

Abstract
Radiation therapy is part of the standard of care for gliomas and kills a subset of tumor cells, while also altering the tumor microenvironment. Tumor cells with stem‐like properties preferentially survive radiation and give rise to glioma recurrence. Various techniques for enriching and quantifying cells with stem‐like properties have been used, including the fluorescence activated cell sorting (FACS)‐based side population (SP) assay, which is a functional assay that enriches for stem‐like tumor cells. In these analyses, mouse models of glioma have been used to understand the biology of this disease and therapeutic responses, including the radiation response. We present combined SP analysis and single‐cell RNA sequencing of genetically‐engineered mouse models of glioma to show a time course of cellular response to radiation. We identify and characterize two distinct tumor cell populations that are inherently radioresistant and also distinct effects of radiation on immune cell populations within the tumor microenvironment.
Keywords: glioma, glioma stem cells, myeloid cells, radiation response, radioresistance, single‐cell RNA sequencing, SP analysis
Main Points
Early after irradiation of murine glioma, FACS‐analysis shows stem‐like cells are enriched, and scRNA‐seq shows the radioresistant tumor cells are progenitors and stem‐like cells. Also, microglia and macrophage populations shift.
1. INTRODUCTION
Glioblastoma (GBM) is the most common primary malignant brain tumor and has a median survival of less than 2 years (Stupp et al., 2017). Radiation therapy is a mainstay of treatment, but its effectiveness is limited by a subset of tumor cells that survive irradiation and lead to recurrence. A reliable method for identifying these radioresistant cells in a mixed population of tumor and non‐tumor cells is required to allow their biological characterization and subsequent advances in treatments for this aggressive disease.
The radioresistant subpopulation of GBM tumor cells has characteristics of stem cells, including therapeutic resistance and high tumorigenicity when transplanted into recipient mice (Bao et al., 2006). Stem‐like glioma cells have been shown to reside in the perivascular niche, physically contacting endothelial cells (Calabrese et al., 2007; Charles et al., 2010; Venere, Fine, Dirks, & Rich, 2011). Multiple approaches have been used to enrich for these stem‐like cells including “side population” (SP) analysis. In this technique, fluorescence‐activated cell sorting (FACS) is used to quantify the “side population” (SP) phenotype, which occurs in cells that use the ABCG2 efflux pump to exclude the fluorescent dye Hoechst 33342. SP cells have been shown to reside in the perivascular niche and to be self‐renewing and highly tumorigenic, confirming their stemness (Bleau et al., 2009). This SP study was performed in the RCAS/tv‐a based, PDGF‐driven genetically engineered mouse glioma model, which has been used extensively for studying the biology of the radiation therapeutic response (Halliday et al., 2014; Leder et al., 2014; Ozawa et al., 2014). To date, SP analysis has not been used to characterize the effects of radiation, in vivo.
Recently, single‐cell RNA sequencing (scRNA‐seq) has been applied to human GBM and low grade glioma tumor samples obtained prior to, but not after, therapy (Darmanis et al., 2017; Müller et al., 2017; Patel et al., 2014; Venteicher et al., 2017). Since these studies were published, scRNA‐seq technologies have advanced and now allow the use of frozen tissues for sequencing nuclear RNA, which results in similar transcriptomic profiles to cellular RNA (Cao et al., 2017). A type of scRNA‐seq, single‐cell combinatorial indexing RNA sequencing (sciRNA‐seq), along with Uniform Manifold Approximation and Projection (UMAP) embedding and Louvain clustering for visualization and analysis of sequencing data have resulted in extensive and detailed mappings of cell types in mouse organogenesis (Cao et al., 2019). UMAP, in contrast to the more commonly used t‐Distributed Stochastic Neighbor Embedding (t‐SNE), consistently places similar cell types next to each other, making it easier to visualize the relationships between them (McInnes, Healy, Saul, & Großberger, 2018).
In this study, to characterize the effects of radiation at early time points, we applied SP analysis followed by sciRNA‐seq, as complementary approaches to the same samples from the aforementioned PDGF‐driven mouse glioma model (Figure S1). Mouse models were chosen because in glioma patients radiation therapy is never followed by immediate surgery and, therefore, human samples are not available. We found that at 72 hr after 10 Gy of ionizing radiation, SP cells are enriched, and by sciRNA‐seq, the bulk of tumor cells are lost, with two populations of radioresistant tumor cells remaining. One is slowly proliferating neuronal and oligodendrocyte progenitors, and the second is rapidly proliferating cells with stem‐like characteristics. Finally, while most non‐tumor cell‐types appear unaffected by radiation, the myeloid cell lineage shows radiation‐induced shifts in microglia and bone marrow‐derived macrophage populations.
2. MATERIALS AND METHODS
2.1. Mice, generation of murine gliomas, radiation treatment, and dissection
All mouse experiments were approved by Memorial Sloan Kettering Cancer Center's Institutional Animal Care and Use Committee (IACUC) and followed NIH guidelines for animal welfare. Gliomas were generated by intracranial injection of RCAS‐PDGFB producing DF1 chick fibroblast cells into neonatal Nestin (Nes) tv‐a (N‐tva) Ink4a/Arf −/− Pten fl/fl pups as previously described (Helmy et al., 2012; Shih et al., 2004). Pten is still expressed in this model because Cre recombinase was never expressed. In this model, mice develop high grade gliomas with near 100% penetrance at around 4–5 weeks post‐injection. For radiation treatment, mice were followed daily for development of glioma symptoms (weight loss, lethargy, gait or behavioral abnormalities, head tilt, swelling, or seizures). Upon symptom development mice were sedated with isoflurane and irradiation of the head was done using an X‐RAD 320 from Precision X‐Ray at 115 cGy/min to a total of 10 Gy. Tumor and contralateral normal brain tissue were identified and dissected at the indicated times after radiation or without radiation. Tumor and normal brain tissue that was not used immediately for SP analysis was snap‐frozen in liquid nitrogen and archived at −80°C.
2.2. Hoechst staining and SP (side population) analysis
Hoechst staining and SP analysis was done as previously described (Bleau et al., 2009). Dissected tumor tissue was sliced into 1 mm coronal sections. Cells were dissociated using papain digestion and were resuspended at 1 × 106 cells/ml in neural basal media, pre‐incubated at 37°C for 30 min with or without 100 μM verapamil (Sigma–Aldrich, St. Louis) and incubated with 5 μg/ml Hoechst 33342 (Sigma–Aldrich) for 90 minutes at 37°C. Hoechst dye was excited at 407 nm by trigon violet laser, and its dual wavelengths were detected using 450/40 (Hoechst 33342‐Blue) and 695/40 (Hoechst 33342‐Red) filters. Dead cells were excluded by gating on forward and side scatter and eliminating the propidium iodide positive population. SP and MP (main population) cells were then identified in Hoechst red and blue channels by comparing to verapamil incubated controls. The data were analyzed by FlowJo (Ashland, OR).
2.3. Statistics
Comparisons between two groups were made using Student's t test. Log‐rank (Mantel‐Cox) test was used to compare groups in the Kaplan–Meier graph. Analysis of covariance (ANOVA) was used when more than two groups were analyzed and Tukey's multiple comparison test was used for post hoc comparisons. Data represent the mean ± SE of the mean (SEM). p‐values <.05 were considered statistically significant.
2.4. Nuclei extraction and fixation for single‐cell combinatorial indexing RNA sequencing (sciRNA‐seq)
The nuclei extraction was performed as previously described in Cao et al. (2017). Previously snap frozen tissue was diced into a fine suspension in cell lysis buffer with a clean razor blade in a sterile dish placed on ice. Cell lysis buffer was composed of stock nuclei buffer (10 mM Tris–HCl, 10 mM NaCl, 3 mM MgCl2, pH 7.4) with 0.1% IGEPAL CA‐630 (Sigma–Aldrich), 1% SUPERase‐in RNase inhibitor (20 U/μl; ThermoFisher Scientific, #AM2696), 2% protease inhibitors (Sigma–Aldrich, #P8340), and 1% Bovine Serum Albumin (BSA, 20 mg/ml; New England BiosLabs, Inc, #B9000S). The nuclei suspension was transferred to an Eppendorf tube and, after being incubated on ice for approximately 1 hr, was put through a 20 μm strainer (pluriStrainer, PluriSelect). The suspension was then pelleted by centrifugation in a table top centrifuge at 4°C for 5 min at 500g. Cell lysis buffer was aspirated and nuclei were resuspended in nuclei buffer (stock nuclei buffer with 1% SUPERase‐in RNase inhibitor, 2% protease inhibitors, and 1% Bovine Serum Albumin). The nuclei were pelleted at 4°C for 5 min at 500g and the buffer was aspirated. The nuclei were resuspended in nuclei buffer with a final concentration of 4% paraformaldehyde and incubated on ice for 15 min with agitation every 5 min. The fixed nuclei were washed twice. Each wash consisted of centrifugation for pelleting at 4°C for 5 min at 500g, aspirating the buffer, and resuspending in nuclei buffer. Nuclei were counted using a Countess II FL Automated Cell Counter (Invitrogen). The fixed nuclei were then snap‐frozen in liquid nitrogen and stored in liquid nitrogen.
2.5. sciRNA‐seq
sciRNA‐seq was performed as previously described in Cao et al. (2017). For reverse transcription, frozen extracted, fixed nuclei were thawed and resuspended in nuclei buffer at 5000 nuclei per μl. Two microliters of nuclei (10,000 nuclei) and 0.25 μl of 10 mM dNTPs (New England BioLabs, # N0447L) were mixed and distributed to wells of 96‐well LoBind plates (Eppendorf, # 0030129512). Twelve wells were used per experimental sample. Control samples, which consisted of a 50:50 mixture of mouse NIH 3T3 cells and human HEK 293T cells, were included. Additionally, human cells from unrelated experiments were also included as samples, but were not included in the bioinformatic analysis. As a result, a total of 384 wells were used. One microliter of 25 μM indexed oligo‐dT primers (ACGACGCTCTTCCGATCTNNNNNNNN[10 bp index]TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN‐3′, where “N” is any base and “V” is either “A”, “C” or “G”; IDT) were added to each well. The plates were incubated at 55°C for 5 min and then placed on ice. One microliter of 5× Superscript IV First‐Strand Buffer (ThermoFisher Scientific, #18091200), 0.25 μl of 100 mM DTT, 0.25 μl of SuperScript IV Reverse Transcriptase, and 0.25 μl RNaseOUT Recombinant Ribonuclease Inhibitor (ThermoFisher Scientific, #10777019) were mixed and added to each well with nuclei. The plates were incubated at 55°C for 10 min and then placed on ice. Five microliters of 40 mM EDTA and 1 mM Spermidine were added to each well to stop the reverse transcription reactions. Wide bore tips were used for pooling all the wells and the pool was transferred to 15 ml conical for fluorescence‐activated cell sorting (FACS).
Immediately prior to FACS, 300 μM 4′,6‐diamidino‐2‐phenylindole (DAPI; ThermoFisher Scientific, #D1306) was added to the pooled nuclei at a final concentration of 3 μM. DAPI solution was prepared by dissolving the powder in deionized water to form a 14.3 mM solution and then diluted in phosphate‐buffered saline (PBS) to 300 μM. One hundred nuclei were sorted into each well of ten 96‐well plates containing 5 μl of Qiagen elution buffer (EB; Qiagen). Gates were set to include only DAPI positive singlets based on a forward scatter (FSC)/side scatter (SSC) plot and a cell cycle profile. An 85 μm nozzle and 50 μm filter were used on the input line. After sorting, plates were centrifuged briefly to force any droplets on the sides of the wells to the bottom and the plates were stored at −80°C.
For second strand synthesis, a mix of 0.5 μl of second strand synthesis (SSS) buffer and 0.75 μl of SSS enzyme (NEBNext Ultra II Non‐Directional RNA Second Strand Synthesis Module; New England BioLabs, # E6111L) was added to each well of the plates containing the sorted nuclei and incubated at 16°C for 150 min. The reaction was terminated by incubating the plate at 75°C for 20 min. Tagmentation was performed by adding to each well, a mix of 2.5 μl of Nextera TD buffer (Illumina, #FC‐121‐1031) and 0.25 μl of 20 ng/μl human genomic DNA (Promega, #G147A) diluted in water followed by addition of a mix of 2.5 μl of Nextera TD Buffer and 0.5 μl of TDE1 enzyme (Illumina, FC‐121‐1031), and incubating the plates at 55°C for 5 min. The reactions were stopped by adding 12 μl of DNA binding buffer (Zymo, #D4004‐1‐L) to each well and incubating the plates at room temperature for 5 minutes. DNA clean‐ups were performed with 3 μl of Ampure XP beads (Beckman‐Coulter, #A63882) per well and otherwise as per manufacturer's instructions. DNA was eluted in 17 μl of EB per well and 16 μl of the eluate in each well was transferred to new 96‐well LoBind plates.
PCR was performed by adding to each well 2 μl each of 10 μM P5 (5′ AATGATACGGCGACCACCGAGATCTACAC[i5]ACACTCTTTCCCTACACGACGCTCTTCCGATCT‐3′; IDT)and P7 primers (5′‐CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG‐3′; IDT) and 20uL of NEBNext High‐Fidelity 2X PCR Master Mix (New England BioLabs, #M0541L) and using the following cycling parameters: 75°C for 3 min, 98°C for 30 s, 18 cycles of 98°C for 10 s, 66°C for 30 s, and 72°C for 1 min, and ending with 1 cycle of 72°C for 5 min. After PCR, all samples were pooled. DNA was purified using an Zymo DNA Clean & Concentrator‐100 kit (#D4029) with Zymo‐Spin Column with Reservoir (#C1016‐50) and vacuum manifold and a 2:1 ratio of binding buffer to PCR reaction volume, but otherwise as per manufacturer's instructions. DNA was eluted in 150 μl of EB. The DNA underwent an Ampure XP bead clean‐up using 0.8× volume of beads as per manufacturer's instructions and the concentration of the amplicon was estimated by Agilent Bioanalyzer using the region between 200 and 750 base pairs. Finally, the library was sequenced using an Illumina Nextseq 550 with a High Output v2 kit (75 cycles) as per manufacturer's instructions; Read 1 was 18 cycles, Read 2 was 52 cycles, Index 1 was 10 cycles, and Index 2 was 10 cycles.
2.6. Aligning reads and creation of cell by gene expression matrix
Alignment and creation of the gene expression matrix was performed as previously described (Cao et al., 2017). Bcl2fastq (Illumina) was used to convert base calls to FASTQ format, tolerating one mismatched base in barcodes. After demultiplexing, sequencing adapters were removed using trim‐galore (version 0.4.1, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Using STAR (version 2.5.2b) with default settings, trimmed reads were aligned to a reference genome including both the human (hg19) and the mouse (mm10) genome with annotations from GENCODE V19 for human and VM11 for mouse (Dobin et al., 2013). Reads that did not map uniquely were removed, as were duplicates based on unique molecular index (UMI), reverse transcription (RT) index, and read 2 end coordinates. Finally, reads were mapped to cells by further demultiplexing using the RT index. A digital gene expression matrix was created by counting, in a strand‐specific manner, the number of unique sequences based on UMIs that map to either exonic or intronic regions of each gene using bedtools (version 2.26.0) (Dobin et al., 2013; Quinlan & Hall, 2010). RCAS virus reads were added back to the matrix by first aligning previously unaligned reads to the RCAS LTR reference sequence (AATGTAGTCTTATGCAATACTCTTGTAGTCTTGCAACATGGTAACGATGAGTTAGCAACATGCCTTACAAGGAGAGAAAAAGCACCGTGCATGCCGATTGGTGGAAGTAAGGTGGTACGATCGTGCCTTATTAGGAAGGCAACAGACGGGTCTGACATGGATTGACGAACCACTGAATTCCGCATTGCAGAGATATTGTATTTAAGTGCCTAGCTCGATACAATAAACGCCATTTGACCATTCACCACA) using bowtie (version 2.2.3). Aligned reads were deduplicated based on the UMIs, reverse transcription index, and rear 2 end coordinates. Counts were then added for the RCAS LTR transcript to the appropriate cells in the digital gene expression matrix based on the barcode mappings. These counts were added under the gene name “RCAS” and the Ensembl‐like accession number, ENSMUSG00000000LTR.1, both of which were created to fit the format of the data files, but do not represent officially sanctioned identifiers.
The overall collision rate was calculated using the control mouse‐human mixture samples as twice the rate of cells with less than 90% of UMIs mapping to a single species. This overall collision rate was ~10%. Cells from experimental samples with less than 90% of UMIs uniquely mapping to a single species were considered “collisions” and were not included in the final gene expression matrix. In cells with 90% of UMIs mapping to a single species, reads mapping to the other species were removed. Cells with fewer than 100 unique UMIs were also not included in the final gene expression matrix. Custom software written in Bash, Python, and R was used for the above.
2.7. UMAP visualization and Louvain clustering
For all subsequent analyses, custom R scripts were used. For visualization of the digital gene expression matrix, UMAP embedding and Louvain clustering were performed via the Monocle 3 alpha package (Cao et al., 2019). The 3,000 genes with the highest variation were used as the input to principal components analysis (PCA). The top 50 components were then used as input to UMAP for two‐dimensional embedding, with the min_dist parameter set to 0.4 and all other parameters set to default. For Louvain clustering, the res parameter was set to 1.64e−3, resulting in the embedding and clustering in Figure 2a. For sub‐clustering of oligodendrocyte lineage cells (Figure 4a) and macrophage/microglia cells (Figure 6a), the UMAP min_dist parameter was set to 1 and the Louvain clustering parameter was 1e−2.
2.8. Gene set enrichment analysis (GSEA) of the OPC/tumor supercluster
To analyze the left‐to‐right shift in the UMAP embedding of the OPC/tumor cluster between No IR and 8 hr after IR samples, the support vector machine (SVM) algorithm as implemented in the e1071 package (https://cran.r-project.org/web/packages/e1071/index.html) was used to define a line dividing the cluster into left and right parts (Figure 3b). An SVM with a linear kernel was trained on the cells of the No IR samples (left) vs. the cells in the 8 hr after IR samples (right). The OPC/tumor cells of the 8 hr after IR samples were downsampled to the same number of cells as were in the No IR samples for training. Since there was no concern for overfitting because the model would not be used for classification of other datasets, a high value for the cost (1e10) was used to get the best line for separating the cells into right and left parts. All cells in the OPC/tumor cluster across all samples were then classified as either in the right or the left parts based on their UMAP coordinates relative to this best line. Differential expression between the left and right parts of the tumor/OPC cluster was calculated using the FindMarkers function of the Seurat package assuming a negative binomial distribution, and setting the logfc.threshold parameter to 0.15, the min.pct parameter to 0, and pos.only parameter to FALSE with all other parameters set to their defaults (Stuart et al., 2019; Butler, Hoffman, Smibert, Papalexi, & Satija, 2018). GSEA was performed using the fgsea function from the fGSEA package with the nperm parameter set to 100,000 and all other parameters set to their defaults. The biological process GO terms from version 6.2 of the MSigDb database (http://software.broadinstitute.org/gsea/msigdb/download_file.jsp?filePath=/resources/msigdb/6.2/c5.bp.v6.2.symbols.gmt) were used with fGSEA (Sergushichev, 2016). Enriched terms were collapsed to independent ones using the collapsePathways function limiting the adjusted p‐values to less than .1.
2.9. Gene ontology enrichment analysis
To generate a ranked list of genes discriminating each cluster of cells from all others, we performed supervised differential analysis of Clusters 17, 22, and 23. We generated a ranked list of differentially expressed genes between each cluster and all the other clusters according to the Mann–Whitney–Wilcoxon (MWW) statistics. Then, for each cluster we computed the gene ontology enrichment analyses using the MWW‐Gene Set Test (MWW‐GST) as described (Frattini et al., 2018).
2.10. Master regulator analysis
To identify the master regulators (MR) that are most active within each cluster, we implemented an approach similar to that used to identify MRs of the mesenchymal subtype of glioma (Carro et al., 2010). More specifically, we first reconstructed a global context‐specific regulatory network from the scRNA‐Seq data and a list using the RGBM algorithm, a computational tool in which a machine learning framework is based on gradient boosting machines (Mall et al., 2018). RGBM has three major steps. In the first step, a Gradient Boosting Machine ranks transcription factors (TF) that regulate a target gene according to variable importance scores. In the second step, a regularization procedure locates the corner of the variable importance L‐curve to identify the optimal set of TFs for a target gene. In the third step, the boosting procedure re‐iterates with the optimal set of TFs for each target gene to reconstruct the final network between TFs and targets. RGBM has been validated on DREAM3, DREAM4 and DREAM5 network inference datasets and simulated RNA‐Seq datasets showing that it obtains higher performance in terms of AUpr and AUroc than the current state‐of‐the‐art methods on the majority of these datasets. Moreover, RGBM was used to identify the main regulators of the molecular subtypes of brain tumors (Ceccarelli et al., 2016; Mall et al., 2018). To build our network we used a list of 1,138 mouse transcriptional regulators, the networks contain a total of 152,607 transcriptional interactions.
To identify MRs of the Clusters 17, 22, and 23 we first generated a ranked list of differentially expressed genes between each cluster and all the other clusters according to the Mann–Whitney–Wilcoxon statistics. The ranked lists were used to compute the activity of each MR. Master Regulator Analysis (MRA) is an algorithm used to identify transcriptional regulators whose targets are enriched for a particular gene signature. The enrichment is evaluated using a statistical test. We applied the MWW gene set test to evaluate the enrichment of the positive targets of the MRs at the top of the ranked list and the negative targets at the bottom of the ranked list (Frattini et al., 2018). The MWW gene set test (available at https://github.com/miccec/yaGST) provides a Normalized Enrichment Score (NES), the value of the activity, which is calculated as Act = log2(NES/[1‐NES]), and a p‐value of the enrichment. The greater the Act the greater the association between the MR and its target genes in terms of upregulation of positive targets and downregulation of negative targets.
2.11. sciRNA‐seq data availability
All sciRNA‐seq data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE142168.
2.12. H & E, immunohistochemical, and immunofluorescence staining
Mouse brains were paraffin‐embedded, sectioned, and stained with H & E as described previously (Hambardzumyan, Amankulor, Helmy, Becher, & Holland, 2009; Holland et al., 2000). Immunohistochemical staining of mouse brains was performed with an automated staining processor using the Discovery DAB Map Detection Kit according to standard protocols as described previously (Ventana Medical Systems, Tucson, AZ) (Hambardzumyan et al., 2008). The following antibodies were used for the IHC: OLIG2 (Millipore, AB9610) at a 1:300 dilution, Ki67 (Vector Laboratories, VP‐RM04) at a 1:200 dilution, Nestin (BD Biosciences, 556309) at a 1:100 dilution, PDGFRA (Cell Signaling, 3174) at a 1:300 dilution, PDGFRB (Cell Signaling, 3169), and YAP1(Cell Signaling, 14074) at a dilution of 1:300.
For immunofluorescence stainings, formalin‐fixed paraffin‐embedded tissues were subjected to de‐paraffinization and antigen‐retrieval on the DISCOVERY XT platform (Ventana Medical Systems, Inc., Tucson) using standard protocols. Subsequently, slides were rehydrated and stained as described previously (Wirsching et al., 2019). Images were taken on a Leica DM5500B fluorescence microscope. Image analysis was done using TissueQuest software (version 6.0.1, Tissue Gnostics, Vienna, Austria). Primary antibodies used were: YAP1 (D8H1X) XP® Rabbit mAb #14074 (Cell Signaling) at a 1:100 dilution, OLIG2 AF2418 (R&D Systems) at a 1:300 dilution, Ki67 (Cat #14‐5698‐82, Invitrogen) at 1:100 dilution, Nestin (CH23001, Neuromics) at a 1:500 dilution. Secondary antibodies used were Donkey Anti‐rat‐488 (A‐21208, Invitrogen), Donkey Anti‐goat‐647 (705‐605‐147, Jackson ImmunoResearch), Donkey anti‐rabbit‐Cy3 (711‐165‐152, Jackson ImmunoResearch), Donkey anti‐chicken‐488 (703‐545‐155, Jackson ImmunoResearch). All secondary antibodies were used at a 1:200 dilution.
2.13. Bulk RNA‐seq data analysis and flow cytometry of myeloid cell lineage
Bulk RNA‐sequencing was performed previously and the data were deposited in the Sequence Read Archive (SRA) database under the accession number PRJNA349180. Detailed description of the procedure can be found in the online supplemental materials of Chen et al. (2017). Four genes of interests Csf1r, Ptprc, Tmem119, and Ctsb were queried from the database and their FPKM (Fragments Per Kilobase of transcript per million mapped reads) values presented.
Flow cytometry was performed as previously described (Chen et al., 2017). Briefly, mice were anesthetized with ketamine (100 mg/kg, intraperitoneal) and xylazine (10 mg/kg, intraperitoneal), and perfused with cold PBS. The brains were dissected and digested with Neural Tissue Dissociation Kit (Miltenyi Biotec) following the manufacturer's instructions. Cells were passed through a 70 μm cell strainer, centrifuged and resuspended in 30% Percoll (GE Healthcare) solution. Cells were separated by centrifuging at 800g for 30 min at 4°C. Cell pellets were collected and washed with FACS buffer (Dulbecco's phosphate buffered saline with 0.5% bovine serum albumin and 0.1% NaN3) and blocked with 100 μl of 2× blocking solution (2% fetal bovine serum, 5% normal rat serum, 5% normal mouse serum, 5% normal rabbit serum, 10 μg/ml 2.4G2 anti‐FcR, and 0.2% NaN3 in DPBS) on ice for 30 minutes. Cells were then stained on ice for 30 min and washed with FACS buffer. Antibodies used in the study include: CD45‐APC, CD11b‐PerCP‐Cy5.5, Ly6C‐PE‐Cy7, F4/80‐APC‐Cy7 (BD Pharmingen), and Ly6G‐V450 (BioLegend). All data were collected on a BD LSR flow cytometer and analyzed using FlowJo software (version 10, Tree Star Inc.).
3. RESULTS
3.1. Radiation enriches for tumor cells with the stem‐like, SP phenotype
Using SP analysis, we examined the effects of radiation on tumors in vivo in an Ink4a/Arf −/−, PDGF‐induced mouse model of glioma. Gliomas were generated by intracranial injection of RCAS‐PDGFB virus into Nestin‐tva mice (Shih & Holland, 2006). Upon symptom presentation, mice were irradiated with 10 Gy of ionizing radiation (IR) to the whole head and SP analysis was performed at 8 ± 2 hr, approximately 72 hr, and upon tumor recurrence (Figure S1). We chose 10 Gy because a previous radiation dose–response assay in this model, varying dose delivered in a single fraction, showed a plateau in tumor response at 10 Gy while heavily enriching for radioresistant, stem‐like tumor cells (Badri, Pitter, Holland, Michor, & Leder, 2016; Leder et al., 2014). Similar to prior studies, 10 Gy resulted in an increased median survival of approximately 20 days as compared to sham treated mice (p < .0001) (Figure 1a) At 72 hr after IR, the SP percentage was five‐fold greater than that of unirradiated controls (3.4% ± 0.8 vs 16.9% ±3, p < .05), but returned to baseline upon tumor recurrence (4.3% ± 2) (Figure 1b,c). These results include both tumor and non‐tumor cells, which we cannot distinguish in this simple SP analysis.
FIGURE 1.
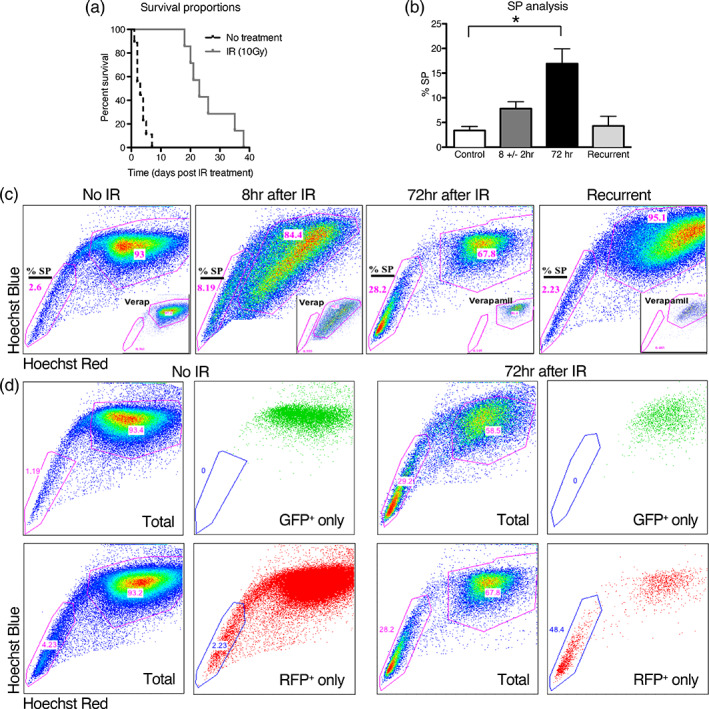
Side population (SP) analysis of a PDGF‐driven mouse model of glioblastoma at early time points after radiation shows that stem‐like cells of the SP are relatively radioresistant and are enriched. (a) A Kaplan–Meier plot showing that mice treated with 10 Gy lived a median of 20 days longer than untreated mice. (b) SP analysis, using Hoechst 33342 dye exclusion assay, of tumors from mice harvested at symptom onset or 8 or 72 hr after IR shows a higher percentage of cells in the SP at 72 hr as compared to control. Error bars represent the SE of the mean. (c) Representative flow cytometry plots, as quantified in (b). SP cells are poorly stained by Hoechst dyes due to efflux pump dye removal, whereas the main population (MP) is highly stained. The percentage of SP cells is highest at 72 hr after IR, but returns to the same level as the control at recurrence. Insets show treatment prior to SP analysis with verapamil as a control, which inhibits the efflux pump, abrogates the SP, and confirms the SP analysis gating strategy. (d) Flow cytometry plots of tumors without and at 72 hr after IR showing SP analysis of Olig2‐expressing tumor cells (GFP+) and tumor cells derived from the earliest tumor cells (RFP+). SP analysis of all tumor cells (Total) are shown prior to gating for GFP positivity (GFP only, top row) or RFP positivity (RFP only, bottom row). GFP+ cells are exclusively in the MP without and at 72 hr after IR, and RFP+ cells are heavily enriched at 72 hr after IR in the SP as compared to control
To determine whether tumor cells fall within the SP or main population (MP) we generated tumors in an Olig2‐GFP genetic background that expresses GFP under the control of the Olig2 promoter. The transcription factor, Olig2, is expressed by oligodendrocyte progenitors and the bulk of tumor cells. Cells expressing it are excluded from the perivascular niche (PVN). However, since not every Olig2‐expressing cell is tumor, and it is possible that not all tumor cells express Olig2, we used the RCAS‐PDGFB‐RFP vector to generate tumors. In this model, in addition to Olig2‐driven GFP‐expression, tumor cells derived from the original RCAS‐infected cells express RFP (Figure S2a). We found that no Olig2‐expressing cells (GFP+) were in the SP in any condition (Figure 1d). In unirradiated tumors, we found that 2–5% of cells derived from the original RCAS‐infected cells (RFP+, Olig2 −) were found in the SP (Figure 1d, Figure S2b). At 72 hr after 10 Gy, the percentage of RFP+, Olig2 − tumor cells in the SP was substantially increased (p < .0001), and at recurrence, the percentage reverted back to that seen in the unirradiated tumors (Figure 1d, Figure S2b).
This binary analysis suggests that the cell‐types in the SP, which include some tumor cells derived from the original RCAS‐infected cells, are relatively radioresistant as compared to the cell types in the MP, including Olig2‐expressing tumor cells and other tumor cells derived from the original RCAS‐infected cells. For a deeper understanding of the tumor cell types involved, we needed to go beyond this binary phenotype analysis.
3.2. Single‐cell RNA sequencing of tumors identifies cell types
We applied sciRNA‐seq to 15 archived, frozen samples that were also used in Figure 1, which consisted of normal brain and tumor‐containing hemisphere for each of three treatment conditions: no IR and 8 ± 2 hr and approximately 72 hr after 10 Gy (Figure S1). Normal brain samples were obtained from the uninjected, non‐tumor‐bearing side of the brain of tumor‐bearing mice. On this sciRNA‐seq data, we applied UMAP embedding for dimensionality reduction and Louvain clustering for assigning clusters, which constitute cell types. The data included 15,648 cells with a median number of unique transcripts of 589.
We first examined the data as a whole, that is, the union of all 6 conditions. UMAP embedding resulted in 5 superclusters, which were well separated suggesting very different cell types (Figure 2a). Using cell type‐specific markers, these superclusters were identified as distinct, major cell types of the brain: astrocytes expressing Gja1, myeloid cells expressing Csf1r, neurons expressing Snap25 and Gad1 (GABAergic) or Slc17a7 (Glutamatergic), oligodendrocytes expressing Mog, and oligodendrocyte precursor cells (OPC)/tumor expressing Pdgfra (Figure 2a,b) (Butovsky et al., 2014; Clarke et al., 2018; Ginhoux et al., 2010; Nishiyama, Komitova, Suzuki, & Zhu, 2009; Scolding et al., 1989; Tasic et al., 2016; Zhang et al., 2016). Adjacent to the OPC/tumor cluster is the endothelial cell cluster expressing Flt1 and Igfbp7, suggesting a similar expression profile. Consistent with the histology of these tumors and human GBM, T cells were not found. The OPC/tumor supercluster could be subdivided into immature oligodendrocytes/OPC expressing Enpp6 and OPC/tumor expressing the RCAS viral transcript (Figure 2a,b) (Nishiyama et al., 2009; Tasic et al., 2016). The assignment of tumor is supported by the much higher representation of the OPC/tumor cluster in tumor as compared to normal brain samples, as expected (Figure 2c). In contrast, normal cell types, such as endothelial cells and immature oligodendrocytes, are fairly evenly represented in normal brain and tumor samples.
FIGURE 2.
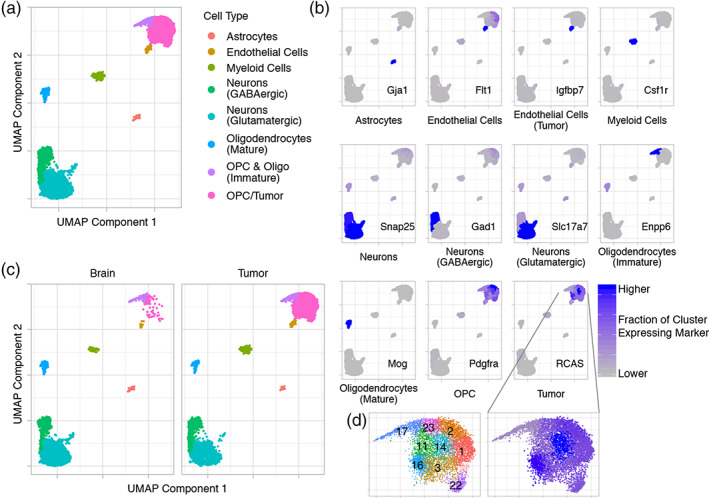
Single‐cell RNA sequencing (scRNA‐seq) identifies tumor and normal brain cell‐types, including normal cells with tumor‐like expression patterns. (a) UMAP embedding and Louvain clustering of scRNA‐seq data including all normal brain and tumor samples across all treatment groups (no IR and 8 ± 2 and 72 hr after 10 Gy) colored by cell‐types. Each dot represents a cell. (b) UMAP plots with each cluster colored by the fraction of cells in the cluster expressing common cell‐type‐specific markers or the RCAS viral transcript confirm cell‐type assignments. (c) Separate UMAP plots of brain and tumor samples show much higher representation of the OPC/tumor cluster in the tumor samples confirming that this cluster includes tumor. Some tumor‐like normal cells are also present in normal brain. (d) Louvain clustering shows multiple expression profiles within the OPCs and Tumor supercluster (left). Clusters are numbered for easier reference. RCAS viral transcript expression varies in this supercluster being lowest in the “OPC & Oligo (Immature)” cluster (Cluster 17) and highest in the OPC/tumor Cluster 14 (right)
The OPC/tumor portion of the supercluster consists of multiple expression profiles (Clusters 1, 2, 3, 11, 14, 16, and 22; Figure 2d), and RCAS expression is different between these clusters. Cluster 14 has the highest fraction of cells expressing RCAS, while the immature oligodendrocyte/OPC cluster has the lowest (Figure 2d). This could be an artifact of random sampling of the RCAS transcript caused by the sparsity of scRNA‐seq data. Alternatively, RCAS expression may affect or reflect the overall expression profile. So, clusters with different expression profiles have varying RCAS transcript levels. However, it is also possible that individual clusters are actually composed of some cells expressing RCAS, which are progeny of the original RCAS‐infected cells, and others that do not express RCAS, but have similar expression profiles to those that do. Consistent with this interpretation, relatively rare cells from normal brain samples, which do not express RCAS, cluster with tumor cells, suggesting similar expression patterns between these cells. (Figure 2c). This is also consistent with our previous finding that tumor cells recruit and cause an expansion of rare normal cell‐types with a tumor‐like phenotype (Fomchenko et al., 2011).
Overall, sciRNA‐seq along with UMAP embedding and Louvain clustering is very effective at identifying different cell‐types and illuminating the relationships between them in our mouse glioma model. Additionally, we found that rare cells with similar expression patterns to tumor cells exist in normal brain. In the context of tumor, there may be an expansion of these tumor‐like cells that are not derived from the original RCAS‐infected cells.
3.3. Tumor cell clusters are differentially sensitive to radiation
Next, we examined the effect of IR on the gene expression of cells in the OPC/tumor supercluster (Figure 3a). In tumor samples, a left‐to‐right shift between no IR (Figure 3a, lower left panel) and 8 hr after IR (Figure 3a, lower middle panel) occurred, suggesting expression changes shortly after IR. This was followed by a reduction in the number of tumor cells recovered at 72 hr after IR, which is likely due to massive cell death (Figure 3a, lower right panel). No clear differences were seen in the normal brain samples (Figure 3a, upper panels).
FIGURE 3.
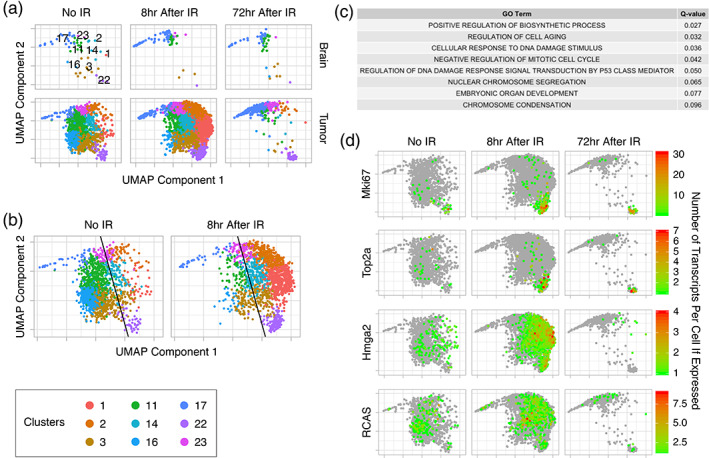
Tumor cell expression at 8 hr after IR shifts toward genes associated with the DNA damage signaling response and cell cycle arrest, followed by cell death leaving two radioresistant groups of cells (Clusters 17 and 23 and Cluster 22) at 72 hr after IR. (a) UMAP plots of the OPC/tumor supercluster by treatment group (No IR and 8 and 72 hr after IR) and sample type (brain and tumor) shows little change in normal brain samples, but a left‐to‐right shift between No IR and 8 hr after IR samples followed by a loss of most tumor cells at 72 hr after IR. Two groups of cells (Clusters 17 and 23 and Cluster 22) remain. Cluster numbers are shown in the upper left panel. (b) UMAP plots of samples without and 8 hr after IR showing the tumor supercluster with a best line drawn by support vector machine (SVM) to divide the cells into right (more similar to 8 hr after IR) and left (more similar to No IR) sides of the shift. (c) GO terms for enriched gene sets, as determined by Gene Set Enrichment Analysis (GSEA) comparing cells on the right of the left‐to‐right shift to those on the left, are associated with processes that are expected after IR. (d) Transcripts per cell of Mki67, Top2a, Hmga2, and RCAS in the OPC/tumor supercluster without (No IR) and 8 and 72 hr after IR are shown. The green to red spectrum represents cells with at least 1 transcript (see scales). Gray represents cells without any transcripts. Rapidly proliferating cells (Cluster 22 expressing Mki67 and Top2a) and slowly proliferating cells (Clusters 17 and 23) are enriched at 72 hr after IR, whereas Hmga2 expressing cells are enriched at 8 hr after IR and then depleted by 72 hr. RCAS viral transcript expression is similar in all treatment groups
To determine what expression changes drive the left‐to‐right shift, we trained a support vector machine (SVM) to determine the best line for dividing the OPC/tumor supercluster into left and right parts. This approach keeps cells with similar expression patterns together irrespective of which treatment group (no IR or 8 hr after IR) they originated from (Figure 3b). We then applied Gene Set Enrichment Analysis (GSEA) to determine which gene sets were enriched at 8 hr after IR (right) as compared to no IR (left). The enriched gene sets were associated with senescence, p53‐mediated DNA damage signaling response, and cell cycle arrest (Figure 3c). This was consistent with our previous study of early effects of IR on this tumor model using bulk polysome RNA sequencing of Olig2‐expressing cells (Halliday et al., 2014).
At 72 hr, most OPC/tumor cells are lost, but three clusters, 17 (immature oligodendrocytes and OPC), 23 and 22, remain significantly populated (Figure 3a). Clusters 17 and 23 are adjacent to each other suggesting similar expression profiles, whereas Cluster 22 is on the opposite side of the supercluster suggesting a different expression profile. Cluster 22 is highly proliferative at baseline as indicated by high expression of the cell cycle genes, Marker of Proliferation Ki67 (Mki67) and Topoisomerase 2a (Top2a), whereas Clusters 17 and 23 are not (Figure 3d). Additionally, IR either induces proliferation or selects for cells that are proliferative since the fraction of cells expressing Mki67 and Top2a in Cluster 22 increases from no IR to 8 and 72 hr after IR. High Mobility Group AT‐Hook 2 (Hmga2), which is involved in DNA damage signaling, is highly upregulated shortly after IR in most OPC/tumor clusters, with the exception of Clusters 17 and 23 (Figure 3d) (Fedele et al., 2006). At 72 hr, clusters previously expressing Hmga2 are lost, with the exception of Cluster 22, which no longer expresses Hmga2. Hmga2 appears to be a marker of impending cell death, suggesting that Clusters 17, 23, and the remaining portion of 22, which do not express Hmga2 at 72 hr are likely to survive to repopulate the tumor. By contrast, RCAS expression remains similar between treatment groups in all three clusters. It is unclear what fraction of these surviving clusters are composed of tumor cells, but the lower bound is the fraction or RCAS‐expressing cells, which is lowest in Cluster 17 and highest in Cluster 23. These 3 clusters fall towards the lower end of the RCAS‐expression spectrum suggesting that they may have a smaller fraction of RCAS‐derived tumor cells than other clusters, such as 14 and 16, which are lost after radiation.
Taken together, IR results in early tumor expression shifts toward the DNA damage signaling response and cell cycle arrest. This is followed by cell death leaving two radioresistant groups of cells at 72 hr after IR. One group is highly proliferative (Cluster 22) and the other (Clusters 17 and 23), which includes immature oligodendrocytes and OPC, is not, but both groups are at least partially composed of RCAS‐expressing tumor cells. These groups are likely to survive and one or both is likely to be the source of recurrent tumor.
3.4. Gene ontology (GO) term enrichment and master regulator analyses reveal biological functions and drivers of the surviving cell clusters
To further characterize the surviving cell clusters, we performed GO term enrichment analysis (GOTEA) to extract the biological properties distinguishing each cluster and Master Regulator Analysis (MRA) for in silico identification of the master regulators activated within each cluster. In this context, master regulators (MR) are proteins, mostly transcription factors (TF), which work as mechanistic regulators of the transcriptional signatures that define distinct tumor phenotypes (e.g., proneural vs. mesenchymal). This MRA approach compensates for the sparse sequence coverage inherent in scRNA‐seq (Methods) (Frattini et al., 2018; Mall et al., 2018).
GOTEA confirmed that Cluster 17 includes OPC and immature oligodendrocytes, as the enriched gene sets were associated with oligodendrocyte development and myelination (Figure S3a). Enriched gene sets for Cluster 23 were associated with neurogenesis and neuronal functions including dendrite and synapse development, suggesting this cluster is neural precursor cells or NG2/polydendrocytes (Figure S3b). Although in the overall UMAP embedding Clusters 17 and 23 are separated from the mature oligodendrocyte cluster, when these three clusters are removed from the context of all other clusters and re‐embedded by UMAP, these three clusters are found to be contiguous (Figures 2a and 4a). This shows a continuum of expression profiles between these clusters and suggests that they belong to the same cell lineage, consistent with the GOTEA. There is also a separate smaller cluster that expresses Mog and Snap25, which are markers of mature oligodendrocytes and neurons, respectively, and likely represents doublets and/or collisions.
FIGURE 4.
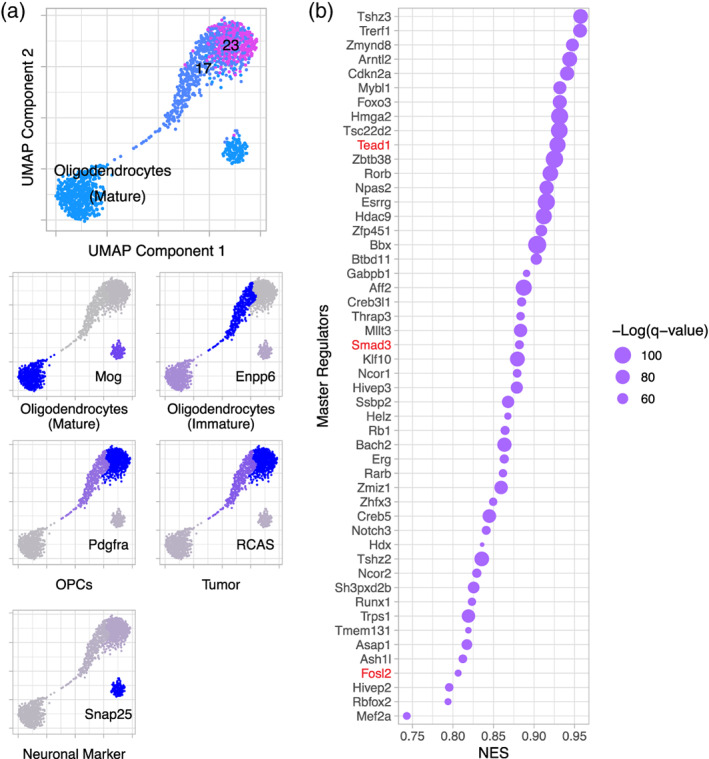
Clusters 17 and 23 belong to the cell lineage leading to mature oligodendrocytes, while Cluster 22 has characteristics of stemness and proliferation. (a) UMAP re‐embedding of Clusters 17, 23, and the mature oligodendrocyte cluster outside the context of all other cells (top panel) shows a continuum of expression between these clusters. The fractions of cells in each cluster expressing cell‐type specific markers (bottom panels) confirm the cell‐types represented by each cluster. The smaller cluster likely represents collisions or doublets. (b) Dot plot showing NES and −Log(q‐value) of master regulators for Cluster 22, including Tead1, Smad3, and Fosl2 (red), which are activated downstream of Hippo signaling and promote oncogenesis, stemness, proliferation, and radioresistance
Consistent with expression of Mki67 and Top2a in Cluster 22, enriched gene sets for this cluster were associated with proliferation, including DNA replication and mitosis (Figure S3c). Importantly, other associations were with stemness and early development (including regulation of stem cell proliferation), DNA damage repair, apoptosis, angiogenesis, and steroid signaling.
MRA showed that the MRs of each cluster were consistent with their enriched biological functions by GOTEA. Among the TFs that exhibit MR activity in Cluster 17, were Sirtuin 2 (Sirt2), an NAD‐dependent histone deacetylase specifically expressed in oligodendrocytes to modulate the cytoskeleton during differentiation and maturation and the NK2 Homeobox 2 (Nkx2‐2) homeodomain TF, a well‐known MR of cell fate determination and differentiation in the oligodendrocyte lineage (Figure S3d) (Qi et al., 2001; Tang & Chua, 2008). The MRs activated in Cluster 23 included, TOX High Mobility Group Box Family Member 3 (Tox3), a TF regulating neural progenitor identity, and Forkhead Box P2 (Foxp2), a TF crucial for specialized neuronal activities (Figure S3e) (Enard, 2011; Sahu et al., 2016). Finally, the TFs acting as specific MRs of Cluster 22 include TEA Domain Transcription Factor 1 (Tead1) and Smad family member 3 (Smad3) (Figure 4b). These TFs are activated downstream of the Hippo signaling pathway by binding YAP/TAZ. Yap1/Tead1 expression has been shown to promote oncogenesis and radioresistance, increase proliferation, and confer cancer stem cell‐like traits, which are the phenotypes associated with the enriched gene sets of this cluster (Fernandez‐L et al., 2012; Zanconato, Cordenonsi, & Piccolo, 2016).
Taken together, Clusters 17 and 23 are pre‐oligodendroglial and pre‐neuronal, respectively, and fall along the cell lineage leading to mature oligodendrocytes. In contrast, Cluster 22 has an expression profile suggesting vascular and stem‐like characteristics, which may at least in part be contributed to by YAP/TAZ signaling via Tead1 and Smad3. Therefore, at least some of Cluster 22 may correspond to the SP. However, it is still unclear if Cluster 22 also includes cells in the MP or if some of the cells in the SP are also in Clusters 17 and 23.
3.5. Histologic analysis identifies the location of the radioresistant tumor cells at 72 hr
For more information about the surviving cell types, we histologically examined tumor without IR, at 72 hr after IR, and at recurrence. At 72 hr after IR, the tumor density was 3 to 4‐fold lower than in unirradiated tumor, consistent with the loss of most cells in OPC/tumor clusters in our scRNA‐seq data at this time point (Figures 3a and 5a). At recurrence, the density returned to similar levels as unirradiated tumor. Expression of OLIG2 and Ki67 followed the same trends as the tumor density with a reduction in cells staining for these proteins at 72 hr, but not complete loss (Figure 5a). Nestin, which is expressed by neural stem and progenitor cells, remained at approximately the same level at all three time points. Nestin staining is primarily perivascular or endothelial, while OLIG2 staining is excluded from this region. Ki67 staining is in both regions, but is higher in the PVN or endothelial cells at 72 hr after IR.
FIGURE 5.
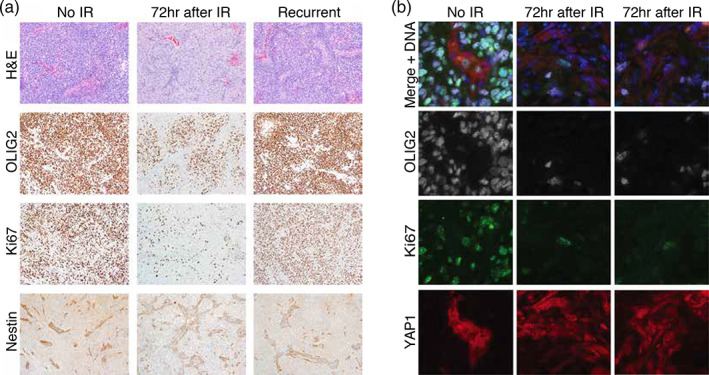
Immunohistochemistry and immunofluorescence microscopy show loss of the bulk of tumor cells, which express OLIG2, but survival of the Cluster 22‐like, YAP1‐expressing, proliferative cells of the perivascular niche at 72 hr after IR. (a) Hematoxylin and eosin (H&E) staining and immunohistochemistry with antibodies to OLIG2, Ki67, and Nestin were performed on tumors without IR (no IR) and at 72 hr and at recurrence after IR. Tumor density and the percentage of OLIG2‐expressing cells, which make up the bulk of tumor cells, are reduced significantly at 72 hr after IR, but return to baseline at recurrence, that is, levels similar to unirradiated tumor. The percentage of Ki67‐expressing cells is also reduced at 72 hr after IR, and those that remain are located in the perivascular niche. Ki67‐expressing cells also return to baseline at recurrence. Nestin‐expressing cells, which are located within the perivascular niche, remain at similar levels between treatment conditions. (b) Immunofluorescence microscopy using antibodies to OLIG2 (white), Ki67 (green), and YAP1 (red) was performed on tumor without and at 72 hr after IR (two examples shown). The top row (Merge + DNA) shows an overlay of the staining by all three antibodies along with DAPI to indicate DNA (blue). At 72 hr after IR, the rare cells expressing Ki67 also express YAP1, which are both specific to Cluster 22, while the OLIG2 expressing cells, which make up the tumor bulk, are distinct from those expressing either Ki67 or YAP1 and are mostly lost
To determine the location of the surviving radioresistant clusters including oligodendrocyte and neuronal progenitors (17 and 23) and the highly proliferative vascular/stem‐like cells (22), we compared tumors without IR and at 72 hr after IR by double immunofluorescence staining using antibodies to OLIG2 and Ki67 (Figure 5b). We found that the most radioresistant population was OLIG2−/Ki67−, distributed throughout the tumor, and likely enriched in cells of the tumor microenvironment such as macrophages and endothelial cells, which is also seen in the scRNA‐seq data (data not shown). At 72 hr, these cells were viable, offsetting tumor cell loss, and made up as much as 45% of total cells (Figure 5b, Figure S4a). The most radiosensitive population was OLIG2+/Ki67+, was excluded from the PVN and vasculature, comprised 40% of total cells in unirradiated tumors, and showed nearly a 95% loss at 72 hr after IR (Figure 5b, Figure S4b). The two remaining populations were relatively radioresistant since they did not follow the general trend towards the loss of OLIG2+ and Ki67+ cells (compare Figure S4c,d to Figure S4e,f). The OLIG2+/Ki67− population, was distributed throughout tumors and comprised 40% of total cells both before and after IR, consistent with the nonproliferating oligodendrocyte and neuronal progenitors of Clusters 17 and 23 (Figure 5b, Figure S4c). The OLIG2−/Ki67+ population was rare (3–4% both before and after RT), preferentially in the PVN or vasculature, and, by virtue of Ki67 levels, was likely to be enriched in the proliferative, vascular/stem‐like cells of Cluster 22 (Figure 5b, Figure S4d). Consistent with this interpretation and the MRA, which suggested that Cluster 22 was enriched in Tead1/Yap signaling, YAP1 staining was enriched in both the nuclei and cytoplasm of cells in the PVN or endothelial where it co‐stains with Nestin, suggesting that YAP1 is actively directing transcription there (Figure S5).
Taken together, at 72 hr after IR, the OLIG2+ tumor bulk is lost, but the OLIG2−/Nestin+/Ki67+ tumor cells of the PVN or endothelium survive. This is consistent with the SP analysis which showed a relative loss of the Olig2‐GFP expressing cells and enrichment of RFP+ cells. Since these cells are Ki67+ and YAP1+, the surviving cells of Cluster 22 are likely to be these cells of the PVN, but are not endothelial cells, which are in Cluster 26. It is not clear, though, that all of Cluster 22 are tumor cells derived from the original RCAS‐infected cells.
3.6. Myeloid cell populations in tumor are affected by radiation
Finally, we examined the early effects of IR on normal cell types. Normal cell clusters were mostly unaffected by radiation, with the exception of the myeloid cell supercluster (Figure 2a). To understand the radiation‐induced changes, this supercluster was removed from the context of all other clusters, re‐embedded by UMAP, and reclustered, resulting in four new clusters (Figure 6a).
FIGURE 6.
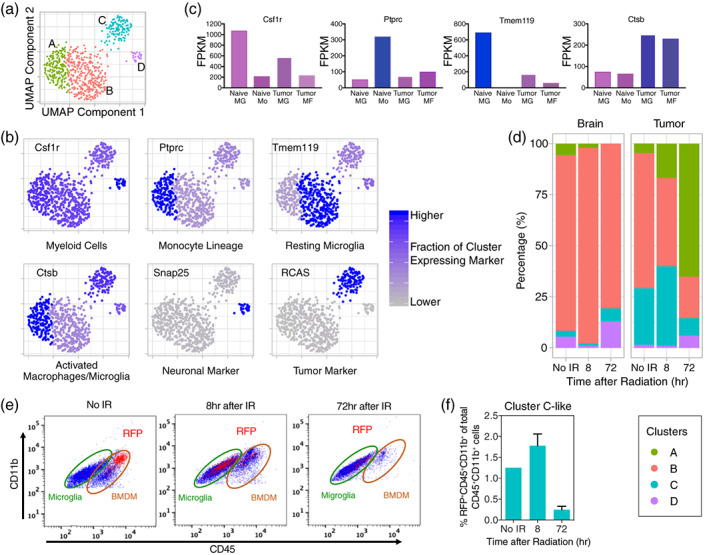
Subclustering of the myeloid cell supercluster reveals four clusters of microglia and bone marrow‐derived macrophages (BMDM) that vary in percentage by sample type and treatment group, some of which have phagocytosed tumor cells. (a) UMAP re‐embedding and Louvain clustering of the myeloid cell supercluster outside the context of all other cells reveals four clusters: Cluster A is tumor‐associated, activated microglia and BMDM that have not phagocytosed tumor cells; Cluster B is non‐tumor‐associated, resting microglia; Cluster C is tumor‐associated microglia and BMDM that have phagocytosed tumor cells; and cluster D is non‐tumor‐associated microglia that have phagocytosed neural cells. (b) UMAP plots with each cluster colored by the fraction of cells in the cluster expressing common cell‐type‐specific markers or the RCAS viral transcript. (c) Fragments per kilobase of transcript per million mapped reads (FPKM) from bulk RNA‐seq of sorted naïve microglia (Naïve MG), blood monocytes (Naïve Mo), tumor‐associated microglia (Tumor MG), and tumor‐associated BMDM (Tumor MF) for common cell type‐specific markers. Combining data from (b) and (c) allows assignment of cell‐types to clusters in (a). (d) Percentages of clusters by treatment group (No IR and 8 and 72 hr after IR) and sample type (brain and tumor) are shown. In normal brain, the percentage of resting microglia (Cluster B), is mostly unchanged between treatment groups. In tumor without IR, the percentage of tumor‐associated, activated microglia and BMDM (Cluster C) is elevated as compared to normal brain. The percentage of tumor‐associated microglia and BMDM (Clusters A and C) increase at the expense of the resting microglia at 8 hr after IR. At 72 hr, the tumor‐associated microglia and BMDM that have not phagocytosed tumor cells (Cluster A) increase further, but those that have phagocytosed tumor cells (Cluster C) decrease consistent with loss of most of the tumor cells at this time. (e) Flow cytometry using cell surface markers (Figure S6 and Methods) was performed on brain with tumor to examine RFP‐positivity in microglia (CD11b+CD45LoLy6g−Ly6c−) and BMDM (CD11b+CD45Hi Ly6g−Ly6c−) without IR and at 8 and 72 hr after IR. RFP‐positivity indicates cells that had phagocytosed RFP‐expressing tumor cells. RFP+ cells are mostly BMDM without IR, but are mostly microglia at 8 hr after IR. (f) Percentages of myeloid cells (CD45+CD11b+) that are RFP+ without IR and at 8 and 72 hr after IR in brain with tumor. Error bars represent the standard deviation. RFP+ cells peak at 8 hr after IR and mostly disappear by 72 hr. These data are consistent with Cluster C in (a)
We used previously published bulk RNA‐seq data from sorted naïve microglia, blood monocytes, tumor‐associated microglia, and bone marrow‐derived macrophages (BMDM) to identify markers to assign cell types to these clusters. We chose four commonly used myeloid markers that are dynamically regulated in the myeloid lineage in the context of tumor, Csf1r, Ptprc, Tmem119, and Ctsb (Figure 6b,c) (Bennett et al., 2016; Butovsky et al., 2014; Chen et al., 2017; Ginhoux et al., 2010; Sevenich, 2018). Csfr1 and Tmem119 are highly expressed by naïve microglia in normal brain, but are downregulated in tumor‐associated microglia, whereas Ptprc is most highly expressed in naïve monocytes, but is expressed at lower levels in the other myeloid cell types (Figure 6c). Expression of Ctsb is low in naïve myeloid cells but increased in both tumor‐associated microglia and BMDM. Based on these observations, we conclude that Cluster A(Csf1r int/Ptprc hi/Ctsb hi/Tmem119 Lo) consists of tumor‐associated microglia and tumor‐infiltrating BMDM. Cluster B, which expresses high levels of Tmem119 and low levels of Ptprc and Ctsb, is resting microglia found outside of the tumor area. Cluster C, which expresses Ctsb and has RCAS transcripts, is activated, phagocytic macrophages and microglia, which have phagocytosed nuclei of RCAS‐expressing tumor cells and thereby contaminated their nuclei with mRNA from those cells. Similarly, cluster D, which expresses high levels of Csfr1 and Ctsb and has neuronal marker (e.g., Snap25) transcripts, is a subpopulation of phagocytic microglia residing outside the tumor, pruning dying or proliferating neural progenitors in the hippocampus or subventricular zone (SVZ).
In the normal brain, resting microglia comprise the vast majority of the myeloid cell population with little variation without IR, or at 8 or at 72 hr after IR, but tumor is more dynamic (Figure 6d). Before IR similarly to normal brain, the majority of myeloid cells in the tumor are resting microglia. However, at 8 hr after IR, there is an influx of activated, but not phagocytic, tumor‐associated microglia and BMDM, and by 72 hr after IR, these cell‐types are the majority of this lineage in the tumor. Cluster C, increases at 8 hr as tumor cells are dying and then decreases again by 72 hr when most tumor cells have been lost from the tumor samples. Cluster D appears to increase by 72 hr after IR. The slower kinetics may be due to neurons or neuronal progenitors having slower kinetics of cell death as compared to tumor cells in response to radiation.
To further delineate whether activated microglia and/or BMDM are the cell‐types in Cluster C phagocytizing tumor cells, we used surface markers to distinguish microglia (CD11b+CD45Lo), macrophages (CD11b+CD45Hi), newly infiltrated monocytes (CD45HiCD11b+Ly6g−Ly6cHi), and neutrophils (CD11b+Ly6g+Ly6cInt) by flow cytometry on tumors from a mouse model in which tumor cells derived from the original RCAS‐infected cells express RFP (Figure 6e, Figure S6a). We examined microglia and BMDM for RFP‐positivity, which would indicate phagocytosis of RFP‐expressing tumor cells. Without IR, the majority of RFP‐positive phagocytizing cells are tumor‐associated BMDM, and account for about 1.2% of total myeloid cells (Figure 6e,f). At 8 hr after IR, these phagocytizing BMDM diminish in number, but microglia appear to increase their phagocytic capability. Consistent with the scRNA‐seq data, the combined percentage of phagocytizing cells from microglia and BMDM populations peaked at 8 hr after IR at about 1.7% of total myeloid cells (Figure 6e,f). At 72 hr, phagocytosis had subsided in both populations.
Taken together, there are at least four myeloid cell subpopulations that are identifiable by scRNA‐seq. These subpopulations consist of tumor‐associated microglia and BMDM that either are not (Cluster A) or are (Cluster C) phagocytosing tumor cells and non‐tumor‐associated resting (Cluster B) and phagocytosing (Cluster D) microglia. In normal brain without or after IR, the percentages of these subpopulations, which are dominated by resting microglia, are constant. In tumor, the percentages of tumor‐associated, activated microglia and BMDM (Clusters A and C) start higher and increase dramatically between samples without IR and at 8 and 72 hr after IR at the expense of resting microglia (Cluster B). Phagocytosis of tumor cells by tumor‐associated, activated microglia and BMDM peaks at 8 hr after IR, but the largest percentage of tumor‐associated, activated microglia and BMDM (phagocytosing or not) is at 72 hr after IR. At this point, most are not phagocytosing tumor cells, likely because very few tumor cells remain.
4. DISCUSSION
Glioma cells with stem‐like character are thought to be radioresistant and give rise to recurrences after therapy. Consistent with previous data, we found that SP cells in our mouse gliomas are relatively resistant to 10 Gy of IR. However, both the SP and MP are mixtures of cell‐types. To dissect out and measure radiation response in these cell‐types we turned to scRNA‐seq, which we applied to nuclear RNA. In this study, we applied nuclear scRNA‐seq to archived samples originally subjected to SP analysis as complementary approaches to better understand the connection between radioresistance and stemness in mouse models of glioma.
Nuclear scRNA‐seq suggests that these mouse gliomas are comprised of multiple expression subtypes, which seem to be similar to rare cell types found in the normal brain. The bulk of tumor cells are predictably radiosensitive based on expression patterns. The radioresistant cells were divided between two distinct expression patterns. The majority are similar to oligodendrocyte and neural progenitors with low proliferation rates (Clusters 17 and 23). A small minority are highly proliferative with vascular and stem‐like expression patterns that preferentially occupy the PVN (Cluster 22). This cluster is characterized by several critical transcriptional networks including that of Yap1/Tead1, which is known to promote oncogenesis, proliferation, and radioresistance. Unfortunately, due to the sparsity of the scRNA‐seq data, we were unable to directly determine the relationship between the radioresistant clusters and the SP, because appropriate markers were not represented in the scRNA‐seq dataset.
Importantly, the results of our study are consistent with human GBM data. The RCAS‐PDGFB‐driven gliomas in this study closely resemble human proneural GBM subtype (Herting et al., 2017). We have previously demonstrated that upon irradiation, murine PDGFB‐driven gliomas undergo a proneural to mesenchymal transition (Halliday et al., 2014). Bhat et al., showed that when RCAS‐PDGFB was overexpressed in combination with TAZ/TEAD overexpression, the resultant tumors had a mesenchymal signature, providing direct evidence of TAZ/TEAD playing a key role in driving the proneural to mesenchymal transition (Bhat et al., 2011). More specifically, recent evidence shows that upon irradiation, human proneural glioma stem cells undergo a mesenchymal transition and upregulate YAP/TAZ pathway genes (Minata et al., 2019). This is very similar to what we report in the current manuscript using our murine model.
The expression patterns of non‐tumor cells are unaffected by 10 Gy of IR over the time course we examined, with the exception of the myeloid cell lineage. In normal brain, this lineage predominantly consists of microglia, and this is not significantly affected by IR. However, tumor within the brain results in the presence of BMDM, and IR activates microglia and increases the number of BMDM significantly. IR results in the presence of phagocytic cells, peaking at 8 hr after IR, that have engulfed tumor cells (by virtue of contaminating RCAS mRNA), and a separate group of phagocytic cells at 72 hr after IR that have engulfed neural progenitors.
The notion that tumor stem cells are relatively nonproliferating is based on normal tissue. In this particular mouse glioma model, the majority of radioresistant cells are nonproliferating. However, a smaller population of radioresistant cells exists that is highly proliferative and, by gene expression patterns, stem‐like. This study does not address what specific cell‐type gives rise to recurrences, but a highly proliferative cell population is likely to contribute. This highly proliferative population may be specifically vulnerable to intervention based on its unique signaling dependencies. Our data suggest that YAP/TEAD signaling may be a candidate target for intervention for gliomas in the context of radiation.
AUTHOR CONTRIBUTIONS
Conceptualization: Jes Alexander, Dolores Hambardzumyan, Antonio Iavarone, Jay Shendure, and Eric C. Holland; software: Jes Alexander, Andrew J. Hill, Francesca Pia Caruso, Pietro Pugliese, and Zhihong Chen; formal analysis: Jes Alexander, Francesca Pia Caruso, Pietro Pugliese, and Zhihong Chen; investigation: Jes Alexander, Quincey C. LaPlant, Frank Szulzewsky, PJC, Francesca Pia Caruso, Pietro Pugliese, Zhihong Chen, Florence Chardon, Cailyn Spurrell, Dakota Ahrendsen, KP, and Alexander Pietras; writing—original draft: Jes Alexander, Quincey C. LaPlant, Frank Szulzewsky, Francesca Pia Caruso, Pietro Pugliese, Zhihong Chen, Dolores Hambardzumyan, Antonio Iavarone, Jay Shendure, and Eric C. Holland; writing—review and editing: Jes Alexander, Quincey C. LaPlant, Siobhan S. Pattwell, Frank Szulzewsky, Francesca Pia Caruso, Pietro Pugliese, Zhihong Chen, Dolores Hambardzumyan, Antonio Iavarone, Jay Shendure, and Eric C. Holland; visualization: Jes Alexander, Quincey C. LaPlant, Frank Szulzewsky, Francesca Pia Caruso, Pietro Pugliese, and Zhihong Chen; supervision: Lea M. Starita, Dolores Hambardzumyan, Antonio Iavarone, Jay Shendure, and Eric C. Holland; funding acquisition: Lea M. Starita, Jay Shendure, and Eric C. Holland.
Supporting information
Appendix S1. Supporting information
ACKNOWLEDGMENT
Sonali Arora provided helpful comments.
Alexander J, LaPlant QC, Pattwell SS, et al. Multimodal single‐cell analysis reveals distinct radioresistant stem‐like and progenitor cell populations in murine glioma. Glia. 2020;68:2486–2502. 10.1002/glia.23866
Funding information NIH Office of the Director, Grant/Award Number: Pioneer Award/DP1HG007811
DATA AVAILABILITY STATEMENT
sciRNA‐seq data availability: All sciRNA‐seq data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE142168.
REFERENCES
- Badri, H. , Pitter, K. , Holland, E. C. , Michor, F. , & Leder, K. (2016). Optimization of radiation dosing schedules for proneural glioblastoma. Journal of Mathematical Biology, 72(5), 1301–1336. 10.1007/s00285-015-0908-x [DOI] [PubMed] [Google Scholar]
- Bao, S. , Wu, Q. , McLendon, R. E. , Hao, Y. , Shi, Q. , Hjelmeland, A. B. , … Rich, J. N. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 444(7120), 756–760. [DOI] [PubMed] [Google Scholar]
- Bennett, M. L. , Chris Bennett, F. , Liddelow, S. A. , Ajami, B. , Zamanian, J. L. , Fernhoff, N. B. , … Barres, B. A. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences, 113(12), E1738–E1746. 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat, K. P. L. , Salazar, K. L. , Balasubramaniyan, V. , Wani, K. , Heathcock, L. , Hollingsworth, F. , … Aldape, K. D. (2011). The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes and Development, 25(24), 2594–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleau, A.‐M. , Hambardzumyan, D. , Ozawa, T. , Fomchenko, E. I. , Huse, J. T. , Brennan, C. W. , & Holland, E. C. (2009). PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem‐like cells. Cell Stem Cell, 4(3), 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, A. , Hoffman, P. , Smibert, P. , Papalexi, E. , & Satija, R. (2018). Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology, 36(5), 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky, O. , Jedrychowski, M. P. , Moore, C. S. , Cialic, R. , Lanser, A. J. , Gabriely, G. , … Weiner, H. L. (2014). Identification of a unique TGF‐β‐dependent molecular and functional signature in microglia. Nature Neuroscience, 17(1), 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese, C. , Poppleton, H. , Kocak, M. , Hogg, T. L. , Fuller, C. , Hamner, B. , … Gilbertson, R. J. (2007). A perivascular niche for brain tumor stem cells. Cancer Cell, 11(1), 69–82. 10.1016/j.ccr.2006.11.020 [DOI] [PubMed] [Google Scholar]
- Cao, J. , Packer, J. S. , Ramani, V. , Cusanovich, D. A. , Huynh, C. , Daza, R. , … Shendure, J. (2017). Comprehensive single‐cell transcriptional profiling of a multicellular organism. Science, 357(6352), 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Spielmann, M. , Qiu, X. , Huang, X. , Ibrahim, D. M. , Hill, A. J. , … Shendure, J. (2019). The single‐cell transcriptional landscape of mammalian organogenesis. Nature, 566(7745), 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro, M. S. , Lim, W. K. , Alvarez, M. J. , Bollo, R. J. , Zhao, X. , Snyder, E. Y. , … Iavarone, A. (2010). The transcriptional network for mesenchymal transformation of brain tumours. Nature, 463(7279), 318–325. 10.1038/nature08712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli, M. , Barthel, F. P. , Malta, T. M. , Sabedot, T. S. , Salama, S. R. , Murray, B. A. , … Verhaak, R. G. W. (2016). Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell, 164(3), 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles, N. , Ozawa, T. , Squatrito, M. , Bleau, A.‐M. , Brennan, C. W. , Hambardzumyan, D. , & Holland, E. C. (2010). Perivascular nitric oxide activates notch signaling and promotes stem‐like character in PDGF‐induced glioma cells. Cell Stem Cell, 6(2), 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Feng, X. , Herting, C. J. , Garcia, V. A. , Nie, K. , Pong, W. W. , … Hambardzumyan, D. (2017). Cellular and molecular identity of tumor‐associated macrophages in glioblastoma. Cancer Research, 77(9), 2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. E. , Liddelow, S. A. , Chakraborty, C. , Münch, A. E. , Heiman, M. , & Barres, B. A. (2018). Normal aging induces A1‐like astrocyte reactivity. Proceedings of the National Academy of Sciences of the United States of America, 115(8), E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis, S. , Sloan, S. A. , Croote, D. , Mignardi, M. , Chernikova, S. , Samghababi, P. , … Quake, S. R. (2017). Single‐cell RNA‐Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Reports, 21(5), 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , … Gingeras, T. R. (2013). STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics, 29(1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard, W. (2011). FOXP2 and the role of cortico‐basal ganglia circuits in speech and language evolution. Current Opinion in Neurobiology, 21(3), 415–424. [DOI] [PubMed] [Google Scholar]
- Fedele, M. , Visone, R. , De Martino, I. , Troncone, G. , Palmieri, D. , Battista, S. , … Fusco, A. (2006). HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell, 9(6), 459–471. [DOI] [PubMed] [Google Scholar]
- Fernandez‐L, A. , Squatrito, M. , Northcott, P. , Awan, A. , Holland, E. C. , Taylor, M. D. , … Kenney, A. M. (2012). Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2‐mediated Akt activation. Oncogene, 31(15), 1923–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomchenko, E. I. , Dougherty, J. D. , Helmy, K. Y. , Katz, A. M. , Pietras, A. , Brennan, C. , … Holland, E. C. (2011). Recruited cells can become transformed and overtake PDGF‐induced murine gliomas in vivo during tumor progression. PLoS One, 6(7), e20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini, V. , Pagnotta, S. M. , Tala Fan, J. J. , Russo, M. V. , Lee, S. B. , Garofano, L. , … Iavarone, A. (2018). A metabolic function of FGFR3‐TACC3 gene fusions in cancer. Nature, 553(7687), 222–227. 10.1038/nature25171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux, F. , Greter, M. , Leboeuf, M. , Nandi, S. , See, P. , Gokhan, S. , … Merad, M. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330(6005), 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday, J. , Helmy, K. , Pattwell, S. S. , Pitter, K. L. , LaPlant, Q. , Ozawa, T. , & Holland, E. C. (2014). In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural‐mesenchymal shift. Proceedings of the National Academy of Sciences of the United States of America, 111(14), 5248–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan, D. , Amankulor, N. M. , Helmy, K. Y. , Becher, O. J. , & Holland, E. C. (2009). Modeling adult gliomas using RCAS/t‐va technology. Translational Oncology, 2(2), 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan, D. , Becher, O. J. , Rosenblum, M. K. , Pandolfi, P. P. , Manova‐Todorova, K. , & Holland, E. C. (2008). PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes and Development, 22(4), 436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmy, K. , Halliday, J. , Fomchenko, E. , Setty, M. , Pitter, K. , Hafemeister, C. , & Holland, E. C. (2012). Identification of global alteration of translational regulation in glioma in vivo. PLoS One, 7(10), e46965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herting, C. J. , Chen, Z. , Pitter, K. L. , Szulzewsky, F. , Kaffes, I. , Kaluzova, M. , … Hambardzumyan, D. (2017). Genetic driver mutations define the expression signature and microenvironmental composition of high‐grade gliomas. Glia, 65(12), 1914–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, E. C. , Celestino, J. , Dai, C. , Schaefer, L. , Sawaya, R. E. , & Fuller, G. N. (2000). Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nature Genetics, 25(1), 55–57. [DOI] [PubMed] [Google Scholar]
- Leder, K. , Pitter, K. , LaPlant, Q. , Hambardzumyan, D. , Ross, B. D. , Chan, T. A. , … Michor, F. (2014). Mathematical modeling of PDGF‐driven glioblastoma reveals optimized radiation dosing schedules. Cell, 156(3), 603–616. 10.1016/j.cell.2013.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall, R. , Cerulo, L. , Garofano, L. , Frattini, V. , Kunji, K. , Bensmail, H. , … Ceccarelli, M. (2018). RGBM: Regularized gradient boosting machines for identification of the transcriptional regulators of discrete glioma subtypes. Nucleic Acids Research, 46(7), e39–e39. 10.1093/nar/gky015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes, L. , Healy, J. , Saul, N. , & Großberger, L. (2018). UMAP: Uniform manifold approximation and projection. Journal of Open Source Software, 3(29), 861 10.21105/joss.00861 [DOI] [Google Scholar]
- Minata, M. , Audia, A. , Shi, J. , Lu, S. , Bernstock, J. , Pavlyukov, M. S. , … Bhat, K. P. (2019). Phenotypic plasticity of invasive edge glioma stem‐like cells in response to ionizing radiation. Cell Reports, 26(7), 1893–1905.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, S. , Kohanbash, G. , Liu, S. J. , Alvarado, B. , Carrera, D. , Bhaduri, A. , … Diaz, A. (2017). Single‐cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biology, 18(1), 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama, A. , Komitova, M. , Suzuki, R. , & Zhu, X. (2009). Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nature Reviews. Neuroscience, 10(1), 9–22. [DOI] [PubMed] [Google Scholar]
- Ozawa, T. , Riester, M. , Cheng, Y.‐K. , Huse, J. T. , Squatrito, M. , Helmy, K. , … Holland, E. C. (2014). Most human non‐GCIMP glioblastoma subtypes evolve from a common proneural‐like precursor glioma. Cancer Cell, 26(2), 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A. P. , Tirosh, I. , Trombetta, J. J. , Shalek, A. K. , Gillespie, S. M. , Wakimoto, H. , … Bernstein, B. E. (2014). Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344(6190), 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, Y. , Cai, J. , Wu, Y. , Wu, R. , Lee, J. , Fu, H. , … Qiu, M. (2001). Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development, 128(14), 2723–2733. [DOI] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu, S. K. , Fritz, A. , Tiwari, N. , Kovacs, Z. , Pouya, A. , Wüllner, V. , … Methner, A. (2016). TOX3 regulates neural progenitor identity. Biochimica et Biophysica Acta, 1859(7), 833–840. [DOI] [PubMed] [Google Scholar]
- Scolding, N. J. , Frith, S. , Linington, C. , Morgan, B. P. , Campbell, A. K. , & Compston, D. A. S. (1989). Myelin‐oligodendrocyte glycoprotein (MOG) is a surface marker of oligodendrocyte maturation. Journal of Neuroimmunology, 22(3), 169–176. 10.1016/0165-5728(89)90014-3 [DOI] [PubMed] [Google Scholar]
- Sergushichev, A. A . (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation . BioRxiv. 10.1101/060012 [DOI]
- Sevenich, L. (2018). Brain‐resident microglia and blood‐borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Frontiers in Immunology, 9, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, A. H. , Dai, C. , Hu, X. , Rosenblum, M. K. , Koutcher, J. A. , & Holland, E. C. (2004). Dose‐dependent effects of platelet‐derived growth factor‐B on glial tumorigenesis. Cancer Research, 64(14), 4783–4789. [DOI] [PubMed] [Google Scholar]
- Shih, A. H. , & Holland, E. C. (2006). Platelet‐derived growth factor (PDGF) and glial tumorigenesis. Cancer Letters, 232(2), 139–147. 10.1016/j.canlet.2005.02.002 [DOI] [PubMed] [Google Scholar]
- Stuart, T. , Butler, A. , Hoffman, P. , Hafemeister, C. , Papalexi, E. , Mauck, W. M. , … Satija, R . (2019). Comprehensive integration of single cell data, 177(7), P1888–1902. 10.1101/460147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp, R. , Taillibert, S. , Kanner, A. , Read, W. , Steinberg, D. M. , Lhermitte, B. , … Ram, Z. (2017). Effect of tumor‐treating fields plus maintenance Temozolomide vs maintenance Temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA: The Journal of the American Medical Association, 318(23), 2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, B. L. , & Chua, C. E. L. (2008). SIRT2, tubulin deacetylation, and oligodendroglia differentiation. Cell Motility and the Cytoskeleton, 65(3), 179–182. [DOI] [PubMed] [Google Scholar]
- Tasic, B. , Menon, V. , Nguyen, T. N. , Kim, T. K. , Jarsky, T. , Yao, Z. , … Zeng, H. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature Neuroscience, 19(2), 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venere, M. , Fine, H. A. , Dirks, P. B. , & Rich, J. N. (2011). Cancer stem cells in gliomas: Identifying and understanding the apex cell in cancer's hierarchy. Glia, 59(8), 1148–1154. 10.1002/glia.21185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher, A. S. , Tirosh, I. , Hebert, C. , Yizhak, K. , Neftel, C. , Filbin, M. G. , … Suvà, M. L. (2017). Decoupling genetics, lineages, and microenvironment in IDH‐mutant gliomas by single‐cell RNA‐seq. Science, 355(6332), eaai8478 10.1126/science.aai8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirsching, H.‐G. , Zhang, H. , Szulzewsky, F. , Arora, S. , Grandi, P. , Cimino, P. J. , … Holland, E. C. (2019). Arming oHSV with ULBP3 drives abscopal immunity in lymphocyte‐depleted glioblastoma. JCI Insight, 4(13), e128217 10.1172/jci.insight.128217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato, F. , Cordenonsi, M. , & Piccolo, S. (2016). YAP/TAZ at the roots of cancer. Cancer Cell, 29(6), 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Sloan, S. A. , Clarke, L. E. , Caneda, C. , Plaza, C. A. , Blumenthal, P. D. , … Barres, B. A. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron, 89(1), 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information
Data Availability Statement
sciRNA‐seq data availability: All sciRNA‐seq data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE142168.