

Abstract
Well‐defined nanofiber scaffold hydrogels made of self‐assembling peptides have found their way into various 3D tissue culture and clinical products. I reflect initial puzzlement of the unexpected discovery, gradual understanding of how these peptides undergo self‐assembly, to eventually translating designer biological scaffolds into commercial products. Peptides are ubiquitous in nature and useful in many fields. They are found as hormones, pheromones, antibacterial, and antifungal agents in innate immunity systems, toxins, as well anti‐inset pesticides. However, the concept of peptides as materials was not recognized until 1990 when a self‐assembling peptide as a repeating segment in a yeast protein was serendipitously discovered. The peptide materials have bona fide materials properties and are made from simple amino acids with well‐ordered nanostructures under physiological conditions. Some current applications include: (a) Real 3D tissue cell cultures of diverse tissue cells and various stem cells; (b) reparative and regenerative medicine as well as tissue engineering; (c) 3D tissue printing; (d) sustained releases of small molecules, growth factors and monoclonal antibodies; and (e) accelerated wound healing of skin and diabetic ulcers as well as instant hemostasis in surgery. Self‐assembling peptide nanobiotechnology will likely continue to expand in many directions in the coming years. I will also briefly introduce my current research using a simple QTY code for membrane protein design. I am greatly honored and humbled to be invited to contribute an Award Winner Recollection of the 2020 Emil Thomas Kaiser Award from the Protein Society.
Keywords: 3D tissue cell cultures, designer peptides, EAK16 & RADA16, membrane protein design, nanofiber scaffold hydrogel, QTY code, regenerative medicine, sustained molecular release, tissue repair
1. INTRODUCTION
Self‐assembling peptide scaffolds can be traced back to the fundamental discoveries made in 1951 by Linus Pauling and his colleagues, who reported the molecular structures of the most fundamental motifs of proteins, namely the α‐helix 1 and the β‐sheet. 2 These secondary structures are the molecular basis of most self‐assembling peptide nanofiber scaffolds. In 1990 a serendipitous discovery of an ionic self‐complementary peptide with a repeating unit in the yeast protein Zuotin, 3 launched peptides as well‐defined materials. Since then, peptide nanofiber scaffolds have been used as 3D scaffolds for the serum‐free culture of animal tissue and stem cells, for reparative and regenerative medicine, for accelerated wound healing and in surgical applications. I highlight the properties and design of a few self‐assembling peptides that are used in clinical applications.
The field of self‐assembling peptide has undergone a substantial growth since the early 1990s. There are numerous groups that are actively pursuing various self‐assembling peptides for a wide range of applications including new biological materials, surface coating materials and semi‐conducting devices, as well as a new class of antibiotics to combat drug resistance. The field is still undergoing expansion at an accelerating pace. It is impossible to capture the entire field in a few pages. The interested reader should read other original reports, more comprehensive reviews in the literature, and search online using “Self‐assembling peptides.” Here I will only focus this article from my own personal experience, my laboratory and a few of my former students' research activities over last 30 years. These activities are summarized in a timeline (Figure 1).
FIGURE 1.
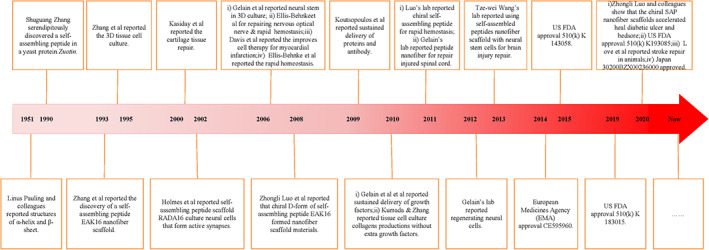
A timeline of progress and diverse applications of designer self‐assembling peptides. It spans over 30 years, from the curiosity‐driven research and serendipitous discovery of the first self‐assembling peptide EAK16‐II in 1990 to successful human clinical trials in 2011. There have been a lot of ups and downs. After gaining full understanding and knowledge of various detailed aspects of amino acid chemistry, peptide structure properties, dynamic molecular self‐assembly behaviors of the self‐assembling peptides, we started to design new peptides with active biological functions that further enhance their usefulness for a wide range of applications including the latest, to repair stroke. MIT Technology Licensing Office (TLO) in 1995 granted an exclusive license to a multi‐billion‐dollar US chemical company, Hercules Inc., to advance the new technology. However, Hercules Inc failed to move it forward 6 years after it took the license, MIT TLO terminated the license in 2001. I was strongly encouraged by MIT TLO in late 2001 to take an exclusive license to found a startup. I then co‐founded 3DMatrix with Alexander Rich and others to translate the technology into new economy. Currently, the self‐assembling peptide nanofiber scaffold hydrogel has been used for all kinds of 3D tissue cell cultures including stem cells and cancer cells and diverse primary tissue cells; surgical applications, slow molecular releases, 3D tissue printing and accelerated wound healing for diabetic ulcers
1.1. Discovery of the first self‐assembling peptide
While I was working on yeast genetics and protein chemistry and trying to understand a left‐handed Z‐DNA structure 4 in the laboratory of Alexander Rich at Massachusetts Institute of Technology in 1989, I identified a protein I named Zuotin (Zuo in Chinese means left and there is a Z in it) for its ability to bind to left‐handed Z‐DNA in the presence of 400‐fold molar excess of sheared salmon DNA that contains ubiquitous right‐handed B‐DNA and other forms of DNA structures. In Zuotin, there is a curious repeat segment with the sequence n‐AEAEAKAKAEAEAKAK‐c, thus I named EAK16 for its amino acid composition and peptide length (Figure 2). 3 , 5 Like some musical melodies that attract attention, repeats in proteins and DNA are not uncommon.
FIGURE 2.

The protein sequence of yeast Zuotin. 3 The positive and negative charges are labeled + or −, respectively. The open and closed circles are predicated phosphorylation sites. The thick underline indicates the alternating sequence that contains the AEAEAKAKAEAEAKAK motif. It was predicted to be a typical α‐helix because of its i, i + 3, i, i + 4 hydrogen bond signature from both the computer algorithm and the publications in 1987. But the reproducible experimental circular dichroism (CD) evidence revealed this motif to be a typical β‐sheet structure 5
In 1990, computer modeling and simulation programs were still quite inadequate and required powerful computers. Luckily, Alex Rich's lab was well equipped for structural biology to analyze X‐ray diffraction data and to determine molecular structures. I initially used computer modeling and simulation to examine this EAK16 sequence. These simulation results showed the EAK16 structure to be a typical α‐helix: its lysines and glutamic acids on the side‐chains with i, i + 3 and i, i + 4 arrangements that could form potential ionic bonds. This prediction is consistent with two previously published papers that similar peptides with A, E, K compositions form stable alpha‐helices under a variety of conditions. 6 , 7
I wondered if this peptide could be synthesized and studied to satisfy my scientific curiosity. I asked Alexander Rich if I can order the EAK16 peptide for further study. This EAK16 peptide then cost over $1,000. Alex Rich asked me “Are you certain you want to pursue these new experiments?” I immediately replied, “Yes” without hesitation. Alex Rich agreed to order the actual EAK16 peptide. I quickly ordered a custom synthesis through the Biopolymers Laboratory at Massachusetts Institute of Technology.
I believe if this discovery had been made in any laboratory other than Alex Rich's lab, my observation perhaps would not have been given such freedom to pursue it further since there was no grant funding to support it. Thus, the entire self‐assembling peptide materials field would have to wait for several more years. After I obtained some reproducible experiments and gradually understand how self‐assembling peptide behaved, Alex remarked to me “You have made an unauthorized discovery.” Alex Rich always had an open mind and allowed people in his lab to pursue unexpected discoveries. Furthermore, his lab was well funded from a variety of sources. For Alex Rich's open‐mindedness, his laboratory made numerous discoveries and contributed a lot of fundamental knowledge to mankind. 8
In 1990, I would never have predicted that my curiosity would lead me into an entirely unexplored field of peptide materials and nanobiotechnology for over a decade. I decidedly made a detour of my research on Z‐DNA biology and went into an uncharted territory to pursue the self‐assembling peptides. When the late Ephraim Katzir of Weizmann Institute of Science invited me to give talks in Israel several times, he remarked during my visit at his home in 1997: “You have opened a new direction for peptide materials research, no one had thought about.” Katzir was a pioneering peptide biochemist in the 1940s in John Edsall's lab at Harvard University. They together contributed enormously to our understanding of peptide science in 1940–1950s.
When the EAK16 peptide was first studied following the reported method using the Aviv circular dichroism (CD) spectroscopy, an unexpected result occurred. Instead of showing a CD spectrum of the computer‐modeled alpha‐helix, to our great surprise, the peptide showed an exceedingly stable β‐sheet CD spectrum (Figure 3). The EAK16 not only was stable in pH 1–11 with little CD spectrum changes, but also was resistent to heat treatment, 1% SDS, 8 M urea and 6 M guanidine HCl. 9 Upon adding a few crystals of sodium phosphate, a membrane‐like transparent material occurred in the petri dish visible to the naked eye. 5 , 9 , 10
FIGURE 3.
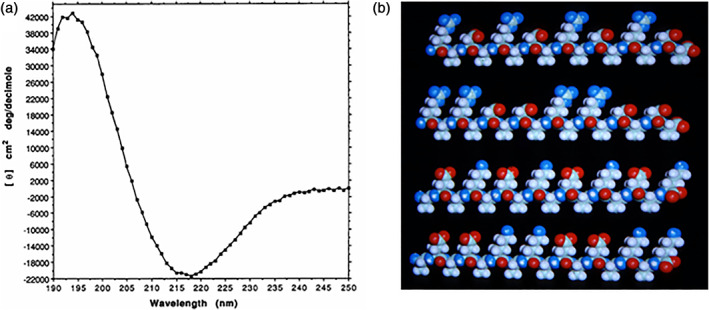
The first self‐assembling peptide. (a) CD spectrum of the first self‐assembling EAK16 peptide (AcN‐AEAEAKAKAEAEAKAK‐CONH2). A typical β‐sheet CD spectrum with a minimum at 218 nm and a maximum at 195 nm is observed. (b) and the molecular models of the designer amphiphilic self‐assembling peptides. These peptides have two distinctive sides, one hydrophobic and the other hydrophilic. The hydrophobic side forms a double sheet inside of the fiber and the hydrophilic side forms the outside of the nanofibers that interacts with water molecules, forming hydrogel that contains as high as 99.9% water. At least three types of molecules can be made, with −, +, −/+ on the hydrophilic side. The individual self‐assembling peptide molecules are ~6 nm long. The first such peptide, EAK16‐II, was discovered from a yeast protein, Zuotin. 5 , 9 This peptide inspired us to systematically design a large class of self‐assembling peptides. When these peptides are dissolved in water in the presence of salt, they spontaneously assemble into well‐ordered nanofibers and then further into nanofiber scaffold hydrogels
Alex Rich's lab is a crystallographic laboratory, I thus attempted to set up conditions to grow single crystals for X‐ray diffraction, but no crystals appeared after numerous attempts. I also tried to fill the capilary with 20 mg/mL (2%) peptide placed on a cusion of frozen butter and centrifuged to pack the peptides and to remove air bubbles. But no clear diffraction pattern emergied. It was later understood that such self‐assembled peptide could not form 3D single crystal, rather it forms a single nanofiber that could only be studied by X‐ray fiber diffraction.
Alex Rich was a very close friend of the legendary Francis Crick since early 1950s. They published several papers on the molecular structure of collagen together in 1955 and 1961. 11 , 12 , 13 Alex Rich introduced me to Francis Crick during Crick's visit to Alex Rich's lab in September 1988, ~3 months after I joined Alex Rich's lab.
In August 1991, I visited Francis Crick at the Salk Institute. During our conversation, I told him about my unexpected discovery. Crick was very curious and suggested doing X‐ray diffraction. I told him this EAK16 peptide did not form a single crystal. Crick then suggested me to look it under an electron microscope (EM). I followed Crick's advice and examined the peptide matrix using both SEM and TEM in late 1991 and early 1992. To my great surprise, I observed under SEM the well‐ordered nanofiber scaffold with nanopores (5–200 nm) (Figure 4). 5 , 10 It took me more than 1 year to understand how the seemingly soluble short peptides undergo self‐assembly to form well‐ordered nanofiber scaffold membranous material that was visible to the naked eye. My colleagues and I reported the first self‐assembling peptide that formed visible nanofiber material. 5 , 9 , 10 Since then the self‐assembling peptide field has expanded in a number of directions, and many medical products have received approval by the US FDA, the European Medicine Agency, the Japan Medical Agency and a medical agency in Chengdu, China (Figure 1). 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24
FIGURE 4.

Discovery of the first self‐assembling peptide scaffold material. (a) The structure was formed in phosphate‐buffered saline and transferred to a glass slide. The colorless membranous structures are isobuoyant; therefore, the image is not completely in focus (×75 Nomarski Phase contrast microscope.) (b) The structure stained bright red with Congo red and can then be seen by the naked eye (×15, each scale unit = 1 mm). (c) A portion of a well‐defined membranous structure with layers is clearly visible; the dimensions of this particular membrane are 2 × 3mm (×20)
1.2. Self‐assembling peptide nanofiber materials
Designer materials that are self‐assembled by numerous individual molecules to produce novel supramolecular architectures is a “bottom‐up” approach, rather than the “top‐down” approach of polymerization of the monomers through catalysis and material processing. The self‐assembly approach requires a deep understanding of individual molecular building blocks, their structures and dynamically assembly properties. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 These self‐assembled materials have become an integral part of a broad spectrum of designer and fine materials.
These self‐assembling peptides have alternating hydrophobic, namely, alanine, valine, leucine, isoleucine, and phenylalanine, and hydrophilic sides, which include positively charged lysine, arginine, histidine, and negatively charged aspartic acids and glutamic acids. 5 , 9 , 10 , 14 , 25 , 26
The complementary ionic sides have been classified into moduli I, II, III, IV and mixed moduli. This classification is based on the hydrophilic surface of the molecules that have alternating + and − amino acid residues, alternating by repeats of 1, 2, 3, or 4 and so on. For example, charge arrangements for the different moduli are as follows: modulus I, − + − + − + − +; modulus II, − − + + − − + +; modulus III, − − − + + +; and modulus IV, − ‐ ‐ − + + + +. 14 , 15 The charge orientation can also be designed in reverse orientations that yield entirely different molecules with distinct molecular behaviors. 15 These well‐defined peptide sequences allow them to undergo ordered self‐assembly, resembling some situations found in well‐studied polymer assemblies. This simple idea is the basis of the self‐assembling peptide building blocks.
1.3. Basic chemical properties of the self‐assembling peptide systems
The process of peptide synthesis using either solid or liquid phase chemistry has become more and more affordable. The cost directly correlates with (a) peptide length, (b) purity, (c) skill of the manufacturer, and (d) the chirality of amino acids. Most self‐assembling peptides are readily soluble in water since their amino acid molecules consist of alternating hydrophilic and hydrophobic that contain 50% charged residues with distinct polar and non‐polar surfaces and periodic repeats. Self‐assembly is accelerated by physiological salt concentration under physiological pH conditions to form ordered nanofiber scaffolds. 27 , 28 , 29
For example, RADA16‐I and RADA16‐II with arginine and aspartate residues replacing lysine and glutamate were designed. The alanines form overlapping hydrophobic interactions in water, while both positive Arg and negative Asp charges are packed together through intermolecular ionic interactions in a checkerboard‐like manner. They self‐assemble to form nanofibers ~10 nm in diameter, and theses nanofibers interweave into scaffolds that retain extremely high hydration, >99% in water (1 mg–10 mg/mL, wt/vol) (Figure 5). 25 , 29
FIGURE 5.

Self‐assembling peptide RADA16 nanofiber scaffold hydrogel. (a) Amino acid sequence of RADA16, molecular model of a single RADA16 peptide. The calculated single peptide dimensions are ~6 nm × 1.3 nm × 0.8 nm, numerous individual peptides self‐assemble into a nanofiber (computer simulation) as confirmed by the SEM image of RADA16 nanofiber scaffold, scale bar = 0.5 mm (SEM image courtesy of Fabrizio Gelain). RADA16‐I peptide forms nanofibers in aqueous solution that further forms a hydrogel with water content of 99.5–99.9% (wt/vol water). (b) The hydrogel gel is used for 3D tissue cell culture and 3D tissue printing. (c) Prefilled peptide hydrogel in a syringe for various surgical uses in hospitals. (d) The self‐assembling peptide RADA16 nanofiber scaffold hydrogel. Image (d) is courtesy of 3DMatrix
The formation of the scaffold and its mechanical properties are influenced by several factors: (a) amino acid sequence, (b) the level of hydrophobicity, (c) length of the peptides, and (d) self‐assembling time. For example, the extent of the hydrophobic residues, Ala, Val, Ile, Leu, Tyr, Phe, Trp (or single letter code, A, V, I, L, Y, P, W) can significantly influence the mechanical properties of the scaffolds and the speed of their self‐assembly. The higher the content of hydrophobicity, the easier it is for scaffold formation, and the better for their mechanical properties. 27 , 28
1.4. The proposed general assembly of nanofiber formations
The detailed mechanism how self‐assembling peptides self‐organize themselves in water at low concentration still remains inadequately understood at present; nevertheless, various self‐assembling peptides as biological materials have been used for diverse applications.
Several plausible ideas for different nanostructures have been proposed. First, for nanofibers, a molecular model to interpret the formation of EAK16 and RADA16 was proposed. These two peptides are representative of a class of peptides that undergo self‐assembly into ordered‐nanofibers, characterized by: (a) numerous intermolecular hydrogen bonds between the peptide —C=O……H—N— in the conventional β‐sheets on the peptide backbones, (b) the side chains of positively and negatively charged residues form intermolecular ionic bonds in a checkerboard manner, (c) hydrophobic interactions between the peptides from the amino acid residues are pushed by water molecules, and (d) alternating polar and nonpolar surface interaction. It is known that salt ions facilitate the self‐assembly, however it is not yet clear where the monovalent ions may coordinate the charged residues in a higher order of geometry. 5 , 29
1.5. Dynamic reassembly of self‐assembling peptides
Self‐assembling peptides form stable β‐sheet structures in water. The interactions between the peptides in β‐sheets are: (a) non‐covalent hydrogen bonds along the backbones, (b) the arrays of ionic + and − charge interactions, (c) alanine hydrophobic interactions and van der Waals interactions, and (d) water‐mediated hydrogen bond formations. The nanofibers can be disrupted mechanically using sonication. 29 The self‐assembling process is reversible and dynamic since these peptides are short and simple. Numerous individual peptides can be readily self‐organized through these weak interactions, and they can undergo dynamic reassembly repeatedly (Figure 6), similar to the material self‐healing processes. The driving energy of the assembly in water is not only through hydrophobic van der Waals interactions, but also the arrays of ionic interactions as well as the peptide backbone hydrogen bonds. This phenomenon can be further exploited for production and fabrication of many self‐assembling peptide materials. 29
FIGURE 6.

Atomic force microscopy (AFM) images of RADA16‐I nanofiber at various time points after sonication. The observations were made using AFM immediately after sample preparation. (a) 1 min after sonication; (b) 2 min; (c) 4 min; (d) 8 min; (e) 16 min; (f) 32 min; (g) 64 min; (h) 2 hr; (i) 4 hr, and (j) 24 hr. Note the elongation and reassembly of the peptide nanofibers over time. By ~1–2 hr, these self‐assembling peptide nanofibers have nearly fully re‐assembled This process of sonication and re‐assembly can be repeated many times, thus truly demonstrating the natural power of self‐assembly. Image courtesy of Hidenori Yokoi 29
Unlike processed polymer microfibers in which the monomers or fragments of polymers cannot undergo reassembly without addition of catalysts or through material processing, the supramolecular self‐assembly and re‐assembly events are without any external instructions. They are likely wide spread in many unrelated fibrous biological materials where numerous weak interactions are involved. Self‐assembly and re‐assembly are a very important property for fabricating novel materials, and it is necessary to fully understand its detailed process in order to design and to improve biological materials.
AFM images revealed that the nanofibers range from several hundred nanometers to a few microns in length before sonication. After sonication, the fragments were broken into ~20–100 nm. The kinetics of the nanofiber reassembly is closely followed at 1, 2, 4, 8, 16, 32, and 64 minutes as well as 2, 4, and 24 hours (Figure 6). The nanofiber length reassembly is as a function of time: by 2 hours, the peptide nanofibers have essentially reassembled to their original length. The β‐sheet structure had little change since the β‐sheets at the molecular level remain unchanged despite the change in nanofiber length. 29
1.6. Molecular modeling of the self‐assembly
We carried out molecular modeling of the self‐assembly process. For molecular modeling clarity, the RADA16‐I β‐sheet is presented as a non‐twisted strand. It is known that these peptides form stable β‐sheet structure in water. They form the intermolecular hydrogen bonding on the peptide backbones, similar as in the conventional β‐sheet. They also have two distinctive sides, one hydrophobic with array of overlapping alanines (Figure 7, green color sandwiched inside), similar to what is found in silk fibroin or spider silk assemblies, the other side of the backbones has negatively charged (−) amino acids, represented as red, and positively charged (+) amino acids, represented as blue.
FIGURE 7.
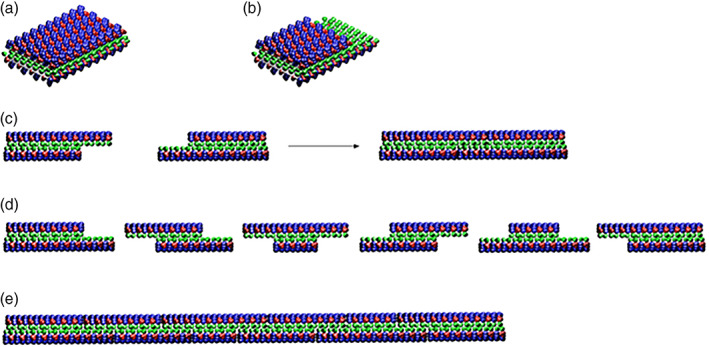
Molecular model for dynamic reassembly. When the peptides self‐assemble into stable β‐sheets in water, they form intermolecular hydrogen bonds along the peptide backbones. The β‐sheet structure has two distinctive sides, one hydrophobic with an array of alanines (green color) and the other with amino acids of negatively charged (red color) and positively charged (blue color). The alanines form overlap packed hydrophobic interactions in water, a structure that is found in silk fibroin from silkworm and spiders. On the charged sides, both positive and negative charges are packed together through intermolecular ionic interactions in a checkerboard‐like manner. When the fragments of nanofiber first meet, the hydrophobic sides may not fit perfectly but with gaps. However, the non‐specific hydrophobic interactions permit the nanofiber to slide diffusion along the fiber in either direction that minimizes the exposure of hydrophobic alanines and eventually fill the gaps. The sliding diffusion phenomenon was also proposed for nucleic acids of polyA and polyU in 1956. 30 , 31 For clarity, these β‐sheets are not presented as twisted strands. Image courtesy of Hidenori Yokoi 29
The alanines form packed hydrophobic interactions in water, which can be disrupted mechanically during sonication. However, these hydrophobic cohesive ends could find each other quickly in water since the exposure of hydrophobic alanine arrays to water is energetically unfavorable. Since the hydrophobic alanines' interaction is non‐specific, they can slide diffuse along the nanofiber, like trains sliding along train tracks. The same sliding diffusion phenomenon was also observed in early studies of nucleic acids where polyA and polyU form complementary base pairings that can slide diffuse along the double helical chains. 30 , 31 If however, the bases are heterogonous, containing A, G, T, C, then the bases cannot undergo sliding diffusion. Likewise, if the hydrophobic side of the peptides does not always contain alanine, containing residues such as valine and isoleucine, it would become more difficult for sliding diffusion to occur due to their structural constraint.
On the charged side, both positive and negative charges are packed together through intermolecular ionic interactions in a checkerboard‐like manner. Likewise, the collectively complementary + and − ionic interactions may also facilitate reassembly. Similar to restriction‐digested DNA fragments, these nanofiber fragments could form various assemblies, like blunt and protruding ends. The fragments with various protruding ends as well as blunt ends can reassemble readily through hydrophobic and ionic interactions (Figure 7).
1.7. Self‐assembling peptide nanofibers
The first self‐assembling peptide EAK16 and the subsequently designed RADA16 are general peptides without any active motifs. Here, the RADA16 made the first milestone for new economic development. RADA16 has become commercial products for various 3D tissue cell cultures. It has also been successfully developed for various applications as medical devices including surgical applications.
These peptides form stable secondary structures including α‐helix and β‐sheet. But some peptides secondary structures are more dynamic depending on several factors. These include: (a) the amino acid sequence arrangements (even with identical composition), (b) the molecular size of the peptide, (c) peptide concentration, (d) pH of the solution, (e) temperature, (f) the medium composition, such as solvent or substrate, (g) ionic strength, and (h) the presence of de‐naturation agents, such as sodium dodecyl sulfate (SDS), urea, and guanidium. HCl. These factors can significantly influence the dynamic behavior of peptide secondary structures, as well as the process of self‐assembly. 5 , 9 , 25 , 26 , 27 , 28 , 29
1.8. Specific self‐assembling peptide materials
The first generation of self‐assembling peptides are promising scaffold materials, with no apparent cell interaction. Like many other polymers, they are not naturally found in living systems. In order to introduce the desired functions targeted for specific cell types, the second generation of designer peptides is designed by directly coupling biologically active and functional peptide motifs onto the generic peptide (Figure 8). This second generation of designer scaffolds have significantly enhanced interactions with cells and tissues as well as other molecules.
FIGURE 8.
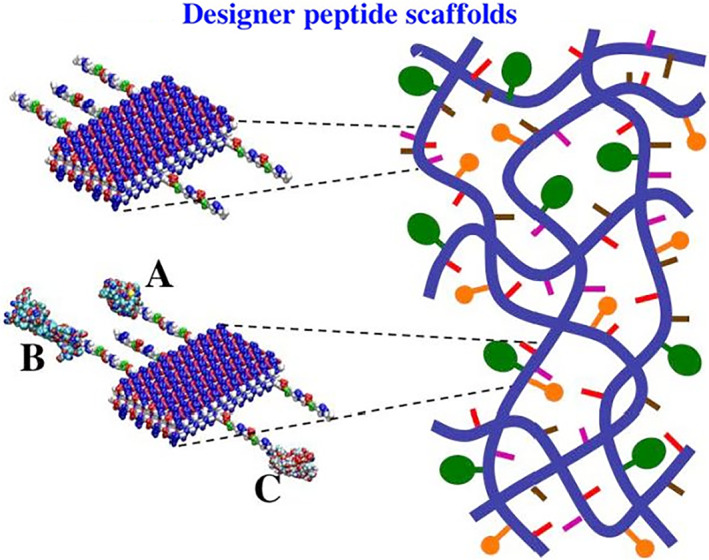
Molecular and schematic models of the designer peptides and of the scaffolds. Direct extension of the self‐assembling peptide sequence by adding different functional motifs. Blue thick lines represent the self‐assembling backbone and the yellow, pink, and tan lines represent various functional peptide motifs. Molecular model of a self‐assembling peptide nanofiber with functional motifs flagging from both sides of the double β‐sheet nanofibers. Either few or more functionalized and active peptide can be mixed at the same time. The density of these functionalized peptides can be easily adjusted by simply mixing them in various ratios, 1:1–1,000,000 or more, before assembling. They will then be part of the self‐assembled scaffold. The left panel shows enlargements of a small part of the peptide nanofibers on the right panel. The enlarged parts show more detail of co‐assembly of peptides with biologically active motifs, either different peptides (upper left panel), or different proteins (lower left panels: a, b, and c)
My lab designed a wide range of designer self‐assembling peptide nanofiber scaffolds for both 3D tissue cell cultures 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 and for sustained molecular release. 42 , 43 , 44 , 45
The designer self‐assembling peptides with bioactive motifs play a much more important biological role, becoming more attractive as biological materials with added functionalities and values. Although additional active motifs can be appended to the generic peptide backbone, the nanofiber structures remain quite similar, as shown by SEM (Figure 9). 35
FIGURE 9.
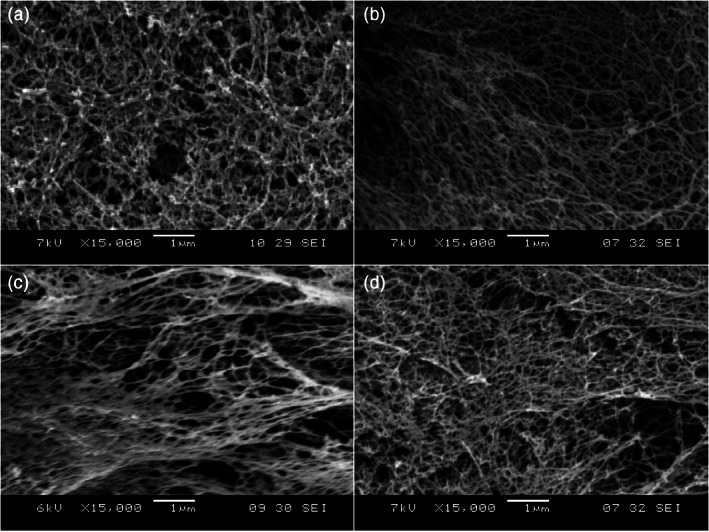
SEM images of Matrigel and various designer peptide nanofiber scaffolds. (a) Matrigel, (b) RADA16, (c) RADA16‐BMHP1, (d) RADA16‐BMHP2 nanofiber scaffolds assembled in PBS solutions. Matrigel nanostructures are comparable in size to nanofibers found after self‐assembly of the designer peptides. Clusters and aggregates of the unidentified naturally derived proteins in Matrigel (a) are absent in the pure peptide scaffolds shown in (b)–(d). The interwoven nanofibers are ~10 nm in diameter in each of the peptide scaffolds with ~5–200 nm pores. The appended functional motifs did not prevent peptide self‐assembly 35
In some cases, when the active sequences become longer, the self‐assembling speed and mechanical properties are affected. 36 , 37 , 38 , 39 , 40 The 3D nanofiber materials are superb scaffolds for diverse tissue cells, not only for differentiation, proliferation, migration, but also for long‐term maintenance. 36 , 37 , 38 , 39 , 40
The self‐assembling peptide nanofiber structure is similar to the widely used Matrigel nanostructure, except the Matrigel is an animal product and not pure. It contains many other substances and growth factors (Figure 9a).
In order to fully understand how cells behave in the 3‐dimensional (3D) microenvironment with respect to 3D gradient diffusion, 3D cell migration, 3D cell–cell contact interactions, and in tissue engineering and regenerative medicine, it is important to develop a well‐controlled 3D tissue culture system where every single ingredient is known.
For over 100 years since the Petri dish was invented and used for cell culture studies, almost all tissue cells have been studied on the 2D Petri dish and various formats of coated 2D surfaces before 1990s. However, this 2D surface is rather unlike 3D tissue and the body's microenvironment. 17 Thus, it is important to develop a true 3D microenvironment to mimic the real tissue and body situation. The commonly used biomaterials are inadequate due to their microfiber and micro‐pore size. Animal derived collagen gel and Matrigel contain other residue materials that are not always adequate for finely controlled studies. The designer‐peptide scaffold can be fine‐tuned to overcome these disadvantages. 17
In order to achieve the fine‐tuning and control, we designed peptides tailored for specific individual cell types, or for needs of studies or applications, by appending selected active motifs onto the basic peptide, such as RADA16‐I or EAK16‐II.
From the point of view of synthetic organic chemistry, peptides with either C‐ or ‐N termini may be attached to the modified motifs. However, the functional motifs should be always located on the C‐terminus because solid phase peptide synthesis initiates from the C‐terminus and proceeds toward the N‐terminus. The longer the peptide sequence is, the more probable a coupling error would occur. Thus, in order to avoid peptide synthesis errors, the biologically active sequence motifs should always be at the C‐terminus without exception (Figure 8). 35 , 36 , 38
Usually a spacer comprising 2‐glycine residues is added to guarantee a flexible and correct exposure of the biologically active motifs to cell surface receptors. If one combines a few designer peptides with different biologically active motifs, these different functional motifs in various ratios can be incorporated in the same scaffold (Figure 8). Upon exposure to solution at neutral pH, the functionalized sequences self‐assemble, leaving the added motifs on both sides of each nanofiber (Figure 8). Nanofibers take part in the overall scaffold, thus providing functionalized microenvironments with specific biological stimuli (Figure 8).
Self‐assembling peptides with functional motifs can now be commercially produced at a reasonable cost. They can be readily adapted for wide spread use, including the study of cell interactions with their local‐ and micro‐environments: cell migration in 3D, tumor and cancer cells interactions with normal cells, cell processes and neurite extensions, cell‐based drug screen assays, to name a few.
My lab and later Dr. Gelain's lab designed a wide range of self‐assembling peptides with a variety of biologically active motifs with different lengths and showed that the addition of motifs to the self‐assembling peptide RADA16‐I did not significantly inhibit self‐assembling properties. Furthermore, one can combine the RADA16‐I nanofiber with biologically active designer self‐assembling peptides by mixing the modified peptides. Although their nanofiber structures are nearly indistinguishable from the RADA16‐I scaffold (Figure 9), the appended functional motifs significantly influence cell behavior (Figure 10).
FIGURE 10.
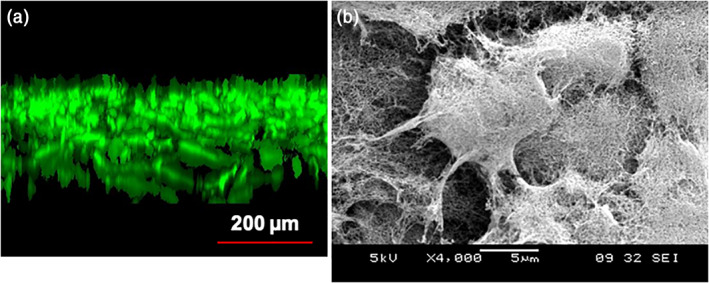
3D cell culture using peptide scaffold hydrogel. (a) confocal image of osteoblast cells in 3D peptide scaffold hydrogel. (b) SEM images of mouse neural stem cells embedded in designer peptide nanofiber scaffold RADA16‐BMHP1 (1%, vol/wt) after 14‐day in vitro cultures. Cluster of three visible mouse neural stem cells embedded in 3‐D self‐assembling RADA16‐BMHP1. It is important to point out that the nanoscales of peptide scaffold and the extracellular matrix made by cells are indistinguishable. This is totally unlike the many processed biopolymer microfibers that are often in 10–50 μm in diameter, which is 1,000–5,000 larger (many more orders of magnitude larger in 3‐dimensions) than the self‐assembling peptide nanofibers. 35 , 36 Images courtesy of Akihiro Horii (a) and Fabrizio Gelain (b)
Using the designer self‐assembling peptide nanofiber system, every ingredient of the scaffold can be defined in the serum‐free media. Furthermore, it can be combined with multiple functionalities including soluble factors (Figure 8). Cells reside in a 3‐D environment where the extracellular matrix receptors on cell membranes can bind to the functional ligands appended to the peptide scaffolds. It has been shown that higher tissue architectures with multiple cell types can be constructed using the designer 3‐D self‐assembling peptide nanofiber scaffolds, such as organoids, organ on a chip or mini‐organs.
Even if only a fraction of functionalized motifs on the 3‐D scaffold are available for cell receptor binding, cells may likely receive more external stimuli than when in contact with coated 2‐D petri dishes or RGD‐coated (or other motifs) polymer microfibers. These latter are often ~10–100 μm in diameter that are substantially larger than the cell surface receptors, and in most cases, larger than the cells themselves. There, cells are not in a real 3‐D environment, but rather, they are on a 2‐D surface wrapping around the microfiber polymers with a curvature that depends on the diameter of the polymers. In a 2‐D environment, where only one side of the cell body is in direct contact with the surface, it is possible that receptor clustering at the attachment site may be induced. On the other hand, the receptors for growth factors, cytokines, nutrients and signals may be on the other sides in direct contact with the culture media. Perhaps cells may become partially polarized. In the 3‐D environment, the functional motifs on the nanofiber scaffold surround the whole cell body in all dimensions. Thus, growth factors may form a gradient in 3‐D nanoporous microenvironment.
In our search for additional functional motifs, we found that a class of bone marrow homing peptides BMHP is one of the most promising active motifs for stimulating adult mouse neural stem cell (NSC) adhesion and differentiation. This observation led to a new class of designer self‐assembling peptides for 3D cell biology studies and for in vivo animal trials. 35
We appended several functional motifs including cell adhesion, differentiation and bone marrow homing motifs on the C‐termini 35 , 36 , 37 , 38 , 39 , 40 as well as a matrix metalloproteinase‐sensitive cleavage motif in the middle of a peptide 41 to modulate the peptide mechanical properties. My lab used them to study neural stem cells. These functionalized peptides underwent self‐assembly into nanofibers structure as well. More functionalized self‐assembling peptides have been shown to promote specific cellular responses, long‐term cell survival, proliferation, migration, morphological differentiation, and the fine‐control of molecular releases. 42 , 43 , 44 , 45 Thus these peptide‐scaffolds are excellent for diverse applications. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24
Designer self‐assembling peptide scaffolds have shown interesting interactions with functional proteins for the study of molecular releases. The release kinetics results suggested that protein diffusion through nanofiber scaffolds primarily depends on the size, shape, and charge of the proteins and other properties. 43 , 45
1.9. Diverse uses of self‐assembling peptide nanofiber scaffold hydrogels
A wide range of diverse uses in various areas has been developed from these self‐assembling peptide hydrogel materials. They include: (a) reparative and regenerative medicine, (b) sustained molecular releases (small molecules, protein growth factors and monoclonal antibodies), and (c) accelerated wound healing in human clinical and surgical applications. Current research activities in animal models include: (d) the repair of spinal cord injuries, and (e) the repair of hemorrhagic stroke injury.
2. REPARATIVE AND REGENERATIVE MEDICINE
Self‐assembling peptide materials are easy to use in tissue repair, wound healing, addressing chronic wound problems, and regenerative medicine (Figure 11). Holmes et al. showed the primary neurites extend a great distance from the cell body and form active synapses with very high density in RADA16‐I nanofiber scaffold (Figure 11a). 25 Inspired by this finding, Ellis‐Behnke asked me to provide him with peptide scaffold material. He and colleagues then used RADA16‐I to repair injured rat and mouse brain structures, and their results showed the peptide scaffold hydrogel was effective not only for axon regeneration through the site of an acute brain injury, but also to knit the injured brain tissue together seamlessly (Figure 11c). 25 This work represents an enabling nanobiomedical technology for tissue brain repair and restoration (Figure 11). 46
FIGURE 11.

From designer peptide to scaffold to tissues. (a) Active synapses on the peptide surface. Primary rat hippocampal neurons form active synapses on peptide scaffolds. The confocal images shown bright discrete green dot labeling indicative of synaptically active membranes after incubation of neurons with the fluorescent lipophilic probe FM‐143. FM‐143 can selectively trace synaptic vesicle turnover during the process of synaptic transmission. The active synapses on the peptide scaffold are fully functional, indicating that the peptide scaffold is a permissible material for neurite outgrowth and active synapse formation. (b) Adult mouse neural stem cells embedded in 3D scaffold (image courtesy of Fabrizio Gelain). (c) Brain damage repair in hamster. The peptide scaffold was injected into the optical nerve area of brain that was first severed with a knife (image courtesy of Rutledge Ellis‐Behnke). The gap was sealed by the migrating cells after a few days. A great number of neurons formed synapses. (d) Peptide KLD12 (KLDLKLDLKLDL), chondrocytes in the peptide scaffold and cartilage. The chondrocytes stained with TB show abundant GAG production (left panel) and antibody to Type II collagen demonstrate abundant Type II collagen production (right panel). A piece of pre‐molded cartilage with encapsulated chondrocytes in the peptide nanofiber scaffold. The cartilage formed over a ~4‐week period after the initial seeding of the chondrocytes (image courtesy of John Kisiday). (e) Von Kossa staining showing transverse sections of primary osteoblast cells on HA‐PHP‐RADA16‐I self‐assembling peptide nanofiber scaffold (image courtesy of Akihiro Horii). Scale bar = 0.1 mm. The intensely stained black areas represent bone nodules forming
Ellis‐Behnke and colleagues found that self‐assembling peptide scaffolds are useful in repairing the damaged brain. 46 They also found that the peptide scaffold hydrogel instantly stopped bleeding in a few seconds during the procedure of repairing injured brain. They then expanded their studies to hemostasis of brain, spinal cord, femoral artery, and liver of rat. 47
The early self‐assembling peptide scaffold work has inspired others to apply it to different kinds of tissues, organs and animals. 48 , 49 , 50 , 51 RADA16‐I functionalized with biologically active motifs also induced reparative activity in injured spinal cords. 52 , 53 , 54
In order to better understand the individual molecular and material building blocks, their structures, assembly properties, dynamic behaviors and application for rapid homeostasis, we also used D‐amino acids, the chiral self‐assembling peptide d‐EAK16 also forms 3‐dimensional nanofiber scaffold. This study not only provided insights into understanding the chiral assembly properties for rapid homeostasis, but also to aid in further design of self‐assembling D‐form peptide scaffolds for clinical trauma emergency. 50 , 55 , 56
2.1. Sustained molecular releases
In 1992 when I first examined the SEM image of EAK16 peptide nanofiber scaffold with numerous nanopores ranging 5–200 nm in size, I recognized the potential application for molecular releases. I was inspired by Robert Langer, the pioneer of controlled and sustained molecular releases. I had already read much about his innovative research on controlled drug delivery and tissue engineering. Before carrying out any experiments, I suggested that such a scaffold could be useful for molecular releases. 5 Since the self‐assembling peptides form a nanofiber scaffold with nanopores, it is possible to load small molecules and large proteins for sustained delivery of various molecules including small molecules, peptides, growth factors, antibodies and more. 42 , 43 , 44 , 45
Using RAD16‐I as a model for the slow release of hydrophobic molecules, we showed that these self‐assembling peptide scaffolds could encapsulate hydrophobic drug. Using various dye molecules including phenol red, bromophenol blue, 8‐hydroxypyrene‐1, 3, 6‐trisulfonic acid tri‐sodium salt, 1, 3, 6, 8‐pyrenetetrasulfonic acid tetra‐sodium salt, and Coomassie Brilliant Blue G‐250, we demonstrated the sustained release of small molecules through RADA16 hydrogels. 42
The RADA16 and its derivatives with appended motifs scaffolds have also been used for the sustained release of proteins including lysozyme, trypsin inhibitor, BSA, MMP‐13, monoclonal antibody IgG (Figure 12), 43 and active growth factors β‐FGF, TGF, VEGF and BDNF. 44 The sustained release continued from a few days to over 100 days (>3 months) when the experiments were terminated. 45 It is likely that the release period for these molecules can be much longer since the content had not decreased significantly when the experiment results were collected (Figure 13). 45
FIGURE 12.
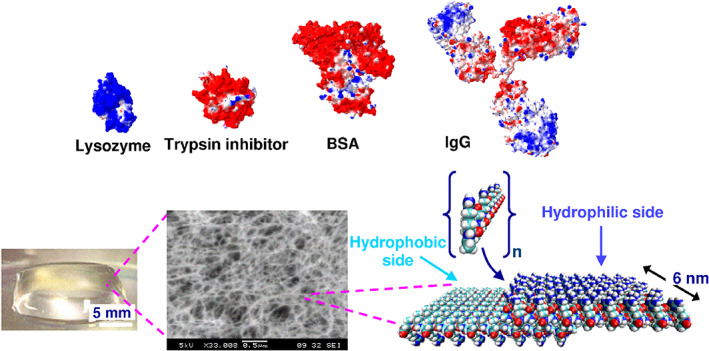
Molecular representation of lysozyme, trypsin inhibitor, BSA, and IgG as well as of the Ac‐N‐(RADA)4‐CONH2 peptide monomer and of the self‐assembling peptide nanofiber. Color scheme for proteins and peptides: positively charged (blue), negatively charged (red) and hydrophobic (light blue). Protein models were based on known crystal structures from PDB (Protein Data Bank). Image courtesy of Sotirios Koutsopolous 43
FIGURE 13.
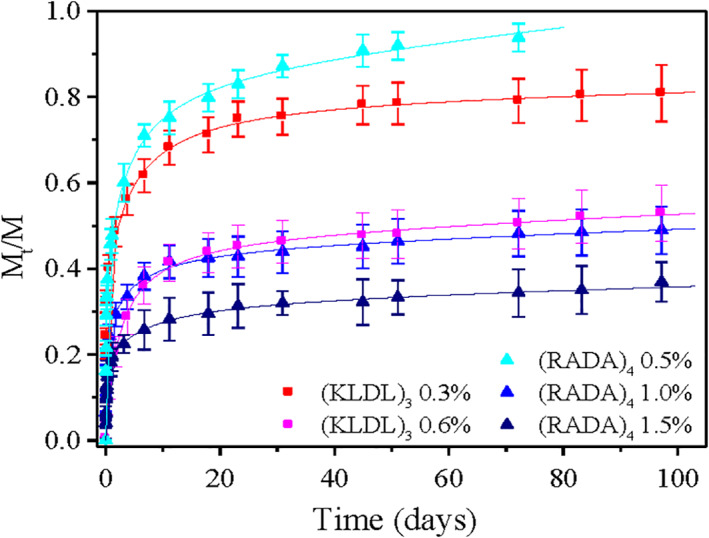
The release profiles during the entire 3‐month period for IgG through hydrogels of different peptides and different peptide nanofiber densities. Hydrogels consisted of the self‐assembling peptides (i) Ac‐N(RADA)4‐CONH2 with concentration 0.5% wt/vol (light blue, ▲), 1.0% wt/vol (blue, ▲), and 1.5% w/v (dark blue, ▲) and of (ii) ac‐(KLDL)3‐CONH2 with concentration 0.3% wt/vol (red, ■) and 0.6% wt/vol (magenta, ■). Release experiments were performed in PBS, pH 7.4 at room temperature. Data points represent the average of five samples. Image courtesy of Sotirios Koutsopolous 45
Our results not only provide evidence for long‐term sustained molecular release from self‐assembling peptide scaffolds, but also it inspires others to design more self‐assembling peptides to control molecular release for clinical applications.
2.2. Self‐assembling peptide scaffold hydrogels for human surgical uses and for accelerated wound‐healing
Discoveries and inventions of new materials should benefit society (Figure 14). It is not good enough to make an exciting discovery, file a patent, publish a scientific paper and move on. Academic researchers should be encouraged to make the new materials widely available for others to use and make them commercially available. Since the discovery of SAP scaffold hydrogels, many efforts have been made to large‐scale produce various commercial products that are both affordable and beneficial to society.
FIGURE 14.
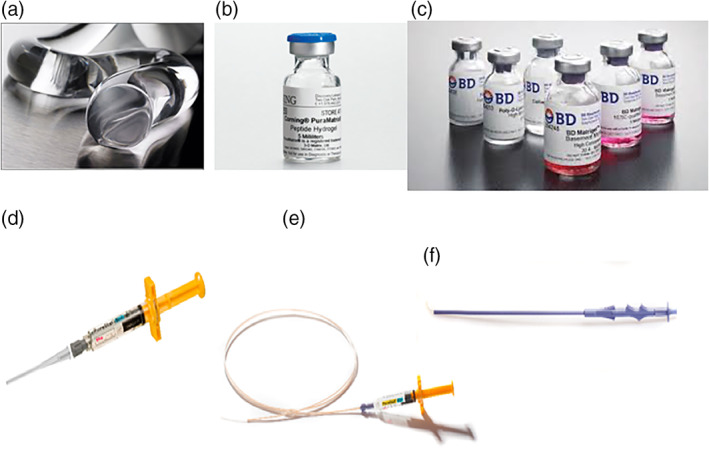
Various commercial products derived from self‐assembling peptide scaffold hydrogel. (a) The image of peptide hydrogel extruded from a needle; (b) peptide hydrogel in a Corning vial (image courtesy of Corning); (c) various products of peptide hydrogel in BD vials (image courtesy of Fisher Scientific); (d) PuraStat® 5 mL syringe with attached nozzle applicator, ready for use as a hemostatic agent during surgery after opening the packaging and attaching the nozzle applicator; (e) endoscopic nozzle (160 mm) attached to a PuraStat® syringe and (f) laparoscopic nozzle. (Images a, d, e, and f are courtesy of 3DMatrix, Inc)
The current clinical practice in wound healing seems to be similar to procedures used many years ago: use of sutures, staples, or tape for wound closure, prevention and mitigation of infection, and other treatment measures. Accelerated wound‐healing will benefit both patients and clinical workers. However, accelerated surgical and traumatic wound healing, especially wounds from chronic diabetic ulcers or bedsores are still problematic. The self‐assembling peptide scaffolds may alleviate the problem if put it to good use to benefit the patients. 23 , 24
The self‐assembling peptide scaffolds have been used for all tissue cell cultures including human cells, and the degradation products of the scaffolds are natural amino acids that pose no harm to the human body. Furthermore, a variety of stringent animal tests with rigorous controls showed that the peptides did not elicit noticeable immune responses, nor cause measurable inflammatory reactions through injections and surgical procedures at various tissue sites. It thus encouraged researchers to carry out human clinical trial for accelerated wound‐healing.
To date, studies have been carried out for various surgical wounds including cardiovascular procedures for coronary artery bypass and synthetic blood vessel replacement, gastrointestinal surgery for partial hepatectomy and gastrointestinal treatment for endoscopic excision of mucosa. The clinical results are very encouraging and beneficial for patients. There were no observed adverse and undesirable side effects so far. This is not surprising since the designer self‐assembling peptide scaffolds are totally pure synthetic amino acid‐based materials; there are no animal‐derived impurities, no chemical and biological contaminations, no organic solvents, and no toxic compounds.
Since the successful human clinical trials, additional trials for other indications have been planned or launched, including human tooth wound healing, skin wound healing from other diseases and injuries, in various parts of the world. Based on previous successful clinical trials, it is anticipated that these clinical trials will likely also be successful. It is hoped that the self‐assembling peptide scaffolds will become an enabling medical technology that will truly benefit society. 23 , 24
2.3. Beyond self‐assembling peptides
After the self‐assembling peptide nanofiber scaffold hydrogels were commercialized in early 2000s, I turned my attention to other interesting areas of research. I had a vivid and lively conversation with my senior colleague, the legendary biochemist John (Jack) M. Buchanan. 57 , 58 In 1953 Jack Buchanan was recruited from University of Pennsylvania Medical School to join MIT to boost MIT biology. The late MIT President James Killian wrote in his autobiography that bringing Buchanan to MIT was among the most important recruitment and most expensive, with six additional appointments during his tenure, as it directly led to raising the biochemistry program to the top ranks in the field very quickly. 59 Buchanan recruited several outstanding young and energetic biochemists, biophysicists and microbiologists to MIT. They set MIT biology on a completely new course focusing on the then‐emerging field of molecular biology. Jack Buchanan was very concerned in 1990s and early 2000s that people focused their attention on other emerging fields of biology and neglected biochemistry and biophysics that he considered to be the bedrock of molecular biology. I promised that I would return to my roots and work on problems directly relevant to biochemistry and biophysics. But what big questions I should ask and to pursue in next 30 years?
After a lot of thinking and reading literature, I finally decided to work on the notoriously difficult membrane proteins. Many people considered me very foolish to tackle such an enormously challenging endeavor. It usually takes years or decades to understand a single membrane protein. Since it is necessary to use detergents (also called surfactants) to work on membrane proteins, before starting on the membrane protein research, I had already designed a class of simple lipid‐like peptides. These peptides behave like lipids. They all have a hydrophilic head containing either aspartate (D), glutamate (E), his (H), lysine (K), or arginine (R), and a hydrophobic tail containing either consecutive glycine (G), alanine (A), valine (V), isoleucine (I), leucine (L), phenylalanine (F), or combination of those hydrophobic amino acids. 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67
My lab first used these lipid‐like peptides to stabilize photosystem I (PSI), 68 , 69 , 70 , 71 glycerol‐3‐phosphate dehydrogenase 72 and bovine rhodopsin, 73 which is a member of G protein‐coupled receptors (GPCRs). Later we expanded to other GPCRs including other sensory receptors. 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82
2.4. A Eureka moment to conceive a simple QTY code
Working on membrane proteins requires systematically screening all kinds of detergents before real work could begin. The detergents screen is time‐consuming, laborious and expensive but absolutely necessary. As Pluto remarked, “Necessity is the mother of invention.” I begin to think of other ways to study membrane proteins. Alex Rich has been my mentor, close colleague and friend. I always discuss with him all kinds of things including research, science, politics, and life in general. On evening of December 20, 2010, we walked out Rich's office toward his car, Alex abruptly stopped during walking during the frozen temperature in totally darkness, turned to me, and asked “Can you convert a hydrophobic alpha‐helix to a hydrophilic one?” I immediately answered, yes! I explained to him that hemoglobin consists most alpha‐helix (>80%) and it is the most water‐soluble protein known (~30%) in red blood cells. Alex's questions further stimulated my thinking.
While writing a grant proposal on Saturday, January 15, 2011, I had a Eureka moment and conceived a very simple QTY code for protein design, particularly for desgins of integral membrane proteins.
Through analyzing the 1.5 Å electron density maps of the 20 amino acids, it is very clear that molecular structures of several hydrophobic amino acids closely resemble several hydrophilic amino acids in structure and electron density (Figure 15). And it has been known in the detailed X‐ray crystallographic studies that all 20 natural amino acids, both soluble and insoluble, are found in α‐helices, 83 , 84 , 85 , 86 although some have higher propensities to form α‐helices than others. 84 , 85 , 86 This suggested that the substitution of some amino acid residues that share similar structures may not significantly alter protein structure or function.
FIGURE 15.
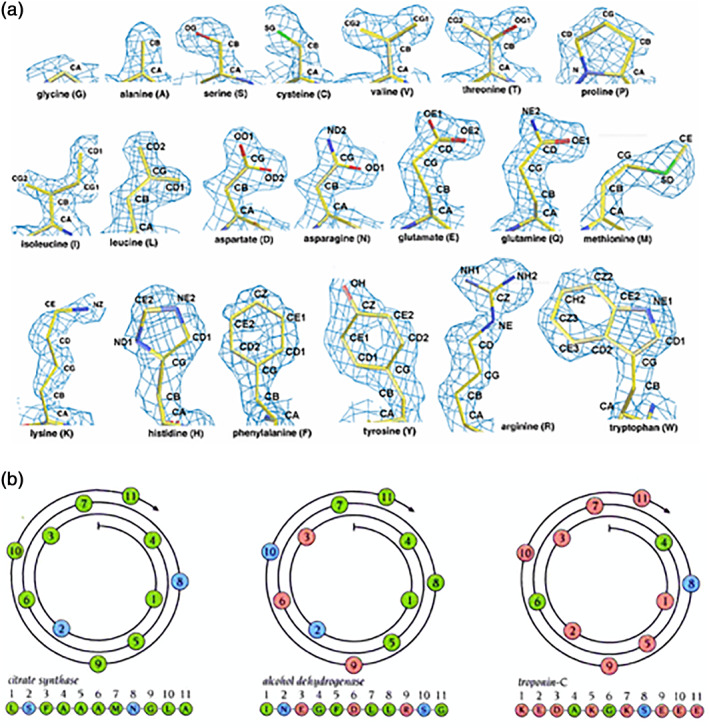
Amino acids in proteins. (a) 1.5 Å resolution electron density maps of the 20 amino acids from X‐ray diffraction. in order to show the individual amino acid electron density maps with high‐resolution and clarity. The density maps clearly show the similarities between V and T; between L, D, N, E, and Q; and between F and Y. In fact, the similarity between V and T is so striking that valine tRNA synthetase (ValRS) mischarges isoleucine and threonine at a rate of one per 200–400. 92 , 93 Courtesy of Dr. Michael R. Sawaya of University of California, Los Angeles, CA, the original website: http://people.mbi.ucla.edu/sawaya/m230d/Modelbuilding/modelbuilding.html. (b) Three types of alpha‐helices with amino acids of different chemical properties. (left panel) the α‐helix is made of nine hydrophobic (green color) and two hydrophilic amino acids (blue), (middle panel) the alpha‐helix is made of six hydrophobic and five hydrophilic amino acids (blue and pink), (right panel) the alpha‐helix is made of nine hydrophilic and two hydrophobic amino acids. However, despite their different composition, the alpha‐helices have nearly identical structure, namely, 1.5 Å/amino acid rise, 100°/amino acid turn, 360°/helical turn, and 5.4 Å/helical turn 86
I reasoned that it may be possible to design a few 7‐transmembrane G protein‐coupled receptor sequences (GPCRs) where the hydrophobic residues are systematically replaced by water soluble residues. I thus coined the systematic replacement as the QTY code.
The QTY code is based on the fact that the 1.5 Å electron density map of the hydrophobic leucine (L) is similar to that of hydrophilic asparagine (N) and glutamine (Q). The electron density maps of the hydrophobic isoleucine (I) and valine (V) are similar to the hydrophilic threonine (T). The electron density map of the hydrophobic phenylalanine (F) is similar to the hydrophilic tyrosine (Y) (Figure 15). Although water molecules also form hydrogen bonds with aspartic acid (−), glutamic acid (−), lysine (+), and arginine (+), these residues introduce charges, thereby altering the surface property of proteins. Thus, they are not introduced in the QTY code. Since the sidechain of asparagine (N) is close to the backbone of the polypeptide chain and sometimes interferes hydrogen bonds on the backbone, asparagine is not used in the code.
Initially, applying the QTY code using the crystal structures of CXCR4 and CCR5 as a guidance, we only replaced the hydrophobic residues L, I, V, and F that face the hydrophobic lipid bilayer of transmembrane domain of these membrane receptors. But we failed to obtain any detergent‐free receptors. Later we replaced more L, I, V, and F in the transmembrane domain, but we still could not obtain water‐soluble QTY variant receptors. Finally, we used the QTY code to replace all hydrophobic residues L, I, V, and F with Q, T, and Y in the transmembrane domain. In a few cases, we also replaced L, I, V, and F with Q, T, and Y the intracellular loops and C‐terminus to further increase water‐solubility since these domains do not expose to external ligands. I initially applied the QTY code (Figure 16) to four chemokine receptors that are members of G protein‐coupled receptors CCR5, CXCR4, CCR10, and CXCR7 87 , 88 , 89 , 90 and later expanded to CXCR2, CXCR5, and CCR9 and cytokine receptors. 91 My laboratory carried out experiments to verify the QTY code. To our great surprise, despite substantial amino acids replacement, ~20–30% overall for the chemokine receptors and 46–62% in the transmembrane helices, these QTY variant receptors still retained their overall structure, with similar amount of α‐helical content, folded into stable proteins, and able to bind their respective ligands, similar to the natural receptors. 87 , 88 , 89 , 90 Currently, determinations of the QTY variant receptor molecular structures are in progress in collaborations with other laboratories.
FIGURE 16.
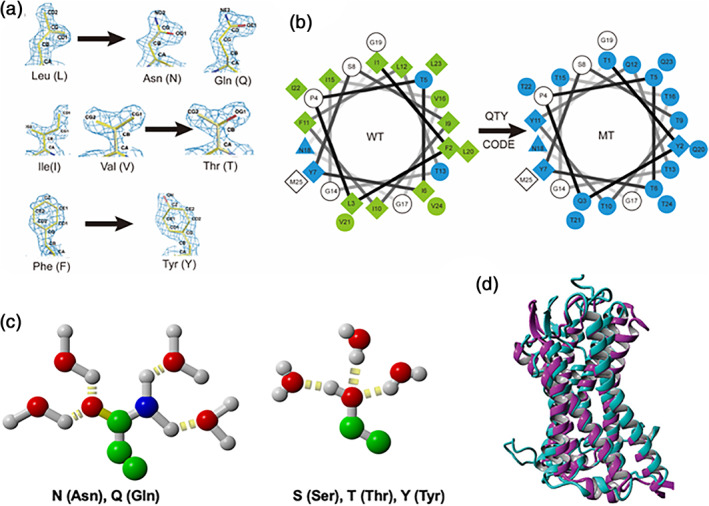
The QTY code and how it replaces L, V, I, and F with Q, T, and Y. (a) Crystallographic electronic density maps of the following amino acids: Leucine (L), Asparagine (N), Glutamine (Q), Isoleucine (I), Valine (V), Threonine (T), Phenylalanine (F) and Tyrosine (Y). The density maps of L, N, and Q are very similar. Likewise, the density maps of I, V, and T are similar, and the density maps of F and Y are similar. The side chains of L, V, I, and F cannot form any hydrogen bonds with water, thus rendering them water‐insoluble. On the other hand, N and Q can form four hydrogen bonds with four water molecules, two as hydrogen donors and two as hydrogen acceptors. Likewise, three water molecules can form hydrogen bonds with the –OH (2 H‐donors and 1 H‐acceptor) of Thr (T) and Tyr (Y). Both L and Q have high tendencies to form α‐helices, but N frequently occurs at turns. Thus, Q was used to replace L, but not N. I, V, and T are all β‐branched amino acids and their density maps are very similar indicating similar shapes. (b) Helical wheels before and after applying the QTY code to transmembrane helical segment 1 (TM1) of CXCR4. Amino acids that interact with water molecules are light blue in color. The QTY code conversions render the α‐helical segment water‐soluble. (c) Hydrogen bond interactions between water and the amino acids. The side chain of glutamine (Q) can form four hydrogen bonds with four water molecules. There are two hydrogen donors from nitrogen and two hydrogen acceptors for oxygen. –OH group of threonine (T) and tyrosine (y) can form three hydrogen bonds with three water molecules (2 H‐acceptors and 1 H‐donor). Color code: Green, carbon; red, oxygen; blue, nitrogen; gray, hydrogen; yellow disks, hydrogen bonds. (d) The superimposed native CCR5 (PDB: 4MBS, magenta color) and computer simulated CCR5QTY variant (teal color) in explicit water environment. The determination of molecular structure of water‐soluble QTY variants are in progress
FIGURE B1.

The photo from the 2003 Crete Workshops on Self‐assembling Peptides and Proteins (not all participants are in the photo). I initiated and co‐organized with Amalia Aggeli, Neville Boden, and Michael Hecht four workshops of “Self‐assembly peptides and proteins” in Capsis Hotel, Crete, Greece in summers of 1999, 2001, 2003, 2005 and passed the torch to Bill DeGrado and Joel Schneider in 2007. We invited people from diverse fields including peptide chemistry, protein science, materials science, mathematics, various engineering, computer science, clinical medicine, food science, industrial research, and more. The invited speakers include Alexander Rich†, Nobel laureate Carleton Gajdusek†, Benoit Mandelbrot†, George Klein†, Carl Brändén†, Chris Dobson†, Susan Lindquist†, Andrew Szent‐Györgyi†, Jack Aviv†, Nobel laureate Martin Karplus, Marci Karplus, Alan Fersht, Carol Robinson, Meir Wilchek, David Eisenberg, Joel Sussman, Eva Klein, Peter Klein, Ingemar Ernberg, Mouad Lamrani, Horst Vogel, Uwe Sleytr, Magdalena Helmer, Jeff Kelly, Jonathan Weissman, Joseph Jacobson, Matthew Tirrell, Reza Ghadiri, Miheal Polymeropoulos, Charlotte Hauser, Stefan Wölfl, Klaus During, Xiaojun Zhao, Alan Windle, Anton Middleberg, Sheena Radford, Tim Deming, Vince Conticello, Anna Mitraki, Aphrodite Kapurniotu, Dek Woolfson, Tom McLeish, Ted Atkins, Alan Cooper, Nick Gay, Deborah Kendall, John Taylor, Bonnie Kaiser, Georgios Archontis, Jianren Lu, Ehud Gazit, Sam Stupp, Phil Messersmith, Joanna Aizenberg, Bruce Tidor, Amy Keating, Roger Kamm, Angie Belcher, Per Westermark, Gunilla Westermark, Joel Schnur, Rutledge Ellis‐Behnke, Yechiel Shai, Hilal Lashuel, Sylvie Blondelle, Donald Hilvert, David Kaplan, Thomas Scheibel, David Lynn, Dan Urry, Matt Tirrell, Dan Kirschner, Joel Schneider, Michal Dadlez, Lars Baltzer, Sandra Burkett, Kenji Yamamoto, Hisakazu Mihara, Shiroh Futaki, Tomi Sasaki, Ikuo Fujii, M. Sisido, Itaru Hamachi, Susumu Yoshikawa, Kei Kamino, Margherita Morpurgo, Miquel Pons, Kees De Kruif, Mark Spector, Edward Timoshenko, Corinne Chevallard, Diego Mantovani, Georgios Archontis, Peter Butko, Yuan Luo, Eser Elcin, Murat Elcin, Thomas Zemb, Pu Chen, Maxim Ryadnov, Mita Desai, S. Yasaman, Stavros Hamodrakas. There were also many young and energetic students and postdocs including Rein Ulijn, Louise Serpell, Cait McPhee, Mark Krebs, Jesús Zurdo, Dominik Rünzler, Meital Reches, Hanna Rapaport, Ronit Sneer, Nikola Kojic, Brian Chow, Sawyer Fuller, Wonmuk Hwang, Steve Yang, Howard Leung, Sloan Kulper, Davide Marini, Patrick Kiley, Andrea Lomander, Hidenori Yokoi, Beau Peelle, Andreas Mershin, Brian Cook, Liselotte Kaiser, Sotirios Koutsopoulos, Joanke Graveland‐Bikker, Cayce Denton, Martin Case, Lisa Crawshaw, James Elliott, Jane Essex, Susan Felton, David Gidalevitz, Nicholas Fischer, Daniel Huemmerich, Margaret McCammon, Salman Rogers, Carlos Silva, Natasha Poklar Ulrich, Vladimir Proks, Katrin Rosenthal‐Aizman, Katerina Papanikolopoulou, Vassili Iconomidou, Roger Kiley, Ken‐Ichi Sano, Shane Scanlon and many more. They presented their research in front of the distinguished scientists, exchanged ideas and received sound advice. These intimate workshops (~70–80 people each time) mixed people from different disciplines and stimulated further interests in pursuing the new field. This self‐assembling peptide material field is not only thriving and advancing at rapid pace but it also generates many novel products that have been commercialized in diverse fields.† These people have passed away.
3. PERSPECTIVE
The path from the discovery to a new medical technology requires: (1) the stimulation, encouragement and support of curiosity‐driven scientific research in order to gain new scientific knowledge, (2) taking calculated risks without fearing a failure to translate scientific knowledge and research into enabling technologies, and (3) persistence in developing discoveries into the knowledge‐based economy for the benefit of mankind.
In science there are numerous examples of curiosity‐driven research and unintentional (unauthorized) discoveries that eventually lead to technological breakthroughs and new economic development. It is extremely difficult to imagine not pursuing curiosity‐driven research and explorations. Curiosity‐driven research not only provides new insights into mysteries of nature and generates new knowledge, but it also can translate the knowledge into new enabling technology that will eventually be developed as knowledge‐based economy. It is clear that who holds new knowledge, also leads the new economy. The lack of knowledge inevitably hinders the new economy development.
There are few examples: the discovery of x‐rays, the structure of DNA double helix, DNA–RNA and RNA–RNA hybridizations, reverse transcription, RNA splicing, RNA as enzymes, telomeres, Natural Killer cells (NK cells), programmed cell death, microRNA, RNA interference, S‐layer proteins, Kuru disease, green fluorescent protein (GFP), carbon 60, carbon nanotubes, carbon graphene and the indispensable worldwide web—www, and the latest CRISPR for simple and elegant method for gene and genome editing. The discovery of the self‐assembling peptide is another an example of an unexpected curiosity‐driven discovery that eventually led to the development of an enabling medical technology and benefited society. The recent example of successful clinical trials of the self‐assembling peptide scaffolds for accelerating wound healings and regenerative medicine and surgical uses and perhaps repair stroke provide a glimpse of what is coming for wide spread uses of self‐assembling peptides. Therefore curiosity‐driven research must be strongly encouraged and fully supported, despite the current emphasis of application‐driven research (Boxes 1 and 2).
BOX 1. Multidisciplinary workshop (1999, 2001, 2003, 2005, 2007): Self‐assembly of peptides and proteins in biology, engineering and medicine at Capsis Hotel in Crete, Greece.
In order to initiate and promote a new research field, it is very important to bring people from diverse fields to be under one roof in order to openly discuss new ideas, to cross‐fertilize a new field in a relaxed and somewhat isolated setting, such as the Capsis Hotel in Crete, Greece. I did precisely that. Through organizing five small biannual workshops with Amaila Aggeli, Neville Boden, Michael Hecht, William DeGrado, and Joel Schneider, by inviting many leading scientists in their own fields to mix them in an intimately marvelous environment in a beautiful Greek island. There, these leading scientists in the world spent lot of time with young and energetic students, postdocs and researchers. These intimate workshops had a big impact and accelerated the development of a new field. All participants enjoyed and had fond memories of such small meetings even after many years (Figure B1). 94
BOX 2. Find as many applications as possible.
In 1993, shortly after I discovered the self‐assembling peptides, I met Marvin Caruthers at a conference and mentioned to him about my unexpected discovery. I was very concerned about the expensive peptide materials for various applications. He gave me a sound advice: “Find as many applications as possible, the economics will take care of itself”. Marvin Caruthers invented the phosphate amide nucleic acid synthesis chemistry, now all nucleic acid synthesis uses his invention. In mid 1980s, each nucleotide base cost $25. In order to make a 10‐base oligonucleotide, it costed $250 in an academic lab! Now each base cost $0.05 and 10 base oligonucleotides costed ~$0.50, a reduction by 500 folds! The peptide synthesis cost at the industrial scale has also reduced significantly since early 1990s.
AUTHOR CONTRIBUTIONS
Shuguang Zhang: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; software; supervision; validation; visualization; writing‐original draft; writing‐review and editing.
ACKNOWLEDGEMENTS
First, I would like to thank my mentor Alexander Rich for providing me freedom to pursue “an unauthorized discovery”. I greatly appreciate my former students, postdocs, visiting scientists and colleagues to make significant contributions to an emerging field. I gratefully acknowledge various funding agencies, the American Cancer Society, the Whitaker Foundation, the John Simmon Guggenheim Foundation, private companies and individuals to sponsor the new field that eventually led to many practical uses. I also like to thank Dorrie Langsley for stimulating discussions and for editing the English and style.
Zhang S. Self‐assembling peptides: From a discovery in a yeast protein to diverse uses and beyond. Protein Science. 2020;29:2281–2303. 10.1002/pro.3951
Shuguang Zhang is the winner of the 2020 Emil Thomas Kaiser Award.
Funding information American Cancer Society
REFERENCES
- 1. Pauling L, Corey RB, Branson HR. The structure of proteins; two hydrogen‐bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci U S A. 1951;37:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pauling L, Corey RB. Configurations of polypeptide chains with favored orientations around single bonds: Two new pleated sheets. Proc Natl Acad Sci U S A. 1951;37:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang S, Lockshin C, Herbert A, Winter E, Rich A. Zuotin, a putative Z‐DNA binding protein in Saccharomyces cerevisiae . EMBO J. 1992;11:3787–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang AH, Quigley GJ, Kolpak FJ, et al. Molecular structure of a left‐handed double helical DNA fragment at atomic resolution. Nature. 1979;282:680–686. [DOI] [PubMed] [Google Scholar]
- 5. Zhang S, Holmes T, Lockshin C, Rich A. Spontaneous assembly of a self‐complementary oligopeptide to form a stable macroscopic membrane. Proc Natl Acad Sci U S A. 1993;90:3334–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marqusee S, Baldwin RL. Helix stabilization by Glu‐…Lys+ salt bridges in short peptides of de novo design. Proc Natl Acad Sci U S A. 1987;84:8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marqusee S, Robinson VH, Baldwin RL. Unusually stable helix formation in short alanine‐based peptides. Proc Natl Acad Sci U S A. 1989;86:5286–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rich A. The excitement of discovery. Annu Rev Biochem. 2004;37:1–37. [DOI] [PubMed] [Google Scholar]
- 9. Zhang S, Lockshin C, Cook R, Rich A. Unusually stable β‐sheet formation of an ionic self‐complementary oligopeptide. Biopolymers. 1994;34:663–672. [DOI] [PubMed] [Google Scholar]
- 10. Zhang S, Holmes T, DiPersio M, Hynes RO, Su X, Rich A. Self‐complementary oligopeptide matrices support mammalian cell attachment. Biomaterials. 1995;16:1385–1393. [DOI] [PubMed] [Google Scholar]
- 11. Crick FHC, Rich A. Structure of polyglycine II. Nature. 1955;176:780–781. [DOI] [PubMed] [Google Scholar]
- 12. Rich A, Crick FHC. The structure of collagen. Nature. 1955;176:915–916. [DOI] [PubMed] [Google Scholar]
- 13. Rich A, Crick FHC. The molecular structure of collagen. J Mol Biol. 1961;3:483–506. [DOI] [PubMed] [Google Scholar]
- 14. Zhang S, Altman M. Peptide self‐assembly in functional polymer science and engineering. Reactive Funct Polym. 1999;41:91–102. [Google Scholar]
- 15. Zhang S (2002) Emerging biological materials through molecular self‐assembly. Biotech Adv; 20:321. [DOI] [PubMed] [Google Scholar]
- 16. Zhang S. Fabrication of novel materials through molecular self‐assembly. Nat Biotechnol. 2003;21:1171–1178. [DOI] [PubMed] [Google Scholar]
- 17. Zhang S. Beyond the petri dish. Nat Biotechnol. 2004;22:151–152. [DOI] [PubMed] [Google Scholar]
- 18. Zhao X, Zhang S. Molecular designer self‐assembling peptides. Chem Soc Rev. 2006;35:1110–1110. [DOI] [PubMed] [Google Scholar]
- 19. Zhang S. Designer self‐assembling peptide nanofiber scaffolds for study of 3‐D cell biology and beyond. Adv Cancer Res. 2007;99:335–362. [DOI] [PubMed] [Google Scholar]
- 20. Zhao X, Pan F, Xu H, et al. Molecular self‐assembly and applications of designer peptide amphiphiles. Chem Soc Rev. 2010;39:3480–3498. [DOI] [PubMed] [Google Scholar]
- 21. Luo Z, Zhang S. Designer nanomaterials using chiral self‐assembling peptide systems and their emerging benefit for society. Chem Soc Rev. 2012;41:4736–4754. [DOI] [PubMed] [Google Scholar]
- 22. Saracino GAA, Cigognini D, Silva D, Caprini A, Gelain F. Nanomaterials design and tests for neural tissue engineering. Chem Soc Rev. 2013;42:225–262. [DOI] [PubMed] [Google Scholar]
- 23. Gelain F, Luo Z, Rioult M, Zhang S. Self‐assembling peptide scaffolds in the clinic. NPJ Regenerat Med. 2020; Revised manuscript under consideration. Accepted (with minor revision). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gelain F, Luo Z, Zhang S. Self‐assembling peptide EAK16 and RADA16 nanofiber hydrogel scaffolds. Chem Rev. 2020. (In press). Revised manuscript under consideration. [DOI] [PubMed] [Google Scholar]
- 25. Holmes T, Delacalle S, Su X, Rich A, Zhang S. Extensive neurite outgrowth and active neuronal synapses on peptide scaffolds. Proc Natl Acad Sci U S A. 2000;97:6728–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kisiday J, Jin M, Kurz B, et al. Self‐assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: Implications for cartilage tissue repair. Proc Natl Acad Sci U S A. 2002;99:9996–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caplan MR, Schwartzfarb EM, Zhang S, Kamm RD, Lauffenburger DA. Effects of systematic variation of amino acid sequence on the mechanical properties of a self‐assembling, oligopeptide biomaterial. J Biomater Sci Polym. 2002;13:225–236. [DOI] [PubMed] [Google Scholar]
- 28. Caplan MR, Schwartzfarb EM, Zhang S, Kamm RD, Lauffenburger DA. Control of self‐assembling oligopeptide matrix formation through systematic variation of amino acid sequence. Biomaterials. 2002;23:219–227. [DOI] [PubMed] [Google Scholar]
- 29. Yokoi H, Kinoshita T, Zhang S. Dynamic reassembly of peptide RADA16 nanofiber scaffold. Proc Natl Acad Sci U S A. 2005;102:8414–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rich A, Davies DR. A new two‐stranded helical structure: Polyadenylic acid and polyuridylic acid. J Am Chem Soc. 1956;78:3548–3549. [Google Scholar]
- 31. Felsenfeld G, Davies DR, Rich A. Formation of a three‐stranded polynucleotide molecule. J Am Chem Soc. 1957;79:2023–2024. [Google Scholar]
- 32. Narmoneva DA, Oni O, Sieminski AL, et al. Self‐assembling short oligopeptides and the promotion of angiogenesis. Biomaterials. 2005;26:4837–4846. [DOI] [PubMed] [Google Scholar]
- 33. Davis ME, Motion JPM, Narmoneva DA, et al. Injectable self‐assembling peptide nanofibers create intramyocardial microenvironments for endothelial cells. Circulation. 2005;111:442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davis ME, Hsieh PCH, Takahashi T, et al. Local myocardial IGF‐1 delivery with biotinylated peptide nanofibers improves cell therapy for myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:8155–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gelain F, Bottai D, Vescovi A, Zhang S. Designer self‐assembling peptide nanofiber scaffolds for adult mouse neural stem cell 3‐dimensional cultures. PLoS One. 2006;1:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horii A, Wang X, Gelain F, Zhang S. Biological designer self‐assembling peptide scaffolds significantly enhance osteoblast proliferation, differentiation and 3‐D migration. PLoS One. 2007;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Horii A, Zhang S. Designer functionalized self‐assembling peptide nanofiber scaffolds for growth, migration, and tubulogenesis of human umbilical vein endothelial cell. Soft Matter. 2008;4:2388–2395. [Google Scholar]
- 38. Kumada Y, Zhang S. Significant type I and type III collagen production from human periodontal ligament fibroblast in 3D peptide scaffolds without extra growth factors. PLoS One. 2010;5:e1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumada Y, Hammond NA, Zhang S. Functionalized self‐assembling short peptide scaffolds significantly promote fibroblasts proliferation and 3‐D migration. Soft Matter. 2010;6:5073. [Google Scholar]
- 40. Koutsopoulos S, Zhang S. Long‐term three‐dimensional neural tissue cultures in functionalized self‐assembling peptide hydrogels, Matrigel and collagen I. Acta Biomater. 2013;9:5162–5169. [DOI] [PubMed] [Google Scholar]
- 41. Chau Y, Luo Y, Cheung ACY, et al. Incorporation of a matrix metalloproteinase‐sensitive substrate into self‐assembling peptides – A model for biofunctional scaffolds. Biomaterials. 2008;29:1713–1719. [DOI] [PubMed] [Google Scholar]
- 42. Nagai Y, Unsworth LD, Koutsopoulos S, Zhang S. Diffusion coefficients in self‐assembling peptide nanofiber scaffold hydrogel. J Control Release. 2006;115:18–25. [DOI] [PubMed] [Google Scholar]
- 43. Koutsopolous S, Unsworth LD, Nagai Y, Zhang S. Slow release of functional proteins through designer self‐assembling peptide nanofiber hydrogel scaffolds. Proc Natl Acad Sci U S A. 2009;106:4623–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gelain F, Unsworth L, Zhang S. Slow and sustained release of active cytokines from designer self‐assembling peptide nanofiber scaffolds for adult mouse neural stem cell cultures. J Control Release. 2010;145:231–239. [DOI] [PubMed] [Google Scholar]
- 45. Koutsopoulos S, Zhang S. Multi‐layered injectable self‐assembling peptide hydrogels for long‐term sustained release of human antibodies. J Control Release. 2012;160:451–458. [DOI] [PubMed] [Google Scholar]
- 46. Ellis‐Behnke R, Liang YX, You SW, et al. Nano neuro knitting: Peptide nanofiber scaffold for brain repair and axon regeneration with functional return of vision. Proc Natl Acad Sci U S A. 2006;103:5054–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ellis‐Behnke RG, Schneider GE. Peptide amphiphiles and porous biodegradable scaffolds for tissue regeneration in the brain and spinal cord. Methods Mol Biol. 2011;726:259–281. [DOI] [PubMed] [Google Scholar]
- 48. Ellis‐Behnke RG, Liang YX, Tay DKC, et al. Nano hemostat solution: Immediate hemostasis at the nanoscale. Nanomed Nanotechnol Biol Med. 2007;2:207–215. [DOI] [PubMed] [Google Scholar]
- 49. Meng H, Chen L, Ye Z, Wang S, Zhao X. The effect of a self‐assembling peptide nanofiber scaffold (peptide) when used as a wound dressing for the treatment of deep second degree burns in rats. J Biomed Mater Res Appl Biomater. 2008;89B:379–391. [DOI] [PubMed] [Google Scholar]
- 50. Luo Z, Wang S, Zhang S. Fabrication of self‐assembling D‐form peptide nanofiber scaffold d‐EAK16 for rapid hemostasis. Biomaterials. 2011;32:2013–2020. [DOI] [PubMed] [Google Scholar]
- 51. Hsu BB, Conway W, Tschabrunn CM, et al. Clotting mimicry from robust hemostatic bandages based on self‐assembling peptides. ACS Nano. 2015;9:9394–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gelain F, Panseri S, Antonini S, et al. Transplantation of nanostructured composite scaffolds results in the regeneration of chronically injured spinal cords. ACS Nano. 2011;5:227–236. [DOI] [PubMed] [Google Scholar]
- 53. Cigognini D, Satta A, Colleoni B, et al. Evaluation of early and late effects into the acute spinal cord injury of an injectable functionalized self‐assembling scaffold. PLoS One. 2011;6:e19782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marchini A, Raspa A, Pugliese R, et al. Multifunctionalized hydrogels foster hNSC maturation in 3D cultures and neural regeneration in spinal cord injuries. Proc Natl Acad Sci U S A. 2019;116:7483–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luo Z, Zhao X, Zhang S. Structural dynamic of a self‐assembling peptide d‐EAK16 made of only D‐amino acids. PLoS One. 2008;3:e2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luo Z, Zhao X, Zhang S. Self‐organization of a chiral D‐EAK16 designer peptide into a 3D nanofiber scaffold. Macromol Biosci. 2008;8:785–791. [DOI] [PubMed] [Google Scholar]
- 57. Chen LB, Hartman S, Zetter B, Zhang S. John M. Buchanan (1917‐2007). Protein Sci. 2007;16:2578–2579. [Google Scholar]
- 58. Hartman S, Zetter B (2009) John M. Buchanan (1917. ‐2007) A biographical memoir, Washington DC, USA: National Academy of Sciences; http://www.nasonline.org/publications/biographical-memoirs/memoir-pdfs/buchanan-john-m.pdf [Google Scholar]
- 59. Killian J Jr. The education of a college president: A memoir. Cambridge, MA: MIT Press, 1985. [Google Scholar]
- 60. Vauthey S, Santoso S, Gong H, Watson N, Zhang S. Molecular self‐assembly of surfactant‐like peptides to form nanotubes and nanovesicles. Proc Natl Acad Sci U S A. 2002;99:5355–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Santoso S, Hwang W, Hartman H, Zhang S. Self‐assembly of surfactant‐like peptides with variable glycine tails to form nanotubes and nanovesicles. Nano Lett. 2002;2:687–691. [Google Scholar]
- 62. von Maltzahn G, Vauthey S, Santoso S, Zhang S. Positively charged surfactant‐like peptides self‐assemble into nanostructures. Langmuir. 2003;19:4332–4337. [Google Scholar]
- 63. Yang S, Zhang S. Self‐assembling behavior of designer lipid‐like peptides. Supramol Chem. 2006;18:389–396. [Google Scholar]
- 64. Nagai A, Nagai Y, Qu H, Zhang S. Self‐assembling behaviors of lipid‐like peptides A6D and A6K. J Nanosci Nanotechnol. 2007;7:2246–2252. [DOI] [PubMed] [Google Scholar]
- 65. Bucak S, Cenker C, Nasir I, Olsson U, Zackrisson M. Peptide nanotube nematic phase. Langmuir. 2009;25:4262. [DOI] [PubMed] [Google Scholar]
- 66. Luo Z, Åkerman B, Zhang S, Nordén B. Structures of self‐assembled amphiphilic peptide‐heterodimers: Effects of concentration, pH, temperature and ionic strength. Soft Matter. 2010;6:2260–2270. [Google Scholar]
- 67. Castelletto V, Nutt DR, Hamley IW, Bucak S, Cenker Ç, Olsson U. Structure of single‐wall peptide nanotubes: In situ flow aligning X‐ray diffraction. Chem Commun. 2010;46:6270–6272. [DOI] [PubMed] [Google Scholar]
- 68. Das R, Kiley PJ, Segal M, et al. Integration of photosynthetic protein molecular complexes in solid‐state electronic device. Nano Lett. 2004;4:1079–1083. [Google Scholar]
- 69. Kiley P, Zhao X, Vaughn M, Baldo M, Bruce BD, Zhang S. Self‐assembling peptide detergents stabilize isolated photosystem I on a dry surface for an extended time. PLoS Biol. 2005;3:1181–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Matsumoto K, Koutsopoulos S, Vaughn M, Bruce BD, Zhang S. Designer lipid‐like peptide surfactants stabilize functional photosystem I membrane complex in solution. J Phys Chem. 2009;115:75–83. [DOI] [PubMed] [Google Scholar]
- 71. Mershin A, Matsumoto K, Kaiser L, et al. Self‐assembled photosystem‐I biophotovoltaics on nanostructured TiO2 and ZnO. Sci Rep. 2012;2:e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yeh JI, Du S, Tordajada A, Paulo J, Zhang S. Peptergent: Peptide detergents that improve stability and functionality of a membrane protein glycerol‐3‐phosphate dehydrogenase. Biochemistry. 2005;44:16912–16919. [DOI] [PubMed] [Google Scholar]
- 73. Zhao X, Nagai Y, Revees P, Kiley P, Khorana HG, Zhang S. Designer lipid‐like peptides significantly stabilize G‐protein coupled receptor bovine rhodopsin. Proc Natl Acad Sci U S A. 2006;103:17707–17712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang X, Corin K, Wienken CJ, et al. Peptide surfactants for cell‐free production of functional G protein‐coupled receptors. Proc Natl Acad Sci U S A. 2011;118:9049–9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang X, Zhang S. Study of a bioengineered G‐protein coupled receptor of human formyl peptide receptor3. PLoS One. 2011;6:e23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang X, Corin K, Rich C, Zhang S. Study of two G‐protein coupled receptor variants of human trace amine‐associated receptor 5. Sci Rep. 2011;1:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Corin K, Baaske P, Ravel DB, et al. A robust and rapid system for studying functional G‐protein coupled receptors. PLoS One. 2011;6:e23036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Corin K, Baaske P, Ravel DB, et al. Designer lipid‐like peptides: A class of detergents for studying functional olfactory receptors using commercial cell‐free systems. PLoS One. 2011;6:e25067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Corin K, Baaske P, Geissler S, et al. Biochemical study of a bioengineered functional G‐protein coupled receptor human vomeronasal type 1 receptor 1. Sci Rep. 2011;1:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Corin K, Pick H, Baaske P, et al. Insertion of T4‐lysozyme (T4L) can be a useful tool for studying olfactory‐related GPCRs. Mol Biosyst. 2012;8:1750–1759. [DOI] [PubMed] [Google Scholar]
- 81. Zhang S. Lipid‐like self‐assembling peptides. Acc Chem Res. 2012;45:2142–2150. [DOI] [PubMed] [Google Scholar]
- 82. Koutsopoulos S, Kaiser L, Eriksson HM, Zhang S. Designer peptide surfactants stabilize diverse functional membrane proteins. Chem Soc Rev. 2012;41:1721–1728. [DOI] [PubMed] [Google Scholar]
- 83. Perutz MF, Rossmann MG, Cullis AF, Muirhead H, Will G, North AC. Structure of haemoglobin: A three‐dimensional Fourier synthesis at 5.5Å resolution, obtained by X‐ray analysis. Nature. 1960;185:416–422. [DOI] [PubMed] [Google Scholar]
- 84. Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem. 1978;47:251–276. [DOI] [PubMed] [Google Scholar]
- 85. Fersht A. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. New York: W.H. Freeman, 1998. pp. 10‐23, 523‐532. [Google Scholar]
- 86. Brändén C‐I, Tooze J. Introduction to protein structure. 2nd ed. London, UK, New York: Garland Publishing, 1999;p. 15–17. [Google Scholar]
- 87. Zhang S, Tao F, Qing R, et al. QTY code enables design of detergent‐free chemokine receptors that retain ligand‐binding activities. Proc Natl Acad Sci U S A. 2018;115:E8652–E8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Qing R, Han Q, Fei T, et al. QTY code designed thermostable and water‐soluble chimeric chemokine receptors with tunable ligand‐binding activities. Proc Natl Acad Sci U S A. 2019;116:25668–25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tegler LT, Corin K, Skuhersky M, Pick H, Vogel H, Zhang S. G protein‐coupled receptor CXCR4 designed by the QTY code becomes more hydrophilic and retains cell‐signaling activity. Sci Rep. 2020; Revision under consideration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Qing R, Tao F, Chatterjee P, et al. Non‐full‐length water‐soluble CXCR4QTY, CCR5QTY chemokine receptors and implication for overlooked truncated membrane receptors. iScience. 2020. (In press). Revision under consideration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hao SL, Jin D, Zhang S, Qing R. QTY code‐designed water‐soluble Fc‐fusion cytokine receptors bind to their respective ligands. QRB Discov. 2020;1:1–9. 10.1017/qrd.2020.4 Cambridge University Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lin L, Hale SP, Schimmel P. Aminoacylation error correction. Nature. 1996;384:33–34. [DOI] [PubMed] [Google Scholar]
- 93. Lin L, Schimmel P. Mutational analysis suggests the same design for editing activities of two tRNA synthetases. Biochemistry. 1996;35:5596–5601. [DOI] [PubMed] [Google Scholar]
- 94. Mershin A, Cook B, Kaiser L, Zhang S. A classic assembly. Nature Biotechnology. 2005;23:1379–1380. [DOI] [PubMed] [Google Scholar]