

Abstract
The regulation and function of cyclin‐dependent kinase 6 (CDK6)‐ and cyclin‐dependent kinase 4 (CDK4)‐cyclin complexes are commonly altered with enhanced kinase activity found in hematopoietic malignancies, breast cancer and melanoma making CDK4 and CDK6 attractive targets for therapeutic interference. Although dual CDK4/6 inhibitors have revolutionized treatment of breast cancer patients and reveal promising results in several solid tumors and hematological malignancies, there is a need for novel compounds targeting the versatile kinase‐independent functions of CDK6 to improve cancer treatment. The following review summarizes the latest findings on CDK6 in cancer development and discusses novel therapeutic approaches to selectively inhibit CDK6s function as a transcriptional regulator.
Keywords: cancer, CDK4, CDK4/6 inhibitors, CDK6
Abbreviations
- AML
acute myeloid leukemia
- ATP
adenosine triphosphate
- AURK
aurora kinase
- BCR/ABL
break point cluster region/Abelson
- Cdc37
cell division cycle 37
- CDK4
cyclin‐dependent kinase 4
- CDK6
cyclin‐dependent kinase 6
- CDKN2A
cyclin‐dependent kinase inhibitor 2A
- CDKs
cyclin‐dependent kinases
- CK2
casein kinase 2
- CKIs
CDK inhibitors
- DN
double‐negative
- DNMT1
DNA methyltransferase 1
- Egr1
early growth response protein 1
- ERV
endogenous retroviral elements
- EZH2
enhancer of zeste 2 polycomb repressive complex 2 subunit
- FDA
Food and Drug Administration
- FLT3
fms‐related tyrosine kinase 3
- FOXM1
forkhead box M1
- HER2
human epidermal growth factor receptor 2
- HR
hormone receptor
- HSCs
hematopoietic stem cells
- Hsp90
heat shock protein 90
- IFN
interferon
- IL2
interleukin 2
- LSCs
leukemic stem cells
- LSK
Lin‐Sca‐1 + c‐Kit+
- Mdm4
transformed mouse 3 T3 cell double minute 4
- MDS
myelodysplastic syndrome
- MEK
mitogen‐activated protein kinase
- MLL
mixed‐lineage leukemia
- NFAT
nuclear factor of activated T‐cells
- NRTKs
nonreceptor tyrosine kinases
- NSCLC
nonsmall cell lung cancer
- PD‐1
programmed cell death 1
- PD‐L1
programmed cell death 1 ligand 1
- PFS
prolonged progression‐free survival
- Ph+
Philadelphia‐positive
- PP5
serine/threonine phosphatase protein phosphatase 5
- Ppm1d
protein phosphatase 1D magnesium‐dependent, delta isoform
- Prmt5
protein arginine methyltransferase 5
- PROTACs
proteolysis‐targeting chimeras
- RB
retinoblastoma
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- SPOP
cullin3‐speckle‐type POZ protein
- STAT3
signal transducer and activator of transcription 3
- T‐ALL
T‐cell acute lymphoblastic leukemia
- T‐LBL
T‐cell lymphoblastic lymphoma
- TReg
regulatory T cells
- TP53
tumor protein 53
- VEGFA
vascular endothelial growth factor A
1. INTRODUCTION
The family of cyclin‐dependent kinases (CDKs) covers 13 different serine/threonine kinases that become catalytically active when bound to their respective regulatory subunits, the cyclins. CDKs, in complex with cyclins, regulate various critical cellular processes including cell‐cycle progression as well as transcription. Disordered cell cycle regulation, due to aberrant kinase activation, often leads to uncontrolled cell proliferation and results in cancer development. The importance of CDKs in promoting cancer initiation as well as progression has made them an attractive target for pharmacological inhibition. 1
The present review gives an overview of the current knowledge on the versatile regulatory functions of CDK6 as a cell cycle‐dependent kinase and transcriptional regulator under homeostatic conditions as well as in malignant transformed settings. (Figure 1) In particular, the role of CDK6 in normal hematopoiesis, hematologic malignancies, breast cancer and melanoma as well as current and novel therapeutic approaches for targeting CDK6 will be discussed.
FIGURE 1.
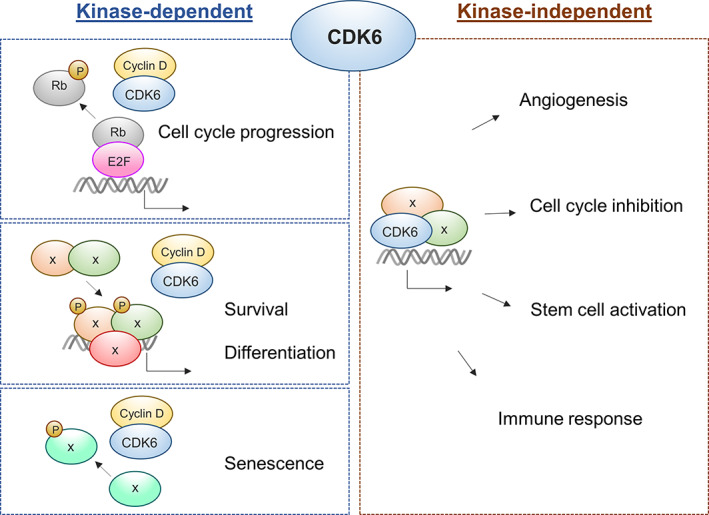
Schematic overview of CDK6‐mediated kinase‐dependent and kinase‐independent functions in cancer. Cyclin‐dependent kinase 6 (CDK6) acts as a chromatin‐bound cofactor that in a kinase‐independent manner induces transcription of genes regulating angiogenesis, cell cycle inhibition, stem cell activation and immune response. The function of CDK6 in cell cycle progression, survival, differentiation and senescence requires binding to D‐type cyclin to enable kinase‐dependent protein phosphorylation
2. REGULATION OF CDK6
CDK6 and the highly homologous enzyme CDK4 are known as classic cell cycle kinases that facilitate the progression of cells through the early G1 phase of the cell cycle by forming complexes with D‐type cyclins (D1, D2 and D3). CDK4/6‐cyclin D complexes enable the phosphorylation of members of the retinoblastoma (RB) protein family resulting in the release of E2F transcription factors from RB‐mediated inhibition. (Figure 1) Subsequently, E2F‐dependent gene activation enables G1 to S phase progression and DNA synthesis. 2 , 3 , 4 The activation and inhibition of CDK4/6‐cyclin D complexes are controlled by two distinct classes of regulatory subunits, the Cip/Kip family, comprising p21Cip1 (CDKN1A), p27Kip1 (CDKN1B) and p57Kip2 (CDKN1C), and the INK4 family, including p15INK4b (CDKN2B), p16INK4a (CDKN2A), p18INK4c (CDKN2C) and p19INK4d (CDKN2D). Cip/Kip family members are able to act on a broader spectrum of CDK‐cyclin complexes, to inhibit CDKs 1, 2, 4 and 6, whereas INK4 proteins exclusively inactivate CDK4/6‐cyclin D complexes. 5 , 6 , 7 , 8
Proteins of the Cip/Kip family are defined as double‐faced regulators based on their ability to operate as both positive and negative regulators of the cell cycle, depending on their phosphorylation status. 6 , 9 Nonreceptor tyrosine kinases (NRTKs) phosphorylate Cip/Kip proteins being part of a trimeric holoenzyme together with CDK4/6 and Cyclin D. Phosphorylation of the Cip/Kip subunit subsequently adapts the mode of action from an inhibitor to a noninhibitor. 6 Binding of the Cip/Kip proteins to the CDK4/6‐containing complex sequesters them from other CDKs, such as CDK2, and thus prevents them from inhibiting. This enables the activation of CDK‐cyclin complexes that act later during cell cycle progression. 4 , 10 , 11 The importance of Cip/Kip proteins as regulatory subunits is underscored by their versatile function contributing to assembly, stabilization and nuclear translocation of CDK4/6‐cyclin D complexes. 5 , 9 , 11 Further studies are needed to fully appreciate the potential of Cip/Kip proteins in terms of CDK regulation.
In contrast to the Cip/Kip family members, INK4 proteins specifically associate with monomeric CDK4 and CDK6, forming a catalytically inactive dimeric complex. Although the INK binding site of CDKs is distant from the cyclin interaction site, INK binding alters the position of the N and C lobes of CDK4 and CDK6. This structural turn distorts the catalytic cleft, leads to allosteric changes in the cyclin binding site and subsequently weakens the CDK‐cyclin interaction. 8 , 12 Consequently, in the absence of D‐type cyclin binding, the INK4‐CDK4/6 complex lacks kinase activity.
In addition to the presence of INK4 proteins in a dimeric complex, binding of INK4 proteins to a preassembled ternary holoenzyme, consisting of CDK4/6‐cyclin D‐Cip/Kip, has been shown.
Thereby INK4 proteins propagate conformational changes, lead to disassembly and consequently inactivation of the ternary CDK4/6‐cyclin D‐Cip/Kip holoenzyme. 8 , 11 , 12 , 13
In the absence of their cognate binding partners, it has been suggested, that CDK6, as well as CDK4, tend to form larger protein complexes consisting of heat shock protein 90 (Hsp90) and its co‐chaperone cell division cycle 37 (Cdc37). 14 Hsp90 is a highly conserved molecular chaperone protein that enables accurate folding, maturation, assembly and stability of a large number of proteins. It has been proposed that the absence of any interaction partner such as cyclins provokes an open and less folded state of the kinase, which can be captured by phosphorylated Cdc37 through binding to the kinase C lobe. 15 The phosphorylation of Cdc37 on Ser13 by the casein kinase 2 (CK2) is required for the appropriate function of Cdc37. 16 Once the binary Cdc37‐kinase complex has formed, it binds to Hsp90. The resulting ternary complex consisting of Hsp90, Cdc37 and the so‐called client protein, finally enables the loading of the client protein onto the Hsp90 ATPase‐coupled chaperone machinery. 17 HSP90 inhibitors, like ganetespib, prevents loading of the client protein onto the chaperone machinery and results in aggregation of the partly unfolded kinase or degradation through the ubiquitin‐proteasome system. 18 The release of the kinase from the ternary chaperone complex requires de‐phosphorylation of Cdc37 on Ser13, facilitated by protein phosphatase 5 (PP5). 19 (Figure 2).
FIGURE 2.
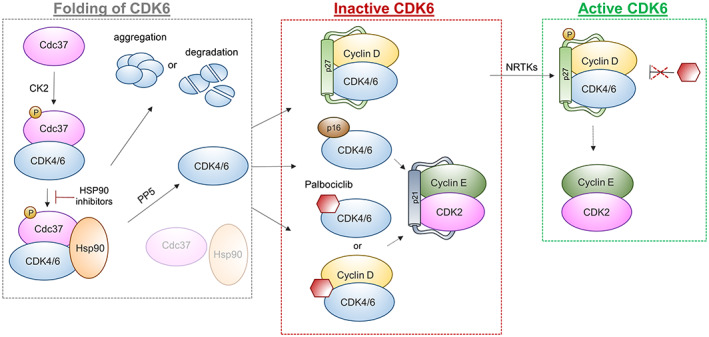
Regulation of CDK4 and CDK6. Phosphorylation of co‐chaperone cell division cycle 37 (Cdc37) by the casein kinase 2 (CK2) enables binding to CDK4/6 and complex formation with heat shock protein 90 (Hsp90), which facilitates accurate protein folding. Inhibition of HSP90 leads to aggregation or degradation of unfolded kinases. Dephosphorylation of Cdc37 by phosphatase protein phosphatase 5 (PP5) releases CDK4/6 and allows complex formation with different regulatory subunits. CDK4/6, Cyclin D and Cip/Kip proteins (p27) form an inactive trimeric complex, which gets activated upon phosphorylation of p27 by nonreceptor tyrosine kinases (NRTKs) and translocates into the nucleus. Association with inhibitors, such as p16 or palbociclib, drives the formation of inactive complexes and indirectly promotes inactivation of CDK2
Recent findings by Hallett et al have shown, that the interaction of CDK6 and CDK4 with Cdc37 and the Cdc37‐Hsp90 chaperone complex occurs with significantly different binding affinities; CDK6 is considered a weak client protein, whereas CDK4 is regarded as a strong interactor. 14 As a consequence D‐type cyclins or INK proteins readily displace the weak client protein CDK6 from Cdc37. The binding of cyclin, INK or Cdc37 to CDK6 is thereby mutually exclusive.
In case of CDK4, only members of the INK family, but not D‐type cyclins alone, are capable of displacing CDK4 from Cdc37 and Cdc37‐Hsp90. The inability of D‐type cyclins to sequester/partition CDK4 from Cdc37‐Hsp90 changes upon binding to CDK inhibitors (CKIs). Formation of a ternary complex of CDK4/6 and cyclin D1 or cyclin D3 together with either p21Cip1 or p27Kip1 is resistant to displacement of the CDK by Cdc37‐Hsp90. 14 Recent data in breast cancer cells by Guiley et al demonstrate that the active phosphorylated trimeric p27‐CDK4‐CycD1 complex is also resistant to the CDK4/6 kinase inhibitor palbociclib. 20 (Figure 2) They propose that palbociclib inhibits the kinase function in a similar manner as INK proteins by primarily targeting kinase‐inactive monomers. This indirectly reduces CDK2 activity by shuttling of Cip proteins to CDK2 complexes. The capability of the small molecule inhibitor palbociclib to imitate p16 function, may partially explain the enhanced sensitivity of cells lacking p16 to this type of inhibitors. 10 , 20 , 21 The specific CDK4/6‐targeting adenosine triphosphate (ATP)‐competitive drug palbociclib has been approved by the US Food and Drug Administration (FDA) for therapeutic application in hormone receptor‐positive breast cancer and is in clinical trials for use in various cancer types. 22 , 23 , 24 More details are discussed below. In summary, these data pinpoint at the highly complex and dynamic regulatory network between CDKs and their interactors that are disturbed under drug treatment. Vice versa the absence or presence of components of this regulatory network will determine the sensitivity of a cell toward inhibitor treatment.
3. CDK6 DURING NORMAL HEMATOPOIESIS
Although CDK6 and CDK4 are described to exert redundant functions as critical regulators in cell cycle progression, findings over the last years have shown that the two kinases differ especially in cell‐cycle independent tissue‐specific functions. While CDK6 plays an important role in hematopoiesis, CDK4's function is crucial for pancreatic beta cells and pituitary glands. 25 , 26 , 27 Despite the fact that concomitant depletion of CDK6 and CDK4 results in late embryonic lethality due to defective erythropoiesis, mice lacking the one or the other are viable. Individual knockouts are hallmarked by distinct tissue‐specific defects. Depletion of the CDK4 gene gives rise to defects in the proliferation of endocrine pancreatic cells and pituitary lactotrophs. Moreover, CDK4‐null mice are smaller in size and infertile. 25 , 26
Loss of CDK6 causes mild anemia, thymic atrophy and delayed G1 progression in lymphocytes. Female mice are marginally smaller but do not exhibit anatomical abnormalities or elevated rates of mortality. 27 In hematopoietic stem cells (HSC), the absence of CDK6 leads to prolonged exit from quiescence resulting in delayed stem cell activation. 28 , 29 Immature Cdk6‐deficient thymocytes fail to undergo ordinary expansion, which is caused by reduced proliferation and increased apoptosis during Notch‐dependent T‐cell development. Additionally, Cdk6 −/− mice are completely resistant to Akt‐driven lymphomagenesis. The critical role of CDK6 in Notch‐Akt‐dependent T‐cell development and tumorigenesis cannot be compensated by CDK4. 30
Analysis of a mouse model expressing a CDK6 kinase‐dead allele (Cdk6 K43M), largely resembled the impact of Cdk6 loss on T‐cell development and resulted in reduced proliferation of thymocytes, hematopoietic stem and progenitor cells as well as defective Notch signaling. Differences between the complete loss of CDK6 and the inactivation of its kinase function were restricted to more mature thymocyte subsets, where the presence of the Cdk6 K43M allele significantly increased double‐negative (DN) T‐cells. In contrast, mice expressing a hyperactive CDK6 knock‐in mutation (R31C), which is resistant to INK4 inhibition, display elevated levels of thymocytes and Lin−Sca‐1+c‐Kit+ (LSK) progenitor cells. 31
4. CDK6 IN HEMATOLOGIC MALIGNANCIES
Components of the CDK6‐Cyclin D complexes are frequently altered in hematological malignancies. Overexpression of CDK6 has been reported in T‐cell lymphoblastic lymphoma and leukemia (T‐LBL or T‐ALL) and in B‐lymphoid malignancies. Although no mutations in CDK6 are documented, sporadic cases with chromosomal translocation involving CDK6 were identified. 32 , 33 , 34 , 35 Lately, a prosurvival impact on T‐ALL cells through regulatory metabolic functions of CDK6 has been demonstrated. 36 Moreover, Jena et al provided evidence that a CDK6 kinase‐dependent repression of CD25 is required to induce and maintain Notch1‐induced T‐ALL. Hematopoietic progenitor cells lacking CDK6 or its kinase‐activity, due to a knock‐in mutation or CDK4/6 inhibitory treatment, were resistant to leukemia induction by activated Notch. This underlines the critical role of CDK6 as a downstream target of Notch in leukemogenesis. 37
In myeloid leukemia CDK6—but not CDK4—has been identified as a critical effector, required for progression of mixed‐lineage leukemia (MLL)‐rearranged acute myeloid leukemia (AML). CDK6 expression is regulated through direct binding of MLL‐AF9 to the Cdk6 locus and the resulting high CDK6 levels block myeloid differentiation and result in an immature phenotype. Pharmacological inhibition of CDK6 by the CDK4/6 inhibitor PD‐0332991 (palbociclib), unlocks the blocked differentiation and reduces the leukemic phenotype in human AML cell lines. These findings proposed that CDK6 inhibition provides a clinically applicable therapeutic opportunity for MLL‐rearranged leukemia. 38 A proliferation advantage of leukemic cells through MLL fusion‐driven upregulation of CDK6 has also been shown in MLL‐rearranged infant ALL. 39
Studies in break point cluster region/ABL (BCR/ABL) transformed murine leukemia/lymphoma models demonstrated that enhanced expression of CDK6 may provoke accelerated as well as reduced proliferation depending on the presence or absence of the tumor suppressor protein p16INK4a. The CDK6 induced p16INK4a expression is considered an internal safeguard mechanism that blocks accelerated cell proliferation to prevent tumor progression. The p16INK4a expression is induced by binding of CDK6, in complex together with signal transducer and activator of transcription 3 (STAT3) and Cyclin D, to the p16 INK4a promoter. Despite the presence of D type cyclins on the promoter, the transcriptional function of CDK6 is independent of its kinase function, can be induced by a kinase‐dead version of CDK6 and is not shared by CDK4. Only in the absence of p16INK4a, CDK6 fulfills its role as a proto‐oncogene and accelerates tumor progression. Consistent with these results, an inverse correlation of CDK6 and p16INK4a has been detected in most human lymphoid malignancies. 40 , 41 , 42
Besides induction of p16INK4A, CDK6 regulates and induces important proto‐oncogenes including vascular endothelial growth factor A (VEGFA), fms‐related tyrosine kinase 3 (FLT3), aurora kinase (AURK) and AKT, that are crucial for survival, proliferation and angiogenesis in ALL and AML. 41 , 42 , 43 , 44 , 45 (Figure 1) A role for CDK6 has been uncovered in hematopoietic stem cells (HSCs) and leukemic stem cells (LSCs) over the last years. Upon stress, CDK6 is required to release stem cells from quiescence by suppression of early growth response protein 1 (EGR1) expression. The CDK6‐mediated downregulation of EGR1 is mediated by binding of CDK6 to the promoter and suppression occurs in a kinase‐independent manner to allow activation of HSC and LSCs. The dependency of LSCs on CDK6 was shown in a bone marrow transplantation model where Cdk6 −/− BCR‐ABLp210+ LSKs were unable to efficiently induce disease. 28 , 46
CDK6 is not only important upon establishment of leukemia but also determines the reaction to oncogene‐induced stress. Bellutti et al showed that CDK6 acts as a prosurvival factor for preleukemic cells and is required to antagonize oncogene‐induced activation of the tumor protein 53 (TP53), also known as p53. During transformation and immortalization, CDK6 induces a complex transcriptional program comprising a variety of genes counteracting p53 functions including protein arginine methyltransferase 5 (Prmt5), transformed mouse 3T3 cell double minute 4 (Mdm4) and protein phosphatase 1D magnesium‐dependent, delta isoform (Ppm1d). In the absence of CDK6, lymphoid cells are forced to mutate or delete p53 to overcome oncogene‐induced stress. Analysis of patient samples suffering from ALL, AML and myelodysplastic syndrome (MDS) confirmed the inverse correlation of CDK6 and p53 mutations. 47 , 48
5. CDK6 IN BREAST CANCER
In breast cancer, as well as normal breast epithelium, D‐type cyclins and their binding partner kinases are key regulators of cell cycle progression, tumor formation and proliferation. The activity and expression of CDK4/6 and cyclin D is regulated and influenced by several mitogenic signaling pathways including the estrogen receptor, receptor tyrosine kinase (RTK) signaling pathways and the downstream PI3K‐AKT‐mTOR or RAS‐RAF‐MEK‐ERK routes. 49 , 50 , 51 , 52
Hyperactivation and dysregulation of components of the cyclin D‐CDK4/6 axis are common in breast cancer rendering them attractive targets for therapeutic interference. 53 , 54 Not surprisingly, the CDK4/6 inhibitors palbociclib (PD0332991), ribociclib (LEE011) and abemaciclib (LY835219) have been approved by the US Food and Drug Administration (FDA) for the treatment of hormone receptor‐positive breast cancer. Treatment of patients with advanced breast cancer is guided by the hormone receptor (HR) status (estrogen and progesterone), and by the expression status of the tyrosine kinase human epidermal growth factor receptor 2 (HER2). In case of metastatic HR‐positive, HER2‐negative breast cancer patients, combinatorial treatment of CDK4/6 inhibitors with endocrine therapy, like letrozole or fulvestrant, significantly prolonged progression‐free survival (PFS). Preclinical and clinical trials proved the beneficial effect of combining CDK4/6 inhibitors with drugs that reduce the estrogen levels, such as aromatase inhibitors like letrozole, or target the estrogen receptor, like tamoxifen or fulvestrant. 55 , 56 , 57 , 58 Strikingly, abemaciclib has also been approved as monotherapy and showed promising results as a single agent in HR+/HER2− metastatic breast cancer patients. 59 The prolonged exposure of MCF‐7 breast cancer cell lines to abemaciclib provokes drug resistance accompanied by increased CDK6 expression. Similarly, the enforced expression of CDK6 in drug‐sensitive cells was sufficient to mediate abemaciblib resistance and was paralleled by reduced ER and PR receptor expression, a phenomenon also observed in patients. Vice versa, knockdown of CDK6 restored sensitivity to the CDK4/6 inhibitor. 54
6. CDK6 AND IMMUNE FUNCTIONS
The effect of CDK4/6 inhibitors goes far beyond cell cycle arrest. One additional effect is that CDK4/6 inhibitors trigger antitumor immunity. Goel et al reported two mechanisms that account for the enhanced antitumor responses in breast cancer. CDK4/6 inhibition reduces expression of the DNA methyltransferase 1 (DNMT1) resulting in a subsequent DNA hypomethylation. 60 (Figure 3) This leads to the upregulation of endogenous retroviral elements, the expression of double‐stranded RNA, a prompt type III interferon (IFN) production and enhanced antigen production resulting in enhanced tumor cell killing. Moreover, CDK4/6 inhibition preferentially reduces the proliferation of immunosuppressive regulatory T cells (TReg). Both effects are mediated by the suppression of the RB‐E2F axis. 60 Inhibition of CDK4/6 increases interleukin 2 (IL2) secretion of effector T‐cells via an upregulation of the nuclear factor of activated T‐cells (NFAT), which further enhances antitumor activity. NFAT4, which belongs to a family of transcription factors critical for T‐cell activation, is a direct substrate of CDK6, but not CDK4. Inhibition of CDK4/6 decreases the phosphorylation of NFAT, which enhances its nuclear translocation and increases its transcriptional activity. 61 (Figure 3) A distinct beneficial mechanism of CDK4/6 inhibition has been reported by Jerby‐Arnon et al in melanoma cells, who showed that inhibition represses a cancer cell‐induced program that is associated with T cell exclusion and immune evasion. 62 On the other hand, Zhang et al showed that CyclinD‐CDK4 together with the cullin3‐speckle‐type POZ protein (SPOP) E3 ligase negatively regulates programmed cell death 1 ligand 1 (PD‐L1) protein stability. PD‐L1 expression levels inversely correlate with CDK4 activity and treatment of tumor cells with CDK4/6 inhibitors increases PD‐L1 protein levels, enabling enhanced tumor evasion. Hence, high PD‐L1 expression on tumor cells goes along with inhibited T cell activation, allowing tumor cells to escape antitumor immune responses. On the other side of the coin, these enforced PD‐L1 levels allow higher sensitivity to immune checkpoint inhibitors (Figure 3). 63 , 64
FIGURE 3.
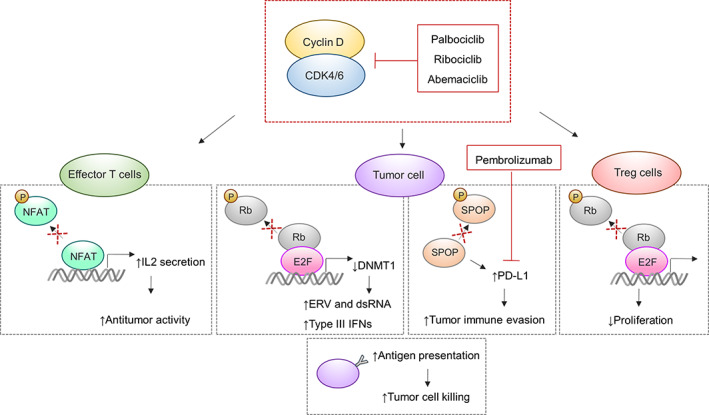
Effects of CDK4/6 inhibition on antitumor immunity. In tumor cells, inhibition of CDK4/6 leads to reduced DNA methyltransferase 1 (DNMT1) expression levels resulting in elevated antigen presentation capacity through upregulation of endogenous retroviral elements (ERV). Additionally, programmed cell death 1 ligand 1 (PD‐L1) levels are upregulated upon inhibition causing sensitivity of tumor cells to immune checkpoint inhibitors, such as pembrolizumab. In effector T cells, CDK4/6 inhibition promotes nuclear factor of activated T‐cells (NFAT) activity, a transcription factor critical for T‐cell activation. Finally, suppression of CDK4/6 causes reduced proliferation of immunosuppressive regulatory T‐cells (Treg)
Collectively, these data point at beneficial effects upon combinatorial treatment of CDK4/6 inhibitors with immune checkpoint inhibitors. in vivo experiments verified synergistic effects of CDK4/6 inhibition with programmed cell death 1 (PD‐1) blockade, which lead to elevated tumor regression and better overall survival rates. 61 , 63 Safety and efficacy of the CDK4/6 inhibitor abemaciclib in combination with pembrolizumab (PD‐1 inhibitor) is currently investigated in a phase 1b study for patients with HR+/HER2‐ metastatic breast cancer or advanced nonsmall cell lung cancer (NSCLC) (NCT02779751). 65
7. CDK6 IN MELANOMA
Melanoma is characterized by a high mutational load, which includes and leads to deregulated expression of a panel of cell cycle regulatory proteins. Inhibitory mutations in the tumor suppressor p16INK4a are a frequent event in primary melanoma samples and melanoma cell lines. Activating germline mutations have been described in CDK4 in families that suffer from hereditary melanoma. These mutation types highly increase the risk of developing melanoma. 66 , 67 , 68 , 69 , 70 Combined overexpression of KIT and CDK4 has been found in a subgroup of melanomas. 71 Several preclinical studies verified the sensitivity for melanoma cells to CDK4/6 inhibition in vivo and in vitro. CDK4, cyclin‐dependent kinase inhibitor 2A (CDKN2A) and RB1 expression levels are predictors of sensitivity. 72 , 73 , 74 A recent study points at CDK4 and CDK6 as regulators of cell‐proliferation, migration and tumor‐angiogenesis in melanoma. The tight balance/equilibrium between CDK4 and CDK6 expression controls the transcriptional activity of CDK6 to regulate protumorigenic genes, including VEGF. The strong correlation between CDK6 levels and enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) target gene expression in human melanoma samples, underscores the proangiogenic role of CDK6 in melanoma as EZH2 is regulated by VEGF. 75 , 76 CDK6 was also found to directly phosphorylate EZH2. 77 We thus postulate a dual influence of CDK6 on VEGF in melanoma.
Anders et al describe the forkhead box M1 (FOXM1) transcription factor as a substrate of CDK4/6‐Cyclin D complexes. CDK4/6 phosphorylate FOXM1 at multiple sites and thereby regulates its stabilization and activation. As FOXM1 protects tumor cells from senescence, by reducing reactive oxygen species (ROS), a link from CDK4/6 kinase activity to senescence induction was postulated (Figure 1). Support for this idea stems from the fact that melanoma cells show increased signs of senescence upon palbociclib treatment. CDK4/CDK6 inhibition in combination with a mitogen‐activated protein kinase (MEK) inhibitor leads to apoptosis as well as cell cycle arrest and show synergistic therapeutic efficacy in a NRAS mutant melanoma mouse model. 78 As a consequence CDK4/CDK6 inhibitors are used in clinical studies including combinations with mitogen‐activated protein kinase (MEK) and BRAF inhibitors. 79 , 80
8. CONCLUSION AND OUTLOOK
Initially identified as cell cycle regulating kinases that enable cell cycle progression, CDK6 and its close homolog CDK4 have attracted substantial attention in cancer research over the last years. Altered expression and dysregulated function have made them attractive targets for pharmacological inhibitors in various cancer types. Dual CDK4/6 inhibitors, that target the ATP‐binding pocket of the kinase, have revolutionized treatment of patients with breast cancer and showed first promising results in hematological malignancies and melanoma. None of these approved small molecule inhibitors distinguishes between CDK6 and CDK4, as they share 94% homology in their ATP‐binding pocket. 81
Moreover, kinase‐independent functions, as described for the transcriptional regulator CDK6, are not targeted by dual CDK4/6 inhibitors. 28 , 42 To address this issue, different selective CDK6 degraders have been designed lately and showed promising in‐vitro results. 81 , 82 , 83 , 84 , 85 , 86 CDK6‐selective proteolysis‐targeting chimeras (PROTACS) remarkably reduced leukemia burden in mice injected with patient‐derived Philadelphia‐positive (Ph+) ALL. 83
The possibility to selectively degrade CDK6 in cancer patients could be a strategy to restore drug sensitivity or even prevent drug resistance, which is seen after prolonged CDK4/6 inhibitory treatment. 54 We also propose that compounds, that distinguish between CDK6 and CDK4, would be less harmful to normal hematopoietic progenitor cells and offer novel therapeutic options. 28 , 83 CDK6 specific drugs may interfere with transcriptional responses downstream of CDK6 while leaving proliferation intact which might be exploited in drug combinations using chemotherapy. First studies propose a benefit of chemotherapy and CDK4/6 inhibition depending on the sequential administration. 87 Although the versatile function of CDK6 in disease and tumor progression has been investigated in depth over the last decades, further studies are needed to improve selective cancer treatment and find novel effective synergistic combinations to prevent drug tolerance and resistance.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENT
This work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program grant agreement No 694354.
Nebenfuehr S, Kollmann K, Sexl V. The role of CDK6 in cancer. Int. J. Cancer. 2020;147:2988–2995. 10.1002/ijc.33054
Funding information European Research Council (ERC), Grant/Award Number: 694354
REFERENCES
- 1. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153‐166. [DOI] [PubMed] [Google Scholar]
- 2. Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910‐917. [DOI] [PubMed] [Google Scholar]
- 3. Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D‐type cyclin‐dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066‐2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tigan A‐S, Bellutti F, Kollmann K, Tebb G, Sexl V. CDK6—a review of the past and a glimpse into the future: from cell‐cycle control to transcriptional regulation. Oncogene. 2016;35:3083‐3091. [DOI] [PubMed] [Google Scholar]
- 5. Labaer J, Garrett MD, Stevenson LF, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847‐862. [DOI] [PubMed] [Google Scholar]
- 6. James MK, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin D‐cdk4 inhibitory activity. Mol Cell Biol. 2008;28:498‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blain SW, Montalvo E, Massagué J. Differential interaction of the cyclin‐dependent kinase (CDK) inhibitor p27(Kip1) with cyclin A‐Cdk2 and cyclin D2‐Cdk4. J Biol Chem. 1997;272:25863‐25872. [DOI] [PubMed] [Google Scholar]
- 8. Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin‐dependent kinase Cdk6 by the tumour suppressor p16(INK4a). Nature. 1998;395:237‐243. [DOI] [PubMed] [Google Scholar]
- 9. Cheng M, Olivier P, Diehl JA, et al. The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D‐dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571‐1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev. 1999;13:1501‐1512. [DOI] [PubMed] [Google Scholar]
- 11. Choi YJ, Anders L. Signaling through cyclin D‐dependent kinases. Oncogene. 2014;33:1890‐1903. [DOI] [PubMed] [Google Scholar]
- 12. Brotherton DH, Dhanaraj V, Wick S, et al. Crystal structure of the complex of the cyclin D‐dependent kinase Cdk6 bound to the cell‐cycle inhibitor p19(INK4d). Nature. 1998;395:244‐250. [DOI] [PubMed] [Google Scholar]
- 13. Jeffrey PD, Tong L, Pavletich NP. Structural basis of inhibition of CDK‐cyclin complexes by INK4 inhibitors. Genes Dev. 2000;14:3115‐3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hallett ST, Pastok MW, Morgan RML, et al. Differential regulation of G1 CDK complexes by the Hsp90‐Cdc37 chaperone system. Cell Rep. 2017;21:1386‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verba KA, Wang RYR, Arakawa A, et al. Atomic structure of Hsp90‐Cdc37‐Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science. 2016;352:1542‐1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shao J, Prince T, Hartson SD, Matts RL. Phosphorylation of serine 13 is required for the proper function of the Hsp90 co‐chaperone, Cdc37. J Biol Chem. 2003;278:38117‐38120. [DOI] [PubMed] [Google Scholar]
- 17. Li T, Jiang HL, Tong YG, Lu JJ. Targeting the Hsp90‐Cdc37‐client protein interaction to disrupt Hsp90 chaperone machinery. J Hematol Oncol. 2018;11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of Hsp90‐client interactions reveals principles of substrate recognition. Cell. 2012;150:987‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oberoi J, Dunn DM, Woodford MR, et al. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc Natl Acad Sci USA. 2016;113:9009‐9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guiley KZ, Stevenson JW, Lou K, et al. p27 allosterically activates cyclin‐dependent kinase 4 and antagonizes palbociclib inhibition. Science. 2019;366:eaaw2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Young RJ, Waldeck K, Martin C, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27:590‐600. [DOI] [PubMed] [Google Scholar]
- 22. Finn RS, Crown JP, Lang I, et al. The cyclin‐dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first‐line treatment of oestrogen receptor‐positive, HER2‐negative, advanced breast cancer (PALOMA‐1/TRIO‐18): a randomised phase 2 study. Lancet Oncol. 2015;16:25‐35. [DOI] [PubMed] [Google Scholar]
- 23. Fröhling S, Agrawal M, Jahn N, et al. CDK4/6 inhibitor Palbociclib for treatment of KMT2A‐rearranged acute myeloid Leukemia: interim analysis of the AMLSG 23‐14 trial. Blood. 2016;128(22):1608‐1608. [Google Scholar]
- 24. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin‐deficient diabetes and Cdk4 activation results in β‐islet cell hyperplasia. Nat Genet. 1999;22:44‐54. [DOI] [PubMed] [Google Scholar]
- 26. Moons DS, Jirawatnotai S, Parlow AF, Gibori G, Kineman RD, Kiyokawa H. Pituitary hypoplasia and Lactotroph dysfunction in mice deficient for cyclin‐dependent Kinase‐4. Endocrinology. 2002;143:3001‐3008. [DOI] [PubMed] [Google Scholar]
- 27. Malumbres M, Sotillo R, Santamaría D, et al. Mammalian cells cycle without the D‐type cyclin‐dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493‐504. [DOI] [PubMed] [Google Scholar]
- 28. Scheicher R, Hoelbl‐Kovacic A, Bellutti F, et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 2015;125(1):90‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laurenti E, Frelin C, Xie S, et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. 2015;16(3):302‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu MG, Deshpande A, Enos M, et al. A requirement for cyclin‐dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009;69:810‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu MG, Deshpande A, Schlichting N, et al. CDK6 kinase activity is required for thymocyte development. Blood. 2011;117:6120‐6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chilosi M, Doglioni C, Yan Z, et al. Differential expression of cyclin‐dependent kinase 6 in cortical thymocytes and T‐cell lymphoblastic lymphoma/leukemia. Am J Pathol. 1998;152:209‐217. [PMC free article] [PubMed] [Google Scholar]
- 33. Brito‐Babapulle V, Gruszka‐Westwood AM, Platt G, et al. Translocation t(2;7)(p12;q21‐22) with dysregulation of the CDK6 gene mapping to 7q21‐22 in a non‐Hodgkin's lymphoma with leukemia. Haematologica. 2002;87:357‐362. [PubMed] [Google Scholar]
- 34. Chen D, Law ME, Theis JD, et al. Clinicopathologic features of CDK6 translocation‐associated B‐cell lymphoproliferative disorders. Am J Surg Pathol. 2009;33:720‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hayette S, Tigaud I, Callet‐Bauchu E, et al. In B‐cell chronic lymphocytic leukemias, 7q21 translocations lead to overexpression of the CDK6 gene. Blood. 2003;102:1549‐1550. [DOI] [PubMed] [Google Scholar]
- 36. Wang H, Nicolay BN, Chick JM, et al. Suski JM1,2, Keibler MA, Sicinska E, Gerdemann U, Haining WN, Roberts TM, Polyak K, Gygi SP, Dyson NJ, Sicinski P. the metabolic function of cyclin D3‐CDK6 kinase in cancer cell survival. Nature. 2017;546:426‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jena N, Sheng J, Hu JK, et al. CDK6‐mediated repression of CD25 is required for induction and maintenance of Notch1‐induced T‐cell acute lymphoblastic leukemia. Leukemia. 2016;30:1033‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Placke T, Faber K, Nonami A, et al. Requirement for CDK6 in MLL‐rearranged acute myeloid leukemia. Blood. 2014;124:13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van der Linden MH, Willekes M, van Roon E, et al. MLL fusion‐driven activation of CDK6 potentiates proliferation in MLL‐rearranged infant ALL. Cell Cycle. 2014;13:834‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Otto T, Sicinski P. The kinase‐independent, second life of CDK6 in transcription. Cancer Cell. 2013;24:141‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kollmann K, Sexl V. CDK6 and p16INK4A in lymphoid malignancies. Oncotarget. 2013;4:1858‐1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kollmann K, Heller G, Schneckenleithner C, et al. A kinase‐independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013;24:167‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uras IZ, Walter GJ, Scheicher R, et al. Palbociclib treatment of FLT3‐ITD 1 AML cells uncovers a kinase‐dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood. 2016;27:2890‐2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hydbring P, Malumbres M, Sicinski P. Non‐canonical functions of cell cycle cyclins and cyclin‐dependent kinases. Nat Rev Mol Cell Biol. 2016;17:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Uras IZ, Maurer B, Nebenfuehr S, Zojer M, Valent P, Sexl V. Therapeutic vulnerabilities in FLT3‐mutant aml unmasked by palbociclib. Int J Mol Sci. 2018;19:3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guzman ML. CDK6 is a regulator of stem cells ‘Egr’ to wake up. Blood. 2015;125:7‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bellutti F, Tigan A‐S, Nebenfuehr S, et al. CDK6 antagonizes p53‐induced responses during tumorigenesis. Cancer Discov. 2018;8:884‐897. 10.1158/2159-8290.CD-17-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nebenfuehr S, Bellutti F, Sexl V. Cdk6: at the interface of Rb and p53. Mol Cell Oncol. 2018;5:e1511206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase‐3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499‐3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vora SR, Juric D, Kim N, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goel S, Wang Q, Watt AC, et al. Overcoming therapeutic resistance in HER2‐positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29:255‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Foster JS, Wimalasena J. Estrogen regulates activity of cyclin‐dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol Endocrinol. 1996;10:488‐498. [DOI] [PubMed] [Google Scholar]
- 53. Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23‐32. [DOI] [PubMed] [Google Scholar]
- 54. Yang C, Li Z, Bhatt T, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36:2255‐2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Finn RS, Martin M, Rugo HS, et al. Palbociclib and Letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925‐1936. [DOI] [PubMed] [Google Scholar]
- 56. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor‐positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone‐receptor‐positive, HER2‐negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA‐3): final analysis of the multicentre, double‐blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17:425‐439. [DOI] [PubMed] [Google Scholar]
- 58. Pernas S, Tolaney SM, Winer EP, Goel S. CDK4/6 inhibition in breast cancer: current practice and future directions. Ther Adv Med Oncol. 2018;10:1758835918786451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dickler MN, Tolaney SM, Rugo HS, et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, n patients with refractory HR+/HER2‐ metastatic breast cancer. Clin Cancer Res. 2017;23:5218‐5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goel S, Decristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti‐tumour immunity. Nature. 2017;548:471‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Deng J, Wang ES, Jenkins RW, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T‐cell activation. Cancer Discov. 2018;8:216‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jerby‐Arnon L, Shah P, Cuoco MS, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang J, Bu X, Wang H, et al. Cyclin D‐CDK4 kinase destabilizes PD‐L1 via cullin 3‐SPOP to control cancer immune surveillance. Nature. 2018;553:91‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.A Study of Abemaciclib (LY2835219) in Participants With Non‐Small Cell Lung Cancer or Breast Cancer [Full Text View]. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/study/NCT02779751. Accessed February 18, 2020.
- 66. Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15‐21. [DOI] [PubMed] [Google Scholar]
- 67. Kamb A, Shattuck‐Eidens D, Eeles R, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:22‐26. [DOI] [PubMed] [Google Scholar]
- 68. Sharpless NE, Chin L. The INK4α/ARF locus and melanoma. Oncogene. 2003;22:3092‐3098. [DOI] [PubMed] [Google Scholar]
- 69. Read J, Wadt KAW, Hayward NK. Melanoma genetics. J Med Genet. 2015;53:1‐14. [DOI] [PubMed] [Google Scholar]
- 70. Meyle KD, Guldberg P. Genetic risk factors for melanoma. Hum Genet. 2009;126:499‐510. [DOI] [PubMed] [Google Scholar]
- 71. Bartkova J, Lukas J, Guldberg P, et al. The p16‐cyclin D/Cdk4‐pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 1996;56:5475‐5483. [PubMed] [Google Scholar]
- 72. Mælandsmo GM, Flørenes VA, Hovig E, et al. Involvement of the pRb/p16/cdk4/cyclin D1 pathway in the tumorigenesis of sporadic malignant melanomas. Br J Cancer. 1996;73:909‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smalley KSM, Contractor R, Nguyen TK, et al. Identification of a novel subgroup of melanomas with KIT/cyclin‐dependent kinase‐4 overexpression. Cancer Res. 2008;68:5743‐5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen P, Lee NV, Hu W, et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol Cancer Ther. 2016;15:2273‐2281. [DOI] [PubMed] [Google Scholar]
- 75. Kollmann K, Briand C, Bellutti F, et al. The interplay of CDK4 and CDK6 in melanoma. Oncotarget. 2019;10:1346‐1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lu C, Han HD, Mangala LS, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18(2):185‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kwong LN, Costello JC, Liu H, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Romano G, Chen PL, Song P, et al. A preexisting rare PIK3CA e545k subpopulation confers clinical resistance to MEK plus CDK4/6 inhibition in NRAS melanoma and is dependent on S6K1 signaling. Cancer Discov. 2018;8:556‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sosman JA, Kittaneh M, Lolkema MPJK, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS ‐mutant melanoma: early encouraging clinical activity. J Clin Oncol. 2014;32:9009‐9009. [Google Scholar]
- 81. Brand M, Jiang B, Bauer S, et al. Homolog‐selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem Biol. 2019;26:300‐306.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhao B, Burgess K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun. 2019;55:2704‐2707. [DOI] [PubMed] [Google Scholar]
- 83. de Dominici M, Porazzi P, Xiao Y, et al. Selective inhibition of Ph‐positive ALL cell growth through kinase‐dependent and independent effects by CDK6‐specific PROTACs. Blood. 2020;135:1560‐1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rana S, Bendjennat M, Kour S, et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett. 2019;29:1375‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Su S, Yang Z, Gao H, et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J Med Chem. 2019;62:7575‐7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jiang B, Wang ES, Donovan KA, et al. Development of dual and selective degraders of cyclin‐dependent kinases 4 and 6. Angew Chem Int Ed. 2019;58:6321‐6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Salvador‐Barbero B, Álvarez‐Fernández M, Zapatero‐Solana E, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;37:340‐353. [DOI] [PubMed] [Google Scholar]