 177–201. 10.1016/B978-0-12-396548-6.00007-X. [DOI] [PubMed] [Google Scholar]
- Liu H.; Irvine D. J. Guiding Principles in the Design of Molecular Bioconjugates for Vaccine Applications. Bioconjugate Chem. 2015, 26, 791–801. 10.1021/acs.bioconjchem.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhagen F.; Kinjo T.; Bode C.; Klinman D. M. TLR-Based Immune Adjuvants. Vaccine 2011, 29, 3341–3355. 10.1016/j.vaccine.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J. K.; Mansell A. Toll-like Receptors: The Swiss Army Knife of Immunity and Vaccine Development. Clin. Transl. Immunol. 2016, 5, e85 10.1038/cti.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle P. M.; Toth I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. ChemMedChem 2013, 8, 360–376. 10.1002/cmdc.201200487. [DOI] [PubMed] [Google Scholar]
- Ignacio B. J.; Albin T. J.; Esser-Kahn A. P.; Verdoes M. Toll-like Receptor Agonist Conjugation: A Chemical Perspective. Bioconjugate Chem. 2018, 29, 587–603. 10.1021/acs.bioconjchem.7b00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingale S.; Wolfert M. A.; Gaekwad J.; Buskas T.; Boons G.-J. Robust Immune Responses Elicited by a Fully Synthetic Three-Component Vaccine. Nat. Chem. Biol. 2007, 3, 663–667. 10.1038/nchembio.2007.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S.; Weterings J. J.; Britten C. M.; de Jong A. R.; Graafland D.; Melief C. J. M.; van der Burg S. H.; van der Marel G.; Overkleeft H. S.; Filippov D. V.; Ossendorp F. Chirality of TLR-2 Ligand Pam3CysSK4 in Fully Synthetic Peptide Conjugates Critically Influences the Induction of Specific CD8+ T-Cells. Mol. Immunol. 2009, 46, 1084–1091. 10.1016/j.molimm.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Abdel-Aal A.-B. M.; Zaman M.; Fujita Y.; Batzloff M. R.; Good M. F.; Toth I. Design of Three-Component Vaccines against Group A Streptococcal Infections: Importance of Spatial Arrangement of Vaccine Components. J. Med. Chem. 2010, 53, 8041–8046. 10.1021/jm1007787. [DOI] [PubMed] [Google Scholar]
- Khan S.; Bijker M. S.; Weterings J. J.; Tanke H. J.; Adema G. J.; van Hall T.; Drijfhout J. W.; Melief C. J. M.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V.; van der Burg S. H.; Ossendorp F. Distinct Uptake Mechanisms but Similar Intracellular Processing of Two Different Toll-like Receptor Ligand-Peptide Conjugates in Dendritic Cells. J. Biol. Chem. 2007, 282, 21145–21159. 10.1074/jbc.M701705200. [DOI] [PubMed] [Google Scholar]
- Zom G. G.; Willems M. M. J. H. P.; Khan S.; Van Der Sluis T. C.; Kleinovink J. W.; Camps M. G. M.; Van Der Marel G. A.; Filippov D. V.; Melief C. J. M.; Ossendorp F. Novel TLR2-Binding Adjuvant Induces Enhanced T Cell Responses and Tumor Eradication. J. Immunother. Cancer 2018, 6, 146 10.1186/s40425-018-0455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems M. M. J. H. P.; Zom G. G.; Khan S.; Meeuwenoord N.; Melief C. J. M.; van der Stelt M.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A.; Ossendorp F.; Filippov D. V. N-Tetradecylcarbamyl Lipopeptides as Novel Agonists for Toll-like Receptor 2. J. Med. Chem. 2014, 57, 6873–6878. 10.1021/jm500722p. [DOI] [PubMed] [Google Scholar]
- Weterings J. J.; Khan S.; van der Heden G. J.; Drijfhout J. W.; Melief C. J. M.; Overkleeft H. S.; van der Burg S. H.; Ossendorp F.; van der Marel G. A.; Filippov D. V. Synthesis of 2-Alkoxy-8-Hydroxyadenylpeptides: Towards Synthetic Epitope-Based Vaccines. Bioorg. Med. Chem. Lett. 2006, 16, 3258–3261. 10.1016/j.bmcl.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Fujita Y.; Hirai K.; Nishida K.; Taguchi H. 6-(4-Amino-2-Butyl-Imidazoquinolyl)-Norleucine: Toll-like Receptor 7 and 8 Agonist Amino Acid for Self-Adjuvanting Peptide Vaccine. Amino Acids 2016, 48, 1319–1329. 10.1007/s00726-016-2190-7. [DOI] [PubMed] [Google Scholar]
- Kramer K.; Young S. L.; Walker G. F. Comparative Study of 5′- and 3′-Linked CpG–Antigen Conjugates for the Induction of Cellular Immune Responses. ACS Omega 2017, 2, 227–235. 10.1021/acsomega.6b00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daftarian P.; Sharan R.; Haq W.; Ali S.; Longmate J.; Termini J.; Diamond D. J. Novel Conjugates of Epitope Fusion Peptides with CpG-ODN Display Enhanced Immunogenicity and HIV Recognition. Vaccine 2005, 23, 3453–3468. 10.1016/j.vaccine.2005.01.093. [DOI] [PubMed] [Google Scholar]
- Alving C. R.; Peachman K. K.; Rao M.; Reed S. G. Adjuvants for Human Vaccines. Curr. Opin. Immunol. 2012, 24, 310–315. 10.1016/j.coi.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella C. R.; Mitchell T. C. Putting Endotoxin to Work for Us: Monophosphoryl Lipid A as a Safe and Effective Vaccine Adjuvant. Cell. Mol. Life Sci. 2008, 65, 3231–3240. 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garçon N.; Di Pasquale A. From Discovery to Licensure, the Adjuvant System Story. Hum. Vaccines Immunother. 2017, 13, 19–33. 10.1080/21645515.2016.1225635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhou Z.; Tang S.; Guo Z. Carbohydrate-Monophosphoryl Lipid A Conjugates Are Fully Synthetic Self-Adjuvanting Cancer Vaccines Eliciting Robust Immune Responses in the Mouse. ACS Chem. Biol. 2012, 7, 235–240. 10.1021/cb200358r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Mondal M.; Liao G.; Guo Z. Synthesis and Evaluation of Monophosphoryl Lipid A Derivatives as Fully Synthetic Self-Adjuvanting Glycoconjugate Cancer Vaccine Carriers. Org. Biomol. Chem. 2014, 12, 3238–3245. 10.1039/C4OB00390J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Liao G.; Mandal S. S.; Suryawanshi S.; Guo Z. A Fully Synthetic Self-Adjuvanting Globo H-Based Vaccine Elicited Strong T Cell-Mediated Antitumor Immunity. Chem. Sci. 2015, 6, 7112–7121. 10.1039/C5SC01402F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G.; Zhou Z.; Suryawanshi S.; Mondal M. A.; Guo Z. Fully Synthetic Self-Adjuvanting α-2,9-Oligosialic Acid Based Conjugate Vaccines against Group C Meningitis. ACS Cent. Sci. 2016, 2, 210–218. 10.1021/acscentsci.5b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Feng S.; Wang S.; Li H.; Guo Z.; Gu G. Synthesis and Immunological Comparison of Differently Linked Lipoarabinomannan Oligosaccharide–Monophosphoryl Lipid A Conjugates as Antituberculosis Vaccines. J. Org. Chem. 2017, 82, 12085–12096. 10.1021/acs.joc.7b01817. [DOI] [PubMed] [Google Scholar]
- Zamyatina A. Aminosugar-Based Immunomodulator Lipid A: Synthetic Approaches. Beilstein J. Org. Chem. 2018, 14, 25–53. 10.3762/bjoc.14.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y.; Shimoyama A.; Saeki A.; Kitayama N.; Kasamatsu C.; Tsutsui H.; Fukase K. Innate Immunomodulation by Lipophilic Termini of Lipopolysaccharide; Synthesis of Lipid As from Porphyromonas gingivalis and Other Bacteria and Their Immunomodulative Responses. Mol. BioSyst. 2013, 9, 987. 10.1039/c3mb25477a. [DOI] [PubMed] [Google Scholar]
- Li Q.; Guo Z. Recent Advances in Toll Like Receptor-Targeting Glycoconjugate Vaccines. Molecules 2018, 23, 1583 10.3390/molecules23071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöver A. G.; Da Silva Correia J.; Evans J. T.; Cluff C. W.; Elliott M. W.; Jeffery E. W.; Johnson D. A.; Lacy M. J.; Baldridge J. R.; Probst P.; Ulevitch R. J.; Persing D. H.; Hershberg R. M. Structure-Activity Relationship of Synthetic Toll-like Receptor 4 Agonists. J. Biol. Chem. 2004, 279, 4440–4449. 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- Cluff C. W.; Baldridge J. R.; Stover A. G.; Evans J. T.; Johnson D. A.; Lacy M. J.; Clawson V. G.; Yorgensen V. M.; Johnson C. L.; Livesay M. T.; Hershberg R. M.; Persing D. H. Synthetic Toll-Like Receptor 4 Agonists Stimulate Innate Resistance to Infectious Challenge. Infect. Immun. 2005, 73, 3044–3052. 10.1128/IAI.73.5.3044-3052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin H. G.; Bess L. S.; Livesay M. T.; Ryter K. T.; Johnson C. L.; Arnold J. S.; Johnson D. A. New Synthesis of Glycolipid Immunostimulants RC-529 and CRX-524. Tetrahedron Lett. 2006, 47, 2087–2092. 10.1016/j.tetlet.2006.01.137. [DOI] [Google Scholar]
- Bazin H. G.; Murray T. J.; Bowen W. S.; Mozaffarian A.; Fling S. P.; Bess L. S.; Livesay M. T.; Arnold J. S.; Johnson C. L.; Ryter K. T.; Cluff C. W.; Evans J. T.; Johnson D. A. The ‘Ethereal’ Nature of TLR4 Agonism and Antagonism in the AGP Class of Lipid A Mimetics. Bioorg. Med. Chem. Lett. 2008, 18, 5350–5354. 10.1016/j.bmcl.2008.09.060. [DOI] [PubMed] [Google Scholar]
- Dupont J.; Altclas J.; Lepetic A.; Lombardo M.; Vázquez V.; Salgueira C.; Seigelchifer M.; Arndtz N.; Antunez E.; von Eschen K.; Janowicz Z. A Controlled Clinical Trial Comparing the Safety and Immunogenicity of a New Adjuvanted Hepatitis B Vaccine with a Standard Hepatitis B Vaccine. Vaccine 2006, 24, 7167–7174. 10.1016/j.vaccine.2006.06.053. [DOI] [PubMed] [Google Scholar]
- Park B. S.; Lee J.-O. Recognition of Lipopolysaccharide Pattern by TLR4 Complexes. Exp. Mol. Med. 2013, 45, e66 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarco M. L.; Woods R. J. From Agonist to Antagonist: Structure and Dynamics of Innate Immune Glycoprotein MD-2 upon Recognition of Variably Acylated Bacterial Endotoxins. Mol. Immunol. 2011, 49, 124–133. 10.1016/j.molimm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the Regulation of Innate Resistance and Adaptive Immunity. Nat. Rev. Immunol. 2003, 3, 133–146. 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- van Duikeren S.; Fransen M. F.; Redeker A.; Wieles B.; Platenburg G.; Krebber W.-J.; Ossendorp F.; Melief C. J. M.; Arens R. Vaccine-Induced Effector-Memory CD8 + T Cell Responses Predict Therapeutic Efficacy against Tumors. J. Immunol. 2012, 189, 3397–3403. 10.4049/jimmunol.1201540. [DOI] [PubMed] [Google Scholar]
- Butler N. S.; Nolz J. C.; Harty J. T. Immunologic Considerations for Generating Memory CD8 T Cells through Vaccination. Cell. Microbiol. 2011, 13, 925–933. 10.1111/j.1462-5822.2011.01594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman M. S.; Peyerl F. W.; Jackson S. S.; Lifton M. A.; Gorgone D. A.; Schmitz J. E.; Letvin N. L. Subsets of Memory Cytotoxic T Lymphocytes Elicited by Vaccination Influence the Efficiency of Secondary Expansion In Vivo. J. Virol. 2004, 78, 206–215. 10.1128/JVI.78.1.206-215.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski A. B.; Boesteanu A. C.; Mueller Y. M.; Carafides C.; Topham D. J.; Altman J. D.; Jennings S. R.; Katsikis P. D. Memory CD8 + T Cells Require CD28 Costimulation. J. Immunol. 2007, 179, 6494–6503. 10.4049/jimmunol.179.10.6494. [DOI] [PubMed] [Google Scholar]
- Mousavi S. F.; Soroosh P.; Takahashi T.; Yoshikai Y.; Shen H.; Lefrançois L.; Borst J.; Sugamura K.; Ishii N. OX40 Costimulatory Signals Potentiate the Memory Commitment of Effector CD8 + T Cells. J. Immunol. 2008, 181, 5990–6001. 10.4049/jimmunol.181.9.5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock D. J.; Shen H. Requirement for CD4 T Cell Help in Generating Functional CD8 T Cell Memory. Science 2003, 300, 337–339. 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- Janssen E. M.; Lemmens E. E.; Wolfe T.; Christen U.; Von Herrath M. G.; Schoenberger S. P. CD4+ T Cells Are Required for Secondary Expansion and Memory in CD8+ T Lymphocytes. Nature 2003, 421, 852–856. 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- Ahrends T.; Busselaar J.; Severson T. M.; Bąbała N.; de Vries E.; Bovens A.; Wessels L.; van Leeuwen F.; Borst J. CD4+ T Cell Help Creates Memory CD8+ T Cells with Innate and Help-Independent Recall Capacities. Nat. Commun. 2019, 10, 5531 10.1038/s41467-019-13438-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet A. Introduction of 9-Fluorenylmethyloxycarbonyl, Trichloroethoxycarbonyl, and Benzyloxycarbonyl Amine Protecting Groups into O-Unprotected Hydroxyamino Acids Using Succinimidyl Carbonates. Can. J. Chem. 1982, 60, 976–980. 10.1139/v82-146. [DOI] [Google Scholar]
- Winzler C.; Rovere P.; Zimmermann V. S.; Davoust J.; Rescigno M.; Citterio S.; Ricciardi-Castagnoli P.. Checkpoints and Functional Stages in DC Maturation. In Dendritic Cells in Fundamental and Clinical Immunology; Ricciardi-Castagnoli P., Eds.; Advances in Experimental Medicine and Biology; Springer, 1997; Vol. 417, pp 59–64. [DOI] [PubMed] [Google Scholar]
- Sanderson S.; Shastri N. LacZ Inducible, Antigen/MHC-Specific T Cell Hybrids. Int. Immunol. 1994, 6, 369–376. 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- Schuurhuis D. H.; Ioan-Facsinay A.; Nagelkerken B.; van Schip J. J.; Sedlik C.; Melief C. J. M.; Verbeek J. S.; Ossendorp F. Antigen-Antibody Immune Complexes Empower Dendritic Cells to Efficiently Prime Specific CD8 + CTL Responses In Vivo. J. Immunol. 2002, 168, 2240–2246. 10.4049/jimmunol.168.5.2240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.