

Abstract
Bacterial trans‐acyltransferase polyketide synthases (trans‐AT PKSs) are multimodular megaenzymes that biosynthesize many bioactive natural products. They contain a remarkable range of domains and module types that introduce different substituents into growing polyketide chains. As one such modification, we recently reported Baeyer–Villiger‐type oxygen insertion into nascent polyketide backbones, thereby generating malonyl thioester intermediates. In this work, genome mining focusing on architecturally diverse oxidation modules in trans‐AT PKSs led us to the culturable plant symbiont Gynuella sunshinyii, which harbors two distinct modules in one orphan PKS. The PKS product was revealed to be lobatamide A, a potent cytotoxin previously only known from a marine tunicate. Biochemical studies show that one module generates glycolyl thioester intermediates, while the other is proposed to be involved in oxime formation. The data suggest varied roles of oxygenation modules in the biosynthesis of polyketide scaffolds and support the importance of trans‐AT PKSs in the specialized metabolism of symbiotic bacteria.
Keywords: bacterial natural products, biosynthesis, marine natural products, polyketides
O where art thou: In the classic model for modular polyketide synthases (PKSs), a small set of reactions generate combinatorially substituted carbon chains. Architecturally diverse PKS modules were identified that diversify polyketides by oxygenation. An orphan PKS with two oxygenase modules from a plant symbiont was found to produce lobatamides, which are in‐chain‐oxygenated oxime‐bearing cytotoxins that were previously only known from a tunicate.
Bacterial complex polyketides belong to the most important natural product classes of therapeutic value.1 Two distinct families of multimodular polyketide synthases (PKSs), termed cis‐ and trans‐acyltransferase (trans‐AT) PKSs, generate most of these compounds. The textbook biosynthetic model is represented by the cis‐AT PKS family, such as the erythromycin PKS.1, 2 These enzymes are usually composed of a limited set of functionally different biosynthetic multidomain modules that elongate and modify intermediates, and give rise to contiguous carbon chains carrying keto, hydroxy, double‐bond, and carbon branch modifications. In contrast, the highly complex trans‐AT PKSs can accommodate a remarkable diversity of modules (>150 module architectures are currently known3) and employ integrated domains as well as free‐standing, trans‐acting enzymes with often poorly understood functions.4 The large functional range of trans‐AT PKSs suggests high biosynthetic diversity outside the scope of canonical polyketides. Another phenomenon of trans‐AT PKSs is their dominant role in the specialized metabolism of non‐actinomycete symbiotic bacteria, with examples reported from fungi,5 insects,6 marine invertebrates,7 and plants.8 To fully access this biosynthetic potential, insights into the function of trans‐AT PKS assembly lines and their components are needed to improve our ability to predict their products,1 identify alternative producers to invertebrate sources,9 and provide novel tools for biosynthetic engineering.
In previous work, we identified the insertion of oxygen into growing polyketide chains as a non‐canonical reaction by which trans‐AT PKSs diversify product skeletons.9 As shown for the trans‐acting flavin adenine dinucleotide (FAD)‐dependent oxygenase OocK from the oocydin pathway, the enzyme performs a Baeyer–Villiger (BV) oxidation on a β‐ketothioester intermediate to generate a malonyl derivative (Figure 1 A) that is then further elongated. Preservation or hydrolysis of the new ester moiety gives rise to oxygen atoms within polyketide backbones (oocydin B and haterumalides), carboxylate pseudostarters (oocydin A), or terminal alcohols (introduced by the OocK homologues in the pederin6 and toblerol pathway,8 Figure 1 B). Mid‐chain oxygen atoms also occur in other polyketides (Figure 1 C).10 Although these moieties could principally result from various processes,11 the presence of OocK homologs in diverse orphan PKS biosynthetic gene clusters (BGCs; Figures S1–S4) suggests that nature employs oxygen insertion more widely to construct unusual polyketide scaffolds. In this work, we applied a genome‐mining strategy to investigate the wider scope of flavoprotein monooxygenases acting during polyketide elongation. The data revealed an architecturally distinct oxygen insertion module that generates a glycolyl intermediate, as well as evidence for a third module type involved in oxime formation. Both new modules are used in the biosynthesis of lobatamides, potent cytotoxins that were previously only known from marine tunicates, but identified here from a culturable plant symbiont.
Figure 1.
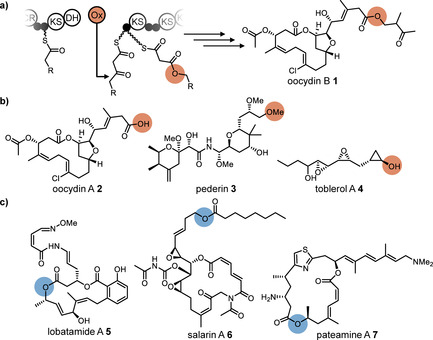
Oxygen incorporation into polyketide backbones. Moieties derived from oxygen insertion are highlighted in orange. Oxygen atoms of unknown biosynthetic origin are highlighted in blue. a) Mechanism for oxygen insertion into growing polyketide backbones, exemplified by OocK, a trans‐acting BV monooxygenase from the oocydin (oocydin B, 1) pathway. The enzyme converts a β‐ketothioester substrate into a malonyl derivative. b) Oocydin A (2), pederin (3), and toblerol A (4) with carboxylate‐ and alcohol‐type termini arising from oxygen insertion and ester cleavage. c) Biosynthetically unassigned polyketides harboring mid‐chain oxygens: lobatamide A (5) from a tunicate, and salarin A (6) and pateamine A (7) from marine sponges.
We initiated our work by identifying OocK homologues and other PKS‐associated flavoprotein monooxygenases encoded in orphan BGCs using GenBank, genome neighborhood,12 phylogenetic, and manual analyses (status November 2019; Figures S1–S4 in the Supporting Information). These revealed 69 candidate trans‐AT PKS systems harboring such enzymes (Figures S1–S4). The phylogram contained a large clade containing the functionally related OocK, PedG, and TobD, as well as homologues from uncharacterized PKSs. Another functionally assigned clade bears module‐integrated oxygenase (Ox) domains for which the variant from Burkholderia sp. FERM BP‐3421 was shown to introduce an epoxide unit in spliceostatin (Figure S1).8 In addition, other non‐OocK clades mostly belong to Ox domains integrated in various module types, thus suggesting further biosynthetic diversity. Focusing on uncharacterized oxygenases (Figure S1), an orphan trans‐AT system (termed lbm PKS, Figure 2 A) in the Gram‐negative bacterium Gynuella sunshinyii (NZ_CP007142) appeared an intriguing candidate, since the PKS contains oxygenase modules from two different unassigned clades (Figure S1, Table S1). G. sunshinyii is an unusual halophilic root‐associated plant symbiont13 that was recently recognized as a talented producer of diverse natural products.14 Its genome contains six trans‐AT PKS BGCs, the highest known number for any organism. The two integrated Ox domains are located in the PKS proteins LmbA and LmbC, and will be referred to as LmbA‐Ox and LmbC‐Ox, respectively. Additional features in these modules are a methyltransferase (MT) in the LmbC‐Ox module, which suggests α‐C‐methylation, and a predicted non‐elongating KS (KS0) in the LmbA‐Ox module, which suggests that a moiety introduced by the upstream module is further modified. This upstream module is located at the N terminus of LbmA and resembles an NRPS loading module with predicted15 glycine specificity. A C‐terminal TE domain is encoded on the PKS gene lmbE at the downstream end, which overall suggests collinearity between the PKS gene order and biosynthetic events.
Figure 2.
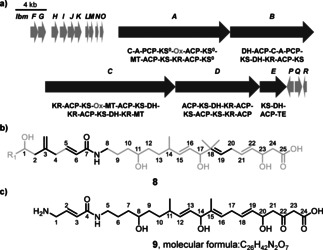
The lbm BGC and predicted polyketide structures. a) BGC architecture with core PKS and accessory genes marked in dark and light grey, respectively. The domain architecture of the core PKSs is shown using the following abbreviations: A=adenylation domain; C=condensation domain; DH=dehydratase; ER=enoylreductase; KR=ketoreductase; KS=ketosynthase; KS0=non‐elongating KS; MT=methyltransferase; Ox=oxygenase; TE=thioesterase. Ox domains are shown in grey. b) TransATor‐predicted structure 8 of the lbm product. High‐ and low‐confidence regions are shown in black and grey, respectively. c) Manually refined structure 9 used to find the natural product.
As guidance for the targeted isolation of the lbm polyketide, we analyzed the PKS using the recently developed automated prediction tool TransATor.3 This web application suggests chemical structures for trans‐AT PKSs from phylogenetically inferred15 KS substrates. In agreement with the unusual PKS architecture, the TransATor output 8 (Figure 2 B) contained extended regions of low confidence. After removal of a methyl group from the five‐bonded C18, the structure was refined by closer inspection of the modular architecture. Since TransAtor can predict intermediates only for modules that have a downstream KS, no elongation was predicted for the terminal module. A terminal C2 unit was therefore added based on cis‐AT PKS rules. To account for the N‐terminal NRPS module of LbmA, we suspected glycine for biosynthetic initiation. This replacement also removed an exomethylene group in the TransATor structure that was unlikely because polyketide β‐branching components16 were not found in the BGC. These modifications resulted in the hypothetical structure 9 (Figure 2 C) as a basis for the analytical work.
To search for the predicted compound, G. sunshinyii was cultivated, and the extract was analyzed by ultra‐high‐performance liquid chromatography/high‐resolution mass spectrometry (UHPLC–HRMS). Manual inspection of metabolite‐related MS features and the molecular formulae suggested from the high‐resolution masses showed a candidate ion peak at m/z 513.2230 [M+H]+ (Figure S5). This corresponds to a molecular formula of C27H32N2O8 (calc. 513.2231). This formula was closest to that of the predicted structure (C26H42N2O7) and appeared a good candidate for the lbm product. No other orphan PKSs in the genome were predicted to account for a similar chemical formula that contains two nitrogen atoms (i.e., PKSs that contain two NRPS modules).14 To isolate the compound, bacterial pellets from a 2 L culture of G. sunshinyii culture were extracted with acetone. MS‐guided fractionation using reversed phase‐high performance liquid chromatography (RP‐HPLC) provided pure compound 5. The 1H NMR and HSQC data showed the presence of an aliphatic methyl coupling with a vicinal proton, one methyl connected to an sp2 carbon, one methoxy group, three oxymethines, and 11 methines connected to sp2 carbons (Figure S6). Analysis of 2D NMR data including COSY, HMBC, and HSQC revealed the structure as that of lobatamide A (Figure 3 A, Figures S7–S9). Lobatamides are potent vacuolar (H+)‐ATPase inhibitors that interfere with tumor metastasis.17 Since they were previously known only from a marine invertebrate, the tunicate Aplidium lobatum,18 G. sunshinyii represents the first culturable source for these compounds. Lobatamides belong to a diverse group of benzolactone (H+)‐ATPase inhibitors (Figure 3 A) with as‐yet unknown PKSs and isolated from a remarkable range of organisms, including a sponge (salicylihalamides),19 a fungus (CJ‐12,950),20 myxobacteria (apicularens),21 and a Pseudomonas strain (oximidines).22 Their related structures and the identification of bacterial sources suggest that all of these compounds are of prokaryotic origin. For an expansion of this family, see a study on necroximes that was conducted concurrently with this work.23
Figure 3.
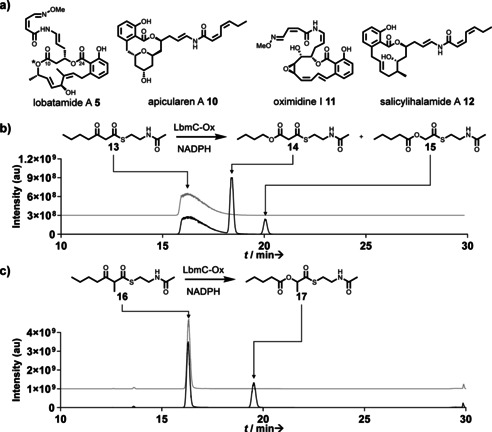
Selected benzolactone enamide polyketides and enzymatic assays with LbmC‐Ox. a) Benzolactone enamides. The moiety derived from oxygen insertion is highlighted with an asterisk. b,c) UHPLC–HRMS data showing the extracted ion chromatograms (EIC) of assay mixtures, including a boiled‐enzyme negative control (upper/grey) and a test reaction using all components (lower/black). b) EIC for 13 and 13+[16O] (calc. for [M+H]+ as 246.1158, and 262.1108, respectively). For the mass spectrum of the new product 15 see Figure S11. c) EIC for 16 and 16+[16O] (calc. for [M+H]+ as 260.1315 and 276.1264, respectively). Mass spectrum of product 17 is shown in Figure S19.
Structure 5 features an internal ester that is reversed in comparison to oocydin, pederin, and toblerol biosynthesis. To investigate the role of the LmbC‐Ox module in its formation, we cloned the region encoding the Ox domain into a variant of the pET28a expression vector (see the Supporting Information) for expression in E. coli as an N‐terminally His6‐tagged protein (Figure S10). Purified LbmC‐Ox was obtained as a soluble yellow protein, thus indicating bound FAD. Since the LmbC‐Ox module contained an additional MT domain, we suspected that the Ox domain accepts either a β‐ketothioester or an α‐methyl‐β‐ketothioester, depending on whether LmbC‐Ox acts before or after C‐methylation. We initially assayed the enzyme with the unmethylated thioester 13 as a simplified surrogate of an acyl carrier protein (ACP)‐bound 4′‐phosphopantetheinyl intermediate. Assay mixtures with test substrate, LbmC‐Ox, and NADPH were incubated for 90 minutes at room temperature and then extracted with ethyl acetate. UHPLC–HRMS analysis suggested two new products in the assay mixture, both with a mass difference of +16 Da relative to the substrate (Figure 3 B). Isolation and NMR‐based structure elucidation of the products (Figures S12–17, Table S4) revealed that the two isomers carry the inserted oxygen either at the γ (Figure 3 B, product 14) or the β position (product 15). The less abundant thioester 15 is noteworthy because it exhibits a reversed ester moiety compared to the OocK/PedG/TobD products and corresponds to the topology present in lobatamides. Suspecting that the major isomer 14 was an aberrant byproduct due to the choice of the surrogate substrate, we also synthesized thioester 16, which harbors the predicted methyl group at the α position (Figure 3 C, Figure S18). Repetition of the enzyme assay with this new test substrate produced one product peak (Figure 3 C). Isolation and structure elucidation identified it as thioester 17 (Figure 3 C, Figure S19–24, Table S5). The result from the biochemical study matches the internal ester topology at C‐10 of lobatamides, that is, facing the same direction as the C‐24 ester created by macrolactonization. In lieu of knock‐out studies that were so far unsuccessful or of heterologous whole‐cluster expression, the unusual function of LmbC‐Ox and its location within the PKS reasonably link the lbm BGC to lobatamide.
The results suggest an overall biosynthetic model for lobatamides as shown in Figure 4. A glycine starter would be the source of the oxime moiety, and oxygen insertion and macrolactonization generate the ester groups at C‐10 and C‐24, respectively. The salicylate group generated by the terminal four modules is also a feature of all other members of the benzolactone family, which currently lack known BGCs. However, similar tetramodular series that generate benzene moieties are also known from the otherwise unrelated psymberin and legioliulin PKSs.24
Figure 4.
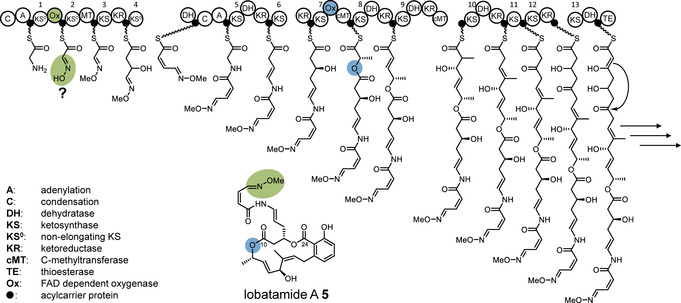
Biosynthetic model for lobatamide A. Ox domains and the corresponding polyketide moieties (proposed for the oxime) are highlighted in color. Consecutive KS numbers are shown above the domains.
Comparing the PKS‐associated enzymes OocK and LbmC‐Ox to a wider range of previously characterized class B flavoprotein monooxygenases (Figure S25), BV oxidation activity appears more prevalent in the LmbC‐Ox than the OocK clade.25 To further explore their biocatalytic potential, we tested 37 substrates that do not resemble polyketide intermediates and harbor diverse functional groups (Figures S26–S27, Table S6). When comparing the free‐standing OocK with the excised LbmC‐Ox, the latter exhibited greater thermostability and a broad substrate scope, accepting 12 of the 37 test compounds (18–26, Figure 5). Both enzymes accept NADPH as hydride donor, while OocK also displays activity with NADH (Table S7). These results suggest that the two enzymes could be used as biocatalysts for various oxidations, in addition to showing potential as PKS engineering tools to introduce non‐standard moieties into polyketides.
Figure 5.
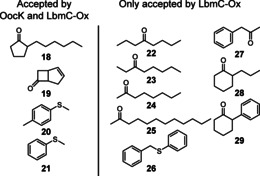
Structures of compounds that deviate from polyketide intermediate but were accepted by LbmC‐Ox and OocK or by LbmC‐Ox alone. For products, see Figure S26.
In conclusion, our data support the existence of at least two further module types in trans‐AT PKSs that permit oxidative modifications of polyketide core structures. In addition to the topologically related oxygen insertions catalyzed by modules utilizing trans‐acting OocK/PedG/TobD‐type enzymes (clade I modules, Figure S1), we identified a distinct, module‐integrated Ox domain that installs esters of the opposite orientation (clade II modules, Figure S1). It is unknown whether all members of one oxygenase clade generate the same ester topology. However, in line with a potentially predictive uniform product pattern for clade II, an uncharacterized oxygenase from the mandelalide trans‐AT PKS7c that is analogous to LmbC‐Ox has been suggested to insert oxygen in the same orientation as for lobatamides.7c A second type of oxygenase‐containing module newly identified in this work is not involved in oxygen insertion but seems to play a role in glycine oxidation to generate the unusual oxime moiety of lobatamides. This hypothesis is currently based on collinearity logic and needs to be experimentally tested. These examples demonstrate the staggering functional diversity of trans‐AT PKSs, which not only introduce substituents but also modify the carbon backbone itself. Our results also exemplify how genome mining can pave the way to finding microbial sources for bioactive compounds that had previously been described only from marine invertebrates. The production of the tunicate metabolite lobatamide A by G. sunshinyii further validates this plant symbiont as a rich source of bioactive specialized metabolites.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Young Ryun Chung and Dmitri Mavrodi for discussions on the biology of G. sunshinyii. This project was funded by the SNSF (grant IDs 205321_165659 and 205320_185077), and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 742739).
R. Ueoka, R. A. Meoded, A. Gran-Scheuch, A. Bhushan, M. W. Fraaije, J. Piel, Angew. Chem. Int. Ed. 2020, 59, 7761.
The copyright line for this article was changed on 8 October 2020 after original online publication.
References
- 1. Hertweck C., Angew. Chem. Int. Ed. 2009, 48, 4688–4716; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4782–4811. [Google Scholar]
- 2. Khosla C., Tang Y., Chen A. Y., Schnarr N. A., Cane D. E., Annu. Rev. Biochem. 2007, 76, 195–221. [DOI] [PubMed] [Google Scholar]
- 3. Helfrich E. J., Ueoka R., Dolev A., Rust M., Meoded R. A., Bhushan A., Califano G., Costa R., Gugger M., Steinbeck C., Nat. Chem. Biol. 2019, 15, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Piel J., Nat. Prod. Rep. 2010, 27, 996–1047; [DOI] [PubMed] [Google Scholar]
- 4b. Helfrich E. J., Piel J., Nat. Prod. Rep. 2016, 33, 231–316. [DOI] [PubMed] [Google Scholar]
- 5. Partida-Martinez L. P., Hertweck C., ChemBioChem 2007, 8, 41–45. [DOI] [PubMed] [Google Scholar]
- 6. Piel J., Proc. Natl. Acad. Sci. USA 2002, 99, 14002–14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Ueoka R., Uria A. R., Reiter S., Mori T., Karbaum P., Peters E. E., Helfrich E. J., Morinaka B. I., Gugger M., Takeyama H., Matsunaga S., Piel J., Nat. Chem. Biol. 2015, 11, 705–712; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Hildebrand M., Waggoner L. E., Liu H. B., Sudek S., Allen S., Anderson C., Sherman D. H., Haygood M., Chem. Biol. 2004, 11, 1543–1552; [DOI] [PubMed] [Google Scholar]
- 7c. Lopera J., Miller I. J., McPhail K. L., Kwan J. C., mSystems 2017, 2, e00096-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ueoka R., Bortfeld-Miller M., Morinaka B. I., Vorholt J. A., Piel J., Angew. Chem. Int. Ed. 2018, 57, 977–981; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 989–993. [Google Scholar]
- 9. Meoded R. A., Ueoka R., Helfrich E. J., Jensen K., Magnus N., Piechulla B., Piel J., Angew. Chem. Int. Ed. 2018, 57, 11644–11648; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11818–11822. [Google Scholar]
- 10.
- 10a. Bishara A., Rudi A., Aknin M., Neumann D., Ben-Califa N., Kashman Y., Org. Lett. 2008, 10, 153–156; [DOI] [PubMed] [Google Scholar]
- 10b. Northcote P. T., Blunt J. W., Munro M. H., Tetrahedron Lett. 1991, 32, 6411–6414. [Google Scholar]
- 11.
- 11a. Dunn Z. D., Wever W. J., Economou N. J., Bowers A. A., Li B., Angew. Chem. Int. Ed. 2015, 54, 5137–5141; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5226–5230; [Google Scholar]
- 11b. Biggins J. B., Ternei M. A., Brady S. F., J. Am. Chem. Soc. 2012, 134, 13192–13195; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Franke J., Ishida K., Hertweck C., Angew. Chem. Int. Ed. 2012, 51, 11611–11615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11779–11783. [Google Scholar]
- 12. Zallot R., Oberg N., Gerlt J. A., Biochemistry 2019, 58, 4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung E. J., Park J. A., Jeon C. O., Chung Y. R., Int. J. Syst. Evol. Microbiol. 2015, 65, 1038–1043. [DOI] [PubMed] [Google Scholar]
- 14. Ueoka R., Bhushan A., Probst S. I., Bray W. M., Lokey R. S., Linington R. G., Piel J., Angew. Chem. Int. Ed. 2018, 57, 14519–14523; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14727–14731. [Google Scholar]
- 15. Nguyen T., Ishida K., Jenke-Kodama H., Dittmann E., Gurgui C., Hochmuth T., Taudien S., Platzer M., Hertweck C., Piel J., Nat. Biotechnol. 2008, 26, 225–233. [DOI] [PubMed] [Google Scholar]
- 16. Calderone C. T., Kowtoniuk W. E., Kelleher N. L., Walsh C. T., Dorrestein P. C., Proc. Natl. Acad. Sci. USA 2006, 103, 8977–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pérez-Sayáns M., Somoza-Martín J. M., Barros-Angueira F., Rey J. M. G., García-García A., Cancer Treat. Rev. 2009, 35, 707–713. [DOI] [PubMed] [Google Scholar]
- 18. McKee T. C., Galinis D. L., Pannell L. K., Cardellina J. H., Laakso J., Ireland C. M., Murray L., Capon R. J., Boyd M. R., J. Org. Chem. 1998, 63, 7805–7810. [Google Scholar]
- 19. Xie X.-S., Padron D., Liao X., Wang J., Roth M. G., De Brabander J. K., J. Biol. Chem. 2004, 279, 19755–19763. [DOI] [PubMed] [Google Scholar]
- 20. Dekker K. A., Aiello R. J., Hirai H., Inagaki T., Sakakibara T., Suzuki Y., Thompson J. F., Yamauchi Y., Kojima N., J. Antibiot. 1998, 51, 14–20. [DOI] [PubMed] [Google Scholar]
- 21. Kunze B., Jansen R., Sasse F., Höfle G., Reichenbach H., J. Antibiot. 1998, 51, 1075–1080. [DOI] [PubMed] [Google Scholar]
- 22. Kim J. W., Shin-ya K., Furihata K., Hayakawa Y., Seto H., J. Org. Chem. 1999, 64, 153–155. [DOI] [PubMed] [Google Scholar]
- 23. Niehs S. P., Dose B., Richter S., Pidot S. J., Dahse H., Stinear T. P., Hertweck C., Angew. Chem. Int. Ed. 2020, 10.1002/anie.201916007; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 10.1002/ange.201916007. [DOI] [Google Scholar]
- 24.
- 24a. Fisch K. M., Gurgui C., Heycke N., van der Sar S. A., Anderson S. A., Webb V. L., Taudien S., Platzer M., Rubio B. K., Robinson S. J., Crews P., Piel J., Nat. Chem. Biol. 2009, 5, 494–501; [DOI] [PubMed] [Google Scholar]
- 24b. Ahrendt T., Miltenberger M., Haneburger I., Kirchner F., Kronenwerth M., Brachmann A. O., Hilbi H., Bode H. B., ChemBioChem 2013, 14, 1415–1418. [DOI] [PubMed] [Google Scholar]
- 25. Riebel A., Fink M. J., Mihovilovic M. D., Fraaije M. W., ChemCatChem 2014, 6, 1112–1117. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary