

Abstract
In spite of improving life expectancy over the course of the previous century, the health of the US population is now worsening. Recent increasing rates of type 2 diabetes, obesity and uncontrolled high blood pressure predict a growing incidence of cardiovascular disease and shortened average lifespan. The daily >$1billion current price tag for cardiovascular disease in the USA is expected to double within the next decade or two. Other countries are seeing similar trends. Current popular explanations for these trends are inadequate. Rather, increasingly poor diets in young people and in women during pregnancy are a likely cause of declining health in the US population through a process known as programming. The fetal cardiovascular system is sensitive to poor maternal nutritional conditions during the periconceptional period, in the womb and in early postnatal life. Developmental plasticity accommodates changes in organ systems that lead to endothelial dysfunction, small coronary arteries, stiffer vascular tree, fewer nephrons, fewer cardiomyocytes, coagulopathies and atherogenic blood lipid profiles in fetuses born at the extremes of birthweight. Of equal importance are epigenetic modifications to genes driving important growth regulatory processes. Changes in microRNA, DNA methylation patterns and histone structure have all been implicated in the cardiovascular disease vulnerabilities that cross generations. Recent experiments offer hope that detrimental epigenetic changes can be prevented or reversed. The large number of studies that provide the foundational concepts for the developmental origins of disease can be traced to the brilliant discoveries of David J.P. Barker.
Keywords: fetal programming, roots of cardiovascular disease, epigenetics, worsening health, heart disease
Introduction
This overview of the developmental programming of cardiovascular disease is dedicated to the memory of my friend and colleague, Professor David J.P. Barker. While others over the last century have linked developmental adversity with later postnatal consequences, David Barker and colleagues were the first to demonstrate the graded pattern over which prenatal growth predicts risk of acquiring chronic diseases in adulthood. He refuted the still popular notion that coronary heart disease is mostly the product of genetic predisposition and careless lifestyle. He showed, rather, that propensity for disease arises from organ-based vulnerabilities in the fetus/neonate which lead to disease, especially under adverse conditions in later life.
Fortunately, he was able to enjoy many of the fruits of his labor before his untimely death. He saw the establishment of the International Society of Developmental Origins of Health and Disease and numerous national satellite societies. He saw the endorsement of developmental origins of disease by Institutes at the US National Institutes of Health. He was pleased to see universities across the globe pouring resources into new centers for developmental health for the purpose of driving new understandings of the early origins of chronic disease.
Declining Health in the USA
While most of the U.S. population seems oblivious to the many health issues facing the American public, many disparate forms of evidence suggest that the overall health of the population is rapidly worsening. The data are in plain sight; one does not have to seek secret government files to discover the growing prevalence of many chronic diseases. Data from the US Center for Disease Control and Prevention (CDC) report that the average prevalence of obesity across the United States continues to rise. Data over the past 5 years show that 35% of adults over 20, 18% of adolescents (age 12–19 y) and 12% of children (age 2–5 y) are obese (www.cdc.gov/nchs/fastats/obesity-overweight.htm). In 2013 alone, two states, Mississippi and West Virginia, crossed the 35% obesity prevalence mark for people of all ages. If historical trends hold (www.cdc.gov/nchs/data/hestat/obesity_adult_11_12/obesity_adult_11_12.htm) other states will soon follow suit. Even now, the vast majority of Americans are overweight. Thus, the rise in obesity prevalence that began in earnest during the 1990s continues well into the 21st century.
Likewise, the prevalence of type 2 diabetes mellitus is on a sustained upward trajectory. CDC records show that while less than 1% of the population of Americans had diabetes in 1958, the prevalence gradually increased until the mid-1990s when the numbers began to rise more steeply. Current CDC estimates suggest that some 12% of the current population has diabetes (http://www.cdc.gov/nchs/fastats/diabetes.htm). In the mid ‘90s some 10 million people were diabetic. Now, in 2015 that number has tripled. The breathtaking rise has prompted the Center for Disease Control to predict that one third of the population will be afflicted by 2050 if the current rate of rise continues (http://www.cdc.gov/media/pressrel/2010/r101022.html).
The number of people in the US who have uncontrolled hypertension has also risen. Over the period 1988–1994 it was estimated that some 37 million people had elevated uncontrolled blood pressure even while many were under provider care. That number increased to 42 million during the period of 1998–2004. [1]. This rise occurred paradoxically at a time when professional societies were supporting public health messages urging adults to get their blood pressure measured and proclaiming the vascular harms associated with chronic hypertension. As the population grows and ages, the number of people whose blood pressure remains above recommended levels continues to rise [2, 3].
The three disease conditions that are most rapidly worsening, obesity [4], diabetes [5] and hypertension [6], are each independent powerful risk factors for cardiovascular disease. It is, therefore, reasonable to expect that the incidence of heart disease will rise over the next few decades. Indeed, estimates by cardiovascular experts suggest that the costs associated with cardiovascular disease will double between the current price tag which costs the nation in excess of $1billion/day to greater than $2 billion per day by 2026, expressed in constant 2010 dollars [7]. Thus US citizens will likely see a substantial surge in the costs of medical care at a time when containing medical costs has become a high national priority. Unfortunately, the need for medical care dollars may escalate to the point where current standards of medical care can no longer be consistently applied across the population. A declaration that the current health condition of the US population is “in crisis” should not be considered hyperbolic.
There have been many theories about the timing of the dramatic worsening US health trend beginning in the mid-90s. One popular news magazine argued that rapid childhood growth, lack of vitamin D, too clean an environment, too much cow’s milk and excess chemical pollution are causes of increases in Type 1 diabetes mellitus (January W. Payne US News and World Report: Health, April 26, 2010). Other dietary trends have also been implicated [8]. Others have argued for genetic causes [9], the lack of exercise among children in school systems [10] [11] and a lack of personal discipline in the lifestyle among members of the populace [12]. While several of these factors are likely contributors to the declining health in the US, some of the most important reasons for the rapid worsening of the US population have yet to be appreciated by government and the US population.
The genetic argument for rapid change is not sustainable if, by genetic, one means changes in population health that derive from recently arising mutated genes that impart vulnerability to the population. Lacking evidence or precedence, one could not seriously argue that the recent 25 year increases in chronic disease could be primarily due to augmented gene mutation rates. Even if by genetic, one means variations in the form of polymorphisms [13] underlying responses to poor lifestyle, the case is little easier to argue. The geographic distribution of diabetes, heart disease, hypertension and obesity which is concentrated in the South (seen on CDC Maps) would demand that nefarious gene polymorphisms causal to each of these conditions be concentrated primarily in one geographical region among unrelated individuals. Thus the geographic distribution of cardiovascular disease together with the rapidity in which the “epidemic” has occurred suggests, if not demands, an additional environmental explanation.
Developmental Programming
The work of David Barker, beginning in the late 1980s [14–16], along with the work of hundreds of others since, offers one compelling explanation for the deteriorating health of Americans: increasing vulnerability for disease in early development due to worsening of the American diet over the last 3 generations. Americans have increased their consumption of processed foods since the ‘60s [17]. Today, a larger portion of daily calorie intake (1kcal, called a calorie in the US = ~4 kjoules) comes from refined carbohydrates and sweetened beverages. According to government census data, total calories consumed in the US population increased from ~3200 calories per capita per day in the 1970s to 3900 calories per day in 2006 even as people became more sedentary. (http://www.census.gov/compendia/statab/2012/tables/12s0215.pdf). As a result, an increasing portion of the population suffers from “high calorie malnutrition.” This high energy form of malnourishment is the result of easy access to energy-rich processed food that has been engineered to stimulate tasteful bliss but lacks nutrients. The result is an ever increasing number of people in the US population with metabolic disease.
David Barker and colleagues reported the clear link between the birthweights of 15,000 English men and women from Hertfordshire [18] and their mortality rate from ischemic heart disease. They showed a 3 to 5 fold elevated risk graded, across the birthweight scale, with smallest babies having the highest risk. Since that time, numerous studies have discovered similar relationships in other countries including Finland [19, 20], Sweden [21], China [22], India [23] and US [24]. The Barker team also found powerful inverse relationships between birthweight and adult blood pressure, insulin resistance and/or type 2 diabetes, stroke and obesity. Large babies born to diabetic mothers have similar risks. The link between the intrauterine environment and the later risk for disease is known by insiders as “developmental programming.”
Both stress and poor nutrition appear to be devastating culprits in the programming process. For unknown reasons, the heart, blood vessels and kidney are among the structures in the body most affected by inadequate nutrition and conditions of toxic stress. The primary causes of cardiac related deaths include myocardial infarction (heart attack), heart failure of various types and sudden cardiac death due to asynchronous myocardial excitation (ventricular fibrillation). Figure 1 shows that these factors are all related to early life events that derive from the combinations of birthweight [14–16, 25, 26], maternal body phenotype as well as placental size and shape [27, 28]. Epidemiological studies, especially those from Helsinki and India, have demonstrated the transgenerational nature of nutritional flow and the intricacies of interactive factors that lead to cardiovascular disease risk in future generations. It is also becoming evident that there are sex-specific pathways by which programming effects lead to cardiovascular disease.
Figure 1.
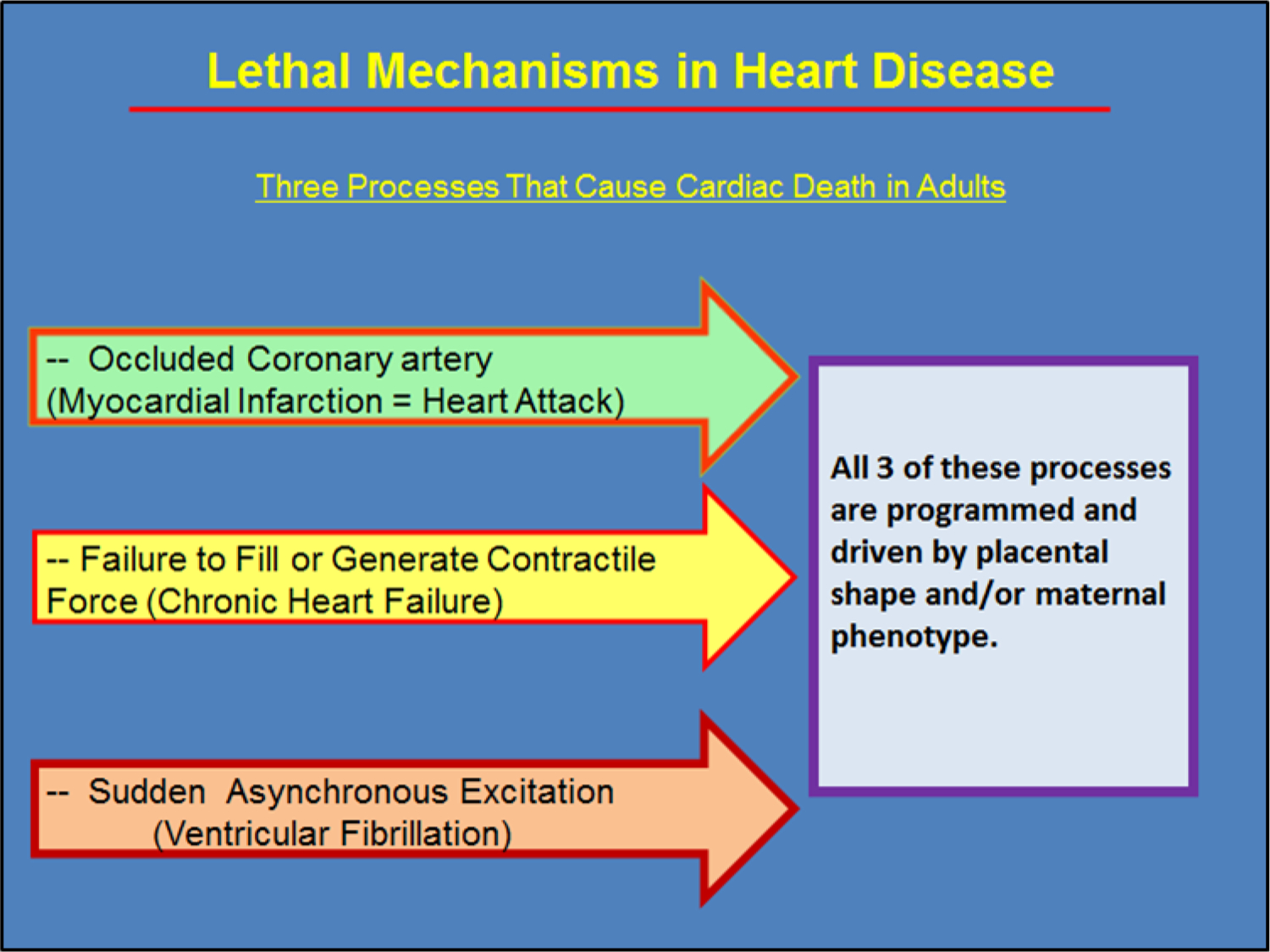
Cardiac disease usually causes death in one of three ways. 1) The well-known heart attack usually occurs when a lipid laden plaque in the wall of a major coronary artery ruptures and a thrombus is formed at the site which occludes blood flow to working heart muscle causing an infarction. 2) The heart is unable to pump adequately either because the heart muscle is weak or because the heart cannot properly fill with blood during diastole. 3) An electrical event occurs which causes cardiomyocytes in the myocardium to lose their ability to contract in a synchronous fashion. This ventricular fibrillation is generally lethal unless reversed by electrical cardioversion. The substrate for each of these lethal scenarios is derived from early life development. Most of the risk factors proclaimed by the American Heart Association originate before birth. Furthermore, while birthweight is a well-documented risk factor for cardiovascular death[15], myocardial infarction [26] and heart failure [27] are related to maternal phenotype including maternal height or BMI in combination with placental shape. However, the degree of placental thinness without a maternal phenotypic modifier predicts sudden cardiac death[28].
As powerful as the early epidemiological evidence was in the early ‘90s, many fetal biologists were skeptical of the birthweight-adult disease link in those early days, not because they doubted the skill of the Barker team, but because they could not envision familiar mechanistic links to explain the associations. Ultimately, it was a series of basic biological studies from around the world that brought light to the genetic, molecular, physiological and population underpinnings of programming biology. Langley Evans et al.[29], were among the first to show that the prenatal nutritional environment could have a powerful impact on cardiovascular function in the offspring in an animal model. Thus, it became clear that the primary determinant of human chronic disease is a biological mechanism that runs through the animal kingdom and has deep evolutionary roots, through a well described mechanism known as developmental plasticity. It is now certain that the unique growth pattern of an individual before birth is largely dictated by the level of nutritional bounty afforded by the nutritional pipeline from mother to baby.
Transgenerational Programming
The progression of evidence that explains the biological processes that underlie programmed cardiovascular disease has expanded rapidly. Figure 2 shows an approximate sequence of understanding the underlying factors related to the developmental origins of cardiovascular disease. Birthweight as the sole indicator of fetal tissue quality has given way to more complex but powerful combinations of placental size, shape and function, maternal metabolic state and body phenotype as well as newborn phenotype in the prediction of adult onset cardiovascular disease [30].
Figure 2.
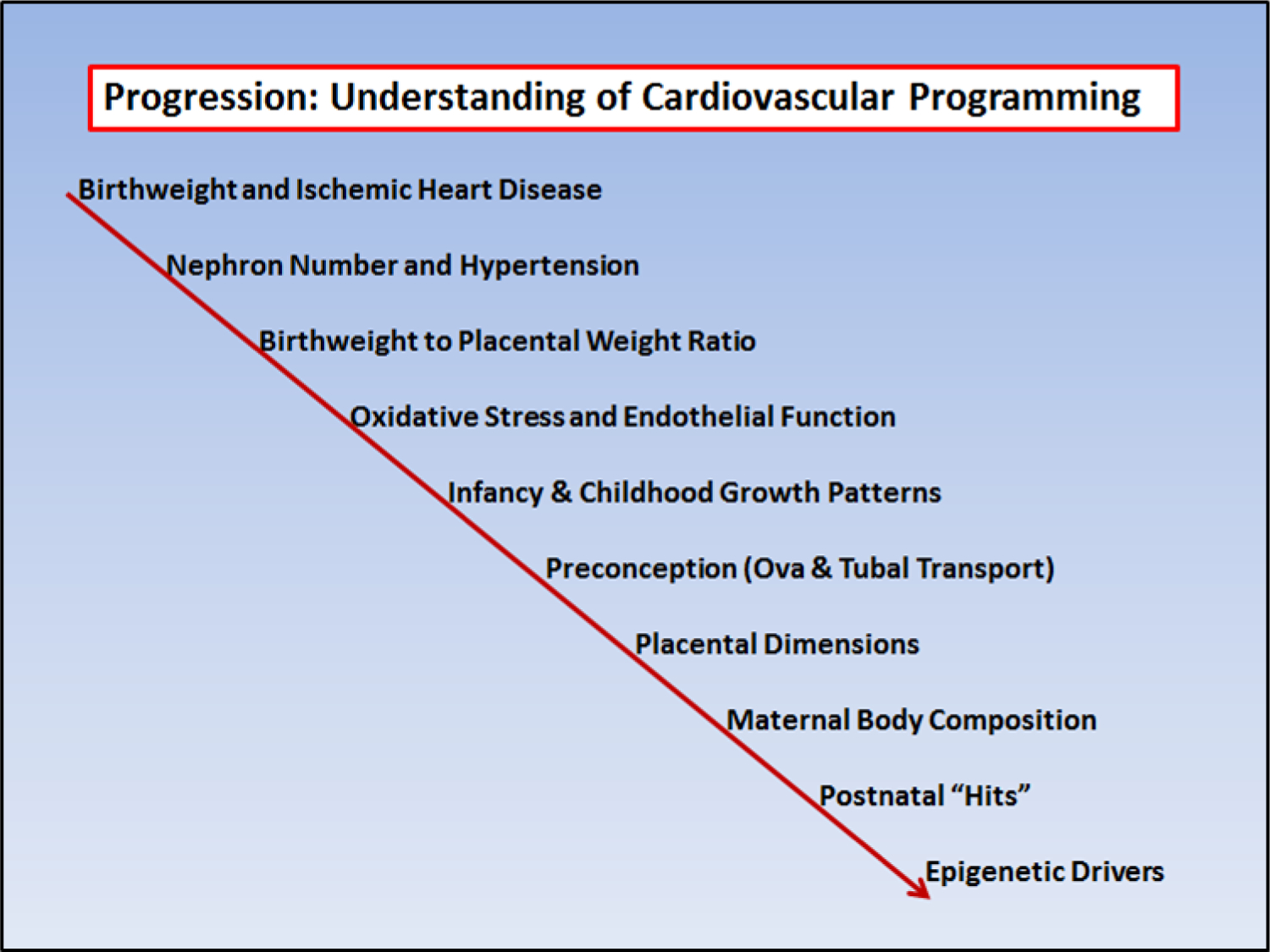
The link between birthweight and mortality from ischemic heart disease was reported in 1989 by Barker’s team. [14]. Since that time layers of knowledge have allowed an ever increasing understanding of the biological underpinnings of the finding. This list shows the approximate evolution of thought.
One new discovery that has surprised scientists across all disciplines is the finding that programming extends beyond influencing a child in a single generation. Because there is now a clear pathway by which nutrients theoretically affect gene expression patterns from grandmother to mother to offspring, the concept of transgenerational nutritional flow can generate mechanistic hypotheses that extend beyond the original Barker Hypothesis. In this case the egg that makes a fetus was likely to have arisen in the ovary of its mother while she was developing in the grandmother’s womb. That egg was then, nourished by nutrients provided by the grandmother’s diet and her tissue turnover. The multi-generational effects of programming in animal experiments were recently reviewed [31].
Periconceptional Programming
Another innovative link to cardiovascular disease was made by the Fleming team which showed that during an early stage embryo’s journey through the oviduct, it is exquisitely sensitive to the nutrient environment as it progresses from zygote to blastocyst [32–34]. In 2011, Watkins and collaborators showed that a poor nutritional environment just before fertilization and during embryogenesis leads to permanent changes in body weight and elevated blood pressure. A low protein isocaloric murine diet across gestation led to lighter female offspring at one year compared to control offspring; when the diet was in effect only during the early stages of embryo development, the females were heavier [35]. Both sexes had elevated systolic blood pressure, compared to controls if they experienced a low protein diet during oocyte maturation [36] or during early embryogenesis or over the whole of gestation.
The extent to which humans are sensitive to the same nutritional stressors as are rodents is not known precisely. However, it is well known that children born to pregnancies from embryos that were fertilized in vitro have more cardiovascular complications compared to those from spontaneous conceptions [37] [38]. A few studies have concluded that different incubation media for embryos fertilized in vitro (IVF) can lead to different body weights at birth and beyond [39] [40]. This finding requires confirmation by additional studies; fortunately, the relationship between embryo growth and later cardiovascular disease will be better established when current cohorts of IVF offspring become aging adults. A review of current knowledge of in vitro fertilization and later cardiovascular disease was recently published [41].
The Placenta and Programming Cardiovascular Disease
The placenta has long been known to be a programming agent for cardiovascular disease [42–44]. Recently it has been discovered that placental size and shape predict a number of disorders in later life including several cancers and different aspects of cardiovascular disease [30]. Many of these relationships were found in the Helsinki Birth Cohort [26, 45], the Dutch Hunger Winter cohort [46, 47], in the Riyadh, Saudi Arabia cohort [48–52] and studies from Mysore, India [53]. Furthermore, among other factors, the size and shape of the placenta often have greater predictive value when associated with a particular maternal phenotype (height, weight, BMI), birthweight, or fetal phenotype. For example, both heart failure [27] and hypertension [45] in men are associated with a small placenta but only to those born to mothers who were below the median in height. Sudden cardiac death is associated with a thin placenta [28] and coronary heart disease is associated combinations of maternal BMI and height and placental size and shape [26].
Cardiovascular Programming in the Embryo
In addition to strong epidemiological evidence for programming, enormous strides have been made in our understanding of environmental influences on processes in heart development that are likely to impart risk for later disease. Several environmental stressors, including nutrient excesses or hemodynamic forces, alter the growth of the heart and make it vulnerable for later disease. The two most vulnerable time points for the heart appear to be the early embryonic and the late fetal periods. These two periods will be discussed below.
The early embryo is sensitive to environmental influences that change the structure and function of the heart. It is in these early periods that most heart defects arise. There are now many hundreds of genes whose expression patterns are associated with structural heart defects in animal models [54], yet fewer than 10% are known to underlie human congenital heart defects [55–57]. We suspect that most human heart defects arise not because of heritable abnormal gene sequences but because abnormal placentation alters hemodynamic forces and cause inappropriate biochemical signaling within nascent myocardial structures [58–60].
Hemodynamic cues may lead to cardiac malformations in the embryo. When the murine gene encoding (HOXA13) is rendered dysfunctional the vascular endothelium of the fetal placenta is disrupted which causes placental insufficiency [61]. This condition leads to a lethal cardiac phenotype between day e14 and e15. The surprising feature is that the HOXA13 gene is not expressed in the heart and yet histological evidence suggests that changes in hemodynamic forces wrought by placental vascular changes lead to lethal embryonic cardiac wall thinning and cardiac rupture (personal communication). Genetic susceptibility may, of course, be also important in determining the sensitivity of a given heart to abnormal hemodynamic cues. Thus, the combination should always be considered.
Programming Fetal Systems
In late fetal life (or early postnatal life in altricial animals), the heart makes a dramatic transition from a prolific myocardium composed of small single working myocytes containing little contractile protein to a robust myocardium consisting of larger non-proliferative cardiomyocytes loaded with contractile protein capable of generating robust contractile force [62]. During this transition the heart is vulnerable because an inadequate endowment of cardiomyocytes offers risk for later cardiovascular disease and heart failure in particular. The size of the cardiomyocyte endowment and the degree of maturation of cardiomyocytes is determined over the period of a few weeks at the end of pregnancy. The fetal markers that correlate with poor cardiovascular outcomes actually represent complex changes in the structure, physiology and epigenetic status of the individual. For example, low birthweight babies may have one or more abnormalities including reduced nephron number, reduced cardiomyocyte number, small coronary vascularization, reduced mass of liver and pancreas as well as compromised brain vascularity. These structural changes cannot be reversed in postnatal life using current technology. In addition, there are physiological systems that are altered in fetuses and infants in response to inadequate nutritional flow during critical periods of development. These lead to disease risk in adults. See Table 1. Compromised systems may include those that provide protection from oxidative stress and inflammation, stem cell number and function, blood lipid regulators, coagulation systems, autonomic traffic, hypothalamic pituitary adrenal axis and epigenetic regulatory systems.
Table 1.
Pathological Changes that Lead to Cardiovascular Disease
| Programming Effect | Example References | ||
|---|---|---|---|
| Reduced nephron number | [1] [2](Brenner see additional refs) | ||
| Small coronary arteries | [3] | ||
| Increased autonomic outflow/ Exaggerated cortisol response |
[4] [5] | ||
| Endothelial dysfunction | [6] | ||
| Coagulopathy | [7] | ||
| Fewer cardiomyocytes | [8] | ||
| Cardiomyocte maturity | [9] | ||
| Cardiac remodelling | [10] | ||
| Vascular stiffness | [11] [12] | ||
| Dyslipidemias | [13, 14] | ||
| HPA axis reprogrammed | [15] | ||
| Oxidative stress | [16, 17] |
Environmental Cues for Late Fetal Heart Development
A large number of factors influence the maturation of the heart but there are two primary categories: Hemodynamic factors and hormonal factors. The placenta plays a role in regulating both. The structural architecture of the placental vasculature determines the impedance that the placenta offers to the pulsatile pressure and flow generated by the fetal heart. It also determines the capacity of the fetus to acquire oxygen and nutrients required for normal cardiac and vascular development. It generates factors that influence cardiovascular growth and maturation. Placental function influences several cardiac features at birth which serve as predictors of later life disease risk. These include cardiomyocyte number and maturation, coronary artery architecture and function and extracellular matrix composition.
The weight of a fetus at birth is by itself a crude indicator of cardiomyocyte number [63]. As in the embryo, placental insufficiency underlies maladaptive growth patterns in the heart. This has been demonstrated in several different laboratories [64–66]. The primary feature of the heart grown under conditions of placental insufficiency is a lower rate of cardiomyocyte proliferation. From the early embryonic period in large mammals when cardiac looping occurs until the last 3rd of gestation, the heart enhances its muscle mass primarily through proliferation. In sheep this proliferation slows dramatically in the last weeks of gestation as cells undergo a maturation phase known as terminal differentiation. This maturation process is characterized by the formation of a second nucleus in the cardiomyocyte through karyokinesis without cytokinesis. By the end of gestation some 2/3 of ovine cardiomyocytes carry a second nucleus and are unable to undergo mitosis [67]. Thus, the composition of the cardiomyocyte changes with maturation. As they mature, the cardiomyocytes manufacture layers of sarcomeres with mitochondria sandwiched between (Figure 3). A second major effect of placental insufficiency is the suppression of cardiomyocyte maturation processes. Fetuses that grew poorly because of placental insufficiency carry a much larger portion of cells that did not undergo terminal maturation and that remain in an immature state [65].
Figure 3.

A: Electron micrograph of 5 immature cardiomyocytes found at birth in cats. [62]. Cells have thin fibers of contractile protein just beneath the surface of the sarcolemma.
B: A single mature cardiomyocyte. Note thick layers of contractile material with darker mitochondria sandwiched between. This maturation process is driven, in part, by thyroid hormone which increases near term. N=nucleus; SL= sarcolemma. A and B have same magnification. (From 61 with permission)
Changes in hemodynamic load, as found when the undergrown placenta offers high impedance to ventricular ejection, stimulate a burst of proliferative cardiomyocyte growth that later gives way to premature terminal differentiation, severe suppression of further proliferative growth and ventricular wall thickening [68–70]. When blood pressure is lowered by blocking the renin-angiotensin system, cardiomyocyte proliferation falters [71]. In both cases, the heart is born with fewer cardiomyocytes than optimal for life long performance. Such hearts are susceptible to failure in late life.
The fetal heart is characterized by high levels of perfusion through the coronary tree. Resting flows are about twice those found in the adult [72]. What is surprising, however, is the degree to which the coronary tree is highly plastic during the fetal period but not thereafter. Davis et al., showed that a relatively short period of anemia leads to a doubling of coronary vascular conductance [73]. Conductance is the amount of blood flow a gram of heart per unit driving pressure when the arterial elements are fully dilated. In Davis’ experiments young adult sheep, previously made anemic as near term fetuses, had coronary conductances that were twice the normal value. Thus the doubling of conductance in the anemic fetus was maintained into adulthood. The unexpected finding in these studies is that the increase in coronary conductance is permanent [74]. One might predict that a “super” coronary tree would be beneficial to counteract later ischemic events. Indeed, these hearts were better able to resist the depressive effects of acute hypoxemia. However, the down side of the condition was a much higher susceptibility to infarction under conditions of ischemia reperfusion [74, 75]. Thus, a short term gain in function is offset by an increased vulnerability to ischemia. This vulnerability to ischemia reperfusion injury parallels that found in male rats that were hypoxic in the womb [76].
Figure 4 shows circulating factors that either stimulate or suppress cardiomyocyte proliferation during the maturation phases of development. Cortisol [77], insulin like growth factor-1 (IGF-1) [78, 79] and angiotensin II are the best studied. Two that suppress cardiomyocyte proliferation include the thyroid hormone, tri-iodo-L-thyronine (T3) [80, 81] and atrial natriuretic peptide [82]. Thus, it appears that for any given hemodynamic load, the ultimate determinant of cardiomyocyte endowment is the balance between pro-proliferation factors and anti-proliferation factors. Of these, the two most powerful opposing agents are IGF-1 and T3. IGF-1 is a powerful stimulant of proliferation and T3 an even more powerful suppressant. Fetal levels of IGF-1 are depressed under conditions of placental insufficiency and fetal undergrowth [83] which leads to an immature myocardium [66]. Thus, poor nutrient delivery leads to a compromised growth support and a less capable myocardium. T3 levels increase in the last few weeks of ovine gestation under the influence of cortisol; the latter deiodinates thyroid hormone and stimulates the conversion of T4 to T3 [84]. The effect of T3 is so overpowering that it suppresses the proliferation effects of any and all hormonal growth stimulants. Excess levels of T3 during the last few weeks of gestation lead to reduced numbers of cardiomyocytes (Figure 5). In addition, T3 drives the maturation of the cardiomyocyte by stimulating terminal differentiation, enlarging the cell and stimulating calcium signaling [80]. Thus, the long term vulnerability of the developing myocardium is determined in part by maternal thyroid hormone levels and placental thyroid transport systems.
Figure 4.
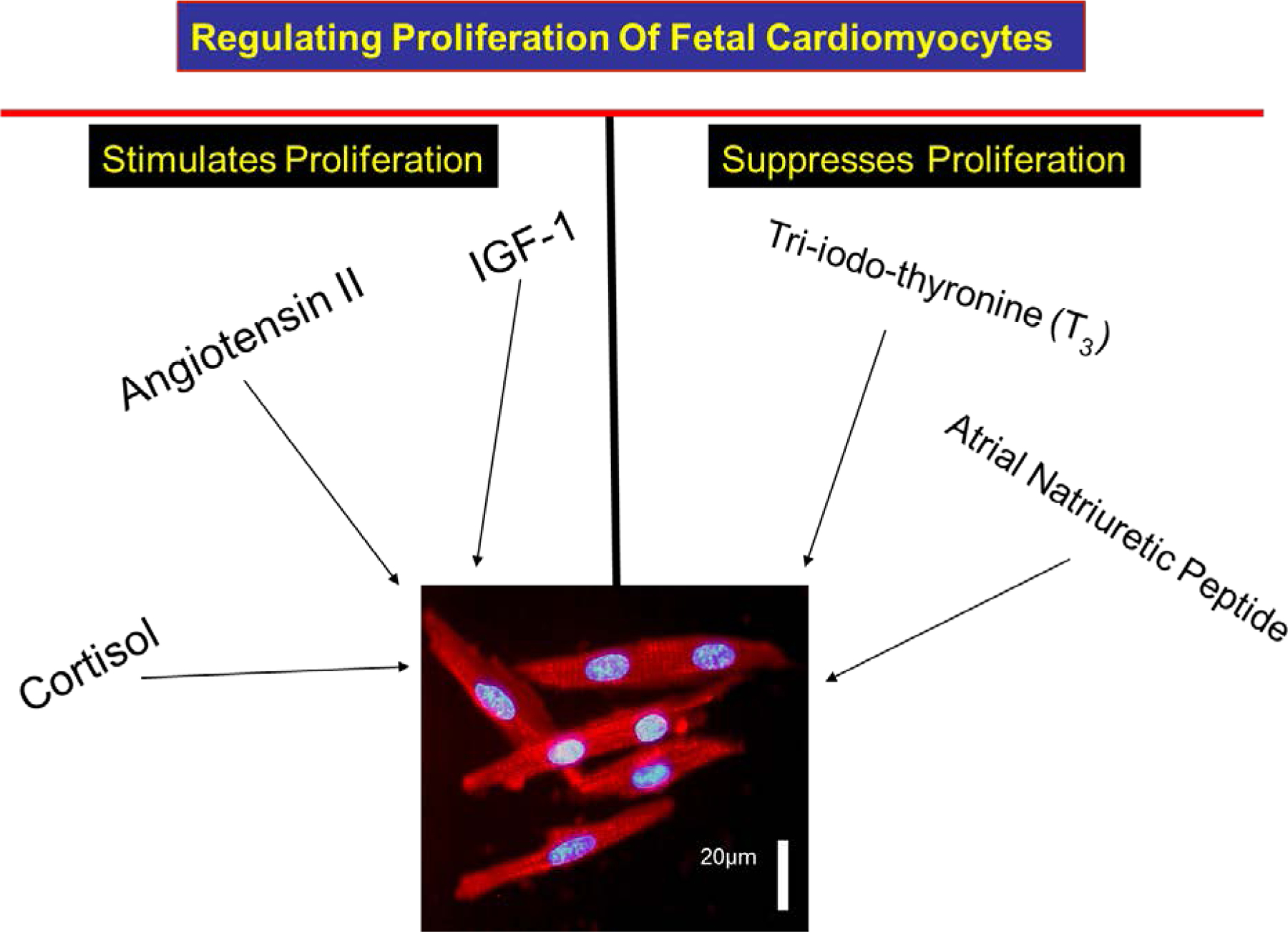
Three protein hormones are powerful stimulants of cardiomyocyte proliferation in the fetal heart: Cortisol, insulin-like growth factor-1 (IGF-1), and angiotensin II. Each works through a separate signaling cascade. Of these, IGF-1 is the most powerful and the most important. Two hormones inhibit IGF-1 production and proliferation: atrial natriuretic peptide and tri-iodo-L-thyronine (T3). These two hormones also work through different signaling pathways. With increasing levels near term, T3 not only suppresses proliferation but works in a synergistic fashion with IGF-1 to activate ERK and the AKT pathways [106].
Figure 5.
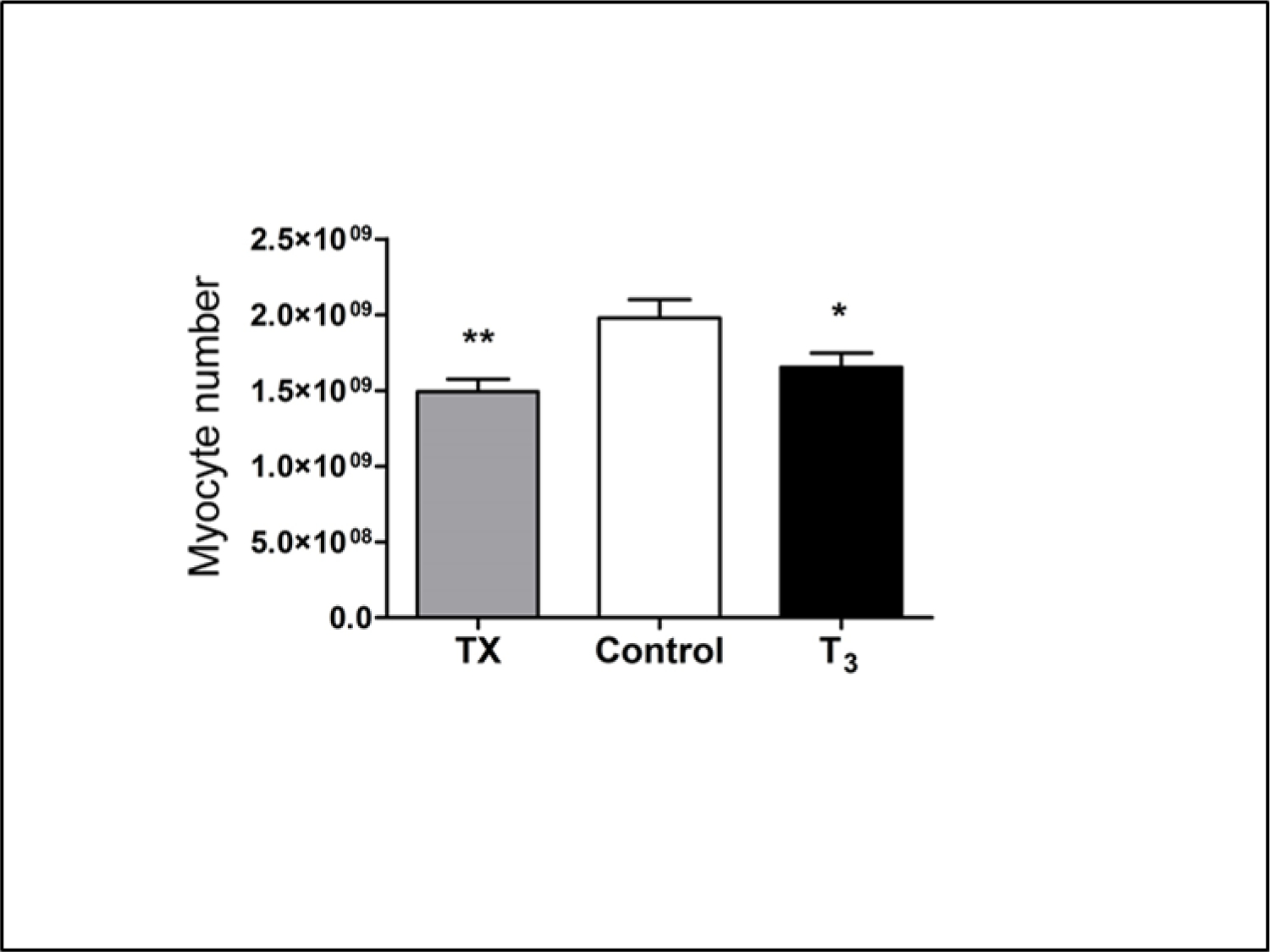
Cardiomyocyte numbers were estimated from average cell volumes of mono- and bi-nucleated cells and the total mass of right, left free walls and septum. T3 was infused in near term fetal sheep for 5 days (beginning on day 125 gestation) to mimic plasma levels at term (∼1.0 ng/ml). Cardiomyocyte sizes and numbers were compared to un-infused control fetuses and thyroidectomized fetuses (Tx). High levels of T3 in infusion fetuses suppressed proliferation and led to premature terminal differentiation; low levels (TX) prevented normal proliferation to occur. [80]. The changes in cardiomyocyte numbers show that a narrow window of T3 concentration is required to ensure an adequate number of cardiomyocytes at birth.
Epigenetic Influences on Cardiovascular Health
There is increasing evidence that many of the stressors that lead to a compromised myocardium also alter the regulatory mechanisms that underlie gene activity in the offspring through epigenetic mechanisms. Thus, a fetal insult may have enduring effects across the lifespan. If genes residing in the germ cells are also epigenetically modified, future generations may also be affected. The epigenetic changes include gene promoter methylation, histone modifications and expression of non-coding mirco-RNAs. While clear examples of “epigenetic programming” in the fetal heart are sparse, there are examples that prove the principle. Rat dams exposed to low oxygen during the latter days of pregnancy give birth to male offspring that appear normal at rest but suffer extensive myocardial damage under conditions of ischemia reperfusion stress [76]. The Zhang laboratory has shown that the protein kinase C, epsilon variant (PKC€) expression pattern is suppressed in the once hypoxic adult offspring, thereby enhancing myocardial disease vulnerability. His group also demonstrated that in males the CpG islands in the promoter of the gene are heavily methylated compared to control animals or females exposed to the same hypoxic environment in utero [85]. The group recently demonstrated that 1) once-hypoxic females are protected from methylation of the promoter sites by estrogen receptor interactions with the ERG-1 promoter site [86] and 2) in a different set of experiments, norepinephrine treatment of adult male rats caused hypertrophy and the associated hypermethylation could be reversed with 5-aza-2′-deoxycytidine treatment over the last 6 days of norepinephrine infusion. Thus, norepinephrine-induced hypermethylation was reversed as was the hypertrophic phenotype.
While there are many aspects of epigenetic regulation in cardiac development and ongoing functional maintenance as well as cardiac regeneration, the discovery of active microRNA species has been particularly exciting. In a very short period of time a large number of miRNAs have been associated with vascular and cardiac biology. Figure 6 shows a number of miRNAs associated with vascular elements. Many more affect the heart directly [87, 88]. Each is a potential target for regulation under conditions of early life programming. In addition, a number of miRNAs are associated with regenerative processes. These various RNA species offer hope that now irreversible changes in gene expression patterns can one day be modified to the therapeutic benefit of people who were undergrown before birth. Histone modifications are also important in the genetic regulation of heart development and adult heart function, especially during compensatory remodeling [89].
Figure 6.
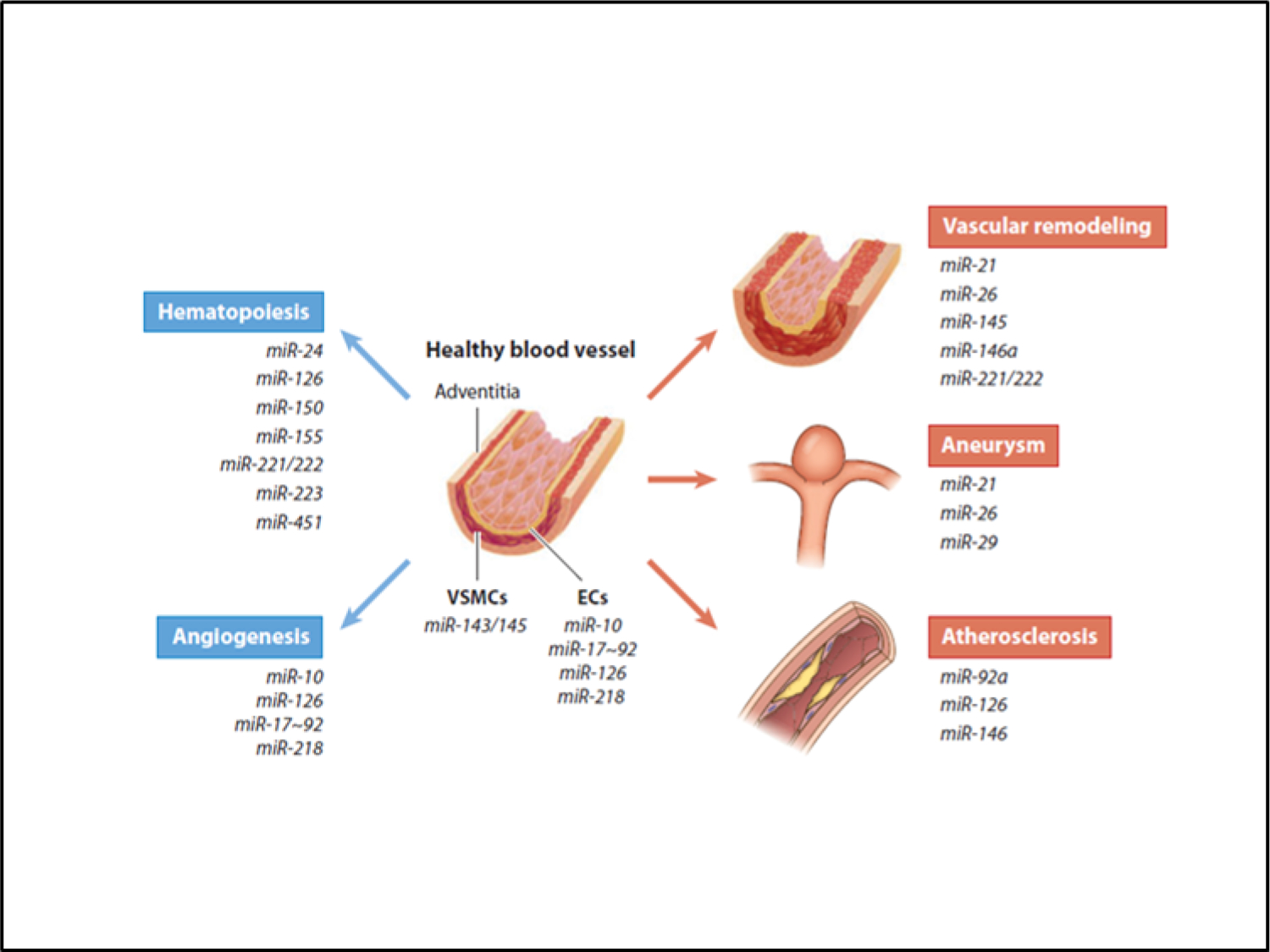
An ever increasing number of microRNA species are associated with normal and pathological changes in vascular elements. Some are associated with specific developmental processes while other are expressed only under stressful conditions. (With permission) [88].
The 2nd Hit and Social Responsibility
While the programming field continues to suffer alongside the proverbial blind man seeking to explain the elephant, the mechanistic underpinnings of programming are slowly emerging. In spite of this progress, however, a better understanding of gene-environment interactions will be required to explain the large gaps in knowledge that remain. New discoveries will necessarily include interactions between nutrients, the microorganisms that colonize the human body and pathophysiological patterns that lead down disease-specific pathways that persist beyond the first 1000 days. It appears that the earliest periods of development, reaching back to follicles in the ovary through infancy, are developmentally plastic and set the stage for vulnerability for disease throughout the lifespan. The degree to which programmed vulnerability manifests as disease depends not only on genetic background but also on later life stressors, so-called “2nd hits,” that drive early life susceptibility into outright adult onset life threatening disease. The 2nd hit concept, while herein purposefully over simplified, is nevertheless, helpful. We have modeled second hit theory in hopes that it will provide a foundation for the links between the social environment and chronic disease[90].
Even when armed with a relatively primitive understanding of the biological underpinnings of programmed disease, scientists have a responsibility to participate in the social dialog regarding how to reduce the burden of chronic disease in countries across the globe. Both early life and late life targets need to be addressed including: 1) Innovative ways to prevent programming in the womb must be implemented. These should include pre-pregnancy health measures and healthy diet and stress levels for pregnant mothers as well as sound breast feeding and weaning practices. 2) Addressing the second hit. Stressors later in life ensure that vulnerable people will be afflicted with the chronic diseases that are currently on the rise. As chronic diseases rapidly overtake communicable diseases in prevalence and cost, scientists and implementers of governmental and non-governmental organizations should work together to bring health and wealth to all citizens of the world. If scientists see this as their responsibility it is much more likely that governmental organizations will listen and be willing to tackle the monumental global nutrition problems that need to be solved.
Conclusions
Since the early Barker reports, we have understood that inadequate growth before birth leads to structural and functional changes that underlie cardiovascular disease. The list of examples is long. But now, the once explosive descriptive phase of programming must be expanded to uncover molecular mechanisms and support social activism. For example, the mechanistic links between phenotype and functional genotype must be discovered; we must acknowledge that the developing cardiovascular system is sensitive to maternal body type and associated placental growth patterns. Maternal phenotype is influenced by the deteriorating US food culture in which girls and young women find themselves, a problem that global societies also increasingly face. The task of the upcoming generation of pioneer scientists is to reveal stunning new discoveries that take the basic lab to the world, as did Barker and colleagues only a few short decades ago. There could be no better way to honor the memory of the original pioneer, Professor David JP Barker.
References:
- 1.Chobanian AV, Shattuck Lecture. The hypertension paradox--more uncontrolled disease despite improved therapy. N Engl J Med, 2009. 361(9): p. 878–87. [DOI] [PubMed] [Google Scholar]
- 2.Wang TJ and Vasan RS, Epidemiology of uncontrolled hypertension in the United States. Circulation, 2005. 112(11): p. 1651–62. [DOI] [PubMed] [Google Scholar]
- 3.Sarafidis PA, Georgianos P, and Bakris GL, Resistant hypertension--its identification and epidemiology. Nat Rev Nephrol, 2013. 9(1): p. 51–8. [DOI] [PubMed] [Google Scholar]
- 4.Eckel RH and Krauss RM, American Heart Association call to action: obesity as a major risk factor for coronary heart disease. AHA Nutrition Committee. Circulation, 1998. 97(21): p. 2099–100. [DOI] [PubMed] [Google Scholar]
- 5.Grundy SM, et al. , Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation, 1999. 100(10): p. 1134–46. [DOI] [PubMed] [Google Scholar]
- 6.Rosendorff C, et al. , Treatment of hypertension in the prevention and management of ischemic heart disease: a scientific statement from the American Heart Association Council for High Blood Pressure Research and the Councils on Clinical Cardiology and Epidemiology and Prevention. Circulation, 2007. 115(21): p. 2761–88. [DOI] [PubMed] [Google Scholar]
- 7.Go AS, et al. , Heart disease and stroke statistics−-2014 update: a report from the American Heart Association. Circulation, 2014. 129(3): p. e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gross LS, et al. , Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: an ecologic assessment. Am J Clin Nutr, 2004. 79(5): p. 774–9. [DOI] [PubMed] [Google Scholar]
- 9.Lyon HN and Hirschhorn JN, Genetics of common forms of obesity: a brief overview. Am J Clin Nutr, 2005. 82(1 Suppl): p. 215S–217S. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Ventura AL, et al. , Barriers to lose weight from the perspective of children with overweight/obesity and their parents: a sociocultural approach. J Obes, 2014 2014: p. 575184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herman KM, et al. , Combined Physical Activity/Sedentary Behaviour Associations With Indices of Adiposity in 8 to 10 Year Old Children. J Phys Act Health, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Teo K, et al. , Prevalence of a healthy lifestyle among individuals with cardiovascular disease in high-, middle- and low-income countries: The Prospective Urban Rural Epidemiology (PURE) study. JAMA, 2013. 309(15): p. 1613–21. [DOI] [PubMed] [Google Scholar]
- 13.Talmud PJ, et al. , Sixty-five common genetic variants and prediction of type 2 diabetes. Diabetes, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker DJ, et al. , Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ, 1989. 298(6673): p. 564–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barker DJ, Osmond C, and Law CM, The intrauterine and early postnatal origins of cardiovascular disease and chronic bronchitis. J Epidemiol Community Health, 1989. 43(3): p. 237–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barker DJ, et al. , Weight in infancy and death from ischaemic heart disease. Lancet, 1989. 2(8663): p. 577–80. [DOI] [PubMed] [Google Scholar]
- 17.Smith LP, Ng SW, and Popkin BM, Trends in US home food preparation and consumption: analysis of national nutrition surveys and time use studies from 1965–1966 to 2007–2008. Nutr J, 2013. 12: p. 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Syddall HE, et al. , Cohort profile: the Hertfordshire cohort study. Int J Epidemiol, 2005. 34(6): p. 1234–42. [DOI] [PubMed] [Google Scholar]
- 19.Andersen LG, et al. , Birth weight, childhood body mass index and risk of coronary heart disease in adults: combined historical cohort studies. PLoS One, 2010. 5(11): p. e14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barker DJ, et al. , Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol, 2002. 31(6): p. 1235–9. [DOI] [PubMed] [Google Scholar]
- 21.Leon DA, et al. , Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915–29. BMJ, 1998. 317(7153): p. 241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan Z, et al. , Relationship between birth size and coronary heart disease in China. Ann Med, 2010. 42(8): p. 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein CE, et al. , Fetal growth and coronary heart disease in south India. Lancet, 1996. 348(9037): p. 1269–73. [DOI] [PubMed] [Google Scholar]
- 24.Rich-Edwards JW, et al. , Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ, 1997. 315(7105): p. 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martyn CN, Barker DJ, and Osmond C, Mothers’ pelvic size, fetal growth, and death from stroke and coronary heart disease in men in the UK. Lancet, 1996. 348(9037): p. 1264–8. [DOI] [PubMed] [Google Scholar]
- 26.Eriksson JG, et al. , Mother’s body size and placental size predict coronary heart disease in men. Eur Heart J, 2011. 32(18): p. 2297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barker DJ, et al. , The early origins of chronic heart failure: impaired placental growth and initiation of insulin resistance in childhood. Eur J Heart Fail, 2010. 12(8): p. 819–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barker DJ, et al. , The placental origins of sudden cardiac death. Int J Epidemiol, 2012. 41(5): p. 1394–9. [DOI] [PubMed] [Google Scholar]
- 29.Langley-Evans SC, Phillips GJ, and Jackson AA, In utero exposure to maternal low protein diets induces hypertension in weanling rats, independently of maternal blood pressure changes. Clin Nutr, 1994. 13(5): p. 319–24. [DOI] [PubMed] [Google Scholar]
- 30.Barker DJ and Thornburg KL, The obstetric origins of health for a lifetime. Clin Obstet Gynecol, 2013. 56(3): p. 511–9. [DOI] [PubMed] [Google Scholar]
- 31.Aiken CE and Ozanne SE, Transgenerational developmental programming. Hum Reprod Update, 2014. 20(1): p. 63–75. [DOI] [PubMed] [Google Scholar]
- 32.Fleming TP, et al. , Nutrition of females during the peri-conceptional period and effects on foetal programming and health of offspring. Anim Reprod Sci, 2012. 130(3–4): p. 193–7. [DOI] [PubMed] [Google Scholar]
- 33.Watkins AJ, Lucas ES, and Fleming TP, Impact of the periconceptional environment on the programming of adult disease. J Dev Orig Health Dis, 2010. 1(2): p. 87–95. [DOI] [PubMed] [Google Scholar]
- 34.Watkins AJ, et al. , Low protein diet fed exclusively during mouse oocyte maturation leads to behavioural and cardiovascular abnormalities in offspring. J Physiol, 2008. 586(8): p. 2231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watkins AJ, et al. , Maternal periconceptional and gestational low protein diet affects mouse offspring growth, cardiovascular and adipose phenotype at 1 year of age. PLoS One, 2011. 6(12): p. e28745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwong WY, et al. , Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development, 2000. 127(19): p. 4195–202. [DOI] [PubMed] [Google Scholar]
- 37.Valenzuela-Alcaraz B, et al. , Assisted reproductive technologies are associated with cardiovascular remodeling in utero that persists postnatally. Circulation, 2013. 128(13): p. 1442–50. [DOI] [PubMed] [Google Scholar]
- 38.Scherrer U, et al. , Systemic and pulmonary vascular dysfunction in children conceived by assisted reproductive technologies. Circulation, 2012. 125(15): p. 1890–6. [DOI] [PubMed] [Google Scholar]
- 39.Eskild A, Monkerud L, and Tanbo T, Birthweight and placental weight; do changes in culture media used for IVF matter? Comparisons with spontaneous pregnancies in the corresponding time periods. Hum Reprod, 2013. 28(12): p. 3207–14. [DOI] [PubMed] [Google Scholar]
- 40.Kleijkers SH, et al. , IVF culture medium affects post-natal weight in humans during the first 2 years of life. Hum Reprod, 2014. 29(4): p. 661–9. [DOI] [PubMed] [Google Scholar]
- 41.Padhee M, et al. , The Periconceptional Environment and Cardiovascular Disease: Does In Vitro Embryo Culture and Transfer Influence Cardiovascular Development and Health? Nutrients, 2015. 7(3): p. 1378–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thornburg KL, O’Tierney PF, and Louey S, Review: The placenta is a programming agent for cardiovascular disease. Placenta, 2010. 31 Suppl: p. S54–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Godfrey KM, The role of the placenta in fetal programming-a review. Placenta, 2002. 23 Suppl A: p. S20–7. [DOI] [PubMed] [Google Scholar]
- 44.Barker DJ, et al. , Fetal and placental size and risk of hypertension in adult life. BMJ, 1990. 301(6746): p. 259–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barker DJ, et al. , The surface area of the placenta and hypertension in the offspring in later life. Int J Dev Biol, 2010. 54(2–3): p. 525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Abeelen AF, et al. , The sex-specific effects of famine on the association between placental size and later hypertension. Placenta, 2011. 32(9): p. 694–8. [DOI] [PubMed] [Google Scholar]
- 47.Roseboom TJ, et al. , Effects of famine on placental size and efficiency. Placenta, 2011. 32(5): p. 395–9. [DOI] [PubMed] [Google Scholar]
- 48.Alwasel SH, et al. , Changes in placental size during Ramadan. Placenta, 2010. 31(7): p. 607–10. [DOI] [PubMed] [Google Scholar]
- 49.Alwasel SH, et al. , The breadth of the placental surface but not the length is associated with body size at birth. Placenta, 2012. 33(8): p. 619–22. [DOI] [PubMed] [Google Scholar]
- 50.Alwasel SH, et al. , Intergenerational effects of in utero exposure to Ramadan in Tunisia. Am J Hum Biol, 2013. 25(3): p. 341–3. [DOI] [PubMed] [Google Scholar]
- 51.Alwasel SH, et al. , The velocity of fetal growth is associated with the breadth of the placental surface, but not with the length. Am J Hum Biol, 2013. 25(4): p. 534–7. [DOI] [PubMed] [Google Scholar]
- 52.Alwasel SH, et al. , Sex differences in regional specialisation across the placental surface. Placenta, 2014. 35(6): p. 365–9. [DOI] [PubMed] [Google Scholar]
- 53.Winder NR, et al. , Mother’s lifetime nutrition and the size, shape and efficiency of the placenta. Placenta, 2011. 32(11): p. 806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersen TA, Troelsen Kde L, and Larsen LA, Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci, 2014. 71(8): p. 1327–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richards AA and Garg V, Genetics of congenital heart disease. Curr Cardiol Rev, 2010. 6(2): p. 91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolf M and Basson CT, The molecular genetics of congenital heart disease: a review of recent developments. Curr Opin Cardiol, 2010. 25(3): p. 192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ransom J and Srivastava D, The genetics of cardiac birth defects. Semin Cell Dev Biol, 2007. 18(1): p. 132–9. [DOI] [PubMed] [Google Scholar]
- 58.Liu A, et al. , Biomechanics of the chick embryonic heart outflow tract at HH18 using 4D optical coherence tomography imaging and computational modeling. PLoS One, 2012. 7(7): p. e40869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Midgett M and Rugonyi S, Congenital heart malformations induced by hemodynamic altering surgical interventions. Front Physiol, 2014. 5: p. 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hogers B, et al. , Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ Res, 1997. 80(4): p. 473–81. [DOI] [PubMed] [Google Scholar]
- 61.Shaut CA, et al. , HOXA13 Is essential for placental vascular patterning and labyrinth endothelial specification. PLoS Genet, 2008. 4(5): p. e1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maylie JG, Excitation-contraction coupling in neonatal and adult myocardium of cat. Am J Physiol, 1982. 242(5): p. H834–43. [DOI] [PubMed] [Google Scholar]
- 63.Stacy V, et al. , The influence of naturally occurring differences in birthweight on ventricular cardiomyocyte number in sheep. Anat Rec (Hoboken), 2009. 292(1): p. 29–37. [DOI] [PubMed] [Google Scholar]
- 64.Morrison JL, et al. , Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol, 2007. 293(1): p. R306–13. [DOI] [PubMed] [Google Scholar]
- 65.Bubb KJ, et al. , Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J Physiol, 2007. 578(Pt 3): p. 871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Louey S, et al. , Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol, 2007. 580(Pt. 2): p. 639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jonker SS, et al. , Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J Appl Physiol (1985), 2007. 102(3): p. 1130–42. [DOI] [PubMed] [Google Scholar]
- 68.Pinson CW, Morton MJ, and Thornburg KL, Mild pressure loading alters right ventricular function in fetal sheep. Circ Res, 1991. 68(4): p. 947–57. [DOI] [PubMed] [Google Scholar]
- 69.Barbera A, et al. , Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol, 2000. 279(4): p. R1157–64. [DOI] [PubMed] [Google Scholar]
- 70.Jonker SS, et al. , Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am J Physiol Regul Integr Comp Physiol, 2007. 292(2): p. R913–9. [DOI] [PubMed] [Google Scholar]
- 71.O’Tierney PF, et al. , Reduced systolic pressure load decreases cell-cycle activity in the fetal sheep heart. Am J Physiol Regul Integr Comp Physiol, 2010. 299(2): p. R573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fisher DJ, Heymann MA, and Rudolph AM, Myocardial oxygen and carbohydrate consumption in fetal lambs in utero and in adult sheep. Am J Physiol, 1980. 238(3): p. H399–405. [DOI] [PubMed] [Google Scholar]
- 73.Davis L, et al. , Augmentation of coronary conductance in adult sheep made anaemic during fetal life. J Physiol, 2003. 547(Pt 1): p. 53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davis L, Thornburg KL, and Giraud GD, The effects of anaemia as a programming agent in the fetal heart. J Physiol, 2005. 565(Pt 1): p. 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Q, et al. , Effect of fetal anaemia on myocardial ischaemia-reperfusion injury and coronary vasoreactivity in adult sheep. Acta Physiol (Oxf), 2008. 194(4): p. 325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li G, et al. , Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Investig, 2003. 10(5): p. 265–74. [DOI] [PubMed] [Google Scholar]
- 77.Giraud GD, et al. , Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology, 2006. 147(8): p. 3643–9. [DOI] [PubMed] [Google Scholar]
- 78.Sundgren NC, et al. , Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am J Physiol Regul Integr Comp Physiol, 2003. 285(6): p. R1481–9. [DOI] [PubMed] [Google Scholar]
- 79.Sundgren NC, et al. , Angiotensin II stimulates hyperplasia but not hypertrophy in immature ovine cardiomyocytes. J Physiol, 2003. 548(Pt 3): p. 881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chattergoon NN, et al. , Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J, 2012. 26(1): p. 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chattergoon NN, Giraud GD, and Thornburg KL, Thyroid hormone inhibits proliferation of fetal cardiac myocytes in vitro. J Endocrinol, 2007. 192(2): p. R1–8. [DOI] [PubMed] [Google Scholar]
- 82.O’Tierney PF, et al. , Atrial natriuretic peptide inhibits angiotensin II-stimulated proliferation in fetal cardiomyocytes. J Physiol, 2010. 588(Pt 15): p. 2879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baschat AA, Fetal responses to placental insufficiency: an update. BJOG, 2004. 111(10): p. 1031–41. [DOI] [PubMed] [Google Scholar]
- 84.Forhead AJ, et al. , Developmental control of iodothyronine deiodinases by cortisol in the ovine fetus and placenta near term. Endocrinology, 2006. 147(12): p. 5988–94. [DOI] [PubMed] [Google Scholar]
- 85.Patterson AJ, et al. , Chronic prenatal hypoxia induces epigenetic programming of PKC{epsilon} gene repression in rat hearts. Circ Res, 2010. 107(3): p. 365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen M, Xiong F, and Zhang L, Promoter methylation of Egr-1 site contributes to fetal hypoxia-mediated PKCepsilon gene repression in the developing heart. Am J Physiol Regul Integr Comp Physiol, 2013. 304(9): p. R683–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eulalio A, et al. , Functional screening identifies miRNAs inducing cardiac regeneration. Nature, 2012. 492(7429): p. 376–81. [DOI] [PubMed] [Google Scholar]
- 88.Hata A, Functions of microRNAs in cardiovascular biology and disease. Annu Rev Physiol, 2013. 75: p. 69–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tingare A, Thienpont B, and Roderick HL, Epigenetics in the heart: the role of histone modifications in cardiac remodelling. Biochem Soc Trans, 2013. 41(3): p. 789–96. [DOI] [PubMed] [Google Scholar]
- 90.Messer L, Developmental Programming: Priming Disease Susceptibility for Subsequent Generations 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Woods LL, Weeks DA, and Rasch R, Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int, 2004. 65(4): p. 1339–48. [DOI] [PubMed] [Google Scholar]
- 92.Luyckx VA and Brenner BM, Birth weight, malnutrition and kidney-associated outcomes-a global concern. Nat Rev Nephrol, 2015. 11(3): p. 135–149. [DOI] [PubMed] [Google Scholar]
- 93.Jiang B, et al. , Birth weight and cardiac structure in children. Pediatrics, 2006. 117(2): p. e257–61. [DOI] [PubMed] [Google Scholar]
- 94.Sominsky L, et al. , Functional programming of the autonomic nervous system by early life immune exposure: implications for anxiety. PLoS One, 2013. 8(3): p. e57700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ward AM and Phillips DI, Fetal programming of stress responses. Stress, 2001. 4(4): p. 263–71. [DOI] [PubMed] [Google Scholar]
- 96.Leeson CP, et al. , Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation, 2001. 103(9): p. 1264–8. [DOI] [PubMed] [Google Scholar]
- 97.Barker DJ, et al. , Relation of fetal and infant growth to plasma fibrinogen and factor VII concentrations in adult life. BMJ, 1992. 304(6820): p. 148–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Crispi F, et al. , Fetal growth restriction results in remodeled and less efficient hearts in children. Circulation, 2010. 121(22): p. 2427–36. [DOI] [PubMed] [Google Scholar]
- 99.Dodson RB, et al. , Increased arterial stiffness and extracellular matrix reorganization in intrauterine growth-restricted fetal sheep. Pediatr Res, 2013. 73(2): p. 147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martyn CN and Greenwald SE, A hypothesis about a mechanism for the programming of blood pressure and vascular disease in early life. Clin Exp Pharmacol Physiol, 2001. 28(11): p. 948–51. [DOI] [PubMed] [Google Scholar]
- 101.Barker DJ, et al. , Growth in utero and serum cholesterol concentrations in adult life. BMJ, 1993. 307(6918): p. 1524–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Napoli C, et al. , Maternal hypercholesterolemia during pregnancy promotes early atherogenesis in LDL receptor-deficient mice and alters aortic gene expression determined by microarray. Circulation, 2002. 105(11): p. 1360–7. [DOI] [PubMed] [Google Scholar]
- 103.Moisiadis VG and Matthews SG, Glucocorticoids and fetal programming part 2: Mechanisms. Nat Rev Endocrinol, 2014. 10(7): p. 403–11. [DOI] [PubMed] [Google Scholar]
- 104.Giussani DA, et al. , Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS One, 2012. 7(2): p. e31017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Giussani DA, et al. , Heart disease link to fetal hypoxia and oxidative stress. Adv Exp Med Biol, 2014. 814: p. 77–87. [DOI] [PubMed] [Google Scholar]
- 106.Chattergoon NN, et al. , Unexpected maturation of PI3K and MAPK-ERK signaling in fetal ovine cardiomyocytes. Am J Physiol Heart Circ Physiol, 2014. 307(8): p. H1216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]