

Abstract
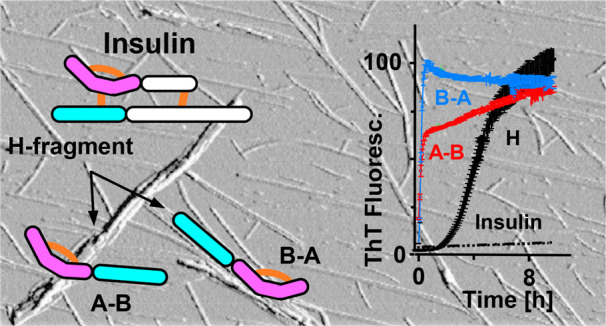
Relatively short amino acid sequences often play a pivotal role in triggering protein aggregation leading to the formation of amyloid fibrils. In the case of insulin, various regions of A- and B-chains have been implicated as the most relevant to the protein’s amyloidogenicity. Here, we focus on the highly amyloidogenic H-fragment of insulin comprising the disulfide-bonded N-terminal parts of both chains. Analysis of the aggregation behavior of single-chain peptide derivatives of the H-fragment suggests that the A-chain’s part initiates the aggregation process while the disulfide-tethered B-chain reluctantly adapts to amyloid structure. Merging of both A- and B-parts into single-chain continuous peptides (A–B and B–A) results in extreme amyloidogenicity exceeding that of the double-chain H-fragment as reflected by almost instantaneous de novo fibrillization. Amyloid fibrils of A–B and B–A present distinct morphological and infrared traits and do not cross-seed insulin. Our study suggests that the N-terminal part of insulin’s A-chain containing the intact Cys6–Cys11 intrachain disulfide bond may constitute insulin’s major amyloid stretch which, through its bent conformation, enforces a parallel in-register alignment of β-strands. Comparison of the self-association behavior of H, A–B, and B–A peptides suggests that A-chain’s N-terminal amyloid stretch is very versatile and adaptive to various structural contexts.
Introduction
Nowadays, formation of amyloid fibrils is recognized as a generic process accessible to various proteins and peptides.1,2 Due to association with a number of degenerative maladies (e.g., Alzheimer’s disease, type 2 diabetes mellitus3−5), as well as benign biological functions essential for numerous organisms,6−8 research on amyloidogenesis has expanded considerably in recent years. Local inter- and intramolecular interactions driving self-assembly of amyloid fibrils are identical with those responsible for folding of the native state. However, formation of thermodynamically stable fibrillar structures imposes specific requirements on amino acid sequence, backbone topology, and conformation of aggregating proteins.9−11 For example, β-sheet-breaking proline residues, uncompensated Coulombic repulsion between ionized side chains, and a main-chain topology restricted by numerous disulfide bonds are all intuitively expected to decrease the tendency to form fibrils. In accordance with this notion, early studies revealed that, for example, proline substitutions in rodent islet amyloid polypeptide (IAPP) prevent its in vivo aggregation and therefore spontaneous-onset type 2 diabetes mellitus.12 On the other hand, it has been shown that protein amyloidogenicity may, in fact, adapt well to the presence of proline residues at some sites in the sequences13 while marginally uncompensated electric charges may even enhance aggregation;14 there is also evidence that disulfide bonds (both intra- and interchain) may accelerate formation of fibrils in specific cases.15−17 Hence the actual impact of even such elementary factors on a protein’s propensity to form fibrils depends on the structural and thermodynamic context. This, in turn, foreshadows the difficulty of accurately predicting amyloidogenic propensities of various amino acid sequences. It has been elegantly demonstrated by Minor and Kim that whether a particular main-chain segment within a globular protein adopts β-sheet conformation depends strongly on the tertiary context, as opposed to being determined solely by the intrinsic secondary structure preference.18,19 From this it follows that an actual amyloidogenic propensity of a given amino acid sequence depends also on the quaternary context; i.e., its accurate estimation would require a priori knowledge of the 3D structure of the fibril. Despite these fundamental challenges, computational algorithms aimed at predicting amyloidogenic tendencies of amino acid sequences (and even of whole folded proteins, e.g., refs (11, 20−24); for a concise review, see ref (10)) have been developed with some success. In part, the underlying rationale is that the abundance of certain hydrophobic amino acid residues, especially when arranged in particular sequence patterns, increases amyloidogenicity.25−29 In a permissive conformational context, a relatively short amyloidogenic segment (so-called amyloid stretch) of the main chain may drive the whole protein to form fibrils.29,30 A distinction should be made between the latter term and a core region which primarily corresponds to a portion of amino acid sequence involved in formation of so-called steric zippers within amyloid fibrils. Mechanisms through which amyloid stretch segments induce their less aggregation-prone surroundings to convert into fibril remain elusive.
Previously, a highly amyloidogenic double-chain fragment released upon partial digestion of bovine insulin (BI) with pepsin was identified.31 This so-called H-fragment consists of N-terminal segments of insulin’s A- and B-chains linked by a Cys7A–Cys7B disulfide bond; the A-chain part is bent due to another Cys6A–Cys11A disulfide bond (Figure 1A). Singly dispersed H-fragment monomers are disordered in aqueous solutions but undergo rapid aggregation into amyloid-like fibrils with the infrared spectral features typical of parallel β-sheet structure.31,32 According to the follow-up study the highly amyloidogenic character of H-fragment arises from the Cys6A–Cys11A disulfide-restricted A-chain part while the B chain part itself is refractory to aggregation.17 Our initial interest that motivated this work was focused on the versatility of A-chain’s N-terminal segment with respect to the B-chain partner. Specifically, we were asking whether different structural arrangements of A- and B-parts (e.g., merging both parts into a single polypeptide chain) would preserve the highly amyloidogenic character of the H-fragment.
Figure 1.

(A) Amino acid sequences of H-fragment (superimposed onto the covalent structure of BI monomer), A–B, and B–A peptides. The sequence portions corresponding to insulin’s chains A and B are marked in violet and blue, respectively. Disulfide bonds are marked with orange lines. In A–B and B–A peptides, Cys residues involved in insulin’s interchain Cys7A–Cys7B disulfide bond were replaced with alanine (marked in red). (B) Prediction of amyloidogenic tendencies of separate A- and B-chain parts of H-fragment and of whole A–B and B–A peptides based on TANGO (0.2 mM peptide concentration, pH 1.9, 310 K, 50 mM ionic strength)34−36 and Waltz (pH 2.6, high sensitivity)21 algorithms. The computational settings were selected to match closely the experimental conditions.
Materials and Methods
Samples
BI, tris(2-carboxyethyl)phosphine (TCEP), and other nondeuterated chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). D2O (“99.8 atom % D” grade) and DCl were from ARMAR Chemicals (Döttingen, Aargau, Switzerland). Insulin fragments listed in Figures 1 and 2 (H, A–B, B–A, ACC, B-7A), as well as the precursor for B-SH peptide (B–B disulfide-bonded covalent dimer17) all without N- or C-terminal modifications, were custom synthesized by Pepscan (Lelystad, The Netherlands). The synthetic peptides were provided by the manufacturer as crude trifluoroacetic acid (TFA) salts (H, ACC, B–B) or were further lyophilized in order to remove TFA (A–B, B–A, B-7A). While powders of H, ACC, B–B, and B-7A could be solubilized according to the previously described protocol17 consisting of dispersion of solid peptide samples in 8 M guanidine hydrochloride (GdnHCl) solution, pH 9.0, crude A–B and B–A proved to be resistant to this approach. We have established an alternative solubilization protocol consisting of the following: (i) initial sonication-assisted dispersion of crude peptide in dimethyl sulfoxide (DMSO) at ∼5.6 mg/mL concentration, followed by (ii) 24-h-long (or 18-h-long in the case of the data presented in Figure 3) agitation at 37 °C, followed by (iii) addition of 1/9 vol of H2O acidified to pH 1.64 and continued vigorous agitation at 37 °C for 24 h. Such a 24 h + 24 h incubation protocol turned out to be extremely effective in solubilizing otherwise insoluble crude A–B, B–A, and H peptides. This holds true even if the incubation period is shortened, as is the case of the preliminary experiment summarized in Figure 3 (18 h + 2 h) in which acidified D2O (pD 1.64; pH-meter readout unadjusted for isotopic effects) was used instead of H2O in order to enable monitoring of the conformational state of peptide in solution with infrared spectroscopy. Thus, prepared concentrated peptide solutions in 90% DMSO/water system were swiftly diluted 10 times with acidified water in order to trigger spontaneous aggregation of disordered monomers into amyloid fibrils (Figure 4). Unless otherwise specified, the typical composition of liquid samples for kinetic experiments was as follows: 0.5 mg/mL peptide in 9 vol/vol % DMSO in H2O, pH 1.9, containing additionally thioflavin T (ThT) at 20 μM concentration and optionally, as indicated in figure captions, NaCl at 50 mM concentration. For the ThT-based kinetic assay of the aggregation behavior of H, ACC, and B-SH reported in Figure 2, the previously described GdnHCl-based protocol17 was employed instead. Portions of H, ACC, and B–B were dissolved at 2 mg/mL concentration in 50 mM NaCl, 1.33 M GdnHCl, 20 μM ThT, pH 1.9; B–B sample contained also the addition of TCEP at 6.8 mg/mL concentration which, by rapid reduction of the disulfide bond, released B-SH peptide in situ;17 B-7A peptide was dissolved at 1 mg/mL concentration in 9 vol/vol % DMSO in 50 mM NaCl, 20 μM ThT, pH 1.9. Measurements were carried out at 37 °C.
Figure 2.

(A) Amino acid sequences of single-chain fragments of H-peptide. In ACC and B-7A, Cys residues involved in the interchain disulfide bond were substituted with alanine (marked in red). (B) Comparison of de novo aggregation kinetics of ACC, B-7A, and B-SH versus H-fragment probed by fluorescence of ThT. Samples of H, ACC, and B-SH were dissolved at 2 mg/mL concentration in 50 mM NaCl, 1.33 M GdnHCl, 20 μM ThT, pH 1.9. B-SH sample contained also TCEP at 6.8 mg/mL concentration. B-7A peptide was dissolved at 1 mg/mL concentration in ∼9 vol/vol % DMSO in 50 mM NaCl, 20 μM ThT, pH 1.9. Measurements were carried out at 37 °C and 300 rpm agitation.
Figure 3.

(A) Normalized ATR FT-IR spectra of raw solid samples of H, A–B, and B–A peptides. (B) Normalized solvent-subtracted transmission FT-IR spectra of H, A–B, and B–A peptides dissolved in acidified DMSO/D2O solution. Solid samples were initially dissolved in pure DMSO at 1.1 wt/vol % peptide concentration (for 18 h at 37 °C) followed by addition of 1/9 vol of D2O, pD 1.64, and 2 h incubation at 37 °C prior to spectral measurements.
Figure 4.

ThT-fluorescence-monitored kinetics of amyloidogenic reassociation of DMSO-dissolved monomers of H, A–B, and B–A in the presence (A) and absence (B) of 50 mM NaCl. Aggregation was initiated by rapid mixing of concentrated solutions of peptides in 90 vol/vol % DMSO/acidified H2O (24 h + 24 h preincubation protocol) with an excess of acidified aqueous solution. At the start of the experiment reported in (A), each peptide was dissolved at 0.5 mg/mL concentration in 9 vol/vol % DMSO in aqueous 20 μM ThT in 50 mM NaCl, pH 1.9. Sample conditions pertaining to (B) were analogous except that no NaCl was added. The green trajectory in (A) depicts typical kinetics of H-fragment reassociation under similar conditions but with DMSO replaced with approximately 500 mM GdnHCl.31 At the end of the (A) kinetic experiment, amyloid precipitates were collected from the plate and after extensive washing with 50 mM NaCl, pH 1.9, were subjected to far-UV CD (C) and ATR FT-IR (D) measurements. CD spectra were collected at 25 °C for 0.04 mg/mL suspensions of aggregates in acidified H2O (pH 1.9) placed in 10 mm quartz cuvette; a CD spectrum of BI amyloid is overlaid for comparison. The inset histogram in (D) presents full width at half-maximum (fwhm) values of the amide I band collected for crude (gray), DMSO-dissolved (green), and collected at the end of the kinetic experiment (orange) peptides.
Insulin seeding experiments were conducted on freshly prepared 0.12 wt/vol % BI solutions in 100 mM NaCl, 20 μM ThT, pH 1.9. Thus, prepared liquid samples of native BI were mixed with preformed and sonicated amyloid fibrils at 1:20 amyloid:native BI mass ratio. Details of the sonication procedure and equipment used were described earlier.33
Fibrillization Kinetics
ThT-fluorescence measurements (λex 440 nm/λem 485 nm) of peptide aggregation kinetics were conducted on a CLARIOstar plate reader from BMG LABTECH (Offenburg, Germany) using 96-well black microplates. Typically, each well was filled with a 150 μL volume of peptide solution containing ThT. Measurements were carried out at 37 °C with or without moderate agitation for 48 h, or for periods of time specified in the figures. Each kinetic trace was calculated as an average of three independently collected trajectories (the error bars correspond to the standard deviations). Once kinetic measurements were completed, aggregates were collected from wells and washed with portions of 50 mM NaCl, pH 1.9, solution in order to remove excess DMSO/denaturants prior to circular dichroism (CD), atomic force microscopy (AFM), and FT-IR (Fourier transform infrared) spectroscopic measurements.
AFM
A small portion of aggregate suspension collected from a plate well was initially washed several times with acidified 50 mM NaCl aqueous solution in order to remove excess DMSO. Subsequently, the suspension of fibrils was diluted with salt-free acidified water (pH 1.9) until the peptide concentration reached 0.1 mg/mL. A small droplet (10 μL) of fibril suspension was swiftly deposited onto freshly cleaved mica and left to dry overnight. AFM tapping-mode measurements were carried out with a Nanoscope III atomic force microscope from Veeco Instruments (Plainview, NY, USA) and TAP300-Al sensors (resonance frequency 300 kHz) from BudgetSensors (Sofia, Bulgaria).
ATR (Attenuated Total Reflectance) FT-IR and Transmission FT-IR Measurements
Centrifuged samples of aggregates collected from the plate reader were washed several times with portions of 50 mM NaCl in H2O, pH 1.9, in order to remove DMSO. Subsequently, aqueous suspensions of fibrils were deposited and allowed to dry up on the diamond surface of a single-reflection diamond ATR accessory of a Nicolet iS50 FT-IR spectrometer from Thermo Fisher Scientific (Waltham, MA, USA) equipped with a DTGS detector. Typically, for a single ATR FT-IR spectrum, 32 interferograms of 2 cm–1 resolution were coadded. Due to ambiguity in determining the real values of the refractive indexes of amyloid aggregates, uncorrected ATR FT-IR data is shown. Spectral data processing was limited to subtracting the water vapor spectrum and adjusting the two-point baseline with the use of GRAMS software (Thermo Fisher Scientific).
Transmission FT-IR spectra shown in Figure 3B were collected on the same Nicolet iS50 FT-IR spectrometer except that a CaF2 transmission cell with a 0.025 mm Teflon spacer was used instead of the ATR accessory. During measurements the cell’s temperature was maintained at 37 °C with a dedicated Peltier accessory while the sample chamber was continuously purged with dry CO2-depleted air. Typically, for a single spectrum, 16 interferograms of 2 cm–1 resolution were coadded. All spectra were corrected by subtracting the proper amount of buffer (DMSO–D2O solution) and water vapor spectra prior to being baseline-adjusted (flat two-point baseline). As neither DMSO nor D2O absorbs appreciably in the amide I band’s infrared region, the subtraction of the solvent spectra multiplied by a factor of 1 gave typically acceptable results. The subtraction factor for the water vapor spectrum was individually selected in order to minimize the rotational–vibrational bands of water vapor overlapping the amide I band region. Data processing was performed with GRAMS software (Thermo Fisher Scientific).
CD Measurements
Aqueous suspensions of aggregates collected from the plate at the end of kinetic experiments were washed several times with 50 mM NaCl, pH 1.9, solution in order to remove residual DMSO, which strongly absorbs UV light. Thus, treated aggregates were suspended in acidified (pH 1.9) water to the final concentration of 0.04 mg/mL and were swiftly transferred to a quartz cuvette with a 10-mm-long optical pathway. Far-UV CD spectra corrected for the buffer signal were acquired at room temperature by the accumulation of five independent spectra (at 200 nm/s scanning rate) on a J-815 S spectropolarimeter from Jasco Corp. (Tokyo, Japan).
Results and Discussion
De Novo Aggregation
We have selected a pair of synthetic single-chain analogues of insulin’s H-fragment in which either the N-terminal section of B-chain was tethered to the C-terminal section of A-chain (“A–B” peptide) or vice versa (“B–A” peptide; see Figure 1A) to study the impact of alternative sequence arrangements on the fragment’s amyloidogenicity. Residues Cys7 in both chains were replaced with alanine, while the intrachain disulfide bond within the A-part which is essential to H-fragment’s amyloidogenicity was retained. Rather unsurprisingly, when removed from an insulin monomer, the H-fragment remains in an unfolded conformation which has been confirmed earlier.31,32 In the absence of a folded globular state, sliding window methods such as TANGO34−36 or Waltz21 may be used for to predict amyloidogenic tendencies of amino acid sequences.10 We used both these tools to assess the amyloidogenicity of separated and merged (as A–B and B–A peptides) N-terminal sections of insulin at low pH, i.e., under conditions favoring aggregation of both insulin and H-fragment (Figure 1B). As an inherent limitation of these methods, the impact of the intrachain disulfide bond is not taken into account. The TANGO and Waltz scores calculated for separate A- and B-parts and B–A peptide are negligible, whereas for A–B peptide heightened scores are observed for the middle section encompassing C-terminal residues of A and N-terminal residues of B-part (TANGO) and for the whole sequence excluding the first nine N-terminal residues of A-part (Waltz). The two algorithms are suited specifically for prediction of strictly amyloidogenic (Waltz) or dehydration/aggregation-prone (TANGO) regions; both tools are consistent in suggesting that swapping the sequential arrangement of A- and B-parts would have a dramatic impact on the amyloidogenic properties of the single-chain peptides. We have also employed FoldAmyloid37 as a complementary bioinformatic tool dedicated to detection of amyloid-forming sequences. The results shown in Figure S1 are in line with the TANGO scores.
Low-pH fibrillization kinetics of individual synthetic peptides corresponding to H-fragment’s A-part (ACC) and B-part (B-7A, and B-SH depending on the substitution state of residue 7, see Figure 2A) were compared with the use of a ThT-fluorescence assay. The de novo aggregation of ACC peptide followed a steep upward trajectory preceded by a very short lag phase. The process was as fast as in the case of intact H-fragment at the same concentration (Figure 2B), which contrasted sharply with the behavior of B peptide while contradicting the predictions based on TANGO and Waltz (Figure 1B).
While commercial freeze-dried samples of H-fragment and ACC peptide could be solubilized following the previously established GdnHCl-based protocol,17 stock A–B and B–A peptides were strongly agglomerated and resistant to this approach. Interestingly, the ATR FT-IR spectra of crude commercial A–B, B–A, and H-fragment exhibit the main spectral component of the amide I band in the range 1623–1631 cm–1 indicative of the β-sheet conformation (Figure 3A) and often observed in the infrared spectra of amyloid fibrils.38 Furthermore, the absence of a minor exciton-split ([v(0,π)]) band above 1680 cm–1 suggests a parallel alignment of β-strands within the aggregate, and this again is in accordance with previous numerous studies on amyloid fibrils (e.g., refs (38 and 39)). These observations indicate that all three peptides formed amyloid-like aggregates already during the final stages of synthesis and purification. The splitting of the main amide I component observed for B–A sample could be interpreted as the presence of two populations of β-sheet varying in the strength of hydrogen bonds (the lower the wavenumber the stronger the bond). The minor bands at 1660 and 1731 cm–1 could be assigned to turns and protonated carboxyl groups, respectively.31,38,39 Moreover, an increased ThT fluorescence was detected when the fluorophore was added to aqueous suspensions of crude peptide powders (data not shown). Thus, it became essential to establish an effective protocol of A–B/B–A peptide solubilization which would allow us to observe bona fide amyloidogenic de novo reassociation of peptide monomers under controlled conditions. This was achieved through the application of the two-step procedure described under Materials and Methods consisting chiefly of prolonged incubation of raw peptide material in neat DMSO and in acidified DMSO–water mixtures. All three peptides dissolved in DMSO within a few hours. Solvent-subtracted transmission FT-IR spectra shown in Figure 3B indicate that the DMSO-dissolved peptides undergo a β → disorder conformational transition: the broad amide I band at ca. 1665 cm–1 is characteristic of various DMSO-denatured proteins40 including amyloid fibrils.41,42 The high frequency shift of the band arises from free amide carbonyl groups which are outcompeted from hydrogen bonding to the main chain’s N–H groups by the stronger hydrogen-bond acceptor (DMSO). The minor sharp component at 1692 cm–1 is likely to originate from residual β-turns; a similar observation was made by Shanmugam and Polavarapu for Aβ25–35 peptide dissolved in DMSO.43
The application of DMSO as a solvent for crude peptides was advantageous due to its effectiveness in disrupting aggregated β-sheets (a necessary condition to study fibrillization of peptide under strictly de novo conditions), compatibility with in situ FT-IR measurements in the amide I band region, and, finally, the fact that while concentrated DMSO dissolves amyloid fibrils, diluted solvent does not have the same effect.41,42 Hence, the reassociation process could possibly be initiated by a rapid mixing of a portion of peptide solution in concentrated DMSO with an excess of aqueous solution. Indeed, this was confirmed in follow-up experiments: kinetic trajectories of de novo fibrillization of A–B, B–A, and H peptides triggered by dilution of DMSO-dissolved peptide samples with acidified aqueous solution of NaCl are shown in Figure 4A.
The explosive rates of aggregation of A–B and B–A peptides are striking: no lag phases are detectable as the self-association begins already within the dead time of mixing (approximately 1 min) This contrasts with the case of H-fragment, whose aggregation is noticeably decelerated in terms of both the lag phase (approximately 80 min) and the less steep elongation phase. Importantly, unlike crude A–B and B–A powders, solid samples of H-fragment may be solubilized without DMSO17,32 and under such conditions the lag phase is negligible (Figure 4A). Hence the observed deceleration of H-fragment’s fibrillization should be attributed to the influence of residual DMSO. At pH 1.9 monomers of A–B and B–A are expected to bear the same positive electric charge (+2) whereas the net charge of H-fragment monomers could be larger (+3) due to additional N-termini. Thus, the Debye screening of repulsive electrostatic interactions by dissolved ions (NaCl) could impact differently aggregation of the A–B/B–A pair and H-fragment. To verify this possibility, we have carried out reassociation of the three peptides also in the absence of NaCl (Figure 4B). At the low ionic strength conditions, fibrillization of A–B and B–A remained very fast although the slope of the elongation phase was less dramatic; surprisingly no self-association of H-fragment was observed during the 24-h-long experiment. At the end of the kinetic experiments conducted in the presence of NaCl, samples of aggregated peptides were collected and, after removal of DMSO, were subjected to CD and infrared spectra measurements (Figure 4, parts C and 4D, respectively). The far-UV CD spectra feature single broad minima approximately at 220 nm (A–B) and ∼224 nm (B–A, H), indicative of aggregated β-sheets (the shift from the canonical 216 nm minimum typically observed for globular β-proteins is often observed for amyloid fibrils32,44). We note that the spectrum of B–A aggregate is flattened, which has been attributed to stronger light scattering on this aggregate. In unison with the CD data, the frequency range (1623–1632 cm–1) of the infrared amide I band spectra shown in Figure 4D points to the β-sheet conformation as the dominating secondary component of aggregates. Furthermore, the absence of a high frequency (above 1680 cm–1) exciton-split band suggests a parallel arrangement of β-strands and this is in agreement with previous studies on insulin fibrils.38,39,42 Pairwise comparison of infrared spectra collected for peptides before dissolution in DMSO (Figure 3A), and after amyloidogenic reassociation (Figure 4D), reveals a high degree of similarity—also in reproducing fine spectral details such splitting of the amide I band observed for exclusively for B–A aggregate. This also confirms that all three peptides were already in an amyloid-like state at the stage of freeze-drying that followed after the synthesis and purification. However, in each case, the reassociation appears to lead to a higher structural homogeneity as compared with crude peptide samples which is reflected by the narrowing of amide I band’s bandwidths (inset in Figure 4D). The sample conditions selected in this study include lowered pH, as is the case of many earlier studies on insulin aggregation (e.g., refs (33 and 39)). The initial data on the H-fragment were also collected at low pH since the peptide was released in situ during digestion of insulin with pepsin active under such conditions. Hence the herein selected pH conditions facilitate comparisons with the previously collected data. However, it must be stressed that all H, A–B, and B–A peptides maintain the highly amyloidogenic properties at close-to-neutral pH (see Figure S2).
Morphologies of aggregates collected at the end of the kinetic experiment are shown in Figure 5. Representative amplitude AFM images presented in the top row confirm the fibrillar nature of these assemblies. Clear differences in appearance of all three aggregates are immediately noticeable: H-fragment amyloid tends to form a straight singly dispersed specimen with a limited tendency to associate laterally into higher order structures. This contrasts with the strongly agglomerated short and straight A–B aggregates. On the other hand, B–A fibrils are long and slightly bent. On the basis of the corresponding height AFM data, we were able to estimate the thickness and periodicity of representative individual fibrils. To this end, cross sections of the selected specimen were analyzed in the directions perpendicular and parallel to the fibril axis. Individual fibrils built of self-associated H-fragment monomers are thinnest with a diameter of approximately 1.5 nm, whereas B–A fibrils are slightly thicker (1.8–2.2 nm).
Figure 5.
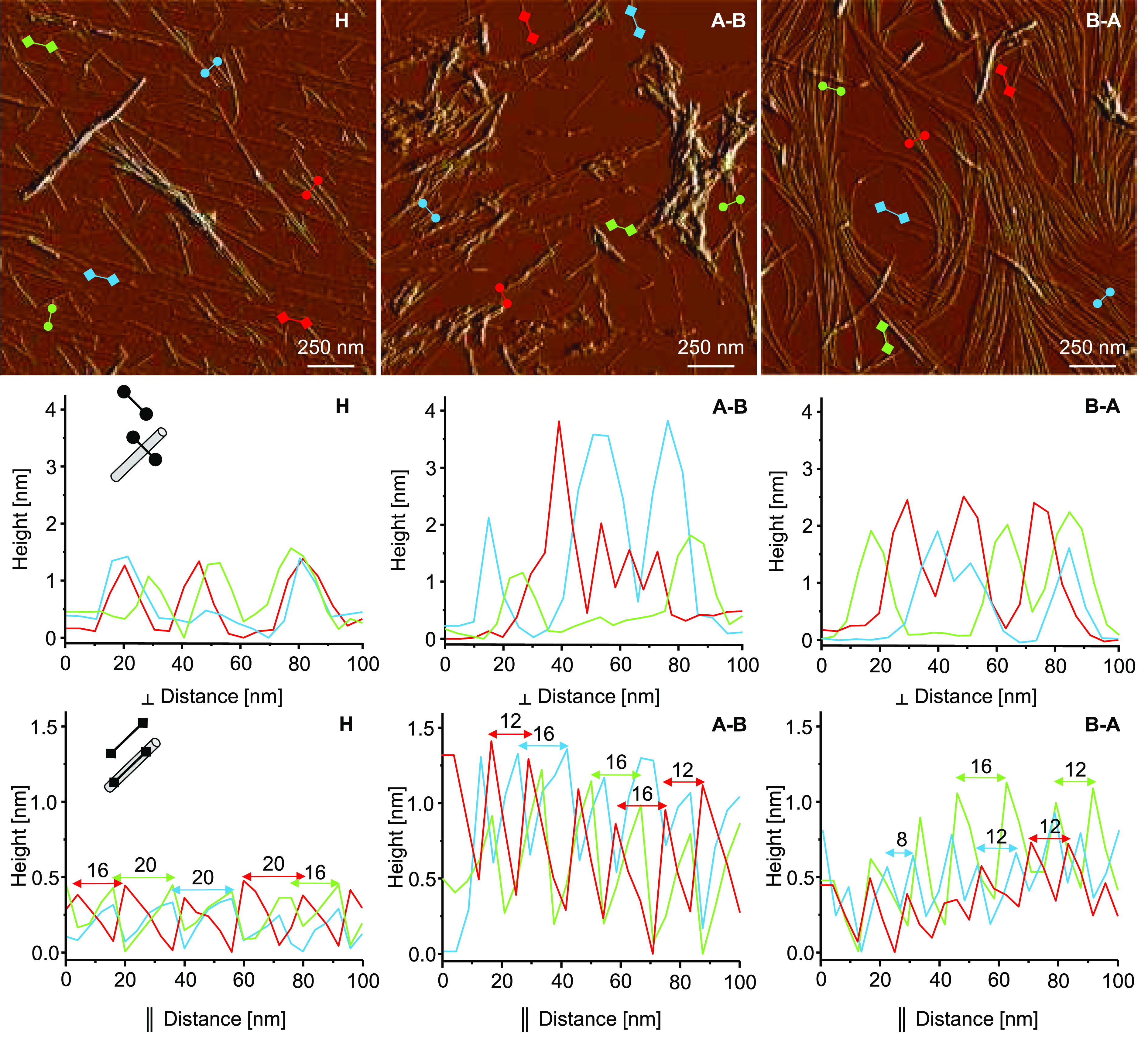
Amplitude AFM images of fibrils formed de novo by H-fragment, A–B, and B–A peptides collected at the end of the kinetic experiment (top row). Cross sections of selected fibrillar specimen obtained in the directions perpendicular (middle row) and parallel (bottom row) to the fibril’s longer axis.
Accurate height measurements of individual A–B fibrils were hampered by the pronounced self-agglomeration. Among thicker superstructures (>3 nm in diameter) there were thinner (∼1.8 nm) forms. Amyloid structures often differ in the periodic twisting pitch, which reflects various ways individual protofilaments are organized into higher order fibrils. The pitch may be easily estimated through a height AFM measurement along the fibrils’ longer axis.45 The periodic gaps between height maxima are typically 16/20 nm measured alongside H-fragment fibrils and 12/16 nm for A–B and B–A fibrils (for the sake of clarity only selected cross sections are shown in Figure 5). All in all, the AFM data indicates that all three peptides form amyloid fibrils with distinct morphological features.
Coaggregation and Cross-Seeding
The data presented so far shows unambiguously that the reorganization of the H-fragment’s primary structure into A–B and B–A single-chain analogues results in even more extreme amyloidogenicity, implying a high degree of versatility of the amyloid stretch segment encompassing the disulfide-bonded N-terminal fragment of insulin’s A-chain. From the distinct fingerprint features visible in the conformation-sensitive amide I band region of A–B and B–A aggregates, and the noticeable morphological differences, it is clear that the two peptides follow different self-assembly pathways. As the interactions between A-chain’s stretch segments appear to be primarily responsible for driving the self-assembly toward parallel β-sheet architectures, we became intrigued as to whether some degree of binding promiscuity between such segments built into A–B and B–A monomers could be observed. A cross-seeding experiment (e.g., seeding of dissolved A–B monomers with preformed B–A amyloid fibrils) is unfeasible due to the explosive kinetics of spontaneous aggregation of either peptide (at a reasonable monomer:seed ratio the aggregation through de novo pathway would outcompete or significantly overlap the seed-dependent pathway). Instead, we have chosen to study coaggregation of A–B and B–A induced by a rapid dilution of mixed DMSO solutions of both peptides with an excess of acidified solution of NaCl in water. The rationale of this approach is that any specific binding interactions between two coaggregating amyloidogenic substrates are expected to impact the kinetic trajectory of the aggregation and possibly result in the formation of new structural variants of aggregates.46−48 However, the data presented in Figure 6 suggests that no actual coaggregation takes place in mixed A–B and B–A solutions: the kinetic trajectories of fibrillization share the same steep appearance irrespective of the mixing stoichiometry.
Figure 6.

De novo amyloid formation in mixed solutions of A–B and B–A. (A) ThT-fluorescence-monitored kinetics of aggregation of mixed A–B and B–A at various mass ratios at 37 °C/300 rpm. Total peptide concentration was maintained at 0.5 mg/mL. Solution composition: 9 vol/vol % DMSO in 50 mM NaCl, 20 μM ThT, pH 1.9. (B) ATR FT-IR spectra of aggregated samples collected at the end of the kinetic experiment; pellets were initially washed with an excess of 50 mM NaCl, pH 1.9.
In unison with this observation, the infrared spectra of fibrils collected afterward lack any evidence that aggregation in mixed A–B/B–A solutions would lead to the emergence of new conformational variants of fibrils. For the series of spectra shown in Figure 6B, the contour of the amide I band changes gradually with the increasing A–B:B–A ratio, and the spectra obtained at 1:9 and 9:1 ratios are very similar to those corresponding to fibrils formed by neat peptides. Hence there is also no evidence of molecular imprinting or conformational memory effect. The fact that A–B and B–A do not coaggregate is consistent with a rather limited conformational space that is accessible to amyloidogenic self-assembly of either peptide. Hypothetically, an in-register parallel β-sheet structure could accommodate the peculiar bent shape of the main chain in the region restricted by the disulfide bridge (Cys6–Cys11 in A–B, Cys17–Cys22 in B–A) allowing for saturation of interstrand hydrogen bonds. Arguably, enforcing such an in-register arrangement at an A–B/B–A interface would leave the B-chain segment frayed and therefore thermodynamically frustrated. A shared amyloidogenic segment is an insufficient condition for effective cross-seeding between two peptides, although it is often the case that a peptide fragment of a larger amyloidogenic precursor can affect (accelerate, or disfavor) aggregation of the latter one (e.g., refs (49 and 50)). Various scenarios of cross-seeding between full-length Aβ1–40 and its Aβ11–40 fragment have been studied by Hao et al.51 Devlin et al. have shown that the lag phase of insulin aggregation is shortened in the presence of fibrils preformed from separated A- and B-chains of the protein.52 A shortened lag phase may imply that amyloid growth is catalyzed through secondary nucleation rather than the direct elongation pathway (a scenario far less demanding in terms of sequence compatibility between the dissolved protein and the seed). We have examined catalytic effects of preformed A–B and B–A amyloid seeds on insulin aggregation (Figure 7).
Figure 7.

(A) Influence of seeding with various amyloid types on aggregation kinetics of BI monitored by ThT fluorescence (normalized intensity) at 37 °C/300 rpm. Solution of 0.12 wt/vol % BI in 100 mM NaCl, 20 μM ThT, pH 1.9, was mixed with sonicated preformed amyloid seeds at 1:20 amyloid:native BI mass ratio. The black and red trajectories correspond to spontaneous and homologously seeded aggregation, respectively. (B) ATR FT-IR spectra of amyloid samples collected at the end of the kinetic experiment.
Only controlled homologous seeding with preformed insulin amyloid results in the complete disappearance of the lag phase. Fibrils of A–B and H-fragment shorten the lag phase (implying possible secondary nucleation of insulin amyloid on side surfaces of seeds), whereas aggregation trajectories in the presence of B–A seeds are very similar to those observed for the nonseeded process. Infrared spectra of daughter fibrils are very similar to those of the de novo formed fibrils. Despite a minor red shift of the amide I band detectable for samples grown in the presence of all nonhomologous seeds, there is no evidence of conformational imprinting (the corresponding far-UV CD spectra are shown in Figure S3). The fact that fibrils of A–B (and of H-fragment) accelerate insulin aggregation, presumably via a secondary nucleation pathway, to a degree that B–A amyloid is incapable of may not necessarily implicate any well-defined molecular recognition between folded insulin and such seeds. For example, solvent-exposed hydrophobic patches alongside A–B fibrils could act a platform for unfolding of insulin monomers and formation of amyloid nuclei. It is unclear at this stage how significant in this context is the N-terminal placement of the GIVEQ sequence in both A–B and H-fragment (as opposed to B–A).
The concept of amyloid stretch is an attractive paradigm explaining how strong selective binding interactions involving relatively small portions of primary structure may drive aggregation of a much larger parent protein which itself may be far less prone to aggregation. In the case of insulin, various regions of its covalent structure have been implicated as the key amyloidogenic sites. Due to the presence of three disulfide bonds, the protein has a complex topology which itself is likely to contribute to and modify the observed amyloidogenicity. However, many of the studies dissecting insulin’s amyloidogenic regions focused on short fragments lacking disulfide bonds (e.g., free A- and B-chains of insulin had either all cysteine residues oxidized to cysteic acid52 or in the fully reduced form53). Ivanova et al. screened amyloidogenic regions of human insulin by analyzing fibrillization of short mostly hexameric fragments, all of which lacked cysteine residues.54 The extensive studies carried out by the same group implicated the segments LYQLEN of A-chain (residues 13–18) and LVEALYL of B-chain (residues 11–17)49,55 as possibly involved in the formation of steric zippers within insulin amyloid fibrils. The earlier serendipitous discovery of the H-fragment31 which preserves two of insulin’s three disulfide bonds pointed to the N-terminal fragment of A-chain as a candidate for an amyloid stretch as long as the intrachain Cys6–Cys11 bridge remains intact.17 The results presented here depict the disulfide-bonded N-terminal fragment of insulin’s A-chain as a powerful and versatile amyloid stretch capable of forcing the aggregation-resistant B-chain’s N-terminal fragment to become a part of fibrillar β-architecture. Our findings resonate with the earlier work by Yang et al.56 pointing to a possible role of fluctuations in the N-terminal part of A-chain in insulin’s amyloidogenesis. The authors hypothesized that a transient detachment of this segment could expose “a hydrophobic surface formed by native-like packing of the remaining α-helices”.56 In light of our work, this segment itself may constitute a hydrophobic patch responsible for self-association of insulin at low pH. The single-chain rearrangements employed in this study appear to only strengthen the amyloidogenic tendencies of H-fragment: parallel β-sheet structure is preserved as the main conformational component of fibrils. It is interesting to note that the very powerful tendency to aggregate both A–B and B–A peptides eludes the TANGO and Waltz algorithms (Figure 1B). On the other hand, in their extensive screening study of various amyloidogenic regions in insulin, Surin et al.57 noted an increased amyloidogenic potential within the A2–A12 region according to the data obtained by application of PASTA 2.058 and ZipperDB algorithms.29,59
The distinct infrared features within the amide I band, various morphologies observed in AFM images, and no coaggregation of A–B and B–A strongly suggest that the self-assembly pathways of both peptides do not overlap. In light of these results, it seems likely that intermolecular interactions between N-terminal parts of A-chain contribute significantly to the thermodynamics of insulin fibrillization. This is significant in the context of desirable modulation of this process. The pronounced versatility of this newly identified amyloid stretch region could be employed as a tool for orderly organization of various chemical groups. More research is needed to illuminate these prospects.
Conclusions
The highly amyloidogenic H-fragment of insulin consists of two disulfide-linked peptide chains varying significantly in amyloidogenic potency. The structural rearrangement of the double-chain peptide into two analogues (A–B, B–A) in which the amyloidogenic A-chain and nonamyloidogenic B-chain parts were merged into continuous single chains resulted in extreme amyloidogenicity. De novo reassociation of A–B and B–A is very fast with no lag phase detectable even under conditions decelerating or preventing aggregation of H-fragment in parallel control experiments. Through the application of infrared spectroscopy and AFM microscopy, structural differences between fibrils of A–B, B–A, and H-fragment were revealed. It appears that the amyloidogenic self-assembly pathways of both peptides are mutually inaccessible: there is no evidence of coaggregation or specific cross-seeding with insulin. These results strongly suggest that the N-terminal fragment of insulin’s A-chain encompassing Cys6–Cys11 disulfide bond has all the characteristics of the amyloid stretch region which may play a significant, if not decisive, role in insulin aggregation. The observed versatility of this segment may be exploited in future in the design of self-assembling nanostructures. Importantly, identification of strongly amyloidogenic regions may facilitate future design of novel insulin analogues resistant to spontaneous aggregation.
Acknowledgments
This work was supported by the National Science Centre of Poland, Grant No. 2015/17/B/NZ1/00832. The study was carried out at the Biological and Chemical Research Centre, University of Warsaw, established within the project cofinanced by the European Union from the European Regional Development Fund under the Operational Programme Innovative Economy, 2007–2013.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.langmuir.0c01747.
Predictions of amyloidogenic tendencies of A–B and B–A based on FoldAmyloid; aggregation of H, A–B, and B–A at a close-to-neutral pH, kinetic data and IR spectra of fibrils; CD spectra of seeded amyloid fibrils (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fändrich M.; Fletcher M. A.; Dobson C. M. Amyloid Fibrils from Muscle Myoglobin. Nature 2001, 410, 165–166. 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- Fändrich M.; Dobson C. M. The Behaviour of Polyamino Acids Reveals an Inverse Side Chain Effect in Amyloid Structure Formation. EMBO J. 2002, 21, 5682–5690. 10.1093/emboj/cdf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Hardy J. A.; Higgins G. A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–186. 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Ittner L. M.; Götz J. Amyloid-β and Tau - a Toxic pas de deux in Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 67–72. 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Fowler D. M.; Koulov A. V.; Alory-Jost C.; Marks M. S.; Balch W. E.; Kelly J. W. Functional Amyloid Formation within Mammalian Tissue. PLoS Biol. 2005, 4, e6. 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein E. A.; Chapman M. R. Polymerizing the Fibre between Bacteria and Host Cells: The Biogenesis of Functional Amyloid Fibres. Cell. Microbiol. 2008, 10, 1413–1420. 10.1111/j.1462-5822.2008.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Avalos R.; King C. Y.; Wall J.; Simon M.; Caspar D. L. Strain-Specific Morphologies of Yeast Prion Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 10165–10170. 10.1073/pnas.0504599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo R.; Ranson N. A.; Radford S. E. Amyloid Structures: Much More than just a Cross-β Fold. Curr. Opin. Struct. Biol. 2020, 60, 7–16. 10.1016/j.sbi.2019.09.001. [DOI] [PubMed] [Google Scholar]
- Trainor K.; Broom A.; Meiering E. M. Exploring the Relationships between Protein Sequence, Structure and Solubility. Curr. Opin. Struct. Biol. 2017, 42, 136–146. 10.1016/j.sbi.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Tartaglia G. G.; Pawar A. P.; Campioni S.; Dobson C. M.; Chiti F.; Vendruscolo M. Prediction of Aggregation-Prone Regions in Structured Proteins. J. Mol. Biol. 2008, 380, 425–436. 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Betsholtz C.; Christmansson L.; Engström U.; Rorsman F.; Svensson V.; Johnson K. H.; Westermark P. Sequence Divergence in a Specific Region of Islet Amyloid Polypeptide (IAPP) Explains Differences in Islet Amyloid Formation Between Species. FEBS Lett. 1989, 251, 261–264. 10.1016/0014-5793(89)81467-X. [DOI] [PubMed] [Google Scholar]
- Lopez de La Paz M.; Serrano L. Sequence Determinants of Amyloid Fibril Formation. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 87–92. 10.1073/pnas.2634884100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de La Paz M.; Goldie K.; Zurdo J.; Lacroix E.; Dobson C. M.; Hoenger A.; Serrano L. De novo Designed Peptide-Based Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 16052–16057. 10.1073/pnas.252340199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou H.; Kanno T.; Hoshino M.; Hagihara Y.; Tanaka H.; Kawai T.; Hasegawa K.; Naiki H.; Goto Y. The Role of Disulfide Bond in the Amyloidogenic State of β2–Microglobulin Studied by Heteronuclear NMR. Protein Sci. 2002, 11, 2218–2229. 10.1110/ps.0213202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei L.; Perrett S. Disulfide Bond Formation Significantly Accelerates the Assembly of Ure2p Fibrils because of the Proximity of a Potential Amyloid Stretch. J. Biol. Chem. 2009, 284, 11134–11141. 10.1074/jbc.M809673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dec R.; Koliński M.; Dzwolak W. Beyond Amino Acid Sequence: Disulfide Bonds and the Origins of the Extreme Amyloidogenic Properties of Insulin’s H–Fragment. FEBS J. 2019, 286, 3194–3205. 10.1111/febs.14849. [DOI] [PubMed] [Google Scholar]
- Minor D. L.; Kim P. S. Context is a Major Determinant of β-Sheet Propensity. Nature 1994, 371, 264–267. 10.1038/371264a0. [DOI] [PubMed] [Google Scholar]
- Minor D. L.; Kim P. S. Context-Dependent Secondary Structure Formation of a Designed Protein Sequence. Nature 1996, 380, 730–734. 10.1038/380730a0. [DOI] [PubMed] [Google Scholar]
- Castillo V.; Graña-Montes R.; Sabate R.; Ventura S. Prediction of the Aggregation Propensity of Proteins from the Primary Sequence: Aggregation Properties of Proteomes. Biotechnol. J. 2011, 6, 674–685. 10.1002/biot.201000331. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S.; Debulpaep M.; Kuemmerer N.; Lopez de La Paz M.; Martins I. C.; Reumers J.; Morris K. L.; Copland A.; Serpell L.; Serrano L.; Schymkowitz J. W.; Rousseau F. Exploring the Sequence Determinants of Amyloid Structure Using Position-Specific Scoring Matrices. Nat. Methods 2010, 7, 237–242. 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- Bemporad F.; Calloni G.; Campioni S.; Plakoutsi G.; Taddei N.; Chiti F. Sequence and Structural Determinants of Amyloid Fibril Formation. Acc. Chem. Res. 2006, 39, 620–627. 10.1021/ar050067x. [DOI] [PubMed] [Google Scholar]
- Tsolis A. C.; Papandreou N. C.; Iconomidou V. A.; Hamodrakas S. J. A Consensus Method for the Prediction of ‘Aggregation-Prone’ Peptides in Globular Proteins. PLoS One 2013, 8, e54175. 10.1371/journal.pone.0054175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano R.; Jamroz M.; Szczasiuk A.; Pujols J.; Kmiecik S.; Ventura S. AGGRESCAN3D (A3D): Server for Prediction of Aggregation Properties of Protein Structures. Nucleic Acids Res. 2015, 43 (W1), W306–W313. 10.1093/nar/gkv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul K. R.; Molliex A.; Cascarina S.; Boncella A. E.; Taylor J. P.; Ross E. D. Effects of Mutations on the Aggregation Propensity of the Human Prion-Like Protein hnRNPA2B1. Mol. Cell. Biol. 2017, 37, e00652-16. 10.1128/MCB.00652-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Nelson A. C.; Paul K. R.; Petri M.; Flores N.; Rogge R. A.; Cascarina S. M.; Ross E. D. Increasing Prion Propensity by Hydrophobic Insertion. PLoS One 2014, 9, e89286. 10.1371/journal.pone.0089286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H.; Buckwalter B. L.; Shieh H. M.; Hecht M. H. Periodicity of Polar and Nonpolar Amino Acids Is the Major Determinant of Secondary Structure in Self-Assembling Oligomeric Peptides. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 6349–6353. 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Chapman M. R. Sequence Determinants of Bacterial Amyloid Formation. J. Mol. Biol. 2008, 380, 570–580. 10.1016/j.jmb.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L.; Teng P. K.; Riek R.; Eisenberg D. Identifying the Amylome, Proteins Capable of Forming Amyloid-Like Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3487–3492. 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteras-Chopo A.; Serrano L.; De La Paz M. L. The Amyloid Stretch Hypothesis: Recruiting Proteins toward the Dark Side. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 16672–16677. 10.1073/pnas.0505905102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piejko M.; Dec R.; Babenko V.; Hoang A.; Szewczyk M.; Mak P.; Dzwolak W. Highly Amyloidogenic Two-Chain Peptide Fragments Are Released upon Partial Digestion of Insulin with Pepsin. J. Biol. Chem. 2015, 290, 5947–5958. 10.1074/jbc.M114.608844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dec R.; Koliński M.; Kouza M.; Dzwolak W. Rapid Self-Association of Highly Amyloidogenic H-Fragments of Insulin: Experiment and Molecular Dynamics Simulations. Int. J. Biol. Macromol. 2020, 150, 894–903. 10.1016/j.ijbiomac.2020.02.153. [DOI] [PubMed] [Google Scholar]
- Surmacz-Chwedoruk W.; Babenko V.; Dzwolak W. Master and Slave Relationship between Two Types of Self-Propagating Insulin Amyloid Fibrils. J. Phys. Chem. B 2014, 118, 13582–13589. 10.1021/jp510980b. [DOI] [PubMed] [Google Scholar]
- Fernandez-Escamilla A. M.; Rousseau F.; Schymkowitz J.; Serrano L. Prediction of Sequence-Dependent and Mutational Effects on the Aggregation of Peptides and Proteins. Nat. Biotechnol. 2004, 22, 1302–1306. 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Linding R.; Schymkowitz J.; Rousseau F.; Diella F.; Serrano L. A Comparative Study of the Relationship between Protein Structure and β-Aggregation in Globular and Intrinsically Disordered Proteins. J. Mol. Biol. 2004, 342, 345–353. 10.1016/j.jmb.2004.06.088. [DOI] [PubMed] [Google Scholar]
- Rousseau F.; Schymkowitz J.; Serrano L. Protein Aggregation and Amyloidosis: Confusion of the Kinds?. Curr. Opin. Struct. Biol. 2006, 16, 118–126. 10.1016/j.sbi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Garbuzynskiy S. O.; Lobanov M. Y.; Galzitskaya O. V. FoldAmyloid: A Method of Prediction of Amyloidogenic Regions from Protein Sequence. Bioinformatics 2010, 26, 326–332. 10.1093/bioinformatics/btp691. [DOI] [PubMed] [Google Scholar]
- Zandomeneghi G.; Krebs M. R.; McCammon M. G.; Fändrich M. FTIR Reveals Structural Differences between Native β–Sheet Proteins and Amyloid Fibrils. Protein Sci. 2004, 13, 3314–3321. 10.1110/ps.041024904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard M.; Zurdo J.; Nettleton E. J.; Dobson C. M.; Robinson C. V. Formation of Insulin Amyloid Fibrils Followed by FTIR Simultaneously with CD and Electron Microscopy. Protein Sci. 2000, 9, 1960–1967. 10.1110/ps.9.10.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.; Mantsch H. H. Beware of Proteins in DMSO. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1991, 1078, 231–235. 10.1016/0167-4838(91)90563-F. [DOI] [PubMed] [Google Scholar]
- Loksztejn A.; Dzwolak W. Noncooperative Dimethyl Sulfoxide-Induced Dissection of Insulin Fibrils: Toward Soluble Building Blocks of Amyloid. Biochemistry 2009, 48, 4846–4851. 10.1021/bi900394b. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Babenko V.; Dzwolak W.; Keiderling T. A. Dimethyl Sulfoxide Induced Destabilization and Disassembly of Various Structural Variants of Insulin Fibrils Monitored by Vibrational Circular Dichroism. Biochemistry 2015, 54, 7193–7202. 10.1021/acs.biochem.5b00809. [DOI] [PubMed] [Google Scholar]
- Shanmugam G.; Polavarapu P. L. Structure of Aβ (25–35) Peptide in Different Environments. Biophys. J. 2004, 87, 622–630. 10.1529/biophysj.104.040907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Kernya L.; Lee Y. H.; Goto Y.; Réfrégiers M.; Kardos J. Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E3095–E3103. 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpatti L. R.; Vendruscolo M.; Dobson C. M.; Knowles T. P. A Clear View of Polymorphism, Twist, and Chirality in Amyloid Fibril Formation. ACS Nano 2013, 7, 10443–10448. 10.1021/nn406121w. [DOI] [PubMed] [Google Scholar]
- Sarell C. J.; Woods L. A.; Su Y.; Debelouchina G. T.; Ashcroft A. E.; Griffin R. G.; Stockley P. G.; Radford S. E. Expanding the Repertoire of Amyloid Polymorphs by Co-Polymerization of Related Protein Precursors. J. Biol. Chem. 2013, 288, 7327–7337. 10.1074/jbc.M112.447524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynes J. K.; Day L.; Crepin P.; Horrocks M. H.; Carver J. A. Coaggregation of κ–Casein and β–Lactoglobulin Produces Morphologically Distinct Amyloid Fibrils. Small 2017, 13, 1603591. 10.1002/smll.201603591. [DOI] [PubMed] [Google Scholar]
- Hu R.; Zhang M.; Patel K.; Wang Q.; Chang Y.; Gong X.; Zhang G.; Zheng J. Cross-Sequence Interactions between Human and Rat Islet Amyloid Polypeptides. Langmuir 2014, 30, 5193–5201. 10.1021/la500632d. [DOI] [PubMed] [Google Scholar]
- Ivanova M. I.; Sievers S. A.; Sawaya M. R.; Wall J. S.; Eisenberg D. Molecular Basis for Insulin Fibril Assembly. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18990–18995. 10.1073/pnas.0910080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B.; Zhang Y.; Zhang M.; Liu Y.; Zhang D.; Gong X.; Feng Z.; Tang J.; Chang Y.; Zheng J. Fundamentals of Cross-Seeding of Amyloid Proteins: An Introduction. J. Mater. Chem. B 2019, 7, 7267–7282. 10.1039/C9TB01871A. [DOI] [PubMed] [Google Scholar]
- Hao X.; Zheng J.; Sun Y.; Dong X. Seeding and Cross-Seeding Aggregations of Aβ40 and Its N-Terminal-Truncated Peptide Aβ11–40. Langmuir 2019, 35, 2821–2831. 10.1021/acs.langmuir.8b03599. [DOI] [PubMed] [Google Scholar]
- Devlin G. L.; Knowles T. P.; Squires A.; McCammon M. G.; Gras S. L.; Nilsson M. R.; Robinson C. V.; Dobson C. M.; MacPhee C. E. The Component Polypeptide Chains of Bovine Insulin Nucleate or Inhibit Aggregation of the Parent Protein in a Conformation-Dependent Manner. J. Mol. Biol. 2006, 360, 497–509. 10.1016/j.jmb.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Babenko V.; Piejko M.; Wójcik S.; Mak P.; Dzwolak W. Vortex-Induced Amyloid Superstructures of Insulin and Its Component A and B Chains. Langmuir 2013, 29, 5271–5278. 10.1021/la400612w. [DOI] [PubMed] [Google Scholar]
- Ivanova M. I.; Thompson M. J.; Eisenberg D. A Systematic Screen of β2-Microglobulin and Insulin for Amyloid-Like Segments. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4079–4082. 10.1073/pnas.0511298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. J. W.; McFarlane H. T.; Madsen A. Ø.; Riekel C.; Eisenberg D. Atomic Structures of Amyloid Cross-β Spines Reveal Varied Steric Zippers. Nature 2007, 447, 453–457. 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Petkova A.; Huang K.; Xu B.; Hua Q. X.; Ye I. J.; Chu Y. C.; Hu S. Q.; Phillips N. B.; Whittaker J.; Ismail-Beigi F.; Mackin R. B.; Katsoyannis P. G.; Tycko R.; Weiss M. A. An Achilles’ Heel in an Amyloidogenic Protein and Its Repair INSULIN FIBRILLATION AND THERAPEUTIC DESIGN. J. Biol. Chem. 2010, 285, 10806–10821. 10.1074/jbc.M109.067850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surin A. K.; Grishin S. Y.; Galzitskaya O. V. Determination of Amyloid Core Regions of Insulin Analogues Fibrils. Prion 2020, 14, 149–162. 10.1080/19336896.2020.1776062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovato A.; Seno F.; Tosatto S. C. The PASTA Server for Protein Aggregation Prediction. Protein Eng., Des. Sel. 2007, 20, 521–523. 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- Thompson M. J.; Sievers S. A.; Karanicolas J.; Ivanova M. I.; Baker D.; Eisenberg D. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4074–4078. 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.