

Summary
By combining genomic data and brain imaging data, a recent study has identified a novel gene named FAM222A that participates in the formation of amyloid-β (Aβ) plaques and brain atrophy in Alzheimer's disease (AD). FAM222A encodes a 47-kDa protein designated Aggregatin that accumulates in the center of amyloid plaques and physically interacts with Aβ to facilitate Aβ aggregation. Aggregatin is expressed predominantly in the central nervous system (CNS) and its levels are increased in brains of the patients with AD and in mouse models of AD. However, at present, the precise cell types that express Aggregatin in the human CNS remain unknown. By immunohistochemistry, we studied Aggregatin expression in the frontal lobe of the patients with AD, Nasu-Hakola disease (NHD), and the subjects who died of non-neurological causes (NNC). We identified the clusters of Aggregatin-positive reactive astrocytes distributed widely in the cerebral cortex of most cases examined. In contrast, small numbers of cortical neurons showed variable immunoreactivities for Aggregatin, whereas microglia and oligodendrocytes did not express Aggregatin. Importantly, amyloid plaques were not clearly labelled with anti-Aggregatin antibody. These results suggest that Aggregatin plays a primarily role in generation of reactive astrocytes in the human CNS.
Keywords: Aggregatin, Alzheimer's disease, amyloid plaque, FAM222A, Nasu-Hakola disease, reactive astrocytes
1. Introduction
Alzheimer's disease (AD) is a progressive intractable dementia that chiefly affects elderly persons. Pathologically, it is characterized by the extracellular plaque deposits of the amyloid-β (Aβ) peptide and the intracellular accumulation of the neurofibrillary tangles composed of the microtubule binding protein tau, resulting in profound activation of microglia, reactive gliosis, and extensive neurodegeneration (1). Recently, by combining genomic data and brain imaging data, a research group has identified a novel gene named FAM222A that contributes to the formation of Aβ plaques and brain atrophy in AD (2). FAM222A encodes a single 47-kDa protein designated Aggregatin that highly accumulates in the center of amyloid plaques. Aggregatin physically interacts with Aβ via its N-terminal Aβ binding domain (NABD), and facilitates Aβ aggregation. Aggregatin cross-seeds Aβ via direct binding. Actually, in a direct binding assay, recombinant Aggregatin (rAggregatin) co-precipitates with Aβ1-40 and Aβ1-42, while immobilized Aβ1-40 or Aβ1-42 binds to rAggregatin. Recombinant NABD of Aggregatin by itself is capable of binding to amyloid deposits of Aβ1-42. Importantly, Aggregatin protein levels were upregulated in the brains of AD and mouse models of AD (2). Overexpression of Aggregatin in neurons enhances amyloid deposition and associated neuroinflammation, whereas deletion of Aggregatin inhibits amyloid deposition and subsequent neuroinflammation. Thus, Aggregatin acts as a potent seeding factor for Aβ oligomerization and aggregation. Reducing aberrant accumulations of Aggregatin might serve as a therapeutic approach for preventing progression of AD. However, at present, the precise cell types that express Aggregatin in AD and non-AD diseases remain unknown.
Nasu-Hakola disease (NHD), also designated polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; OMIM 221770), is a rare autosomal recessive disorder, characterized by progressive presenile dementia and formation of multifocal bone cysts, caused by loss-of-function mutations of either TYROBP(DAP12) or TREM2 (3). Clinically, the patients with NHD show recurrent bone fractures during the third decade of life, a frontal lobe syndrome during the fourth decade of life, and progressive dementia and death until the fifth decade of life (4). Pathologically, NHD brains exhibit extensive demyelination designated leukoencephalopathy, astrogliosis, accumulation of axonal spheroids, and remarkable activation of microglia predominantly in the white matter of frontal and temporal lobes and the basal ganglia (5). Although NHD patients are clustered in Japan and Finland, approximately 200 NHD cases are presently reported worldwide. Because TREM2 and DAP12 constitute a receptor/ adaptor signaling complex expressed exclusively on osteoclasts, dendritic cells, macrophages, and microglia, it is generally supposed that a complete loss of function of the TREM2-DAP12 signaling pathway in microglia induces NHD (6). Notably, a rare variant of the TREM2 gene encoding p.Arg47His causes a 3-fold increase in the risk for late-onset AD (7). Thus, neurodegenerative processes in NHD and AD share a common pathological mechanism of aberrant TREM2 signaling in microglia.
In the present study, we studied Aggregatin expression in the frontal lobe of AD, NHD, and non-neurological controls (NNC) by immunohistochemistry.
2. Materials and Methods
2.1. Human brain tissues
The brain autopsies were performed at the National Center Hospital, National Center of Neurology and Psychiatry (NCNP), Japan, Kohnodai Hospital, National Center for Global Health and Medicine (NCGM), Japan, and affiliated hospitals of Research Resource Network (RRN), Japan. The comprehensive examination by established neuropathologists (YS and TI) validated the pathological diagnosis. In all cases, written informed consent was obtained. The Ethics Committee of the NCNP for the Human Brain Research, the Ethics Committee of the NCGM on the Research Use of Human Samples, and the Human Research Ethics Committee of the Meiji Pharmaceutical University (MPU) approved the present study.
For immunohistochemical studies, serial sections of the frontal cortex were prepared from four subjects who died of non-neurological causes (NNC), composed of three men and one woman with a mean age of 79.0 ± 9.2 years, ten AD patients, composed of five men and five women with a mean age of 69.9 ± 8.2 years, and five NHD patients, composed of three men and two women with a mean age of 40.8 ± 6.1 years. The homozygous mutation of a single base deletion of 141G (c.141delG) in exon 3 of DAP12 was identified in three NHD patients. All AD cases were satisfied with the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria for diagnosis of definite AD (8). They were categorized into the stage C of amyloid deposition and the stage VI of neurofibrillary degeneration, following the Braak's staging (9).
2.2. Immunohistochemistry
After deparaffination, tissue sections were heated in 10 mM citrate sodium buffer, pH 6.0 by autoclave at 110℃ for 15 min in a temperature-controlled pressure chamber (Biocare Medical, Pacheco, CA, USA). They were treated at room temperature (RT) for 15 min with 3% hydrogen peroxide-containing methanol to block the endogenous peroxidase activity. They were then incubated with phosphate-buffered saline (PBS) containing 10% normal goat serum at RT for 15 min to block non-specific staining, followed by incubation in a moist chamber at 4℃ overnight with rabbit polyclonal anti-Aggregatin antibody (C12orf34; ab122626, Abcam, Cambridge, UK). The specificity of anti-Aggregatin antibody was validated by western blot analysis of recombinant human Aggregatin protein expressed in HEK293 cells, which were transfected with the pcDNA4/HisMax TOPO vector (Thermo Fisher Scientific, Carlsbad, CA, USA) containing the full-length Aggregatin sequence. The antigen sequence that anti-Aggregatin antibody recognizes is composed of PSIHSLLYQL NQQCQAPGAA PPACQGMAIP HPSPAKHGPV PSFPSMAYSA AAGLPDCRKG TELGQGATQA LTLAGAAKPA GYADSGLDYL LWPQKP, corresponding to amino acids 197-292 of human Aggregatin.
After washing with PBS, tissue sections were incubated at RT for 30 min with horseradish peroxidase (HRP)-conjugated secondary antibody (Nichirei, Tokyo, Japan), followed by incubation with diaminobenzidine tetrahydrochloride (DAB) substrate (Vector, Burlingame, CA, USA). Enhancement was not applied to DAB staining. They were processed for a counterstain with hematoxylin. Negative controls underwent all the steps except for exposure to primary antibody. In limited experiments, double immunolabeling was performed using rabbit anti-Aggregatin antibody in combination with mouse monoclonal antibodies against glial fibrillary acidic protein (GFAP) (GA5, Nichirei, for an marker of astrocytes), gp91phox (ab139371, Abcam, for a marker of microglia), apolipoprotein E (ApoE; ab1906, Abcam), phospho-tau (AT8, Thermo Fisher Scientific) or Aβ peptide (Aβ11-28; 12B2, Immunobiological Laboratories, Gunma, Japan), followed by incubation with HRP-conjugated or alkaline phosphatase-conjugated anti-rabbit or anti-mouse secondary antibody and exposure to DAB substrate and Warp Red chromogen (Biocare Medical).
2.3. RT-PCR analysis
Total cellular RNA was extracted from human neural cell lines and tissues with TRIZOL (Thermo Fisher Scientific). Pooled human frontal lobe total RNA (#636563, Clontech, Mountain View, CA, USA) was utilized to prepare human brain cDNA. RNA treated with DNase I was processed for cDNA synthesis by using oligo(dT)20 primers and SuperScript II reverse transcriptase (Thermo Fisher Scientific). PCR cocktails were prepared by mixing with sense and antisense primer sets following: 5'agcacggtggccagcaagtcccctgag3' and 5'ttatctgtagacgggtaggcggatgtg3' for detection of a 444 bp product of the FAM222A gene (NM_032829.3). Then, cDNA was amplified by PCR with HotStarTaq DNA Polymerase (Qiagen, Valencia, CA, USA) on a PC815 thermal cycler (Astec, Fukuoka, Japan) at 95℃ for 1 min for denaturation, at 62℃ for 40 sec for annealing, and 72℃ for 50 sec for extension for 35 cycles.
3. Results
First of all, we studied the expression of Aggregatin in human neural cell lines by RT-PCR analysis. We found that Aggregatin is ubiquitously expressed in various human neural cell lines, along with astrocytes in culture (Figure 1, lanes 1, 3-10), while we did not detect Aggregatin in cDNA samples of the human frontal lobe prepared without inclusion of reverse transcription step (Figure 1, lane 2). Next, we validated the specificity of anti-Aggregatin antibody ab122626 by western blot analysis of Xpress-tagged recombinant human Aggregatin protein expressed in HEK293 cells (Figure 2, panels a-c, lane 2). Then, by immunohistochemistry using ab122626, we identified an intense expression of Aggregatin immunoreactivity chiefly in reactive astrocytes forming clusters and surrounding glial scar distributed in the grey matter of the frontal cortex derived from AD and NHD patients (Figures 3, panels a-c and Figure 4, panel a).
Figure 1.
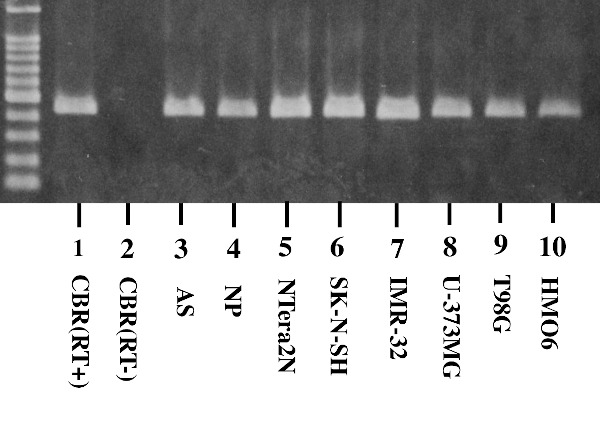
Aggregatin expression in human neural cell lines. The mRNA expression was studied by RT-PCR in human tissues and cultured cells. The lanes (1-10) indicate (1) the frontal cortex of the human cerebrum (CBR) with inclusion of the reverse transcription (RT) step, (2) CBR without inclusion of the RT step, (3) astrocytes (AS), (4) neuronal progenitor (NP) cells, (5) NTera2 teratocarcinoma-derived neurons (NTera2N), (6) SK-N-SH neuroblastoma, (7) IMR-32 neuroblastoma, (8) U-373MG glioblastoma, (9) T98G glioblastoma, and (10) HMO6 immortalized microglia. cDNA was amplified by PCR for 35 cycles.
Figure 2.
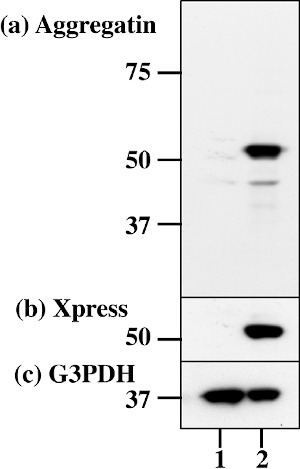
The specificity of Aggregatin antibody. Western blot of non-transfected HEK293 cells (lane 1) and the cells transfected with the vector containing the full-length Aggregatin sequence (lane 2). (a) Aggregatin (ab122626), (b) Xpress tag, and (c) G3PDH as a loading control. The position of molecular weight markers is shown on the left.
Figure 3.
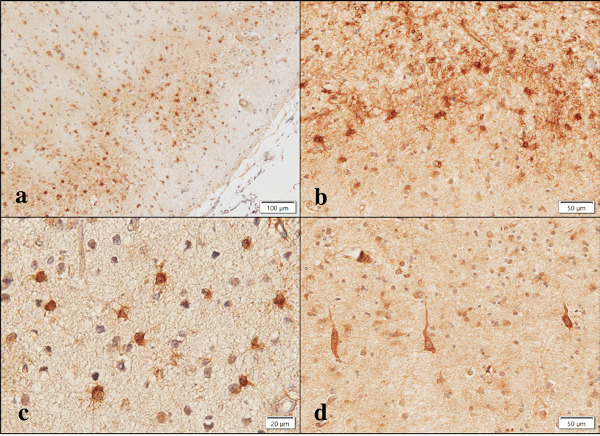
Aggregatin expression in AD brains. Aggregatin is expressed in (a) reactive astrocytes, (b) reactive astrocytes and glial scar, (c) reactive astrocytes at higher magnification, and (d) degenerating neurons.
Figure 4.
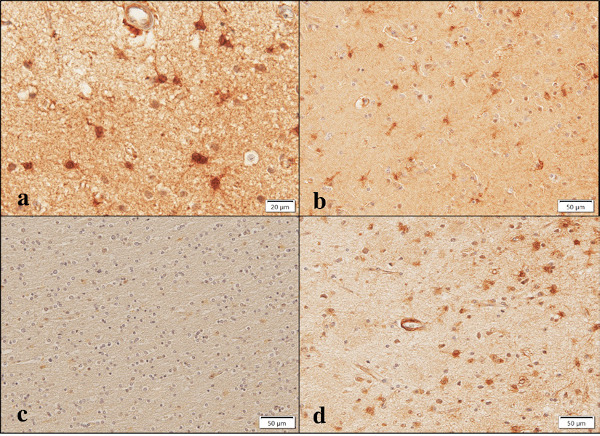
Aggregatin expression in NHD, NNC, and AD brains. Aggregatin is expressed in (a) reactive astrocytes at higher magnification, NHD, (b) reactive astrocytes, NNC, (c) no immunolabeling on oligodendrocytes, AD, and (d) a small blood vessel and reactive astrocytes, AD.
In addition, Aggregatin immunoreactivity was detectable in reactive astrocytes occasionally found in the brains of NNC (Figure 4, panel b). Aggregatin immunoreactivity was located in the cell soma and proximal processes of reactive astrocytes. By double immunolabelling, Aggregatin-positive reactive astrocytes expressed GFAP (Figure 5, panel b). Some of the Aggregatin-expressing astrocytes showed a binuclear morphology, suggesting that they underwent active proliferation (Figure 6, panel a, arrows).
Figure 5.

Aggregatin expression in AD brains. (a) double immunolabeling of Aggregatin (brown) and gp91phox (red), (b) double immunolabeling of Aggregatin (brown) and GFAP (red), (c) the negative control without inclusion of the primary antibody, and (d) double immunolabeling of Aggregatin (brown) and Aβ (red).
Figure 6.
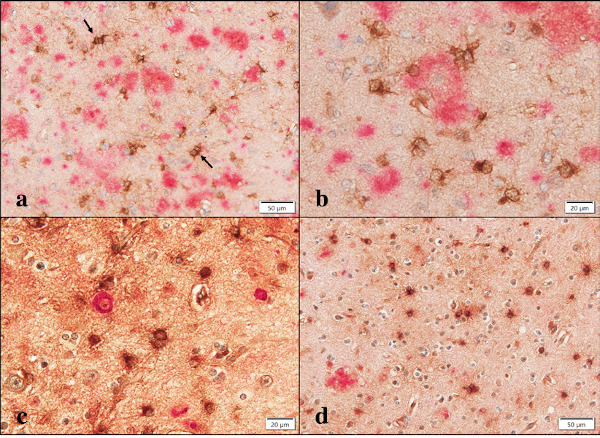
Aggregatin expression in AD brains. (a) double immunolabeling of Aggregatin (brown) and Aβ (red). Arrows indicate reactive astrocytes with a binuclear morphology, (b) double immunolabeling of Aggregatin (brown) and Aβ (red) at higher magnification. The center of amyloid plaque is devoid of Aggregatin immunoreactivity, (c) double immunolabeling of Aggregatin (brown) and AT8 (red) at higher magnification. AT8-expressing intraneuronal inclusions are negative for Aggregatin immunoreactivity, and (d) double immunolabeling of Aggregatin (brown) and ApoE (red). Aggregatin-expressing reactive astrocytes are negative for ApoE immunoreactivity.
In contrast, surviving oligodendrocytes distributed in the white matter and gp91phox-positive microglia in AD brains did not display Aggregatin (Figure 4, panel c and Figure 5, panel a), while we found inconsistent staining of blood vessel walls and variable immunoreactivities for a subset of cortical degenerating neurons (Figure 3, panel d and Figure 4, panel d). No immunoreactivity was detected, when the primary antibody was omitted from the reaction (Figure 5, panel c). Although reactive astrocytes forming clusters were often closely associated with amyloid plaques, the plaques themselves were not clearly labelled with anti-Aggregatin antibody (Figure 5, panel d and Figure 6, panel a, b). Furthermore, the centers of amyloid plaques were devoid of Aggregatin immunoreactivity (Figure 6, panel b). By double immunolabelling, phospho-tau-positive intraneuronal inclusions in AD brains were not stained with anti- Aggregatin antibody (Figure 6, panel c). In addition, the majority of Aggregatin-immunoreactive astrocytes in AD brains did not express ApoE (Figure 6, panel d).
4. Discussion
By combining genomic data and brain imaging data, a recent study has identified a novel gene named FAM222A encoding Aggregatin involved in the formation of Aβ plaques and brain atrophy in AD (2). The authors found that Aggregatin is concentrated on the center of amyloid plaques and physically interacts with Aβ to facilitate Aβ aggregation. However, the previous study did not characterize the precise cell types expressing Aggregatin in the central nervous system (CNS). By immunohistochemistry, we found an intense immunoreactivity for Aggregatin expressed predominantly in reactive astrocytes in AD, NHD, and NNC brains. Therefore, we supposed that Aggregatin plays a pivotal role in generation of reactive astrocytes in the human CNS. A subpopulation of degenerating neurons also expressed Aggregatin in AD and NHD brains, suggesting that Aggregatin expression is not purely specific for astrocytes. In contrast, amyloid plaques and cores were negative for Aggregatin immunoreactivity in most AD cases. An obvious difference in the results of the previous study and the present study is in part attributable to different tissue sections and immunohistochemical protocols utilized, although the primary antibody tested (ab122626) is the same in both studies (2). Importantly, human astrocytes in culture exhibited Aggregatin by RT-PCR, being consistent with our observations.
Astrocytes represent the most numerous cell type uniformly distributed throughout the CNS. Astrocytes play a pivotal role in various physiological processes, including glutamate homeostasis, blood-brain barrier permeability, metabolic support of neurons, and synaptic development and plasticity (10). Reactive astrocytes are induced by exposure to various stressful insults in the brain, such as brain injury, ischemia, inflammation, and neurodegeneration, associated with marked upregulation of GFAP, vimentin, and nestin, those of which represent the markers of astrocyte reactivity (10). Recent studies indicate that reactive astrocytes are composed of two distinct subtypes with different pathological properties designated neurotoxic A1 and neuroprotective A2 astrocytes (11). Numerous A1 astrocytes induced by activated microglia accumulates in AD brains (11), suggesting that Aggregatin-expressing reactive astrocytes surrounding amyloid plaques belong mostly to A1.
In NHD brains, along with AD brains, Aggregatin expression was actively induced in reactive astrocytes. NHD is pathologically characterized by extensive demyelination and profound astrogliosis (5). The molecular mechanisms underlying upregulation of Aggregatin in reactive astrocytes are overlapping between NHD and AD, and even in aged NNC.
Under neurodegenerative and neuroinflammatory circumstances, reactive astrocytes capable of producing a detectable amount of Aβ, constitute an integral component of amyloid plaques in AD (12). We found that Aggregatin-expressing reactive astrocytes are frequently associated with amyloid plaques in AD brains, as previously described (13). These results suggest that Aggregatin-expressing reactive astrocytes might contribute to the continuous production of amyloid plaques in AD.
Acknowledgements
The authors thank Drs. Kenji Jinnai, Nobutaka Arai, Kiyotaka Nakamagoe, Nobutaka Motohashi, and Saburo Yagishita for providing us brain samples. This work was supported by grants from the Research on Intractable Diseases, entitled "Clinicopathological and genetic studies of Nasu-Hakola disease" (H21-Nanchi- Ippan-201; H22-Nanchi-Ippan-136), the Ministry of Health, Labour and Welfare of Japan (JS), grants from the JSPS KAKENHI (C-16K07043 and C-20K07876) (JS), and the Dementia Drug Development Research Center (DRC) project (S1511016), the Ministry of Education, Culture, Sports, Science and Technology ( M E X T ) , J a p a n ( J S , Y K ) , a n d s u p p o r t e d b y JSPS KAKENHI (JP-16H06277) (YS), AMED JP18dm0107103 (YS), and Intramural Research Grant (30-8) for Neurological and Psychiatric Disorders of NCNP (YS).
References
- 1. Chen XQ, Mobley WC. Alzheimer disease pathogenesis: Insights from molecular and cellular biology studies of oligomeric Aβ and tau species. Front Neurosci. 2019; 13:659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yan T, Liang J, Gao J, Wang L, Fujioka H, The Alzheimer Disease Neuroimaging Initiative, Zhu X, Wang X. FAM222A encodes a protein which accumulates in plaques in Alzheimer's disease. Nat Commun. 2020; 11:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005; 64:1502-1507. [DOI] [PubMed] [Google Scholar]
- 4. Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG, da Silva Júnior JP, Neto Evaldo S, Poffo MA, Walz R, Carlotti Júnior CG, Sakamoto AC. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy - PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004; 24:1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Satoh J, Tabunoki H, Ishida T, Yagishita S, Jinnai K, Futamura N, Kobayashi M, Toyoshima I, Yoshioka T, Enomoto K, Arai N, Arima K. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology. 2011; 31:363-375. [DOI] [PubMed] [Google Scholar]
- 6. Xing J, Titus AR, Humphrey MB. The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: a molecular genetics perspective. Res Rep Biochem. 2015; 5:89-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013; 368:107-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991; 41:479-486. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006; 112:389-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Acosta C, Anderson HD, Anderson CM. Astrocyte dysfunction in Alzheimer disease. J Neurosci Res. 2017; 95:2430-2447. [DOI] [PubMed] [Google Scholar]
- 11. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017; 541:481-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol. 2017; 7:170228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang H-Y. Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003; 971:197-209. [DOI] [PubMed] [Google Scholar]