

Abstract
Background:
Tardive dyskinesia (TD) is an iatrogenic involuntary movement disorder occurring after extended antipsychotic use with unclear pathogenesis. CYP2D6 is a liver enzyme involved in antipsychotic metabolism and a well-studied gene candidate for TD.
Materials & methods:
We tested predicted CYP2D6 metabolizer phenotype with TD occurrence and severity in our two samples of European chronic schizophrenia patients (total n = 198, of which 82 had TD).
Results:
TD occurrence were associated with extreme metabolizer phenotype, controlling for age and sex (p = 0.012). In other words, individuals with either increased and no CYP2D6 activity were at higher risk of having TD.
Conclusion:
Unlike most previous findings, TD occurrence may be associated with both extremes of CYP2D6 metabolic activity rather than solely for poor metabolizers.
Keywords: : CYP2D6, metabolizer phenotype, pharmacogenetics, schizophrenia, tardive dyskinesia
Schizophrenia is a debilitating psychiatric disorder that is treated with antipsychotic medication. Like all pharmacological agents, there are side effects that can occur along with the use of antipsychotics, including tardive dyskinesia (TD). TD is an iatrogenic involuntary movement disorder that occurs after prolonged antipsychotic use. These movements are choreiform or athetoid and mainly affect the orofacial and upper limb regions; however, irregular movements have been documented to occur elsewhere as well [1]. The word tardive refers to the ‘delayed onset’, with most cases developing after 3–12 months of antipsychotic use. It is potentially irreversible as discontinuing the medication does not ensure its resolution [2]. Our understanding of TD beyond its clinical presentation is limited, although certain risk factors have been identified. Most studies agree that the use of second-generation antipsychotics such as clozapine and olanzapine results in a lower incidence of TD cases when compared with first-generation antipsychotics (FGA) such as haloperidol and chlorpromazine (20.7 vs 30%, respectively) [3]. The emergence of TD and other extrapyramidal symptoms prompted some individuals to switch from FGA to second-generation antipsychotics (SGA) [4] and it has been hypothesized that this risk difference between FGA and SGA may be due to differences in dopamine receptor occupancy levels [5]. Age is also an identified risk factor, with findings suggesting TD risk triples in patients older than the age of 60 years , possibly related to differences in drug metabolism [2,6,7]. Two other potential risk factors are sex and ethnicity. A review article of 76 studies reported that women tend to have a higher prevalence of TD (26.6–21.6%); however, this relationship has not been reproduced in some other studies [8,9]. Similarly, patients of ‘nonWhite’ (mostly African–American) ethnicity appeared to have twice the risk for TD compared with patients of European ancestry in one study [10], but this also has not been replicated in a meta-analysis or the CATIE sample [3,11]. The risk difference between ethnicities could have been attributed to the difference in average ability to metabolize medications, or different treatments received [10]. Other risk factors include substance use (smoking, alcohol and cocaine use in particular), akathisia, diabetes and intermittent antipsychotic treatment or anticholinergic use, although these require additional investigations [2].
The cause of TD is unclear. Several hypotheses suggest that dopamine D2 receptor sensitivity, dysfunctional gamma-aminobutyric acid (GABA) systems and neuronal damage from free radicals may each play a role in the underlying pathophysiological process [5,12,13]. Part of the variability in the risk for TD could be driven by genetic factors, as demonstrated by the familial occurrence of TD in a number of studies [14–16]. To this end, a number of candidate genes have been investigated for possible association with TD over the last two decades. For example, the DRD2, VMAT2/SLC18A2 and DISC1 genes have all been researched with varying levels of success, but another promising candidate is the CYP2D6 genes. CYP2D6 is expressed primarily in the liver and is involved in the metabolism of 25% of commercial drugs, including the primary or secondary metabolism of almost all antipsychotic drugs [17]. Because of its role in drug metabolism, it is an important gene candidate for pharmacogenetic studies. CYP2D6 is highly polymorphic as there are over 100 known allelic variations for this gene which are categorized into four phenotypes. Alleles may confer normal function, loss of function or no function at all, and can even vary in the number of functional gene copies (copy number) as well. The ultra-rapid metabolizer (UM) possesses more than two functional copies of CYP2D6 (or i.e., gene duplication), the extensive metabolizer (EM) has between one and two functional wild-type CYP2D6 alleles, the intermediate metabolizer (IM) phenotype has one decreased-function allele and one inactive allele and the poor metabolizer (PM) has two inactive alleles. The EM phenotype is the most commonly found in the general population with approximately 72–88%, whereas occurrences of PM and UM are much less common with 1–20% and 1–10%, respectively [18]. The composition of metabolizer phenotype varies across ethnic groups as well. For instance, PM phenotype is observed in 7% of Caucasians but only 1% of the Asian population. The UM phenotype is found in 2% of Caucasians but is present in 25% of Ethiopian ancestry [19].
With regards to previous research on CYP2D6 and TD, there have been somewhat mixed findings. The majority of gene association studies suggest that individuals lacking CYP2D6 activity tend to develop TD at a higher rate [20,21]. For instance: Ivanova et al. found that the nonfunctional CYP2D6*4 allele (1846G>A) is associated with limbotruncal TD in schizophrenia patients [22]. Similarly, a retrospective matched case– control study by Kobylecki et al. found that the PM phenotype was significantly higher among TD cases than non-TD controls [23]. Locatelli had very similar findings, reporting that CYP2D6*3 and *4 alleles were associated with higher TD occurrence than wild-type alleles as well [24]. Even in other ethnic groups that have been less researched, TD is largely associated with lower activity as found by Lam and Liou who looked at the intermediate-activity CYP2D6*10 alleles in an east-Asian sample [25,26]. On the other hand, only a handful of studies have analyzed the UM phenotype, with one study finding TD risk to be higher with the ultra-rapid metabolism phenotype [27].
Materials & methods
To add to the existing literature, we aimed to study the possible association between genotype-predicted CYP2D6 metabolizer phenotype (or CYP2D6 metabolizer status) and TD in two sample sets of European ancestry. The study was approved by individual institutional review boards and is in accordance with the Declaration of Helsinki. Informed consent was given by each study participant. The first sample set (CAUS) included 148 participants from two countries (Canada and USA) whose sample characteristics have been described previously [28–30]. In short, participants were enrolled from four sites throughout North America: Centre for Addiction and Mental Health in ON, Canada (G Remington; n = 97); Case Western Reserve University in OH, USA (DHY Meltzer; n = 41); Hillside Hospital in NY, USA (JA Lieberman; n = 12); and the University of California at Irvine, CA, USA (SG Potkin; n = 8). Participants had either DSM-III-R or DSM-IV diagnoses of schizophrenia/schizoaffective disorder (American Psychiatric Association). Patients recruited in the USA had no prior exposure to atypical antipsychotics, while the chronic patients from Canada may have been on either typical or atypical antipsychotics. All patients, however, had been exposed to typical antipsychotics for at least one year before being assessed for TD. The rate of TD was not significantly different between the USA and Canadian subsamples (p = 0.626), but was significantly lower in males (65%) when compared with females (35%) in the sample (p = 0.034).
Our second sample set (PGx) consisted of schizophrenia or schizoaffective patients from a naturalistic pharmacogenetics study (The Individualized Medicine: Pharmacogenetics Assessment and Clinical Treatment) (n = 58) with 19 TD cases [31,32]. A summary of sample characteristics is shown in Table 1.
Table 1. . Demographic information on the CAUS and pharmacogenomic schizophrenia/schizoaffective disorder patient samples included in the study.
| Samples | CAUS | PGx |
|---|---|---|
| n (TD+/TD-) | 148 (63/85) | 58 (19/39) |
| Sex (male/female) | 97/51 | 45/13 |
| Mean age in years (standard deviation) | 39.01 (10.50) | 41.10 (13.42) |
| CYP2D6 metabolizer phenotype (PM/IM/EM/UM) | 6/11/123/8 | 4/6/48/0 |
EM: Extensive metabolizer; IM: Intermediate metabolizer; PGx: Pharmacogenomic; PM: Poor metabolizer; TD: Tardive dyskinesia; UM: Ultra-rapid metabolizer.
TD classification was based on the Schooler and Kane criteria, using the Abnormal Involuntary Movement Scale (AIMS) or the modified Hillside Simpson Dyskinesia Scale for the 12 patients recruited from the Hillside Hospital [33,34]. As per the aforementioned criteria, presence of TD included at least one moderate rating or at least two mild ratings on the first seven items of the AIMS. AIMS scores were available for 194 European participants in our sample.
Genotyping & analysis
For the CAUS sample, we genotyped nine SNPs (rs1135840, rs16947, rs28371706, rs28371725, rs35742686, rs3892097, rs5030655, rs5030656 and rs1065852) in the CYP2D6 gene; these are key variants for the individual star alleles for CYP2D6. Genotyping was performed by PCR amplification with the TaqMan genotyping assays (Thermo Fisher Scientific, ON, Canada) following the manufacturer’s protocol, followed by genotype determination in the ViiA 7 Real-Time PCR system. We also assessed the copy number of CYP2D6 using a copy-number assay (Thermo Fisher Scientific) following the manufacturer’s protocol in the ViiA 7 Real-Time PCR System. Ten percent of the genotypes were repeated as quality controls, and no mismatches were observed. Genotypes for the PGx sample were extracted from the pharmacogenetic test panel based on the multiplex Luminex xTAG bead array done on cheek swab DNA for all of the SNPs mentioned for CAUS [35]. Using the CYP2D6 nomenclature, all samples were genotyped for CYP2D6*1–5, 2xN, 4xN, *10, *17 and *41. The DNA variants associated with these alleles is provided in Table 2.
Table 2. . CYP2D6 DNA variants that are tested in the present study.
| Variant ID | CYP2D6 Allele | Function | Variant |
|---|---|---|---|
| rs16947 | *2 | Normal (wild type) | 2851C>T (R296C) |
| rs1135840 | *2 | Normal | 4181G>C (S486T) |
| rs35742686 | *3 | None | 2550delA (259 frameshift) |
| rs3892097 | *4 | None | 1847G>A (splicing defect/169 frameshift) |
| Copy number | *5 | Gene deletion/duplication | Gene deletion/duplication |
| rs5030655 | *6 | None | 1708delT (152 frameshift) |
| rs5030656 | *9 | Decreased | 2616delAAG (K281del) |
| rs1065852 | *10 | Decreased | 100C>T (P34S) |
| rs28371706 | *17 | Decreased | 1022C>T (T107I) |
| rs28371725 | *41 | Decreased | 2851C>T (R296C) |
We translated the CYP2D6 genotypes for each participant into an activity score and metabolizer phenotype according to the Clinical Pharmacogenetics Implementation Consortium system [36]. We conducted logistic regression analysis of TD occurrence and linear regression analysis of log-transformed AIMS scores separately for the CAUS and PGx samples, taking into account age and sex as covariates (SPSS).
Results
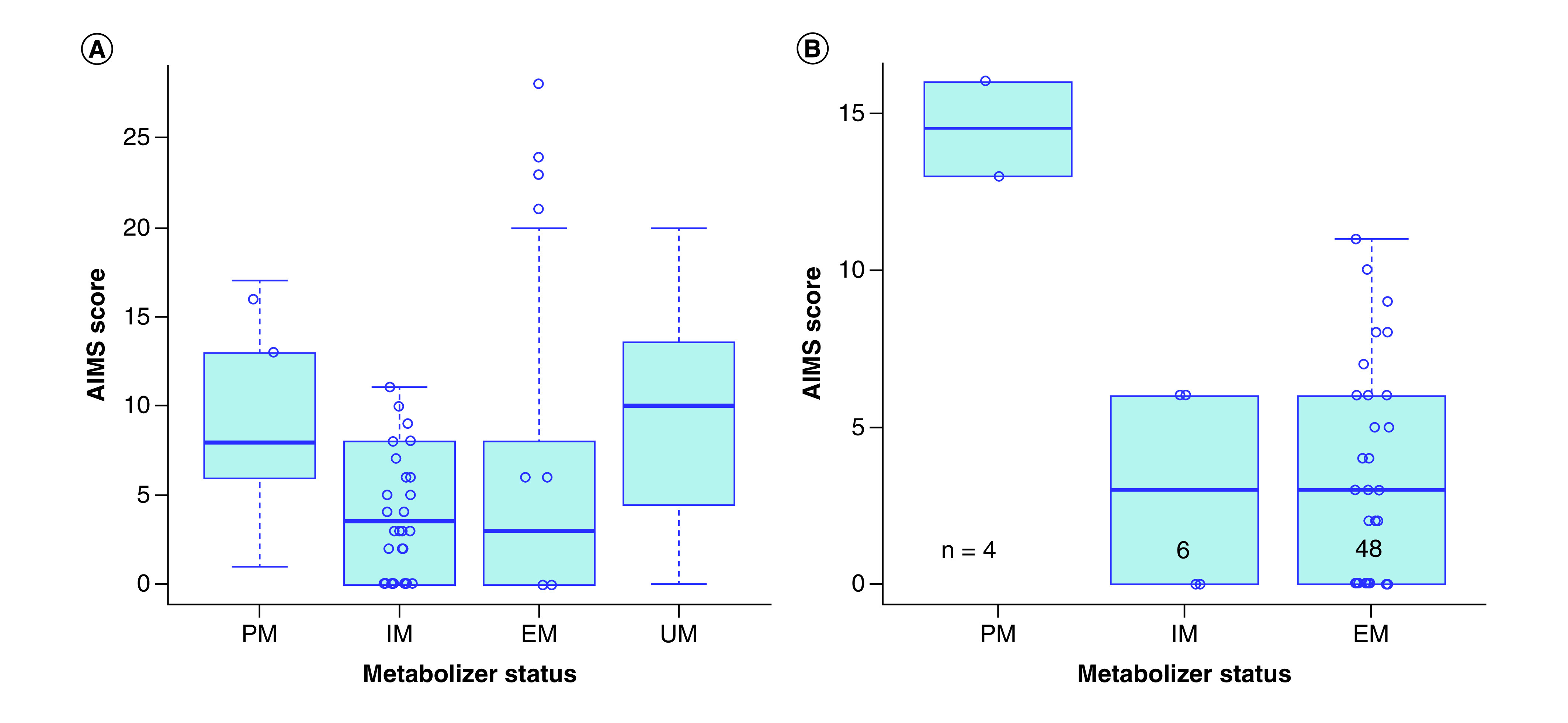
Inspection of the boxplot for the CAUS sample (Figure 1A) revealed a U-shaped trend that shows that individuals categorized as being CYP2D6 PM or UM (each UM having three copies of functional CYP2D6) appear to have higher average total AIMS scores than those categorized as being CYP2D6 EM and IM, controlling of sex and age (beta = 0.277; 95% CI: 0.025–0.530; p = 0.032). Logistic regression analysis further revealed that CYP2D6 extreme metabolizer phenotypes were significantly associated with TD occurrence, controlling for sex and age (odds ratio [OR] = 5.580; 95% CI: 1.454–21.410; p = 0.012). However, activity score did not reveal any significant associations with TD in this comparison, controlling for sex and age (OR = 0.987; 95% CI: 0.604–1.612; p = 0.957). The results are summarized in Table 3.
Figure 1. . Boxplots depicting CYP2D6 metabolizer phenotype versus Abnormal Involuntary Movement Scale score for the (A) CAUS sample and (B) pharmacogenomic sample.
AIMS: Abnormal Involuntary Movement Scale; EM: Extensive metabolizer; IM: Intermediate metabolizer; PM: Poor metabolizer; UM: Ultra-rapid metabolizer.
Table 3. . Summary of analysis of CYP2D6 activity scores, CYP2D6 metabolizer phenotype and CYP2D6 extreme metabolizer phenotype with tardive dyskinesia occurrence and tardive dyskinesia severity (log Abnormal Involuntary Movement Scale scores).
| TD occurrence | CAUS sample | PGx sample | ||
|---|---|---|---|---|
| OR (95% CI) | p-value | OR (95% CI) | p-value | |
| Activity score† | 0.987 (0.604–1.612) | 0.957 | 0.361 (0.139–0.938) | 0.037‡ |
| Metabolizer phenotype† | 0.863 (0.459–1.621) | 0.647 | 0.343 (0.119–0.989) | 0.048‡ |
| Extreme metabolizer phenotype | 5.580 (1.454–21.410) | 0.012‡ | 8.066 (0.646–100.748) | 0.105 |
| AIMS scores | Beta (95% CI) | p-value | Beta (95% CI) | p-value |
| Activity score† | 0.014 (-0.096–0.124) | 0.804 | -0.13 (-0.282–0.016) | 0.079 |
| Metabolizer phenotype† | -0.024 (-0.165–0.118) | 0.741 | -1.43 (-0.309–0.023) | 0.089 |
| Extreme metabolizer phenotype | 0.277 (0.025–0.530) | 0.032‡ | 0.292 (-0.090–0.674) | 0.131 |
CYP2D6 activity scores and CYP2D6 metabolizer phenotypes were defined according to the Clinical Pharmacogenetics Implementation Consortium system.
p-values less than 0.05.
AIMS: Abnormal Involuntary Movement Scale; OR: Oodds ratio; PGx: Pharmacogenomic; TD: Tardive dyskinesia.
Upon visual inspection of the boxplot for the PGx sample (Figure 1B), we noticed individuals with CYP2D6 PM phenotype appeared to have higher AIMS scores than those with other CYP2D6 metabolizer phenotypes. CYP2D6 metabolizer phenotype were associated with TD occurrence, controlling for sex and age (OR = 0.343; 95% CI: 0.119–0.989; p = 0.048; Table 3). A significant association was also found for the logistic regression analysis of activity score and TD, controlling for sex and age (OR = 0.361; 95% CI: 0.139–0.938; p = 0.037; Table 3).
Discussion
We have conducted an association study of the CYP2D6 gene in TD occurrence and severity. The association between extreme metabolizer phenotypes and TD presence supports a modulatory role for this gene in the risk of TD development, and ties in previous genetic findings. We observed patients with UM phenotype which, to our knowledge, was tested in only one previous TD study [27].
The majority of previous CYP2D6 gene association studies with TD suggest that lower activity score, or PMs, incur greater risk for TD [20,21]. We were able to support this association with our significant finding of CYP2D6 metabolizer phenotype and activity scores with TD occurrence. Although we were unable to confirm the association with TD severity in our PGx sample, we observed trends for both low activity scores and CYP2D6 PM phenotype to be associated with TD severity.
In the CAUS sample, our initial logistic regression analysis between metabolizer phenotype and TD occurrence revealed no significant association. Subsequently, investigation of the boxplot revealed that CYP2D6 metabolizer phenotype may have a U-shaped relationship with TD severity. By grouping together the two extreme metabolizer phenotypes, we found the CYP2D6 UM/PM group to have a higher risk for TD and higher TD severity than the IM/EM group. Not only does this confirm previous studies, but also conjoins the two opposing hypotheses regarding CYP2D6 and TD in that both UM and PM are associated with higher TD risk and severity. Establishing that UM individuals may be predisposed to TD may also suggest that future studies will need to assay for CYP2D6 gene deletion/duplication in addition to other DNA variants in CYP2D6 in order to derive more precise metabolizer phenotypes that may unmask the relationship we have observed in this study.
Conclusion
While the findings from the present study are encouraging, a number of limitations should be considered before drawing firm conclusions. First, an important risk factor in TD development is cumulative drug dose that incorporates treatment duration, medication type and treatment adherence; this information was not available for our samples. As mentioned earlier, certain antipsychotics, mainly FGAs, confer a greater likelihood of developing TD. CYP2D6 falls on either the major or minor metabolic pathway for all antipsychotic drugs. We have not accounted for the effects of other liver enzymes; this might have limited our ability to estimate the effect of CYP2D6 on TD risk and severity. A second limitation is the moderate sample size which affects almost all CYP2D6 genetic investigations and is reflected in our study by the low counts for PM and UM participants. As many of the alleles which result in the CYP2D6 extreme metabolizer phenotypes are rare in the general population, larger samples will be needed in order to increase the chance of finding more CYP2D6 PM and UM patients. Third, there were a number of covariates that were not taken into account for our analysis. Smoking, drug and alcohol use were not available for all the participants and could have influenced our findings in light of evidence suggesting their role in TD [2]. Smoking in particular is known to induce CYP1A2 enzyme activity, which may in turn influence the steady-state concentrations of certain antipsychotic drugs [37,38].
Future perspective
Moving forward, there are several avenues that can be explored for future studies of the CYP2D6 gene. First, deep sequencing will identify all CYP2D6 gene variants in order to increase the accuracy of the predicted CYP2D6 metabolizer phenotype that we used in our analysis. Second, testing CYP2D6 metabolizer phenotype with TD across ethnic groups would enhance our understanding of the role of this enzyme, especially with higher frequency of CYP2D6 IMs in East Asians and higher frequency of UMs in individuals of African ancestry [25,26,39]. Third, as we found in our samples, CYP2D6 PMs are not the only individuals who incur greater TD risk. Essentially, in CYP2D6 PMs the inability to lower antipsychotic drug plasma levels might have led to excessively high levels of the drug in the body, resulting in higher TD risk. However, TD risk observed in UMs may not act through the same mechanism. Since CYP2D6 UMs are able to clear the drug at an accelerated rate, the metabolites which result from drug metabolism accumulate in the body as well. This suggests that there may be active metabolites which are involved in the pathophysiology of TD. Additional studies on the effects of drug metabolites on TD and related phenotypes will help us understand the mechanism of the association between CYP2D6 UM and TD. Furthermore, since the levels of most drugs are influenced by more than one liver enzyme and drug transporters, the combined effect of gene panels on drugs of interest should be considered in future pharmacogenetic studies. At last, many chronic schizophrenia patients are often prescribed multiple medications [40], thus drug–drug interactions, which can alter liver enzyme activity, need to be considered [41–43]. Overall though, findings from our current study encourage more in-depth interrogation of liver enzyme genes in TD.
Summary points.
Testing on CYP2D6 metabolizer phenotypes reveals an association with tardive dyskinesia (TD) in schizophrenia patients.
Our findings suggest ultrarapid and poor metabolizers are at increased risk for TD and suffer higher TD severity.
Our study supports the need for more detailed investigations of the CYP2D6 gene and TD risk medications.
Acknowledgments
The authors express gratitude toward Larry and J Tanenbaum for their generous support in creating the Tanenbaum Centre for Pharmacogenetics, which is advancing research for the CAMH Pharmacogenetic Program. The authors also thank the participants in the study.
Footnotes
Author contributions
All authors contributed significantly in the work, including conception of the study (CC Zai, JY Lu, AK Tiwari, N Freeman, N King and JL Kennedy), data collection (N Freeman, M Tampakeras, D Herbert, H Emmerson and SY Cheema), data analysis (CC Zai, JY Lu, AK Tiwari and GC Zai), interpretation (Vd Luca, DJ Müller, AN Voineskos, SG Potkin, JA Lieberman, HY Meltzer and G Remington), manuscript drafting (JY Lu and CC Zai) and review (all).
Financial & competing interests disclosure
The authors thank the Ministry of Research and Innovation of Ontario, for funding the IMPACT project. CC Zai, AK Tiwari, DJ DJ Müller and JL Kennedy have been supported by the Genome Canada Genomic Applications Partnership Program (GAPP) and the CAMH Foundation. DJ Müller was supported by the Canadian Institutes of Health Research (CIHR operating grant MOP 142192), the National Institutes of Health (R01MH085801), the CAMH Foundation (J Murphy Professorship) and received a Brain & Behaviour Research (NARSAD) Independent Investigator Award, the M Smith New Investigator Salary Prize for Research in Schizophrenia (CIHR) and an Early Researcher Award by the Ministry of Research and Innovation of Ontario. G Remington was supported by the Canadian Institutes of Health Research (CIHR), as well as the Research Hospital Fund – Canadian Foundation for Innovation (RHF-CFI). CC Zai, AK Tiwari, DJ Müller and JL Kennedy are investigators in two pharmacogenetic studies where genetic test kits were provided as in-kind contribution by Assurex Health (Myriad Neuroscience) to evaluate the feasibility of pharmacogenetic testing in clinical practice and potential benefits of pharmacogenetic testing compared with treatment as usual. They have not received any payments or received any equity, stocks or options from this company or any other pharmacogenetic companies. CC Zai, AK Tiwari, DJ Müller and JL Kennedy are authors in two filed genetic patents assessing risk for antipsychotic-induced weight gain. JL Kennedy has received honoraria from Novartis, Roche and Eli Lilly corporations, and is an unpaid member of the scientific advisory board of AssureRx Corp. HY Meltzer has received grants or is or was a consultant to: Abbott Labs, ACADIA, Alkemes, Bristol Myers Squibb, DaiNippon Sumitomo, Eli Lilly, EnVivo, Janssen, Otsuka, Pfizer, Roche, Sunovion and BiolineRx. HY Meltzer is a shareholder of ACADIA and Glaxo Smith Kline. In the past three years JA Lieberman reports having received research funding or is a member of the advisory board of Allon, Alkermes, Bioline, GlaxoSmithKline Intracellular Therapies, Lilly, Merck, Novartis, Pfizer, Pierre Fabre, Psychogenics, F Hoffmann-La Roche LTD, Sepracor (Sunovion) and Targacept. JA Lieberman receives no direct financial compensation or salary support for participation in these research, consulting or advisory board activities. SG Potkin: consultancy/board of advisors/honoraria: American Psychiatric Association, Astra Zeneca, Bristol-Myers Squibb, Cortex, Dainippon-Sumitomo, Janssen Pharmaceutica, Novartis, Otsuka, Pfizer, Roche, Schering Plough, Vanda; research grants: Amgen, Bristol-Myers Squibb, Dainippon Sumitomo, Elan, En Vivo, Forest Laboratories, Janssen Pharmaceutica, Merck, Novartis, Otsuka, Pfizer, Solvay Pharmaceuticals, Roche, Sunovion, NIH, Harvard Massachusetts General Hospital, Brigham and Women’s Hospital, Vanda, speakers’ bureau: Lundbeck, Otsuka, ISCTM, Novartis, Pfizer, Sunovion. In the past 3, years G Remington has received consultant fees from HLS Therapeutics and Mitsubishi Tanabe Pharma Corporation, as well as research support from HLS Therapeutics. He holds no commercial investments in any pharmaceutical company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The study was approved by individual institutional review boards and is in accordance with the Declaration of Helsinki. Informed consent was given by each study participant.
References
- 1.van Harten PN, Tenback DE. Tardive dyskinesia: clinical presentation and treatment. Int. Rev. Neurobiol. 98, 187–210 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Solmi M, Pigato G, Kane JM, Correll CU. Clinical risk factors for the development of tardive dyskinesia. J. Neurol. Sci. 389, 21–27 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Carbon M, Hsieh CH, Kane JM, Correll CU. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J. Clin. Psychiat. 78(3), e264–e278 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Eloff I, Esterhuysen W, Odayar K. Antipsychotic use in a resource-limited setting: findings in an Eastern Cape psychiatric hospital. S. Afr. J. Psychiatr. 23, 1093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teo JT, Edwards MJ, Bhatia K. Tardive dyskinesia is caused by maladaptive synaptic plasticity: a hypothesis. Mov. Disord. 27(10), 1205–1215 (2012). [DOI] [PubMed] [Google Scholar]
- 6.O'Brien A. Comparing the risk of tardive dyskinesia in older adults with first-generation and second-generation antipsychotics: a systematic review and meta-analysis. Int. J. Geriatr. Psychiatry 31(7), 683–693 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Caligiuri MR, Jeste DV, Lacro JP. Antipsychotic-Induced movement disorders in the elderly: epidemiology and treatment recommendations. Drugs & Aging 17(5), 363–384 (2000). [DOI] [PubMed] [Google Scholar]
- 8.van Os J, Walsh E, van Horn E, Tattan T, Bale R, Thompson SG. Tardive dyskinesia in psychosis: are women really more at risk? UK700 Group. Acta Psychiatr. Scand. 99(4), 288–293 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Yassa R, Jeste DV. Gender differences in tardive dyskinesia: a critical review of the literature. Schizophr. Bull. 18(4), 701–715 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Morgenstern H, Glazer WM. Identifying risk factors for tardive dyskinesia among long-term outpatients maintained with neuroleptic medications. Results of the Yale Tardive Dyskinesia Study. Arch. Gen. Psychiatry 50(9), 723–733 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Miller DD, McEvoy JP, Davis SM. et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr. Res. 80(1), 33–43 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Lohr JB, Kuczenski R, Niculescu AB. Oxidative mechanisms and tardive dyskinesia. CNS Drugs 17(1), 47–62 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Waln O, Jankovic J. An update on tardive dyskinesia: from phenomenology to treatment. Tremor Other Hyperkinet. Mov. 3, tre-03-161-4138-1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinhold P, Wegner JT, Kane JM. Familial occurrence of tardive dyskinesia. J. Clin. Psychiat. 42(4), 165–166 (1981). [PubMed] [Google Scholar]
- 15.Müller DJ, Schulze T, Knapp M. et al. Familial occurrence of tardive dyskinesia. Acta Psychiatr. Scand. 104, 375–379 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Yassa R, Ananth J. Familial tardive dyskinesia. Am. J. Psychiatry 138, 1618–1619 (1981). [DOI] [PubMed] [Google Scholar]
- 17.Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part II. Clin. Pharmacokinet. 48(12), 761–804 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Risperidone Therapy and CYP2D6 Genotype. Dean L, Pratt V, McLeod H, Rubinstein W, Kattman B, Malheiro A (). Medical Genetics Summaries, MD, USA: (2012). [PubMed] [Google Scholar]
- 19.Roke Y, van Harten PN, Franke B, Galesloot TE, Boot AM, Buitelaar JK. The effect of the Taq1A variant in the dopamine D(2) receptor gene and common CYP2D6 alleles on prolactin levels in risperidone-treated boys. Pharmacogenet. Genomics 23(9), 487–493 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Patsopoulos NA, Ntzani EE, Zintzaras E, Ioannidis JP. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenet. Genomics 15(3), 151–158 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Fleeman N, Dundar Y, Dickson R. et al. Cytochrome P450 testing for prescribing antipsychotics in adults with schizophrenia: systematic review and meta-analyses. Pharmacogenomics J. 11(1), 1–14 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Ivanova SA, Loonen AJ, Bakker PR. et al. Likelihood of mechanistic roles for dopaminergic, serotonergic and glutamatergic receptors in tardive dyskinesia: a comparison of genetic variants in two independent patient populations. SAGE Open Med. 4, 2050312116643673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobylecki CJ, Jakobsen KD, Hansen T, Jakobsen IV, Rasmussen HB, Werge T. CYP2D6 genotype predicts antipsychotic side effects in schizophrenia inpatients: a retrospective matched case–control study. Neuropsychobiology 59(4), 222–226 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Locatelli I, Kastelic M, Koprivsek J. et al. A population pharmacokinetic evaluation of the influence of CYP2D6 genotype on risperidone metabolism in patients with acute episode of schizophrenia. Eur. J. Pharm. Sci. 41(2), 289–298 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Lam LC, Garcia-Barcelo MM, Ungvari GS. et al. Cytochrome P450 2D6 genotyping and association with tardive dyskinesia in Chinese schizophrenic patients. Pharmacopsychiatry 34(6), 238–241 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Liou YJ, Wang YC, Bai YM. et al. Cytochrome P-450 2D6*10 C188T polymorphism is associated with antipsychotic-induced persistent tardive dyskinesia in Chinese schizophrenic patients. Neuropsychobiology 49(4), 167–173 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Koola MM, Tsapakis EM, Wright P. et al. Association of tardive dyskinesia with variation in CYP2D6: is there a role for active metabolites? J. Psychopharmacol. 28(7), 665–670 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zai CC, Hwang RW, De Luca V. et al. Association study of tardive dyskinesia and twelve DRD2 polymorphisms in schizophrenia patients. Int. J. Neuropsychopharmacol. 10(5), 639–651 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Zai CC, Tiwari AK, Mazzoco M. et al. Association study of the vesicular monoamine transporter gene SLC18A2 with tardive dyskinesia. J. Psychiatr. Res. 47(11), 1760–1765 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Lu JY, Tiwari AK, Zai GC. et al. Association study of disrupted-in-schizophrenia-1 gene variants and tardive dyskinesia. Neurosci. Lett. 686, 17–22 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Herbert D, Neves-Pereira M, Baidya R. et al. Genetic testing as a supporting tool in prescribing psychiatric medication: design and protocol of the IMPACT study. J. Psychiatr. Res. 96, 265–272 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Zai CC, Lee FH, Tiwari AK. et al. Investigation of the HSPG2 gene in tardive dyskinesia – new data and meta-analysis. Front. Pharmacol. 9, 974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch. Gen. Psychiatry 39(4), 486–487 (1982). [DOI] [PubMed] [Google Scholar]
- 34.Basile VS, Masellis M, Badri F. et al. Association of the MscI polymorphism of the dopamine D3 receptor gene with tardive dyskinesia in schizophrenia. Neuropsychopharmacology 21(1), 17–27 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Jablonski MR, King N, Wang Y. et al. Analytical validation of a psychiatric pharmacogenomic test. Per. Med. 15(3), 189–197 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Caudle KE, Sangkuhl K, Whirl-Carrillo M. et al. Standardizing CYP2D6 genotype to phenotype translation: consensus recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 13(1), 116–124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carrillo JA, Herraiz AG, Ramos SI, Gervasini G, Vizcaino S, Benitez J. Role of the smoking-induced cytochrome P450 (CYP)1A2 and polymorphic CYP2D6 in steady-state concentration of olanzapine. J. Clin. Psychopharmacol. 23(2), 119–127 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Ellingrod VL, Schultz SK, Arndt S. Abnormal movements and tardive dyskinesia in smokers and nonsmokers with schizophrenia genotyped for cytochrome P450 2D6. Pharmacotherapy 22(11), 1416–1419 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet. Med. 19(1), 69–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher MD, Reilly K, Isenberg K, Villa KF. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry 14, 341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen E, Eriksson B, Oberg K, Bondesson U, Rane A. Selective effects of somatostatin analogs on human drug-metabolizing enzymes. Clin. Pharmacol. Ther. 64(2), 150–159 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Tyson SC, Devane CL, Risch SC. Pharmacokinetic interaction between risperidone and clozapine. Am. J. Psychiatry 152(9), 1401–1402 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Koreen AR, Lieberman JA, Kronig M, Cooper TB. Cross-tapering clozapine and risperidone. Am. J. Psychiatry 152(11), 1690 (1995). [DOI] [PubMed] [Google Scholar]