

Abstract
PfSERA5, a significantly abundant protein present within the parasitophorous vacuole (PV) and essential for normal growth during the blood‐stage life cycle of the malaria parasite Plasmodium falciparum, displays structural similarity to many other cysteine proteases. However, PfSERA5 does not exhibit any detectable protease activity and therefore the role of the PfSERA5 papain‐like domain (PfSERA5E), thought to remain bound to its cognate prodomain, remains unknown. In this study, we present a revised structure of the central PfSERA5E domain at a resolution of 1.2 Å, and the first structure of the “zymogen” of this papain‐like domain including its cognate prodomain (PfSERA5PE) to 2.2 Å resolution. PfSERA5PE is somewhat structurally similar to that of other known proenzymes, retaining the conserved overall folding and orientation of the prodomain through, and occluding, the archetypal papain‐like catalytic triad “active‐site” cleft, in the same reverse direction as conventional prodomains. Our findings are congruent with previously identified structures of PfSERA5E and of similar “zymogens” and provide a foundation for further investigation into the function of PfSERA5.
Keywords: crystal structure, egress, malaria, molecular dynamics, parasite, prodomain, protease
Short abstract
Abbreviations
- ACT
artemisinin combination therapy
- ASU
asymmetric unit
- DPAP‐1
dipeptidyl aminopeptidase 1
- DPAP‐3
dipeptidyl aminopeptidase 3
- MD
molecular dynamics
- MR
molecular replacement
- NCS
non‐crystallographic symmetry
- NPT
isothermal‐isobaric
- PfSERA5
Plasmodium falciparum serine repeat antigen 5
- PfSERA6
Plasmodium falciparum serine repeat antigen 6
- PfSUB1
Plasmodium falciparum subtilisin‐like protease 1
- PMIX
plasmepsin IX
- PMX
plasmepsin X
- PV
parasitophorous vacuole
- r.m.s.d.
root mean square deviation
- RBC
red blood cells
- SASA
solvent accessible surface area
1. INTRODUCTION
The increased prevalence of drug‐resistant Plasmodium falciparum malaria strains, limiting the effectiveness of current clinical artemisinin combination therapy (ACT) interventions, is a global health threat with significant implications to mortality. 1 Transmitted to humans by the female Anopheles mosquito during a blood meal, the protozoan malaria parasite subsequently causes fever, anemia, and respiratory distress that, if left untreated, can lead to death. 2 , 3 Although persistent effort is being focused on the design of a vaccine to prevent the disease, with some promising early success, 4 a highly efficacious vaccine remains elusive. 5
Upon entry into a human host, malaria parasite sporozoites migrate to the liver and invade liver hepatocytes, multiplying within to form mature schizonts. Upon rupturing, release of daughter merozoites leads to invasion of red blood cells (RBC) and replication via asexual reproduction. 2 , 3 , 6 The egress of merozoites is essential for replication, and is protease dependent. 7 Serine, cysteine and most recently aspartic protease inhibitors that are capable of arresting merozoite egress have been identified. 8 , 9 , 10 , 11 This process of schizogony during the asexual blood stage is the primary cause for the symptoms associated with the disease, and therefore inhibiting merozoite egress has been proposed as a drug target. 12 , 13
Cysteine and serine protease and protease‐like proteins, some of which accumulate inside the membrane‐bound parasitophorous vacuole (PV) during the late trophozoite and schizont stages, play integral roles in erythrocytic development and in egress 7 ; these include falcipain cysteine proteases, 14 subtilisin serine proteases, 15 , 16 the upstream plasmepsin aspartic proteases, 17 chiefly plasmepsins IX and X (PMIX and PMX), 10 and the serine repeat antigen (SERA) family of proteins. Named for the presence of consecutive serine residues present within the protein sequence, the occurrence of the multigene SERA family has evolved via instances of gene duplication, 18 resulting in the presence of a number of SERA members existing in all Plasmodium species. Although the size of the family differs between species, all SERA members contain a conserved central papain‐like domain. 18 , 19 Of the nine sequence similar members within P. falciparum (PfSERA1‐9), those of PfSERA6‐8 contain a conventional papain‐like domain, with an archetypal catalytic triad (cysteine, histidine and aspartate residues). The remainder (PfSERA1‐5 and ‐9), however, have this cysteine replaced by a serine. 20 , 21 , 22 Of the nine members, PfSERA5 and PfSERA6 are both essential to the normal blood stage asexual growth of the parasite, 23 with PfSERA5 one of the most abundantly expressed during egress. 24 , 25 The conventional cysteine proteases SERA7 and SERA8 are not required for normal blood‐stage growth. 23 Where deletion of PfSERA6 is lethal due to its essential role in RBC membrane rupture via targeted protease activity leading to cytoskeleton disassembly, 16 disruption of PfSERA5 results in significantly reduced efficiency, accelerated and subsequently premature merozoite egress. 26 It is therefore essential for normal and efficient blood stage growth.
Although the most studied of the family, the exact function of PfSERA5 is still poorly understood with its proteolytic activity being a contentious issue. Despite the high sequence and structural similarity to catalytically active papain‐like proteases, the presence of a serine in place of the cysteine (Ser596) at the putative “catalytic triad” appears to render PfSERA5 catalytically inactive; evidence of weak activity had been observed in early studies, 27 , 28 although more recent studies report a complete lack of activity. 29 Serine‐containing papain‐like proteases are unique to that of the SERA family. Nevertheless, truncation, deletion or significant alterations to either the “catalytic triad,” or the protein itself is not well tolerated. 29 Mutation of Ser596 to a cysteine results in an enzyme with protease activity, indicating PfSERA5 maintains the ability to bind and process peptide substrates, 29 placing yet further confusion as to the possible molecular function.
Initially identified as P126 due to its molecular weight, 30 , 31 PfSERA5 is cleaved via subtilisin‐like protease 1 (PfSUB1) at (least at) two distinct regions (387IKAE–TEDD394 and 883IFGQ–DTAG890) releasing three fragments; an N‐terminal 47 kDa (P47), a central 56 kDa (P56) and a C‐terminal 18 kDa domain (P18). 9 , 29 , 32 Further C‐terminal processing of the central domain occurs via an as yet unidentified protease yielding the 50 kDa (P50) central domain (Thr391‐Leu842). 27 , 29 Very early studies identified modest sequence similarity of the C‐terminal region of P50 with that of the papain‐like domains of cathepsin cysteine proteases, 19 with subsequent presumption that the N‐terminal region comprised an associated prodomain, analogous to that which exist in other papain‐like zymogens. 27 Prediction of the papain‐like domain via sequence comparison paved the way for the structural characterization of this central protease‐like domain (Glu560‐Asn828), determined to a resolution of 1.6 Å. 22 The structure of the 29 kDa, 251 residue, papain‐like domain indeed displays a high degree of structural similarity to other cysteine proteases, however, suggestions of the N‐terminal 22 kDa region comprising a prodomain, or that cleavage of this region occurred as a product of autocatalysis had yet to be determined. 27 There currently exists no structural insight into the 187 residue N‐terminal region (Thr391‐Asp577), although there has been an attempt to computationally model the structure of the zymogen. 33 This region, which is thought to remain bound to the papain‐like domain, has previously been shown to have an inhibitory effect on merozoite egress. 34 Prodomains are known to act with a myriad of functions to ensure correct folding, to prevent premature enzymatic activation by occupation of the active site groove, and in chaperoning. 35
In this study we present a higher resolution crystal structure of the central papain‐like domain (Asp563‐Asn827: PfSERA5E). More importantly, we demonstrate that the N‐terminal region of P50 indeed does comprises a papain‐like prodomain, resembling cathepsin prodomains in overall tertiary structure, and with similar interactions to the associated central papain‐like domain. This is the first structural study to demonstrate the organization of the N‐terminal region of P50, and the first structure of the central and prodomains (Thr391‐Asn828: PfSERA5PE) of PfSERA5, revealing the atomic details of the interaction between the central “enzyme” domain and the prodomain. Despite the conservation in the overall configuration of the two domains, this structure also details significant disparity to that of other cathepsin prodomains. We also investigate, through molecular dynamics (MD) simulations, the structure of PfSERA6; PfSERA6, with high sequence similarity to PfSERA5 likely has a similarly unusual prodomain. This model allows us to suggest potential interactions between these partners.
Our findings provide a basis for further investigation of the role of PfSERA5 by providing greater insight into the structure of this enigmatic protein, in particular the orientation and organization of the prodomain.
2. RESULTS AND DISCUSSION
The three‐dimensional crystal structures of recombinant PfSERA5E and PfSERA5PE have been solved to a resolution of 1.2 and 2.2 Å, respectively. The structures were solved by molecular replacement (MR) with the data collection and refinement statistics presented in Table 1.
TABLE 1.
X‐ray data collection and refinement statistics
| PfSERA5E | PfSERA5PE | |
|---|---|---|
| PDB: 6X42 | PDB: 6X44 | |
| Data collection | ||
| Wavelength (Å) | 0.956619 | 1.03930 |
| Resolution range (Å) | 24.47–1.20 (1.24–1.20) | 47.55–2.20 (2.23–2.20) |
| Space group | R 3 | P 2 2 21 |
| Unit cell parameters | ||
| a, b, c (Å) | 102.29 102.29 71.72 | 77.67 78.94 178.71 |
| α, β, γ (°) | 90 90 120 | 90 90 90 |
| Total reflections | 981,108 (92,030) | 413,403 (41,853) |
| Unique reflections | 87,458 (8714) | 56,459 (5430) |
| Multiplicity | 11.2 (10.6) | 7.3 (7.5) |
| Completeness (%) | 99.9 (99.5) | 99.1 (97.1) |
| Mean I/σ(I) | 17.7 (1.2) | 11.6 (2.0) |
| R merge | 0.0633 (1.901) | 0.1162 (0.917) |
| R pim | 0.0199 (0.610) | 0.0458 (0.356) |
| CC1/2 | 0.999 (0.453) | 0.998 (0.737) |
| Refinement | ||
| No. reflections used | 87,433 (8691) | 56,319 (5429) |
| R work | 0.126 (0.273) | 0.180 (0.239) |
| R free | 0.146 (0.294) | 0.214 (0.281) |
| No. non‐hydrogen atoms | 2,409 | 6,385 |
| Macromolecules | 2,144 | 6,148 |
| Non‐protein | 27 | 7 |
| Solvent | 238 | 230 |
| Protein residues | 265 | 773 |
| RMS (bonds, Å) | 0.008 | 0.011 |
| RMS (angles, degrees) | 1.01 | 1.03 |
| Ramachandran plot | ||
| Favored (%) | 97.34 | 94.86 |
| Allowed (%) | 2.66 | 4.87 |
| Outliers (%) | 0.00 | 0.26 |
| Average B‐factor | 24.39 | 42.34 |
| Macromolecules | 22.88 | 42.45 |
| Ligands | 34.97 | 66.74 |
| Solvent | 36.78 | 38.78 |
Note: Highest‐resolution shell data are shown in parentheses.
3. PFSERA5E
Recombinant PfSERA5E (Val544‐Asn828) with an N‐terminal hexa‐His tag was produced using a method different to that previously described. 22 PfSERA5E was expressed using T7 pLysY SHuffle cells, which promote disulfide bond formation without the requirement for lengthy refolding processes.
The domain was crystallized in the same space group with similar unit cell dimensions to that of previously reported experiments, 22 and the resulting packing and orientation is nearly identical to that of previous structural data (PDB: 2WBF). Data was collected from crystals grown in the presence of either a phage‐derived inhibitor of parasite development, or an eight‐residue synthetic peptide (Acetyl‐NKRGLVLP‐Amide, identified as the C‐terminal stalk of the prodomain lining the active‐site groove – see below). However, there was no evidence of either peptide in the diffraction data with the former data set presented here therefore treated as “native.” The structure was solved by MR using 2WBF as a starting model to a refined R factor of R work/R free 0.126/0.146 (Table 1). The higher resolution structure at 1.2 Å allowed for the identification of numerous sidechains not resolved in previous structures 3CH3, 3CH2, and 2WBF (1.8, 1.8, and 1.6 Å resolution, respectively), including Gln671 and Glu694. No other significant differences in the protein structure were observed with the root mean square deviation (r.m.s.d.) of 0.21 Å for 265 common Ca positions.
4. PFSERA5PE
Crystals of the PfSERA5 central domain in conjunction with the prodomain (PfSERA5PE) were grown from recombinantly derived protein generated in the same manner as that described for PfSERA5E. As for the preceding structure, the structure of PfSERA5PE was solved via MR using 2WBF as a starting model, to refined R factors R work/R free 0.180/0.214 (Table 1 and Figure 1a), with two molecules in the asymmetric unit (ASU). The central domain has a high degree of structural similarity to that of the high‐resolution structure presented herein and with previous studies (2WBF, r.m.s.d. 1.94 Å over 263 Ca positions). The central domain retains the typical cathepsin fold closely resembling that of previously identified catalytic domains of peptidases; cathepsin K (PDB: 1BY8), L (PDB: 1CS8) and caricain (PDB: 1PCI), and demonstrates similarity with the position of the prodomains orientating along the groove harboring the putative catalytic triad. Despite this, the structure of the prodomain is dissimilar to that of the aforementioned proteases (discussed subsequently).
FIGURE 1.
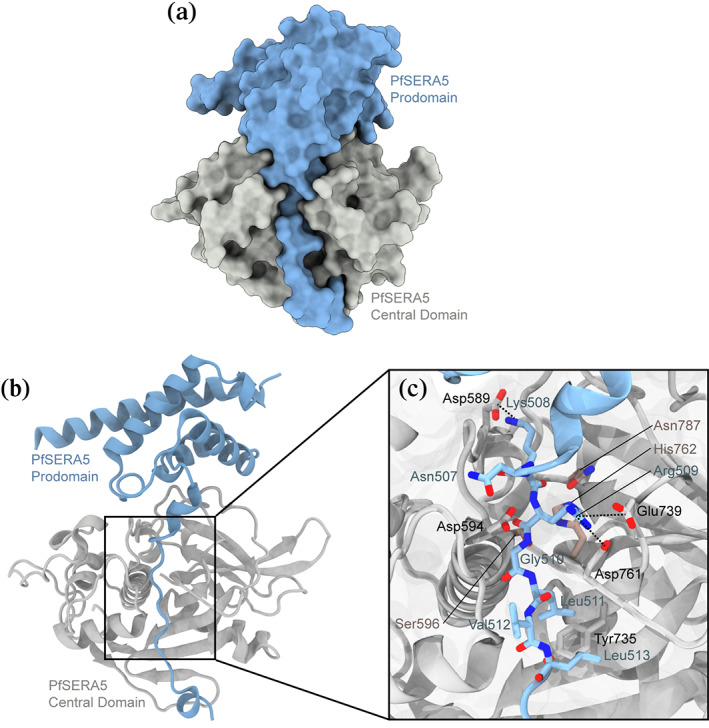
Structures of (a) PfSERA5PE. Structure is displayed in space fill representation (b) PfSERA5PE displayed as ribbon representation. (c) Zoomed view of the architecture of the PfSERA5PE “active‐site” groove interface. The prodomain is shown in blue, the central domain in grey, and the “catalytic triad” residues in dusty pink
The 187 residue prodomain (of which 117 and 126 residues are defined within electron density of each molecule within the ASU) consists of four α‐helical domains (Glu392‐Lys414; Asp435‐Val451; Glu465‐Tyr478; Ile484‐Phe491) and three short 310 helices (Leu430‐Glu432; Ala494‐Cys497; Val501‐Ile505). Electron density for residues forming a loop from Thr418‐Lys424 could not be resolved. The prodomain forms a globular structure from Thr391‐Val506 and a C‐terminal “stalk” from Asn507‐Phe523, similar to the prodomain of PfSUB1 (Figure 2a,b), 36 and of other proenzymes. 37
FIGURE 2.
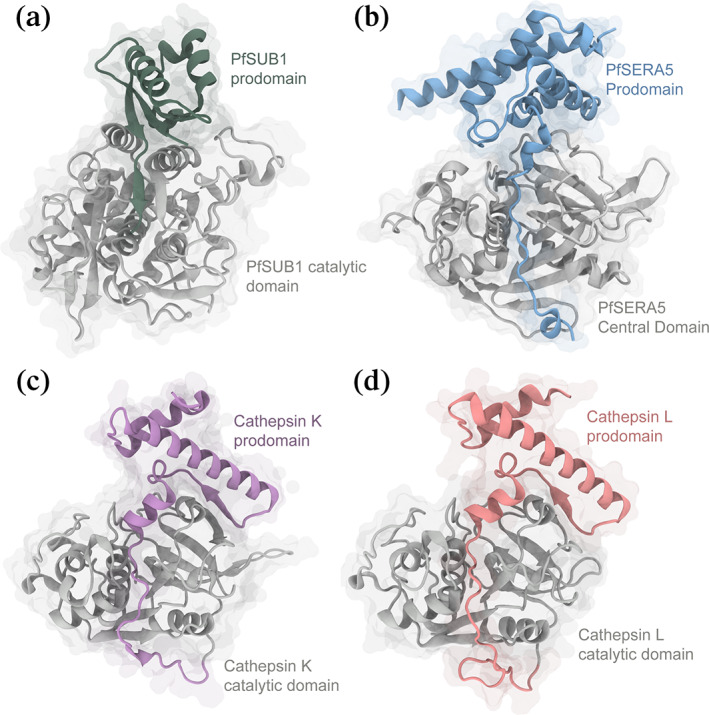
Comparison of zymogen structures. Ribbon diagrams with transparent space‐fill representations of (a) PfSUB1 (PDB: 4LVN), (b) PfSERA5PE (PDB: 6X44), (c) procathepsin K (PDB: 1BY8), and (d) procathepsin L (PDB: 1CS8). In all panels, the enzyme domains are colored grey, with the prodomains of PfSUB1, SERA5, procathepsin K and L colored green, blue, purple and coral, respectively
The globular prodomain makes numerous salt bridges and an extensive hydrogen bonding network with the central domain, in particular associated with the cleft region. The prodomain lines the substrate binding cleft (Figure 1b), resulting in the loop consisting of Lys738‐Ala749 to be displaced with respect to its position in the apo central domain structure. In the absence of the prodomain this loop consists of two short right‐handed 310 helices. No noticeable or significant changes were observed for the catalytic triad residues Ser596, His762, and Asn787. Where both Glu745 and Tyr744 were solvent exposed in the PfSERA5E structure, these residues are instead orientated towards and buried within the prodomain further stabilizing the domain interaction forming a salt bridge between Glu745 and Arg474. The displaced loop maintains the orientation of Phe746 towards the hydrophobic pocket interior of the central domain, bounded by the aliphatic sidechains of Ile736, Leu754, Val785, along with the aromatic sidechains of Phe799, Phe813, and Trp793.
The C‐terminal stalk lining the central domain groove orients Lys508 and Arg509 (occupying the P2′ and P1′ positions, respectively) to the S2′ and S1′ pockets, respectively. 1 These residues form electrostatic interactions with the sidechains of Asp589 and both Glu739 and Asp761, respectively. The aliphatic sidechain of Leu511 occupies the P1 position with its mainchain amide forming a hydrogen bond with the carbonyl oxygen of Asp761. Leu513 further occupies the hydrophobic depression forming the S2‐subsite displacing the sidechain of Tyr735 from its position observed in the apo structure (Figure 1c). The remaining residues of the prodomain for which electron density is visible along the stalk indicates hydrogen bonding between Tyr522 and Glu707, and cation‐π interactions between the sidechains of Tyr522 and Arg710. The presence of these residues and the remainder of the prodomain tether the loop consisting of the central domain residues His690‐Lys701 to pack against the papain‐like domain. Electron density of Asn524‐Asn561 forming the remainder of the central‐prodomain tether was not evident. This unresolved region corresponds to a site of in vivo phosphorylation, at Thr549. 28 Packing of the molecules within the ASU, and the absence of the 20 interconnecting residues between the prodomain and central domain in this crystal, allow the possibility of a domain‐swapped dimer, with the prodomain of one monomer binding the central domain of a second, and vice versa. Specifically, the distance between the last visible C‐terminal residue of the prodomain and the first visible N‐terminal residue of the central domain could be spanned between domains of one zymogen or in trans across two zymogens (Figure S1). Although there is no evidence that the full‐length protein forms dimers in solution, 29 if indeed such an interaction occurs it is possible that this mechanism may facilitate interactions between PfSERA5 and PfSERA6 altering enzymatic activity of the latter.
5. COMPARISON TO PROCATHEPSIN
The mature central domain of PfSERA5 has structural similarity to that of the Clan CA, Family C1 (papain family), 22 closely overlapping that of the prototypical member, papain. Expressed in P. falciparum, members of this family also include that of the falcipains and dipeptidyl aminopeptidases. 14 , 38 Although sharing a similar central domain, there is significantly less structural homology between corresponding prodomains within the family (Figure 2c,d), leading to the classification of two subfamilies. PfSERA5PE shares greatest similarity to that of the longer prodomains of the cathepsin‐L‐like subfamily (L, V, K, S, W, F, and H), which are approximately 100 residues in length and consist of two α‐helices followed by a short beta sheet and a third short helix. 39 , 40 Members of this subfamily contain two distinct conserved sequence motifs within the prodomain, that being ERFNIN and GNFD. 41 Present within falcipains, but not in PfSERA5, these motifs have been identified to contribute to the structure and stability of the prodomain, 42 , 43 and may contribute to their inhibitory function. 43 , 44 In contrast, prodomains of the cathepsin‐B‐like subfamily are significantly shorter, containing approximately 60 residues and consisting of a short beta strand and two α‐helices. 45 , 46
As demonstrated here, the PfSERA5 prodomain is significantly larger with a greater helical content than either subfamily, instead consisting of seven helices across 187 residues; this trait is somewhat similar to that of the P. falciparum falcipains, which contain “unusually” long prodomains. Previous modelling of profalcipain‐2 mimicked that of procathepsin K/L, however ignored ~160 N‐terminal residues. 43 This modelling suggested, at least in principal, that profalcipain‐2 mimics the organization of similar cathepsin L‐like protease prodomains.
6. INTERACTION BETWEEN PRO AND CENTRAL DOMAINS
Notwithstanding the unknown function full‐length PfSERA5 plays within the malaria lifecycle, less is known of the molecular function of the zymogen prodomain. Previous studies suggested the prodomain inhibited the catalytic activity of the central domain, 34 with speculation the prodomain assisted in recognition and binding of the substrate of the central domain. In addition to controlling protease activity, prodomains have been shown to play an important role in folding and trafficking. 35 The mechanism of interaction made by the prodomain, particularly in orientation of the C‐terminal “stalk” region along the “active‐site” groove to occlude the substrate‐binding cleft, is similar in molecular design to comparable proteases (Figure 2). The orientation of the prodomain conventionally serves to ablate catalytic activity by binding to the active site in the reverse direction to that of native substrates; reducing the availability of the proteases to bind and cleave substrate. 35 , 42 Activation of zymogens by removal of the prodomain leads to activation of the catalytic domain. Notably, the PfSERA5 prodomain is orientated through the active‐site cleft in this same reverse direction. Unlike typical zymogens, whereby the precursory cleavage of the prodomain occurs, there is no in vivo evidence to suggest the PfSERA5 prodomain is cleaved from the central domain, therefore precluding it from engaging with any substrate. However, early studies have identified that exogenous protease addition in vitro can induce cleavage of the prodomain from the central domain, 27 suggesting such a dissociation between domains is plausible. This unconventional organization by which the prodomain is retained with the central enzyme domain has been identified in the similar Family C1 P. falciparum papain‐like zymogen dipeptidyl aminopeptidases 1 and 3 (DPAP‐1 and DPAP‐3, respectively), which retain activity without removal of the prodomain. 47 We note again that the data presented here does not preclude a domain‐swapped dimer, with the prodomain of one monomer binding the central domain of a second, and vice versa. Evidence indicates that neither PfSERA5 or PfSERA6 form dimers, 29 , 48 yet nothing is known as to the affinity between the two. The architecture of the domain swapped dimer would allow the central domain of one (say PfSERA5) to bind the prodomain of another (PfSERA6) in trans. This model of dimerization would be in accord with the in vivo evidence indicating an absence of dissociation of the central and prodomains; such an arrangement would protect both stalk domains (on PfSERA5 and PfSERA6) from cleavage by self or other proteases. Alternatively, engagement of the prodomain of PfSERA6 by the central domain of PfSERA5, leaving the enzyme domain of PfSERA6 free, could facilitate separation of the pro‐ and enzyme domains in PfSERA6, and permit access to the substrate binding site by exogenous substrate. Thus, PfSERA5 could act as a chaperone to protect PfSERA6 from proteolysis, or it could prime PfSERA6 for activation; the specific role of PfSERA5 requires further investigation.
In the structure of SERA5E, the “active‐site” groove flanking residues Asp638‐Ser641 form the S3‐subsite and are unusual with regard their orientation when compared to their homologues in cathepsin L. 22 Additionally, the S2‐site, which frequently defines substrate specificity in similar proteases, is occupied by Tyr735, reducing the site to a shallow hydrophobic depression; collectively these features suggest an inherent inability to orient and subsequently bind substrate. However, the catalytically active PfSERA5 Ser596Cys mutant 29 demonstrates the enzyme domain does maintain an ability to bind and process substrate, indicating sufficient plasticity to adopt a suitable conformation. Engagement of the C‐terminal prodomain stalk results in reorientation of the S3‐site flanking residues, with the amide and carbonyl of Ser640 forming hydrogen bonds with prodomain residues Val512 and Gly510. The S2‐site, formed by residues Ser641 and Pro642, is significantly wider and deeper (Figure 3a) due to the rotation of Tyr735 with respect to the orientation observed in the PfSERA5E structure, allowing accommodation of prodomain residue Leu511 (Figure 3b). Similarly, the S1‐site formed by Asp594, and the S1′‐site formed by Trp789, Ala738, and Glu739, are likewise rotated from their orientations observed in the SERA5E structure, allowing Arg509 to occupy the P1′‐site and form interactions with Asp594 and Glu739.
FIGURE 3.
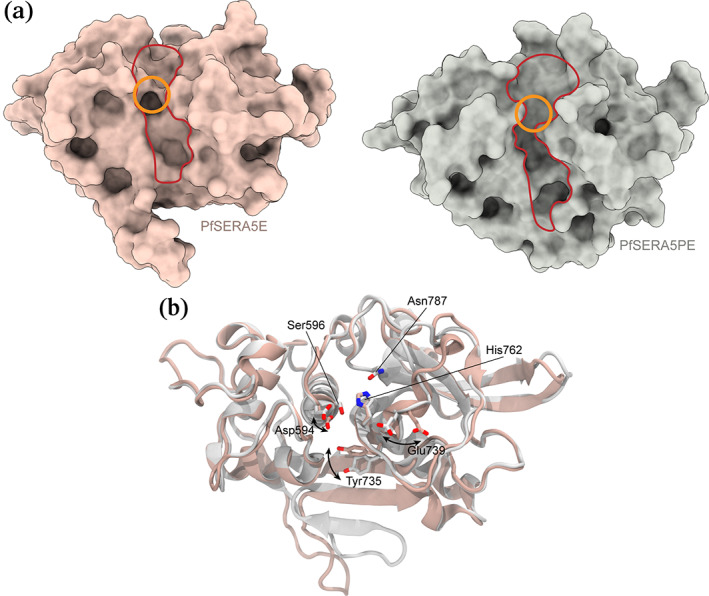
Structures of the central domains of PfSERA5E and PfSERA5PE (the prodomain is removed for clarity) displayed in (a) space fill and (b) as ribbon representation overlaid. The non‐canonical "active‐site” groove is outlined in red and the “catalytic triad” in orange in the upper panel. Sidechains of the “catalytic triad” residues, and residues whose positions differ between PfSERA5E and PfSERA5PE responsible for the change in the molecular surface at the “active site” groove, are displayed in the lower panel in stick representation
The amino acid sequence of the prodomain occupying the substrate binding groove demonstrates significant sequence similarity across all PfSERAs, suggesting all PfSERA members may contain an N‐terminal papain‐like prodomain (Figure S2, with exception of SERA8 due to its vector stage role in oocyst rupture and release of sporozoites) 12 ; in particular between PfSERA5 and PfSERA6 (Figure 4). Residues highly conserved across all SERAs, Lys508, Gly510, and Leu511 (SERA5 numbering), all contact conserved residues in the central domain. Lys508 forms a salt bridge with Asp589; an acidic residue at the equivalent position as Asp589 in SERA5 is conserved in all members. Gly510 abuts the catalytic machinery, residue Ser596, and is strictly conserved across all SERAs; a glycine residue at this position is synonymous with cysteine proteases allowing the prodomain to pack deeply into the active site. 39 The occupation of Leu511, orientated towards the sidechain of Tyr735 also appears conserved, with both residues strictly conserved across all members. In contrast, residues lining the groove which demonstrate greater solvent accessibility, and that do not form any specific interactions with the central domain (Asn507 and Val512) demonstrate low levels of conservation. An exception is Arg509, which is solvent exposed and conserved between only PfSERA5 and ‐6, and forms a salt bridge to Asp761 and Asp739, of which only Asp761 is strictly conserved. Across the remainder of the zymogen, sequence similarity is identified in residues that line the interface between the pro‐ and central domains (Figure S2). Indeed, such high sequence similarity between SERA prodomains, restricted to the interface regions may indicate that the pro‐ and central domains retain an ability to interact in trans across the family. Furthermore, the high sequence similarity of the C‐terminal stalk region between PfSERA5 and ‐6 may also suggest an ability to recognize and bind similar substrates, if indeed the prodomain is cleaved, or if domain flexibility facilitates exposure. Indeed, recent studies conducted on PfSUB1 and on PfSERA5‐absent parasites identified a potential role of PfSERA5, amongst others, to bind and protect substrates of the egress‐related protease PfSERA6. 26
FIGURE 4.

Sequence conservation between PfSERA5 and PfSERA6. Residues are highlighted in accordance with conservation, with strictly conserved residues colored dark blue and dark grey, and conservative replacements colored light blue and light grey. The positions of the “catalytic triad” serine/cystine, histidine, and asparagine residues are highlighted brown
7. COMPARISON AND MODELLING OF PFSERA6
Of the SERA genes, PfSERA6 is the only member indispensable for parasite development, performing an essential enzymatic role mediating cytoskeletal disassembly via target cleavage of β‐spectrin. 16 , 48 The sequences of the PfSERA5 and PfSERA6 central domains share a high degree of similarity (56% identity, 76% similarity; Figure 4), with the sequences of the respective prodomains suggesting PfSERA6 retains a structure more similar to that of PfSERA5 than those of procathepsins or of the proposed falcipain prodomain structures.
In the absence of any structural information regarding PfSERA6, and, owing to the significant importance and similarity between PfSERA5 and ‐6 we constructed a homology model of PfSERA6PE based on the structure and available data presented here, and subjected it to MD simulations. In analysis, we sought to make a comparison of the prodomain–central domain interfaces of the two proteins and sought to address the attractive suggestion by Collins et al. most recently that there may exist a form of interaction between these egress related proteins. 26
MD simulations of a model of PfSERA6PE pro‐ and enzyme domain residues Ala369‐Ser501 and Asn611‐Leu872, respectively, were stable throughout the simulation, with the orientation of the prodomain C‐terminal stalk notably conserved, accommodating residues Lys486‐Thr490 into the S2′‐through S3‐sites. The S2‐site consists of residues Asn689 and Pro690, with Tyr783 similarly rotated with respect to the structure of PfSERA5E. This rotation similarly allows the occupation of residues Leu489 and Leu491. In contrast, the S1‐site is not occupied by the anomalous aspartic acid as found in PfSERA5, instead this position is a glycine in PfSERA6, as found in similar conventional cysteine proteases. 22 Like PfSERA5, the S1′‐site is comprised of Trp837 and the loop Thr786‐Asp788, however, the loop is not strictly conserved (comparable residues PfSERA5: AEN, PfSERA6: TQD). Residues of the prodomain Asn485‐Pro492 mimic those of PfSERA5, with the exception of Thr490 that is solvent exposed. Furthermore, comparison of the surfaces between PfSERA5 and ‐6 indicate that the regions of lower sequence similarity correspond with residues that are surface exposed, and not at the interface between the prodomain and central domain (Figure 5a). The region comprising the substrate binding groove, on both prodomain and central domains of PfSERA5 and ‐6 are strikingly similar. Although the net charges between the “enzyme” domains at neutral pH are markedly different (product of the presence of varying acidic and basic residues) (Figure 5b), the electrostatic surface potential of the two PfSERA members presented here, calculated at their corresponding pIs, demonstrates conserved features. The S2′ through S1 pockets are largely negatively charged, corresponding to significant sequence conservation within this region. In contrast, the difference in surface potential is reflected at the periphery, and distal to the “catalytic” groove.
FIGURE 5.
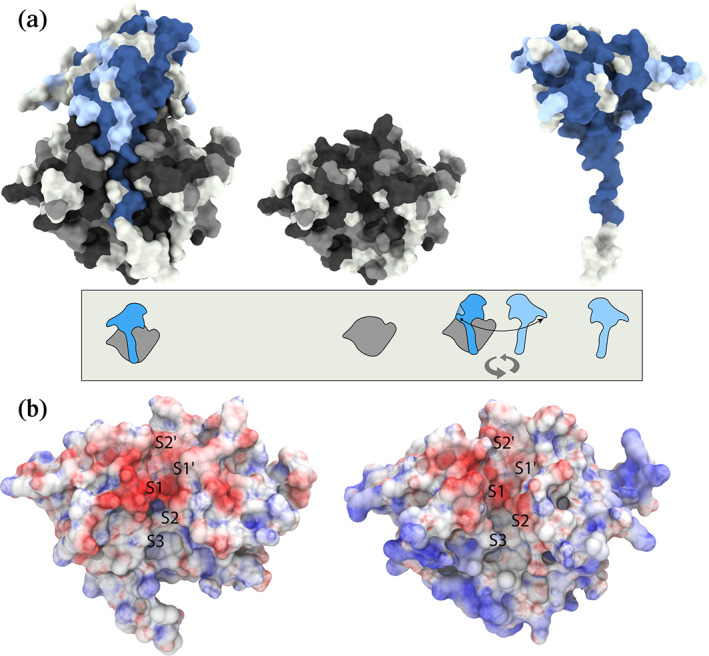
(a) Sequence conservation between PfSERA5 and PfSERA6 from Figure 4 mapped onto the PfSERA5PE protein surface. The surface is colored in accordance with Figure 4, with the interaction faces of the "enzyme” (center) and prodomains (right) displayed. The cartoon representation below describes the molecular views. (b) Electrostatic surface potential of PfSERA5 (left) and PfSERA6 (right) calculated from the APBS web‐server. The surface is colored from red, through white to blue from −10 to +10 kT
8. CONCLUSION
The recent identification of bioavailable small‐molecule compounds which bind to and inhibit the enzymatic activity of PMIX and PMX is promising. 11 The inhibition of PMX reduces the availability of mature PfSUB1 and subsequently PfSUB1‐mediated processing of PfSERA5, demonstrating the suitability of such proteases as drug targets blocking erythrocytic egress. The targeting of these proteases that are essential to independent steps within the parasite blood invasion, development and egress stages, has been demonstrated to result in synergistic effects, 49 suggesting the targeting of a combination of these proteases could yield a therapeutic advantage.
Prior to this work, efforts to understand full length PfSERA5 were unsuccessful. Although the function of PfSERA5 is still unclear, its significant abundance, avoidance by the parasite to significant alteration, and resulting premature egress kinetics on disruption suggests an intrinsic role associated with parasite egress. Here, most importantly, in this study we have confirmed previous speculation as to the presence of a prodomain within the P50 fragment, and that this fragment is orientated with respect to the central domain analogously to other zymogens. Our study provides insight into the interactions between the PfSERA5 pro‐ and central domains and highlights the high degree of similarity between the active sites and prodomain “stalks” of PfSERA5 and of the sequence similar PfSERA6. Such similarity could suggest a role of PfSERA5 in binding and protecting substrates from PfSERA6; we note such a role has been speculated previously. 26 Via modelling, MD, electrostatic surface analysis and sequence comparison of PfSERA6 overlayed on the structure of PfSERA5PE presented here, we have shown that the substrate binding groove of PfSERA6 is highly similar to that of PfSERA5, and that significant dissimilarity between PfSERA5 and ‐6 is localized to the protein periphery and is principally solvent exposed. Indeed, although these results do not preclude the direct targeting of PfSERA5, the issues surrounding potential catalytic activity has yet to be validated and is still debated. It is therefore likely the most immediate outcomes from this study may instead provide a platform for the rational design of compounds to target and inhibit the known actions of PfSERA6 in proteolytic cleavage of cytoskeletal proteins.
9. MATERIALS AND METHODS
9.1. Cloning, expression, and purification
Recombinant P. falciparum PfSERA5E (Val544‐Asn828) and PfSERA5PE (Thr391‐Asn828) (PlasmoDB Gene ID PF3D7_0207500) were inserted into the pProExHTb (Invitrogen) expression vector, as described previously. 22 Where Thr391 represents the PfSUB1 cleavage site, the P50 domain was truncated at Asn828 based on sequence alignments defining the termini of other papain‐like proteases. 27 The proteins were synthesised in E. coli T7 pLysY SHuffle cells [New England Biolabs (NEB)] to facilitate disulfide‐bond formation for 48 hour at 16°C. The cells were lysed by sonication before the solubilized protein was isolated by Immobilized Metal Affinity Chromatography on a Ni–NTA(Qiagen) column. The N‐terminal His tag was removed with TEV protease leaving Val544‐Asn828 and Thr391‐Asn828 with five N‐terminal residues (GAMGS) from the vector. The protein was further purified by size exclusion chromatography using a Superdex 75 10/30 column (GE Healthcare). The protein was concentrated (50 mM NaCl, 5 mM Tris–HCl pH 7.5) and subsequently crystalized.
9.2. Crystallization and data collection
9.2.1. PfSERA5E
PfSERA5E was crystalized in the same conditions as previously described. Briefly, PfSERA5E buffered in 20 mM Bis‐Tris (pH 6.5) and 20 mM NaCl was crystallized by vapor diffusion with 0.2 M KH2PO4 (pH 4.6), 12% polyethylene glycol PEG‐8000, and 5% PEG‐400. The protein was crystalized in a sitting drop set‐up at 20°C in equal volumes of reservoir and protein (10 mg/mL). Glycerol (16%) was added immediately prior to flash cooling in liquid nitrogen.
Diffraction data, processed to 1.2 Å (Table 1), were collected using the MX2 beamline at the Australian Synchrotron.
Peptides derived from the prodomain (507NKRGLVLP514) or a phage‐derived inhibitor of late‐stage parasite development 50 were screened with PfSERA5E, however, there was no evidence of these peptides in the electron density, and therefore this data set (PfSERA5E—PDB: 6X42) has been treated as “native.” Initial screening attempts yielded only conditions consistent with the apo‐form of PfSERA5E, with packing constraints preventing these conditions in yielding co‐crystals.
9.2.2. PfSERA5PE
PfSERA5PE was crystalized using a two‐to‐one ratio volume of reservoir (0.2 M LiSO4, 0.1 M Tris–HCl (pH 8.1), 24% polyethylene glycol (PEG‐4000) and protein, which were added and equilibrated in a hanging‐drop set up at 20°C. A single crystal was extracted and mounted on a polyimide loop after cryoprotection by passing the crystal through a solution containing 15% PEG‐400, 3 M TMAO/HCl (pH 7.0), 0.05 M Tris–HCl (pH 8.9), 0.1 M LiSO4. Diffraction data processed to 2.2 Å was collected from a single crystal using the Advanced Photon Source beamline 24‐ID‐C (NE‐CAT).
9.2.3. Structure determination, refinement, and analysis
Images were processed with the XDS package, 51 with space group assignment by POINTLESS 52 , 53 and scaling and merging with AIMLESS. 54 Both structures were solved by molecular replacement, using a modified version of PfSERA5E as the search model (PDB 2WBF) using PHASER. Two‐fold non‐crystallographic symmetry (NCS) was identified in the PfSERA5PE structure. BUCANEER 55 was used to build an initial model into the density map of the prodomain. Crystallographic refinement was made using PHENIX, 56 iterated with several manual model building cycles within COOT. 57 Anisotropic refinement was applied only to the structure of PfSERA5E. Crystallographic data collection and refinement statistics are summarized in Table 1.
9.2.4. Molecular interaction surface area
The residue solvent accessibility defined by the interaction between the prodomain and the papain‐like domain was calculated using the classical approximation of Lee and Richards, 58 from the difference of the solvent accessible surface area (SASA) of the complex from the individual components using the default atomic van der Waals radii and resolution using the NACCESS (v2.1.1) 59 program.
9.2.5. Sequence alignment
Sequences for all SERA genes were obtained from PlasmoDB. Multiple sequence alignment was performed using Clustal Omega. 60 SERA1: PF3D7_0208000, SERA2: PF3D7_0207900, SERA3: PF3D7_0207800, SERA4: PF3D7_0207700, SERA5: PF3D7_0207600, SERA6: PF3D7_0207500, SERA7: PF3D7_0207400, SERA9: PF3D7_0902800.
9.2.6. Molecular modelling
A comparative model of PfSERA6PE (PlasmoDB gene ID PF3D7_0207500) was generated using the MODELLER (v9.19) program. The crystal structure of PfSERA5PE (PDB: 6X44) presented herein was used as a template, with residues Ala369‐Ser501 and Asn611‐Leu872 of SERA6PE modelled. From 50 models created, the model with the lowest MODELLER objective function was used in subsequent MD simulations.
9.2.7. Molecular dynamics simulation
Molecular dynamics simulations used the gromacs (v2016) 61 software package with the charmm36m force field. 62 The models were solvated using the TIP3P water model in a box extending at least 10 Å beyond all atoms with periodic boundary conditions. The system was neutralized, and an ionic strength of 0.1 M was obtained with sodium and chloride ions.
Temperature coupling was conducted in two groups with the protein and solvent coupled independently to a velocity rescaling thermostat 63 at 300 K, both groups utilizing a time constant of 0.1 ps. Pressure coupling was handled using the Parrinello‐Rahman technique 64 with a reference pressure of 1 bar and a time constant of 2.0 ps. Non‐bonded interactions were applied with a cut off of 12 Å with the particle‐mesh Ewald method 65 for long range electrostatics, with a grid width of 1.0 Å and a sixth‐order spline interpolation. Neighbor searching was updated every 40 steps applying the Verlet grid cut‐off scheme with a short‐range cutoff distance of 12.81 Å. The model was minimized using a steepest descent minimization followed by 50 ps equilibration in the canonical ensemble, and 50 ps in the isothermal‐isobaric (NPT) ensemble before unrestrained MD for 500 ns.
9.2.8. Electrostatic surface potential and visualization
The electrostatic surface potential was calculated using the PDB2PQR 66 and the Adaptive Poisson‐Boltzmann Solver (APBS) web‐server. 67 Charges were assigned based on a pH corresponding to the predicted pI for each protein, with all remaining options set as default. All molecular visualization and surface representations was subsequently performed with the use of UCSF Chimera, 68 UCSF ChimeraX 69 or VMD. 70
CONFLICT OF INTEREST
The authors declare no conflicts of interest with regards to this manuscript.
AUTHOR CONTRIBUTIONS
Nicholas A. Smith, formal analysis, investigation, methodology, visualization, writing‐original draft, writing‐review and editing; Oliver B. Clarke, methodology, formal analysis, investigation, writing‐review and editing; Mihwa Lee, methodology, formal analysis, investigation, writing‐review and editing; Anthony N. Hodder, resources, writing‐review and editing; Brian J. Smith, conceptualization, formal analysis, supervision, writing‐review and editing.
DATA AVAILABILITY STATEMENT
Coordinates for PfSERA5E and PfSERA5PE have been deposited in the Protein Data Bank under the accession code PDB: 6X42 (PfSERA5E) and 6X44 (PfSERA5PE), respectively.
Supporting information
Data S1 Figure S1. Packing of the two PfSERA5PE molecules within the asymmetric unit.
Figure S2. Multiple sequence alignment indicating conservation between PfSERA proteins.
ACKNOWLEDGEMENTS
This work was supported by an Australian Government Research Training Program scholarship to N.A.S. and the Australian Research Council Discovery Early Career Research Award (ARC DECRA) Fellowship (DE150101243) and Tracey Banivanua Mar Fellowship (La Trobe University, Melbourne, Australia) to M.L. Diffraction data were obtained at the Australian Synchrotron (beam line MX2) and from the Advanced Photon Source beamline 24‐ID‐C (NE‐CAT). Infrastructure support from the NHMRC IRIIS (361646) and a Victorian State Government OIS grant is gratefully acknowledged. Part of this work was undertaken using resources from the National Computational Infrastructure, which is supported by the Australian Government and provided through Intersect Australia Ltd. under LIEF grant LE170100032, and through the HPC‐GPGPU Facility, which was established with the assistance of LIEF Grant LE170100200.
Smith NA, Clarke OB, Lee M, Hodder AN, Smith BJ. Structure of the Plasmodium falciparum PfSERA5 pseudo‐zymogen. Protein Science. 2020;29:2245–2258. 10.1002/pro.3956
Funding information DECRA, Grant/Award Number: DE150101243; HPC‐GPGPU Facility, Grant/Award Number: LE170100200; Intersect Australia Ltd., Grant/Award Number: LE170100032; NHMRC IRIIS, Grant/Award Number: 361646
Endnote
In accordance with Schechter and Berger nomenclature 71 , substrate residues Pn and Pn' bind respective sites Sn and Sn' within the protease active site. Bond cleavage of the substrate subsequently occurs between the P1 and P1' positions.
REFERENCES
- 1.World Health Organization. World Malaria Report 2019. 2019.
- 2. Cowman AF, Healer J, Marapana D, Marsh K. Malaria: Biology and disease. Cell. 2016;167:610–624. [DOI] [PubMed] [Google Scholar]
- 3. Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria. Nat Rev Dis Prim. 2017;3:17050. [DOI] [PubMed] [Google Scholar]
- 4. Olotu A, Fegan G, Wambua J, et al. Seven‐year efficacy of RTS, S/AS01 malaria vaccine among young african children. N Engl J Med. 2016;374:2519–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pance A. How elusive can a malaria vaccine be? Nat Rev Microbiol. 2019;17:129–129. [DOI] [PubMed] [Google Scholar]
- 6. Matz JM, Beck JR, Blackman MJ. The parasitophorous vacuole of the blood‐stage malaria parasite. Nat Rev Microbiol. 2020;18:379–391. [DOI] [PubMed] [Google Scholar]
- 7. Blackman MJ. Malarial proteases and host cell egress: An “emerging” cascade. Cell Microbiol. 2008;10:1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosenthal PJ. Cysteine proteases of malaria parasites. Int J Parasitol. 2004;34:1489–1499. [DOI] [PubMed] [Google Scholar]
- 9. Yeoh S, O'Donnell RA, Koussis K, et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell. 2007;131:1072–1083. [DOI] [PubMed] [Google Scholar]
- 10. Nasamu AS, Glushakova S, Russo I, et al. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science. 2017;358:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Favuzza P, de Lera Ruiz M, Thompson JK, et al. Dual plasmepsin‐targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe. 2020;27:642–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aly ASI, Matuschewski K. A malarial cysteine protease is necessary for Plasmodium sporozoite egress from oocysts. J Exp Med. 2005;202:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burns AL, Dans MG, Balbin JM, et al. Targeting malaria parasite invasion of red blood cells as an antimalarial strategy. FEMS Microbiol Rev. 2019;43:223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosenthal PJ. Falcipain cysteine proteases of malaria parasites: An update. Biochim Biophys Acta Proteins Proteomics. 2020;1868:140362. [DOI] [PubMed] [Google Scholar]
- 15. Withers‐Martinez C, Jean L, Blackman MJ. Subtilisin‐like protease of the malaria parasite. Mol Microbiol. 2004;53:55–63. [DOI] [PubMed] [Google Scholar]
- 16. Thomas JA, Tan MS, Bisson C, et al. A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nat Microbiol. 2018;3:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paul AS, Duraisingh MT. Targeting plasmodium proteases to block malaria parasite escape and entry. Trends Parasitol. 2018;34:95–97. [DOI] [PubMed] [Google Scholar]
- 18. Arisue N, Palacpac NMQ, Tougan T, Horii T. Characteristic features of the SERA multigene family in the malaria parasite. Parasites Vectors. 2020;13:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higgins DG, McConnell DJ, Sharp PM. Malarial proteinase? Nature. 1989;340:604. [DOI] [PubMed] [Google Scholar]
- 20. Eakin AE, Higaki JN, McKerrow JH, Craik CS. Cysteine or serine proteinase? Nature. 1989;342:132. [DOI] [PubMed] [Google Scholar]
- 21. Gardner MJ, Hall N, Fung E, et al. Genome sequence of the human malaria parasite Plasmodium falciparum . Nature. 2002;419:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hodder AN, Malby RL, Clarke OB, et al. Structural insights into the protease‐like sntigen Plasmodium falciparum SERA5 and its noncanonical active‐site serine. J Mol Biol. 2009;392:154–165. [DOI] [PubMed] [Google Scholar]
- 23. Miller SK, Good RT, Drew DR, et al. A subset of Plasmodium falciparum SERA genes are expressed and appear to play an important role in the erythrocytic cycle. J Biol Chem. 2002;277:47524–47532. [DOI] [PubMed] [Google Scholar]
- 24. Lasonder E, Ishihama Y, Andersen JS, et al. Analysis of the Plasmodium falciparum proteome by high‐accuracy mass spectrometry. Nature. 2002;419:537–542. [DOI] [PubMed] [Google Scholar]
- 25. Le Roch KG, Johnson JR, Florens L, et al. Global analysis of transcript and protein levels across the Plasmodium falciparum life cycle. Genome Res. 2004;14:2308–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collins CR, Hackett F, Atid J, Tan MSY, Blackman MJ. The Plasmodium falciparum pseudoprotease SERA5 regulates the kinetics and efficiency of malaria parasite egress from host erythrocytes. PLoS Pathog. 2017;13:e1006453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hodder AN, Drew DR, Epa VC, et al. Enzymic, phylogenetic, and structural characterization of the unusual papain‐like protease fomain of Plasmodium falciparum SERA5. J Biol Chem. 2003;278:48169–48177. [DOI] [PubMed] [Google Scholar]
- 28. Iyer GR, Singh S, Kaur I, et al. Calcium‐dependent phosphorylation of Plasmodium falciparum serine repeat antigen 5 triggers merozoite egress. J Biol Chem. 2018;293:9736–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stallmach R, Kavishwar M, Withers‐Martinez C, et al. Plasmodium falciparum SERA5 plays a non‐enzymatic role in the malarial asexual blood‐stage lifecycle. Mol Microbiol. 2015;96:368–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delplace P, Bhatia A, Cagnard M, et al. Protein p126: A parasitophorous vacuole antigen associated with the release of Plasmodium falciparum merozoites. Biol Cell. 1988;64:215–221. [DOI] [PubMed] [Google Scholar]
- 31. Delplace P, Fortier B, Tronchin G, Dubremetz JF, Vernes A. Localization, biosynthesis, processing and isolation of a major 126 kDa antigen of the parasitophorous vacuole of Plasmodium falciparum . Mol Biochem Parasitol. 1987;23:193–201. [DOI] [PubMed] [Google Scholar]
- 32. De Monerri NCS, Flynn HR, Campos MG, et al. Global identification of multiple substrates for Plasmodium falciparum SUB1, an essential malarial processing protease. Infect Immun. 2011;79:1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rahul CN, Shiva Krishna K, Pawar AP, Rajesh V. In silico approach to ascertain the calcium dependent role of Plasmodium falciparum SERA5. J Biomol Struct Dyn. 2017;35:17–25. [DOI] [PubMed] [Google Scholar]
- 34. Alam A, Chauhan VS. Inhibitory potential of prodomain of Plasmodium falciparum protease serine repeat antigen 5 for asexual blood stages of parasite. PLoS One. 2012;7:e30452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boon L, Ugarte‐Berzal E, Vandooren J, Opdenakker G. Protease propeptide structures, mechanisms of activation, and functions. Crit Rev Biochem Mol Biol. 2020;55:111–165. [DOI] [PubMed] [Google Scholar]
- 36. Withers‐Martinez C, Strath M, Hackett F, et al. The malaria parasite egress protease SUB1 is a calcium‐dependent redox switch subtilisin. Nat Commun. 2014;5:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khan AR, James MNG. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 2008;7:815–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deu E. Proteases as antimalarial targets: Strategies for genetic, chemical, and therapeutic validation. FEBS J. 2017;284:2604–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Verma S, Dixit R, Pandey KC. Cysteine proteases: Modes of activation and future prospects as pharmacological targets. Front Pharmacol. 2016;7:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Musyoka TM, Njuguna JN, Bishop ÖT. Comparing sequence and structure of falcipains and human homologs at prodomain and catalytic active site for malarial peptide based inhibitor design. Malar J. 2019;18:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou J, Zhang YY, Li QY, Cai ZH. Evolutionary history of cathepsin L (L‐like) family genes in vertebrates. Int J Biol Sci. 2015;11:1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coulombe R, Grochulski P, Sivaraman J, Ménard R, Mort JS, Cygler M. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996;15:5492–5503. [PMC free article] [PubMed] [Google Scholar]
- 43. Pandey KC, Barkan DT, Sali A, Rosenthal PJ. Regulatory elements within the prodomain of falcipain‐2, a cysteine protease of the malaria parasite Plasmodium falciparum . PLoS One. 2009;4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aich P, Biswas S. Highly conserved Arg residue of ERFNIN motif of pro‐domain is important for pH‐induced zymogen activation process in cysteine cathepsins K and L. Cell Biochem Biophys. 2018;76:219–229. [DOI] [PubMed] [Google Scholar]
- 45. Turk B, Turk D, Turk V. Lysosomal cysteine proteases: More than scavengers. Biochim Biophys Acta Protein Struct Mol Enzymol. 2000;1477:98–111. [DOI] [PubMed] [Google Scholar]
- 46. Turk V, Stoka V, Vasiljeva O, et al. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim Biophys Acta Proteins Proteomics. 2012;1824:68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lehmann C, Tan MSY, de Vries LE, et al. Plasmodium falciparum dipeptidyl aminopeptidase 3 activity is important for efficient erythrocyte invasion by the malaria parasite. PLoS Pathog. 2018;14:e1007031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ruecker A, Shea M, Hackett F, et al. Proteolytic activation of the essential parasitophorous vacuole cysteine protease SERA6 accompanies malaria parasite egress from its host erythrocyte. J Biol Chem. 2012;287:37949–37963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arastu‐Kapur S, Ponder EL, Fonović UP, et al. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum . Nat Chem Biol. 2008;4:203–213. [DOI] [PubMed] [Google Scholar]
- 50. Fairlie WD, Spurck TP, McCoubrie JE, et al. Inhibition of malaria parasite development by a cyclic peptide that targets the vital parasite protein SERA5. Infect Immun. 2008;76:4332–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Evans P. Scaling and assessment of data quality. Acta Crystallogr. 2006;D62:72–82. [DOI] [PubMed] [Google Scholar]
- 53. Evans PR. An introduction to data reduction: Space‐group determination, scaling and intensity statistics. Acta Cryst D. 2011;67:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Cryst D. 2013;69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cowtan K. The buccaneer software for automated model building. 1. Tracing protein chains. Acta Cryst D. 2006;62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 56. Liebschner D, Afonine PV, Baker ML, et al. Macromolecular structure determination using X‐rays, neutrons and electrons: Recent developments in Phenix. Acta Cryst D. 2019;75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee B, Richards FM. The interpretation of protein structures: Estimation of static accessibility. J Mol Biol. 1971;55:379–400. [DOI] [PubMed] [Google Scholar]
- 59. Hubbard SJ, Thornton JM. NACCESS Computer Program 1993. University College London: Department of Biochemistry and Molecular Biology. [Google Scholar]
- 60. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol. 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abraham MJ, Murtola T, Schulz R, et al. Gromacs: High performance molecular simulations through multi‐level parallelism from laptops to supercomputers. SoftwareX. 2015;1‐2(1–2):19–25. [Google Scholar]
- 62. Huang J, Rauscher S, Nawrocki G, et al. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. [DOI] [PubMed] [Google Scholar]
- 64. Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 65. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 66. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: An automated pipeline for the setup of Poisson‐Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 69. Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. [DOI] [PubMed] [Google Scholar]
- 71. Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Figure S1. Packing of the two PfSERA5PE molecules within the asymmetric unit.
Figure S2. Multiple sequence alignment indicating conservation between PfSERA proteins.
Data Availability Statement
Coordinates for PfSERA5E and PfSERA5PE have been deposited in the Protein Data Bank under the accession code PDB: 6X42 (PfSERA5E) and 6X44 (PfSERA5PE), respectively.