

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to a global pandemic of coronavirus disease 2019 (COVID-19). The spike protein expressed on the surface of this virus is highly glycosylated and plays an essential role during the process of infection. We conducted a comprehensive mass spectrometric analysis of the N-glycosylation profiles of the SARS-CoV-2 spike proteins using signature ions-triggered electron-transfer/higher-energy collision dissociation (EThcD) mass spectrometry. The patterns of N-glycosylation within the recombinant ectodomain and S1 subunit of the SARS-CoV-2 spike protein were characterized using this approach. Significant variations were observed in the distribution of glycan types as well as the specific individual glycans on the modification sites of the ectodomain and subunit proteins. The relative abundance of sialylated glycans in the S1 subunit compared to the full-length protein could indicate differences in the global structure and function of these two species. In addition, we compared N-glycan profiles of the recombinant spike proteins produced from different expression systems, including human embryonic kidney (HEK 293) cells and Spodoptera frugiperda (SF9) insect cells. These results provide useful information for the study of the interactions of SARS-CoV-2 viral proteins and for the development of effective vaccines and therapeutics.
Introduction
The global pandemic of coronavirus disease 2019 (COVID-19) caused by the severe acute respiratory syndrome-2 coronavirus (SARS-CoV-2) emerged in late 2019 and has led to considerable economic and social disruption throughout the globe.1−3 The disease has also led to considerable morbidity and mortality; according to the data compiled by the World Health Organization (September 7th Weekly Epidemiological Update) as of early September 2020, there are approximately 27.4 million diagnosed cases of infection and 895 000 estimated deaths from the disease.4 Analyzing molecular mechanisms of viral cellular entry and infectivity will help guide research and development of therapeutic countermeasures and treatment and treatment.
The SARS-CoV-2 viral genome encodes four structural proteins present in the mature virion—spike protein (S), envelope protein (E), membrane protein (M), and nucleoprotein (N).5 The spike protein of SARS-CoV-2 is a class I viral fusion protein, employed by the virus for host cell entry, binding through interaction with angiotensin-converting enzyme 2 (ACE2) protein present on the cell surface.6,7 The spike protein is similar to the structures of the spike protein in other coronaviruses, such as those that cause SARS (SARS-CoV-1) and Middle East respiratory syndrome (MERS);8−11 recent cryo-electron microscopy (cryoEM) studies12 indicate that the S protein is a homotrimer, each monomer consisting of two covalently linked functional subunits, an S1 subunit, that contains the receptor-binding domain (RBD), and an S2 subunit, which is responsible for membrane fusion, with a furin-like cleavage site at the boundary between the two domains. The S1 and S2 subunits combined have a monomeric, unmodified molecular weight of ca. 142 kDa. Because the spike protein is one of the primary targets for the development of vaccines and therapeutics against COVID-19, a more thorough understanding of its structure and function will be vital to mitigating the impact of SARS-CoV-2 on global health.
Viral proteins used in cell entry are often extensively glycosylated (often described as a “glycan shield”) for several reasons: to assist in protein folding, provide stability, and most importantly, shield the virus from immune recognition by its host, as has been reported for other coronavirus species.10,11 The SARS-CoV-2 S protein is a prime example, having 22 potential sites of glycosylation per protein monomer, as predicted from the primary sequence.13 Development of vaccines and therapeutics for SARS-CoV-2 requires an understanding of the overall composition of the spike protein “glycan shield”, both in naturally occurring isolates and vaccine formulations containing this protein. Observed variances in glycan site occupation and speciation might affect antigenicity as well as vaccine safety and efficacy.14,15
In the past several decades, mass spectrometry has become one of the primary tools for the analysis of protein glycosylation,16−18 both on a proteome-wide scale19−21 and in targeted analyses of single protein therapeutics, most notably monoclonal antibodies.22 Mass spectrometry has also been employed extensively for viral protein characterization. Seasonal and pandemic influenza, with its substantial morbidity and mortality, has been a primary focus of many of these efforts, for antigen quantification,23−28 vaccine potency determination,29,30 and analysis of protein glycosylation.31−34 As the COVID-19 pandemic has progressed, several reports on the use of mass spectrometry for the analysis of SARS-CoV-2 have been made. These investigations have focused on both targeted quantitative analysis for potential diagnostic development35 and structural/post-translational modification analyses of the virus protein complement.13,36−40
Electron-transfer dissociation (ETD), developed by Hunt and co-workers,41−44 and subsequently introduced hybrid ETD and higher-energy collisional dissociation (HCD) fragmentation regimes such as electron-transfer/higher-energy collisional dissociation (EThcD), developed by Heck et al.,45 have been extensively employed for mass spectrometric post-translational modification analysis of phosphorylated46 and glycosylated47,48 proteins and peptides. A key advantage of ETD-based approaches is that they provide more comprehensive fragment ion coverage than conventional collision-induced dissociation (CID) for post-translational modification of larger peptides due to the less biased nature of the fragmentation mechanism. This allows the sites of peptide modification to be unambiguously determined in many cases.49
The glycan shield on the SARS-CoV-2 S protein has been recently characterized using mass spectrometry by several laboratories.13,37,38,50,51 Their results show significant differences in the glycan occupancy and abundance of some sequons. For instance, all 22 potential glycosylation sites were determined to be occupied on a stabilized extracellular domain (ectodomain) S protein expressed in human HEK cells in one report by Crispin and co-workers,13 whereas in another report, only 17 sites were found occupied based on the analysis of individual HEK cell expressed S1 and S2 subunits.50 A third report analyzed an insect cell expressed full-length SARS-CoV-2 protein, with unambiguous determination of 21 of 22 glycosylation sites on this construct. A most recent investigation38 analyzed a full-length ectodomain of SARS-CoV-2 protein of similar composition to the Crispin report. They also observed occupation of all 22 potential sites, with some significant differences. Interestingly, O-glycosylation and/or N-linked sulfated glycans on the S protein and subunits were also observed by several groups,13,39,50,52 albeit at relatively low levels of site occupation. These variations could be attributed to differences in protein structure, expression systems, sample preparation, instrumentation, acquisition methods, or data analysis. The use of manual inspection of the mass spectra for the confirmation of identities could be subjective as well.
In this report, we describe a comprehensive, high-fidelity mass spectrometric approach to the glycosylation analysis of SARS-CoV-2 S protein, applied to multiple recombinant spike protein sources and constructs (S1 domain and ectodomain) with identical analytical methods. In contrast to other reports on SARS-CoV-2 S protein glycoproteomic analysis that used conventional CID, HCD,13,37,50 or stepped collision energy HCD (sceHCD),38 we employed an approach based on glycan reporter ion-triggered EThcD, which allowed the sites of glycosylation to be unambiguously determined with a greater proportion of fragment ions observed, which increased the degree of confidence in the observed search results.
Experimental Section
Materials
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) except where otherwise indicated. Endoproteases including trypsin, chymotrypsin, Asp-N, and Lys-C were purchased from Promega (Madison, WI). Recombinant SARS-CoV-2 prefusion stabilized spike protein was provided by Dr. Jason McLellan at University of Texas at Austin. The protein was expressed in human FreeStyle293F cells.12 Recombinant SARS-CoV-2 spike protein expressed in baculovirus insect cells (S1 + S2 ECD, Cat. no. 40589-V08B1), SARS-CoV-2 spike protein S1 subunit (Cat. no. 40591-V08H), and MERS-CoV spike protein S1 subunit, both expressed in human-derived HEK cells (Cat. no. 40069-V08H) were purchased from Sino Biological (Wayne, PA). SARS-CoV-1 spike protein (Cat. no. NR-686) prepared in insect cells was obtained from BEI Resources (Manassas, VA).
One-Dimensional (1D) Gel Electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed on a NuPAGE Novex Bis–Tris gel following manufacturer’s instructions (Invitrogen). Protein solution or pellets from acetone precipitation were mixed with 4× sample buffer and deionized water (1:3 v/v) and heated at 80 °C for 10 min. The proteins were loaded on a 4–12% gradient gel, and the gel was run in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer at 200 V for 45 min. The gel was stained with GelCode Blue Safe Protein Stain (Cat. no. 243596, Thermo Scientific, Waltham, MA).
Size-Exclusion High-Performance Liquid Chromatography (HPLC) Analysis
Size-exclusion HPLC of recombinant spike protein samples was performed using an Agilent Technologies (Santa Clara, CA) 1200 HPLC System equipped with a quaternary pump, diode array detector, and fraction collector, coupled to an Agilent BioSEC-3 size-exclusion column (4.6 mm ID × 150 mm OD). The column flow rate was 200 μL/min with a sample injection size of 15 μL. The sample was eluted isocratically over 45 min using a commercially prepared Dulbecco’s phosphate-buffered saline mobile phase (Thermo/Life Technologies). Detection was performed by monitoring the UV wavelengths of 216 and 280 nm using a diode array detector, and 1 min fractions were collected for subsequent mass spectrometric identification. The column was calibrated using commercially available size-exclusion chromatography (SEC) standards (Acquity BEH 450 Protein Standards) obtained from Waters Corporation (Milford, MA) that spanned the molecular weight range of 112–700 kDa. A plot of log MW vs retention time of the standards was used to determine a calibration curve fit to a quadratic model, which was employed to calculate the estimated relative molecular weight of the spike protein samples. The UV traces were baseline-corrected.
Digestion of Spike Proteins
The aliquots of full-length spike protein or S1 subunit (0.4–0.7 μg) were denatured and reduced at 90 °C for 20 min in a solution containing 50 mM ammonium bicarbonate (pH 7.8), 0.015% RapiGest SF surfactant (Waters Corporation) and 1.5 mM dithiothreitol (DTT). Samples were alkylated using iodoacetamide for 30 min in the dark at room temperature with gentle mixing. Nine identical aliquots of each protein were prepared for three digestion methods, and each digestion was performed with three replicates. The digestions with trypsin were conducted at 37 °C overnight at an enzyme-to-protein ratio of 1/20 (w/w). For the samples digested sequentially by two enzymes, the first digestion was performed at 52 °C for 60 min with Asp-N or Lys-C and the second digestion was conducted at 37 °C overnight using chymotrypsin at an enzyme/substrate ratio of 1/5 (w/w). The proteolytic reactions were quenched, and the RapiGest was precipitated by adding 5% trifluoroacetic acid to decrease the pH to below 3. The mixture was then incubated at 37 °C for 30 min. The solutions were centrifuged at 4000 rpm for 10 min, and the supernatants (12 μL) were transferred into new sample vials.
Mass Spectrometry Analysis
Nanoflow liquid chromatography coupled to electrospray ionization tandem mass spectrometry (LC-MS/MS) analysis was performed on an Orbitrap Eclipse Tribrid mass spectrometer connected to an UltiMate 3000 RSLCnano chromatography system (Thermo Scientific). The protein digest was separated on an integrated separation column/nanospray device (Thermo Scientific EASY-Spray PepMap RSLC C18, 75 μm ID × 15 cm length, 3 μm 100 Å particles) coupled to an EASY-Spray ion source. The mobile phase was 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in 80% acetonitrile/20% water (mobile phase B) using the following gradient: 4% B (0–8 min); 4–10% B (8–10 min); 10–35% B (10–43 min); 35–60% B (43–45 min); 60–95% B (45–46 min); 95% B (46–53 min); 95–4% B (53–53 min); 4% B (53–63 min). The flow rate was 300 nL/min, and 9 μL of samples was injected. The spray voltage was set to 1.8 kV, and the temperature of the integrated column/nanospray device was set at 55 °C. The temperature of the ion transfer tube was set at 275 °C.
Mass spectrometric data acquisition was performed using a signature ion-triggered EThcD method.53 MS precursor scans were acquired by the orbitrap at a resolution of 120 000 (measured at m/z 200), from m/z 375 to 2000 with the automatic gain control (AGC) target setting as “standard” and the maximum injection time as “auto”. An initial data-dependent MS/MS scan was acquired using HCD at a resolution of 30 000, mass range of m/z 120–2000, and a normalized collision energy (NCE) of 28%. Signature ions representing glycan oxonium fragments were used to trigger the ETD fragmentation. If one of three common glycan signature ions (m/z 204.0867 (HexNAc), 138.0545 (HexNAc fragment), or 366.1396 (HexNAcHex)) was detected in the HCD spectrum within 15 ppm mass accuracy, additional precursor isolation and EThcD acquisition were performed at a resolution of 50 000 (measured at m/z 200), scan range of m/z 150–2000 with normalized AGC target = 500% and maximum injection time = 150 ms. The supplemental activation NCE was set to 35%.
Data Analysis
MS/MS data were processed using PMi-Byonic (version 3.7) and PMi-Byologic (version 3.7, Protein Metrics, Inc.). Data were searched using the Protein Metrics 182 human N-glycan library (for proteins expressed in HEK cells) or 38 insect N-glycan library (for proteins expressed in insect cells). The search parameters for enzyme digestion were set to fully specific, three allowed missed cleavage sites, and 6 and 20 ppm mass tolerance for precursors and fragment ions, respectively. Carbamidomethylation of cysteine was set as a fixed modification with variable modifications set to include deamidation at Asn and Gln and oxidation of Met. Tandem mass spectra of identified glycopeptides with a Byonic score54 of higher than 300 were considered valid identifications and were examined by manual inspection for further validation based on the presence of predicted peptide fragments and diagnostic N-glycan fragment ions. The precursor ion peak areas of identified glycopeptides and unoccupied peptides were obtained through the Byologic algorithm, with the relative abundance of each glycan at each site calculated as the area ratio of the peptides bearing a particular glycan over the total peptides of the same peptide sequence. The glycan abundance was represented as the mean of either two or three replicates, depending upon the number of observations, along with standard deviation of the mean.
Results and Discussion
To compare the glycan profiles of SARS-CoV-2 S protein and its subunits prepared from different expression systems, we examined three SARS-CoV-2 recombinant S protein constructs, including SARS-CoV-2 prefusion stabilized spike protein (S2P), unmutated spike protein (SF), and the S1 subunit (S1), using bottom-up LC-MS/MS techniques on a Thermo Orbitrap Eclipse mass spectrometer. In addition, SARS-CoV-1 spike protein (SN) and MERS-CoV spike protein S1 subunit (MS1) were analyzed for comparison. Among these five proteins, S2P, S1, and MS1 were expressed in human-derived HEK cells, and SF and SN were prepared in insect cells using a baculovirus-SF9 expression system. To generate high-quality MS/MS spectra for potential glycopeptides, HCD-triggered EThcD was used because it focused more data acquisition time on analyzing glycan bearing peptide ions. The EThcD fragmentation regime favored the formation of both glycan and peptide backbone fragment ions. A recent report by Bertozzi and co-workers55 compared the analytical figures of merit of ETD, EThCD, HCD, and stepped collision energy HCD (sceHCD) for analysis of N- and O-linked glycopeptides. Our results corroborate their findings that ETD-based methods provide better spectral quality—superior peptide and glycan sequence coverage to HCD methods alone. It is important to note that they observed sceHCD methods provided a greater number of peptide spectral matches (PSMs) with similar peptide and glycan sequence coverages than EThCD from a standard glycopeptide mixture at a compromise in spectral quality.
The heterogeneity of the five samples was demonstrated by SDS-PAGE gel (Figure 1A); a single band of S2P, S1, and MS1 revealed the high purity of these proteins, whereas an additional band observed in the SF sample indicated a spike protein fragment or contaminating protein was present in the sample. A band was barely visible in the lane loaded with SN, suggesting that protein had degraded; this reagent was manufactured in 2005 as a vaccine candidate for SARS-CoV-1 and held in long-term storage. The size-exclusion chromatogram obtained under native conditions, shown in Figure 1B, illustrated the difference in structure between the stabilized prefusion protein (S2P) and wild-type version of the S protein (SF), where the former was observed to be predominantly a trimer of apparent molecular mass 700 kDa, peak eluting at 10.5 min, with a relatively lower amount of monomer eluting at 13 min. The SF sample was observed to be present as a mixture of trimer (700 kDa, 10.4 min) and monomer (230 kDa, 13.5 min) with a degradation product (100 kDa) at 15 min that presumably corresponds to the species observed in SDS-PAGE at this approximate mass. The identities of the chromatographic peaks observed in the SEC peaks were confirmed to be SARS-CoV-2 S protein by LC-MS/MS analysis of collected fractions.
Figure 1.

(A) SDS-PAGE gel of the unfractionated S proteins (full-length and S1 subunit). Lane 1: molecular markers; lanes 2–6: S2P, S1, MS1, SF, and SN, respectively. (B) Size-exclusion chromatogram (UV detection at 216 nm) of the recombinant SARS-CoV-2 S proteins expressed in human (S2P, blue trace) and insect cells (SF, red trace). (C) Representative MS/MS spectra of the glycopeptides with a complex glycan, HexNAc4Hex4Fuc, at amino acid position 1074. The two peptides were cleaved from S2P by trypsin (top) and Asp-N/chymotrypsin (bottom), respectively.
The unambiguous characterization of glycoproteins by mass spectrometric analysis of proteolytically cleaved glycopeptides presents several significant analytical challenges. Compared to the typical molecular mass of unmodified peptides (1–5 kDa), the size of the glycan moiety (typically >1 kDa) can lead to a higher prevalence of false-positive spectrum matches even at high mass measurement accuracies. In addition, a lower population of fragment ions containing the glycan moiety is often observed in MS/MS spectra of glycopeptides.16 To address these issues, the samples were digested using three protease/protease combinations (trypsin, Lys-C + chymotrypsin, and Asp-N + chymotrypsin) with each digestion condition conducted in triplicate. The identification of the same glycan at a sequon by two or more peptides derived from the cleavage by a different enzyme or enzyme combination can cross-validate each other. For example, a complex glycan, HexNAc4Hex4Fuc, modified at N1074 of S2P, was determined with high confidence by two glycopeptides of different lengths, one was S2P(1046-1086) derived from trypsin proteolysis and another was S2P(1068-1087) generated from the digestion by Asp-N/chymotrypsin (Figure 1C).
To obtain accurate quantitative results, we processed the data of specific proteolytic digestion experiments separately. This resulted in a majority of the peptides bearing a specific sequon to have identical amino acid lengths and sequences because they were produced from the digestion by identical enzyme(s). This is particularly important because the LC-MS/MS peak intensity or integrated peak area derived from a given peptide is dependent on its amino acid composition and sequence, assuming that it is not affected by the associated glycans. Summing the areas of a group of peptides with the same glycan site but different amino acid sequences could lead to an inaccurate calculation of relative abundance of individual glycans on a specific modification site. In addition, a 6 ppm precursor ion tolerance was employed in database searching, and the criteria for positive identification of a glycopeptide were a Byonic score of ≥300 and validation of MS/MS data by manual inspection. Moreover, any glycopeptides that were detected in only one of the three replicates were not included for the quantification of glycan distribution, even if their identities were unambiguously determined by the criteria described previously.
Of the 22 possible N-linked glycosylation sites on the S2P protein, 21 sites were detected as glycosylated by this comprehensive approach (Data S-1). No peptides containing the sequon N17 were detected from any of data meeting the selection criteria described above. In addition to the categories of high mannose (HexNAc2Hex>4X), hybrid containing three HexNAc, and complex glycans with more than three HexNAc, we also added a truncated species type including paucimannose and other small glycans consisting of only one HexNAc or two HexNAc but less than five Hex groups in their compositions, due to the presence of large amounts of such glycans in some samples. The relative abundances of different types of N-glycosylation on the prefusion S2P proteins are listed in Table 1. The results generated from various experimental conditions showed high consistency within two or three datasets, demonstrating the reproducibility of this approach. Although the glycosylation on the N74 and N1158 sequons was not detected from the experiment using Lys-C/chymotrypsin digestion, their modifications were determined from Asp-N/chymotrypsin digests, revealing that complementary outcomes can be gained by the combination of multiple enzyme digestions.
Table 1. Relative Abundance of the Glycans on the Glycosylation Sites of the Prefusion Spike Protein (S2P) of the SARS-CoV-2 Virus Determined by LC/MS Analysis of Protein Digests under Various Experimental Conditionsa.
| Lys-C/chymotrypsin digestion |
Asp-N/chymotrypsin digestion |
trypsin
digestion |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| glyco-site | complex | high mannose | hybrid | truncated | unoccupied | complex | high mannose | hybrid | truncated | unoccupied | complex | high mannose | hybrid | truncated | unoccupied |
| 61 | 0.47 | 0.41 | 0.11 | 0.01 | 0.47 | 0.37 | 0.15 | 0.01 | |||||||
| 74 | 0.78 | 0.22 | |||||||||||||
| 122 | 0.31 | 0.30 | 0.39 | 0.38 | 0.24 | 0.38 | |||||||||
| 149 | 1.00 | 1.00 | 1.00 | ||||||||||||
| 165 | 0.76 | 0.21 | 0.01 | 0.01 | 0.73 | 0.23 | 0.02 | 0.01 | 0.69 | 0.31 | |||||
| 234 | 0.01 | 0.98 | 0.01 | 0.01 | 0.97 | 0.01 | 0.01 | 0.01 | 0.98 | ||||||
| 282 | 0.93 | 0.02 | 0.04 | 0.01 | 0.99 | 0.01 | 0.97 | 0.01 | 0.02 | ||||||
| 331 | 0.99 | 0.01 | 0.98 | 0.02 | |||||||||||
| 343 | 0.96 | 0.04 | 0.96 | 0.04 | |||||||||||
| 603 | 0.45 | 0.40 | 0.14 | 0.40 | 0.42 | 0.18 | |||||||||
| 616 | 0.94 | 0.03 | 0.03 | 0.81 | 0.04 | 0.14 | 0.01 | ||||||||
| 657 | 1.00 | 0.99 | 0.01 | ||||||||||||
| 709 | 0.06 | 0.94 | 1.00 | ||||||||||||
| 717 | 0.11 | 0.89 | 0.12 | 0.88 | |||||||||||
| 801 | 0.15 | 0.60 | 0.21 | 0.04 | 0.07 | 0.70 | 0.20 | 0.02 | 0.21 | 0.54 | 0.21 | 0.04 | |||
| 1074 | 0.45 | 0.35 | 0.20 | 0.66 | 0.21 | 0.13 | 0.41 | 0.36 | 0.23 | ||||||
| 1098 | 0.72 | 0.02 | 0.27 | 0.57 | 0.31 | 0.12 | 0.69 | 0.03 | 0.28 | ||||||
| 1134 | 1.00 | 0.91 | 0.01 | 0.09 | |||||||||||
| 1158 | 1.00 | ||||||||||||||
| 1173 | 1.00 | 0.93 | 0.07 | ||||||||||||
| 1194 | 1.00 | 0.00 | 1.00 | 0.98 | 0.02 | 0.01 | |||||||||
The values were determined by summing the mean area ratio of individual glycans of three biological replicates then normalizing at each glycosylation site.
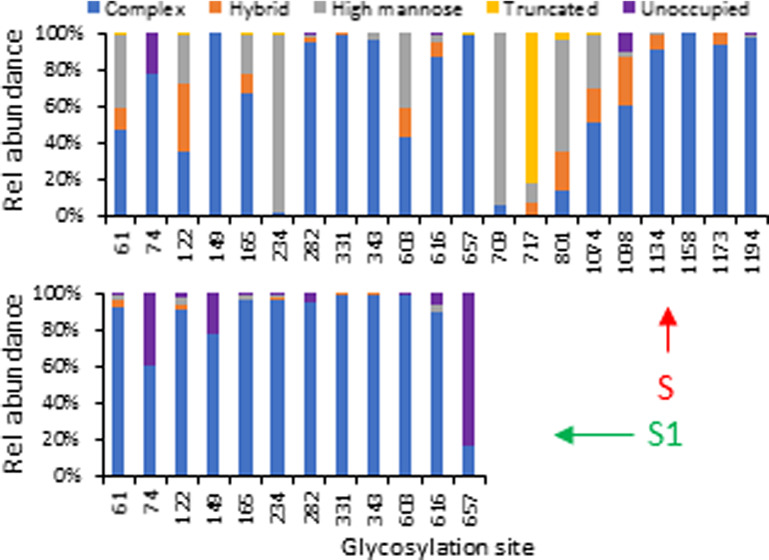
There were 19 of 21 detected sequons on the prefusion stabilized ectodomain of the SARS-CoV-2 S protein (S2P) that were fully occupied by various glycans (Table 1 and Figure 2A). The unoccupied fraction of sequons N74 and N1098 was relatively low (10–20%). This is consistent with the results reported for other glycoproteins. Among four types of glycans, complex glycans were the most frequently observed within most of the individual N-glycosylation sites. There were 17 sites that contained more than 40% complex glycans, of which 11 sites contained 80% or higher relative abundance of complex glycans including N74, N149, N282, N331, N343, N616, N657, N1134, N1158, N1173, and N1194, suggesting this protein was modified with extensively processed N-glycans. This observation is similar to a recent report on a full-length S protein13 but different from that reported on two subunits of the spike protein expressed separately, where a larger proportion of high-mannose glycans was determined.50
Figure 2.

N-Glycosylation profile of recombinant S proteins including (A) stabilized SARS-CoV-2 S protein expressed in human cells (S2P); (B) SARS-CoV-2 S protein S1 subunit expressed in human cells; (C) MERS-CoV S protein S1 subunit expressed in human cells; (D) SARS-CoV-2 S protein expressed in insect cells; and (E) SARS-CoV-1 S protein expressed in insect cells. The data represent the sum of the mean values of three replicates for each type of glycans. The majority of the glycan data was obtained with Asp-N/chymotrypsin digestion.
Two glycosylation sites (N234 and N709) were almost fully occupied by high-mannose glycans, while the sites of N61, N122, N603, N801, and N1074 were also modified by both high-mannose and complex glycans in comparable abundance. It was not surprising that this trend agreed well with the results reported on the same full-length S protein construct.13 A significant difference between the two studies lies on the glycans attached to N717. The glycans detected at the N717 site were paucimannosidic or truncated small N-glycans in this report, but mainly high-mannose glycans in the other study. This inconsistency could be attributed to the differences in sample preparation, data acquisition, or data processing.
The difference in the N-glycosylation profiles on the SARS-CoV-2 S proteins reported recently by several laboratories13,37,38,50,51 led us to investigate the effect of the S protein structure (full-length vs S1 subunit alone) on the glycan complement. We examined the N-glycosylation pattern of the SARS-CoV-2 S1 subunit prepared from HEK cells. The major glycan type in the available sequons of the S1 protein was still complex (Figure 2B). In comparison to the complete S2P construct, significant variations occurred. For instance, site N234 was almost fully occupied by complex glycans in S1 while the same site contained high-mannose glycans in S2P. This suggests that N234 may represent a location of a trimer-associated mannose patch, a phenomenon observed for HIV-1 envelope glycoprotein where underprocessed oligomannose-type glycans dominate some sites on the trimeric protein to stabilize its conformation.56 In addition, N657 was solely modified with complex glycans on S2P, but less than 20% of the sites were occupied by complex glycans on S1 with the remainder of sites left unoccupied. Four sequons including N61, N122, N165, and N603 on the full-length spike contained a mixture of complex, hybrid, and high-mannose glycans, while complex glycans almost exclusively occupied the sites observed on the S1 subunit.
The composition of individual glycans of the full-length version and the S1 subunit of SARS-CoV-2 is displayed in Figure 3, illustrating the quantitative occupancy of the glycans with the highest and the second highest relative abundance on each glycosylation sites (referred to as the primary glycans thereafter). The reproducibility of our approach was demonstrated by the results obtained from two independent sample preparation methods, Asp-N/chymotrypsin digestion (Figure 3A upward) and Lys-C/chymotrypsin (Figure 3A, downward). As shown in the representative plots for S2P protein, the speciation and the relative abundance of the top 2 occupied N-glycans were comparable for majority of the modification sites. Although heterogeneity is a common phenomenon of N-linked glycosylation, our data show that many of the sites on SARS-CoV-2 S protein were mainly occupied by only one or two primary glycans. For example, more than half of each of the sites including N61, N234, N343, N603, N709, N717, N801, N1074, and N1134 were modified primarily by one or two glycans. Another interesting observation was some of the glycan species were predominant in multiple sites. For example, the biantennary complex glycan with the composition of HexNAc5Hex3Fuc occurred as the highest or the second highest abundant N-glycan on N74, N149, N282, N331, N343, N657, and N1134. Among those sites, N331 and N343 are located within the receptor-binding domain (RBD) of the SARS-CoV-2 S protein. Meanwhile, the high-mannose glycan Man5HexNAc2 HexNAc2Hex5 appeared on six modification sites, as one of the two primary modifiers. These observations revealed that only a few primary glycan species might play an important role in shielding the virus’ immunogenic epitopes. In other words, the glycan density of viral spike protein was dominated by a few major glycan species although hundreds of distinct glycan species were identified on this protein.
Figure 3.

Relative abundance of the top 2 N-glycans on individual glycosylation sites of (A) SARS-CoV-2 prefusion ectodomain spike protein (S2P) derived from the digestion by Asp-N/chymotrypsin (upward) and Lys-C/chymotrypsin (downward). Note that the reflected y-axis represents positive values as per the labeled y scale; (B) SARS-CoV-2 S protein S1 subunit (S1); (C) MERS-CoV S protein S1 subunit (MS1); (D) SARS-CoV-2 S protein (SF); and (E) SARS-CoV S protein (SN). The data represent the average values of three replicates, and the error bars represent the standard deviation of the mean. Most of the data were obtained from the Asp-N/chymotrypsin digests.
The variation between the full-length and S1 subunit recombinant proteins was further demonstrated by the distribution of primary glycans (Figure 3A,B). In contrast to the ectodomain protein, S2P, the combined abundance of the top two glycans did not exceed 50% at any site of the S1 subunit, revealing a higher degree of microheterogeneity (different glycans at the same site) in the S1 protein than in S2P. In addition, the predominantly occurring glycans in these two proteins were not the same. On the S1 protein, a possible biantennary complex glycan with the composition of HexNAc4Hex5FucNeuAc containing one fucose and one sialic acid modified 9 of 12 identified sites (N61, N74, N122, N149, N165, N234, N331, N343, and N603) including two sites on the RBD domain. In contrast, only three N-glycan sites (N165, N331, and N657) on the S2P were dominated by the complex type of glycans.
Another significant difference between the S2P and S1 proteins was the content of sialic acid glycans. Among 24 of the top 2 glycans detected on the 12 sequons in the sequence of the S1 subunit, 19 were sialylated glycans for S1 protein while only three N-glycans on the full-length S2P had sialic acid groups (Figure 4A,B). Increased sialylation on S1 subunit protein was further confirmed by comparing the relative ratio of sialylated glycans to nonsialylated ones on the two proteins (Figure 4). On each of the 12 sites of S2P protein, there were approximately 60% or higher total detected glycans containing no sialic acids; no sialylated glycans at all were detected on N74 and N234 (Figure 4A). In contrast, 9 of 12 sites on the S1 protein were sialylated by greater than 75%. Less than 50% of the glycans on the other three sites (N61, N149, N343) were unsialyated. In addition, there was a range in the degree of sialylation of S1 protein, including mono- and disialylated forms with some tri- and tetrasialylated glycans. The sialylation on the glycans of S2P was predominately the monosialylated glycoforms. For the sequons located in the RBD domain of the spike protein, for N331 on the S1 protein, a total of 80% of glycans were sialylated (60% were monosialylated). In contrast, on the S2P protein, only 40% of the glycans were sialylated at sequon N331. On the other hand, only ∼10% of the glycans on N343 of S2P were sialylated but approximately half of the glycoforms on the same residue of S1-contained terminal sialic acids.
Figure 4.

Distribution of sialylated N-glycans on the glycosylation sites within the sequence range of the S1 subunit of (A) the SARS-CoV-2 ectodomain S protein and (B) its S1 subunit recombinant protein, (C) and the S1 subunit of the MERS-CoV spike protein, MS1. “0 SA” indicates glycans without sialic acid attached, and “1–4 SA” represents mono-, bi-, tri-, and tetrasialylated glycans, respectively.
Sialylated N-glycans play an important role in the immune system, pathogen recognition, protein–protein interactions, and cancer.57 The substantial variation in sialic acid content between ectodomain and S1 subunit of SARS-CoV-2 S protein observed in our results suggests careful consideration in data interpretation is needed when a construct derived from a partial protein sequence is used to study ligand–receptor binding, antibody recognition, or other structure/function relationships. The existence of a large quantity of negatively charged branch-terminal sialic acids, particularly in a smaller protein, could affect the isoelectric point of this protein, and this may cause differences in the three-dimensional structure or conformation between full-length and reduced-size proteins.
To understand the effect of the primary sequence of the protein on the glycosylation profile and the formation of sialylated N-glycans, we examined the glycan profile of the S1 subunit (MS1) of the spike protein of MERS coronavirus (MERS-CoV), which belongs to the same coronavirus family as SARS-CoV-2. The recombinant MS1 has a similar molecular weight to the S1 protein (Figure 1B) and was prepared in HEK cells by the same manufacturer that produced the SARS-CoV-2 S1 recombinant protein examined here. The two proteins, S1 and MS1, differed significantly in the distribution of N-glycan types (Figure 2C) and also displayed clear variation in the composition of the primary glycans on each site (Figure 3C), presumably due to their variation in primary amino acid sequences. One-third of the N-glycosylation sites of MS1 (N125, N166, N222, N410) were high-mannose glycans with high occupancy (>80%), and 10–20% of sites N66, N104, and N236 were occupied by this glycan type. While complex glycans were still the major N-glycan type on the remaining sites, relatively low abundant hybrid glycans were also observed on some of these sites. Although the same biantennary complex glycan (HexNAc4Hex5FucNeuAc) occupied multiple sites in MS1 and 14 sialylated glycans were among 24 of the most frequently observed glycans, the high preference for nonsialylated glycans on most of the sequons implies that reduced size of a recombinant protein might not necessarily cause enhanced sialylation (Figure 4C). The amino acid sequence of the S1 subunit of SARS-CoV-2 S protein may play a role in its elevated sialic acid content when expressed as a recombinant protein. Further study is needed to confirm this speculation and its biological significance.
The baculovirus insect cell system has been widely utilized to produce functional, post-translationally modified recombinant proteins.58 Since the beginning of the COVID-19 pandemic, this system has been used by different manufacturers and laboratories to produce many SARS-CoV-2 recombinant proteins for research and therapeutic purposes. A recent preprint has reported the N-glycosylation mapping on the surface of SARS-CoV-2 S protein expressed in insect cells, but no quantitative results regarding the relative abundance of glycans are included.37 It is well known that expression hosts affect the glycosylation of a recombinant protein. Expression in baculoviral-insect cells is expected to form smaller or truncated glycans including pausimannosidic and other smaller species because the glycosylation pathway in insect cells is far simpler than in higher eukaryotes.59 Characterizing the glycan profile of this protein under identical analytical conditions as the spike proteins expressed in the HEK expression systems was useful. When this protein construct (SF) was analyzed, not surprisingly, a vastly different N-glycosylation profile was observed (Figure 2D). Most of the N-glycosylation sequons were modified by truncated glycans with high abundances. For example, 15 Asn residues were modified with 80% or higher truncated glycans. There were, however, a few exceptions including N61, N234, and N717, where the main glycan type was high mannose and their relative abundances were comparable to those that were measured in human expressed S2P protein. A similar pattern was observed on a SARS-CoV S protein (SN) that was prepared in insect cells, although some sites such as N256, N1142, and 1163 were modified by complex glycans of comparable densities (Figure 2E).
O-glycosylation has been reported in SARS-CoV-2 full-length S protein constructs38,52 and subunits.50 In all cases, the relative degree of occupation for these observed O-glycosylation sites was relatively low—less than 11% in the report on full-length construct. In addition, Wells and colleagues have reported the observation of sulfonated glycans on a recombinant full-length S protein.38 Prompted by these reports, we also searched our data for O-linked glycans and sulfoglycans in Byonic using a similar approach and searched identification criteria as we employed for N-glycosylation. Given our relatively strict and conservative search criteria (Byonic score ≥300), we observed only a few peptides modified by sulfoglycans with a lower variety and diversity of modification sites than Wells and colleagues (Table S-1). On the other hand, we did not observe any occupied O-glycosylation sites that passed our filters. The relatively low amount (<1 μg) of proteins used in this study might hinder the detection of high-quality tandem mass spectra for low-abundance O-glycan or sulfoglycan peptides. In the future, we will more thoroughly characterize the O-linked and sulfonated glycan complement on SARS-CoV-2 proteins.
Conclusions
We conducted a comprehensive characterization of the N-glycosylation profiles of several SARS-CoV-2 S protein constructs and contrasted them with the glycan profiles from SARS-CoV-1 and MERS. The combination of multiple enzyme digestions, glycan signature-triggered EThcD analysis, and rigorous data processing parameters allowed the detection and relative quantification of N-glycan distribution on the proteins with high confidence and reproducibility. The patterns of the N-glycosylation between two versions of the recombinant spike proteins, ectodomain and S1 subunit, were carefully examined, and significant variations were observed on the distribution of glycan types and specific individual glycans on various sequons of entire and partially expressed proteins. Our data demonstrate that the relative abundance of sialylated glycans was significantly elevated in the reduced-size S1 subunit than in the full-length protein. This observation could possibly point to differences in the structure and function of these two species. In addition, we compared N-glycan profiles of the recombinant S proteins produced from different expression systems including human cells and insect cells. These results should provide useful information for the study of the interactions of SARS-CoV-2 viral proteins and the development of effective vaccines and therapeutics.
Acknowledgments
The authors thank Daniel Wrapp, Nianshuang Wang, and Jason McLellan for providing recombinant SARS-CoV-2 prefusion stabilized spike protein. They also thank Eric Wang for processing some data using tools developed in R. The following reagent was obtained through BEI Resources, NIAID, NIH: SARS-CoV spike (S) protein with C-terminal Histidine Tag, Recombinant from Baculovirus, NR-686. References in this paper to any specific commercial products, process, service, manufacturer, or company do not constitute an endorsement or a recommendation by the U.S. Government or the Centers for Disease Control and Prevention. The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03301.
The authors declare no competing financial interest.
Supplementary Material
References
- Bassetti M.; Vena A.; Giacobbe D. R. The novel Chinese coronavirus (2019-nCoV) infections: Challenges for fighting the storm. Eur. J. Clin. Invest. 2020, 50, e13209 10.1111/eci.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Wang Y.; Li X.; Ren L.; Zhao J.; Hu Y.; Zhang L.; Fan G.; Xu J.; Gu X.; Cheng Z.; Yu T.; Xia J.; Wei Y.; Wu W.; Xie X.; Yin W.; Li H.; Liu M.; Xiao Y.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Yu Y.; Xu J.; Shu H.; Xia J.; Liu H.; Wu Y.; Zhang L.; Yu Z.; Fang M.; Yu T.; Wang Y.; Pan S.; Zou X.; Yuan S.; Shang Y. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COVID-19 Weekly Epidemiological Update; World Health Organization: Geneva, 2020. (Sept 7, 2020). [Google Scholar]
- Walls A. C.; Park Y. J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281.e6–292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Shang J.; Ye G.; Shi K.; Wan Y.; Luo C.; Aihara H.; Geng Q.; Auerbach A.; Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa V. G.; Moreli M. L.; Saivish M. V. The emergence of SARS, MERS and novel SARS-2 coronaviruses in the 21st century. Arch. Virol. 2020, 165, 1517–1526. 10.1007/s00705-020-04628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul D. An overview of coronaviruses including the SARS-2 coronavirus - Molecular biology, epidemiology and clinical implications. Curr. Med. Res. Pract. 2020, 10, 54–64. 10.1016/j.cmrp.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Tortorici M. A.; Bosch B. J.; Frenz B.; Rottier P. J. M.; DiMaio F.; Rey F. A.; Veesler D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. 10.1038/nature16988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Tortorici M. A.; Frenz B.; Snijder J.; Li W.; Rey F. A.; DiMaio F.; Bosch B. J.; Veesler D. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 2016, 23, 899–905. 10.1038/nsmb.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C. L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B.; Qing X.; Liao J.; Zhuo K. Role of Protein Glycosylation in Host-Pathogen Interaction. Cells 2020, 9, 1022 10.3390/cells9041022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Bowden T. A.; Wilson I. A.; Crispin M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta, Gen. Subj. 2019, 1863, 1480–1497. 10.1016/j.bbagen.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Khatri K.; Klein J.; Leymarie N.; Zaia J. A review of methods for interpretation of glycopeptide tandem mass spectral data. Glycoconjugate J. 2016, 33, 285–296. 10.1007/s10719-015-9633-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J. Liquid chromatography-tandem mass spectrometry-based fragmentation analysis of glycopeptides. Glycoconjugate J. 2016, 33, 261–272. 10.1007/s10719-016-9649-3. [DOI] [PubMed] [Google Scholar]
- Hever H.; Darula Z.; Medzihradszky K. F. Characterization of Site-Specific N-Glycosylation. Methods Mol. Biol. 2019, 1934, 93–125. 10.1007/978-1-4939-9055-9_8. [DOI] [PubMed] [Google Scholar]
- Hoffmann M.; Marx K.; Reichl U.; Wuhrer M.; Rapp E. Site-specific O-Glycosylation Analysis of Human Blood Plasma Proteins. Mol. Cell. Proteomics 2016, 15, 624–641. 10.1074/mcp.M115.053546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert P.; Halim A.; Zauser M.; Essig A.; Joshi H. J.; Zatorska E.; Larsen I. S.; Loibl M.; Castells-Ballester J.; Aebi M.; Clausen H.; Strahl S. Mapping the O-Mannose Glycoproteome in Saccharomyces cerevisiae. Mol. Cell. Proteomics 2016, 15, 1323–1337. 10.1074/mcp.M115.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K. K.; Wilson G. M.; Kirkemo L. L.; Riley N. M.; Coon J. J.; Wells J. A. Broad and thematic remodeling of the surfaceome and glycoproteome on isogenic cells transformed with driving proliferative oncogenes. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 7764–7775. 10.1073/pnas.1917947117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Sun Z.; Zhang L.; Cai Y.; Peng Y.; Cao T.; Zhang Y.; Lu H. Chemical labeling for fine mapping of IgG N-glycosylation by ETD-MS. Chem. Sci. 2019, 10, 9302–9307. 10.1039/C9SC02491C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T. L.; Luna L.; Guo Z.; Cox N. J.; Pirkle J. L.; Donis R. O.; Barr J. R. Quantification of influenza virus hemagglutinins in complex mixtures using isotope dilution tandem mass spectrometry. Vaccine 2008, 26, 2510–2520. 10.1016/j.vaccine.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Pierce C. L.; Williams T. L.; Moura H.; Pirkle J. L.; Cox N. J.; Stevens J.; Donis R. O.; Barr J. R. Quantification of immunoreactive viral influenza proteins by immunoaffinity capture and isotope-dilution liquid chromatography-tandem mass spectrometry. Anal. Chem. 2011, 83, 4729–4737. 10.1021/ac2006526. [DOI] [PubMed] [Google Scholar]
- Williams T. L.; Pirkle J. L.; Barr J. R. Simultaneous quantification of hemagglutinin and neuraminidase of influenza virus using isotope dilution mass spectrometry. Vaccine 2012, 30, 2475–2482. 10.1016/j.vaccine.2011.12.056. [DOI] [PubMed] [Google Scholar]
- Santana W. I.; Williams T. L.; Winne E. K.; Pirkle J. L.; Barr J. R. Quantification of viral proteins of the avian H7 subtype of influenza virus: an isotope dilution mass spectrometry method applicable for producing more rapid vaccines in the case of an influenza pandemic. Anal. Chem. 2014, 86, 4088–4095. 10.1021/ac4040778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A.; Chen L. M.; Winne E.; Santana W.; Metcalfe M. G.; Mateu-Petit G.; Ridenour C.; Hossain M. J.; Villanueva J.; Zaki S. R.; Williams T. L.; Cox N. J.; Barr J. R.; Donis R. O. Identification of Influenza A/PR/8/34 Donor Viruses Imparting High Hemagglutinin Yields to Candidate Vaccine Viruses in Eggs. PLoS One 2015, 10, e0128982 10.1371/journal.pone.0128982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridenour C.; Johnson A.; Winne E.; Hossain J.; Mateu-Petit G.; Balish A.; Santana W.; Kim T.; Davis C.; Cox N. J.; Barr J. R.; Donis R. O.; Villanueva J.; Williams T. L.; Chen L. M. Development of influenza A(H7N9) candidate vaccine viruses with improved hemagglutinin antigen yield in eggs. Influenza Other Respir. Viruses 2015, 9, 263–270. 10.1111/irv.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern K.; Xie Y.; Palladino G.; Barr J. R.; Settembre E. C.; Williams T. L.; Wen Y. Reference antigen-free and antibody-free LTD-IDMS assay for influenza H7N9 vaccine in vitro potency determination. Vaccine 2018, 36, 6144–6151. 10.1016/j.vaccine.2018.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce C. L.; Williams T. L.; Santana W. I.; Levine M.; Chen L. M.; Cooper H. C.; Solano M. I.; Woolfitt A. R.; Marasco W. A.; Fang H.; Donis R. O.; Barr J. R. Immunocapture isotope dilution mass spectrometry in response to a pandemic influenza threat. Vaccine 2017, 35, 5011–5018. 10.1016/j.vaccine.2017.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake T. A.; Williams T. L.; Pirkle J. L.; Barr J. R. Targeted N-linked glycosylation analysis of H5N1 influenza hemagglutinin by selective sample preparation and liquid chromatography/tandem mass spectrometry. Anal. Chem. 2009, 81, 3109–3118. 10.1021/ac900095h. [DOI] [PubMed] [Google Scholar]
- Chang D.; Zaia J. Why Glycosylation Matters in Building a Better Flu Vaccine. Mol. Cell. Proteomics 2019, 18, 2348–2358. 10.1074/mcp.R119.001491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipollo J. F.; Parsons L. M. Glycomics and glycoproteomics of viruses: Mass spectrometry applications and insights toward structure-function relationships. Mass Spectrom. Rev. 2020, 39, 371–409. 10.1002/mas.21629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargett A. A.; Renfrow M. B. Glycosylation of viral surface proteins probed by mass spectrometry. Curr. Opin. Virol. 2019, 36, 56–66. 10.1016/j.coviro.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezstarosti K.; Lamers M. M.; Haagmans B. L.; Demmers J. A. A. Targeted Proteomics for the Detection of SARS-CoV-2 Proteins. bioRxiv 2020, 1–20. 10.1101/2020.04.23.057810. [DOI] [Google Scholar]
- Davidson A. D.; Williamson M. K.; Lewis S.; Shoemark D.; Carroll M. W.; Heesom K.; Zambon M.; Ellis J.; Lewis P. A.; Hiscox J. A.; Matthews D. A. Characterisation of the transcriptome and proteome of SARS-CoV-2 using direct RNA sequencing and tandem mass spectrometry reveals evidence for a cell passage induced in-frame deletion in the spike glycoprotein that removes the furin-like cleavage site. bioRxiv 2020, 1–39. 10.1101/2020.03.22.002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhao W.; Mao Y.; Wang S.; Zhong Y.; Su T.; Gong M.; Du D.; Lu X.; Cheng J.; Yang H. Site-specific N-glycosylation Characterization of Recombinant SARS-CoV-2 Spike Proteins. bioRxiv 2020, 1–37. 10.1101/2020.03.28.013276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P.; Praissman J. L.; Grant O. C.; Cai Y.; Xiao T.; Rosenbalm K. E.; Aoki K.; Kellman B. P.; Bridger R.; Barouch D. H.; Brindley M. A.; Lewis N. E.; Tiemeyer M.; Chen B.; Woods R. J.; Wells L. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28, 586.e6–601.e6. 10.1016/j.chom.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J. A.; Zaia J. Assignment of coronavirus spike protein site-specific glycosylation using GlycReSoft. bioRxiv 2020, 1–39. 10.1101/2020.05.31.125302. [DOI] [Google Scholar]
- Chalk R.; Greenland W.; Moreira T.; Coker J.; Mukhopadhyay S. M. M.; Williams E.; Manning C.; Bohstedt T.; McCrorie R.; Fernandez-Cid A.; Burgess-Brown N. A. Identification, Mapping and Relative Quantitation of SARS-CoV-2 Spike Glycopeptides by Mass-Retention Time Fingerprinting. bioRxiv 2020, 1–13. 10.1101/2020.07.24.217562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka J. E.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon J. J.; Ueberheide B.; Syka J. E.; Dryhurst D. D.; Ausio J.; Shabanowitz J.; Hunt D. F. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 9463–9468. 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia B. A.; Shabanowitz J.; Hunt D. F. Analysis of protein phosphorylation by mass spectrometry. Methods 2005, 35, 256–264. 10.1016/j.ymeth.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Mikesh L. M.; Ueberheide B.; Chi A.; Coon J. J.; Syka J. E.; Shabanowitz J.; Hunt D. F. The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1811–1822. 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J.; Mohammed S. Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 2012, 84, 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; Zhou H.; Taus T.; Altelaar A. F.; Mechtler K.; Heck A. J.; Mohammed S. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 2013, 12, 1520–1525. 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q.; Wang B.; Chen Z.; Urabe G.; Glover M. S.; Shi X.; Guo L. W.; Kent K. C.; Li L. Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28, 1751–1764. 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čaval T.; Zhu J.; Heck A. J. R. Simply Extending the Mass Range in Electron Transfer Higher Energy Collisional Dissociation Increases Confidence in N-Glycopeptide Identification. Anal. Chem. 2019, 91, 10401–10406. 10.1021/acs.analchem.9b02125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lermyte F.; Valkenborg D.; Loo J. A.; Sobott F. Radical solutions: Principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure. Mass Spectrom. Rev. 2018, 37, 750–771. 10.1002/mas.21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A.; Supekar N. T.; Gleinich A. S.; Azadi P. Deducing the N- and O- glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, cwaa042 10.1093/glycob/cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Berndsen Z. T.; Raghwani J.; Seabright G. E.; Allen J. D.; Pybus O. G.; McLellan J. S.; Wilson I. A.; Bowden T. A.; Ward A. B.; Crispin M. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat. Commun. 2020, 11, 2688 10.1038/s41467-020-16567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda M.; Morrison L.; Goldman R. N and O glycosylation of the SARS-CoV-2 spike protein. bioRxiv 2020, 1–6. 10.1101/2020.07.05.187344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba J.; Dutta S.; Hemenway E.; Viner R. Increasing the productivity of glycopeptides analysis by using higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation. Int. J. Proteomics 2012, 2012, 560391 10.1155/2012/560391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M.; Cai Y.; Goldberg D. Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem. 2007, 79, 1393–1400. 10.1021/ac0617013. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Malaker S. A.; Driessen M. D.; Bertozzi C. R. Optimal Dissociation Methods Differ for N- and O-Glycopeptides. J. Proteome Res. 2020, 19, 3286–3301. 10.1021/acs.jproteome.0c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A. J.; Harvey D. J.; Milne E.; Cupo A.; Kumar A.; Zitzmann N.; Struwe W. B.; Moore J. P.; Crispin M. Molecular Architecture of the Cleavage-Dependent Mannose Patch on a Soluble HIV-1 Envelope Glycoprotein Trimer. J. Virol. 2017, 91, e01894-16 10.1128/JVI.01894-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide G. P.; Colley K. J. Sialylation of N-glycans: mechanism, cellular compartmentalization and function. Histochem. Cell Biol. 2017, 147, 149–174. 10.1007/s00418-016-1520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra V. A Comprehensive Guide to the Commercial Baculovirus Expression Vector Systems for Recombinant Protein Production. Protein Pept. Lett. 2020, 27, 529–537. 10.2174/0929866526666191112152646. [DOI] [PubMed] [Google Scholar]
- Shi X.; Jarvis D. L. Protein N-glycosylation in the baculovirus-insect cell system. Curr. Drug Targets 2007, 8, 1116–1125. 10.2174/138945007782151360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.