

Porcine reproductive and respiratory syndrome virus (PRRSV) is a major economic concern worldwide. There are currently large data sets available about the ORF5 gene of the virus, with thousands of sequences available, but little data are currently available on the full-length genome of PRRSV. We hypothesized that whole-genome sequencing (WGS) of the PRRSV genome would allow better epidemiological monitoring than ORF5 gene sequencing. PRRSV PCR-positive serum, oral fluid, and tissue clinical samples submitted to the diagnostic laboratory for routine surveillance or diagnosis of PRRSV infection in Québec, Canada, swine herds were used.
KEYWORDS: animal viral disease, swine virus, porcine reproductive and respiratory syndrome virus, PRRSV, next-generation sequencing, NGS, whole-genome sequencing, WGS, recombinant, coinfection, classification
ABSTRACT
Porcine reproductive and respiratory syndrome virus (PRRSV) is a major economic concern worldwide. There are currently large data sets available about the ORF5 gene of the virus, with thousands of sequences available, but little data are currently available on the full-length genome of PRRSV. We hypothesized that whole-genome sequencing (WGS) of the PRRSV genome would allow better epidemiological monitoring than ORF5 gene sequencing. PRRSV PCR-positive serum, oral fluid, and tissue clinical samples submitted to the diagnostic laboratory for routine surveillance or diagnosis of PRRSV infection in Québec, Canada, swine herds were used. The PRRSV reverse transcription-quantitative PCR Cq values of the processed samples varied between 11.5 and 34.34. PRRSV strain genomes were isolated using a poly (A)-tail method and were sequenced with a MiSeq Illumina sequencer. Ninety-two full-length PRRSV genomes were obtained from 88 clinical samples out of 132 tested samples, resulting in a PRRSV WGS success rate of 66.67%. Three important deletions in ORF1a were found in most wild-type (i.e., not vaccine-like) strains. The importance of these deletions remains undetermined. Two different full-length PRRSV genomes were found in four different samples (three serum samples and one pool of tissues), suggesting a 4.55% PRRSV strain coinfection prevalence in swine. Moreover, six PRRSV whole genomes (6.52% of PRRSV strains) were found to cluster differently than they did under the ORF5 classification method. Overall, WGS of PRRSV enables better strain classification and/or interpretation of results in 9.10% of clinical samples than ORF5 sequencing, as well as allowing interesting research avenues.
INTRODUCTION
Porcine reproductive and respiratory syndrome (PRRS) is a major economic concern worldwide, costing $663 million yearly to the American swine industry (1) and over $150 million yearly to the Canadian swine industry (2). Part of this cost is due to reproductive troubles in sows (characterized by late abortion, increased incidence of stillbirth, mummified fetuses, and weaker newborn piglets) (3–6) and retarded growth in piglets, mainly caused by respiratory problems (characterized by interstitial pneumonia) (5–10) and increased animal susceptibility to other pathogens (11–13). Besides this direct loss in productivity, a lot of effort and resources are allocated to controlling and containing this infectious disease. Among those efforts, vaccination and epidemiological surveillance are the most prominent (14, 15).
The etiological agents of PRRS are one of two viruses of the Arteriviridae family and the Porartevirus genus that are aptly named porcine reproductive and respiratory syndrome virus (PRRSV) type 1 and type 2 (officially Betaarterivirus suid 1 and Betaarterivirus suid 2). Both PRRSV are enveloped viruses with a diameter of around 50 to 65 nm (16). They contain a single-stranded positive-sense RNA genome of around 15 kb in length, capped at the 5′ end and polyadenylated at the 3′ end, and contain at least 11 known open reading frames (ORF) (17, 18). The first two ORFs (ORF1a and ORF1b), which constitute about 75% of the viral genomes and contain at least three ribosome shifting sites, encode replicase polyproteins that are posttranslationally cleaved into at least 16 distinct nonstructural proteins (nsp) (18–20). The remaining 25% of the genomes code for at least 8 known structural proteins (GP2, GP3, GP4, GP5, M, N, E, and GP5a) (17, 18, 33). Both PRRSV species share a nucleotide identity of around 60% (21, 22).
Many genotypes of both PRRSV exist, and the virulence of each strain is highly variable (34–36). PRRSV strains found in Canadian swine herds belong to type 2 PRRSV (37–39). To our knowledge, indigenous PRRSV type 1 strains have never been reported in Canada but were found on one occasion in European imported piglets kept in quarantine (40). Nine distinct monophyletic lineages of PRRSV type 2 have been described worldwide (37, 38). These lineages are genetically distinct, with a nucleotide identity between lineages of under 89%. Of these 9 lineages, lineages 1, 2, 5, and 8 are the most prominent in Canada, with lineage 9 showing up sporadically. The vast majority of lineage 5 and 8 strains are thought to be vaccine related. These lineages have been determined using Bayesian phylogenies of the ORF5 gene encoding the membrane structural glycoprotein protein GP5 of the virion. Interestingly, recent studies have investigated the genomic diversity and relatedness of PRRSV Canadian strains, but all those reports used PRRSV ORF5 nucleotide sequences in their genomic analyses (38, 39). However, there is much less information and data available about the whole viral genome of PRRSV strains both in Canada and around the globe.
The interstrain genetic variability of PRRSV is very high (41). It is believed that this enables the virus to better evade the immune system and potentially diminish vaccine efficacy (42–44). In an ongoing effort in Québec, Canada, as many PRRSV ORF5 sequences as possible are added to an ever-growing database, with more than 4,695 ORF5 sequences to date. The goal of this initiative was to better understand the epidemiological links between various PRRSV strains versus outbreaks and determine the proximity of a given strain to commercially available vaccinal strains as a strategy to fight PRRS disease. This database is also used to find genetically similar strains, enabling veterinarians to better predict the severity and outcome of a current outbreak based on the severity and outcome of past outbreaks with ORF5 genetically similar strains. The Molecular Diagnostic Laboratory of the Diagnostic Service (Faculté de médecine vétérinaire of Université de Montréal) is the depository of that Québec ORF5 sequence data bank. The PRRSV ORF5 gene was selected for molecular epidemiological surveillance, because it was considered hypervariable and encodes the GP5 protein that acts as the main target of neutralizing antibodies (45–47).
However, ORF5 is only 603 nucleotides (nt) long, representing only 4% of the viral genome, and it has been shown that other genes are also hypervariable and that pathogenicity of PRRSV is determined by multiple genes (48, 49). The GP5 antigenicity property (i.e., its recognition by neutralizing antibodies) has contributed to the selection of ORF5 for PRRSV surveillance despite the fact that other viral proteins are involved in virion recognition by neutralizing antibodies (23). Given the high prevalence of PRRSV infections, concomitant infections with different strains are expected to occur quite frequently, as is known to happen already in other swine respiratory viruses (31). It was previously reported that PRRSV recombination occurs prominently during coinfection with two different strains (24, 50) . All of these facts suggest that in some clinical cases of PRRSV infections, PRRSV recombinant strains could be misclassified, affecting the interpretation of the data at hand and the subsequent intervention by veterinarian practitioners. Thus, we hypothesized that whole-genome sequencing of PRRSV strains from clinical samples would enable a better classification of PRRSV strains compared to the current surveillance method of ORF5 sequencing. Therefore, whole-genome sequencing of PRRSV strains could lead to more appropriate interventions by veterinarians and swine producers, in addition to improving our understanding of the pathogenicity and the epidemiology of this important swine pathogen.
MATERIALS AND METHODS
Swine samples.
Convenience swine samples that tested positive for PRRSV type 2 by a reverse transcription-quantitative PCR (RT-qPCR) diagnostic assay were selected for whole-genome sequencing (WGS) of the entire PRRSV viral genome. The samples were submitted to and tested by the Molecular Diagnostic Laboratory (MDL) of Faculté de médecine vétérinaire (FMV), Université de Montréal (UdeM), for the identification of PRRSV after an outbreak of the disease in swine herds and, to a lesser extent, to conduct surveillance of the virus in swine herds. The PRRSV RT-qPCR diagnostic assay, conducted by MDL (American Association of Veterinary Laboratory Diagnosticians, AAVLD, accreditation), was an in-house assay (protocol PON-MOL-029). The swine samples originated from different herds and types of production throughout the province of Québec, Canada. Those clinical samples were submitted between December 2015 and November 2018 (most of them were collected in 2017 [n = 62] and 2018 [n = 66]). A total of 132 samples were used and included, 70 serum samples (composed of 43 pooled serum samples), 2 oral fluid samples (OF), 32 lung samples, and 28 pools of tissues (PoT; mainly lungs with several other types of tissues, such as lymph nodes, spleen, liver, intestine, etc.), with PRRSV RT-qPCR diagnostic assay Cq values between 11.5 and 34.34. The PRRSV viral genome was considered complete if at least 98% of the coding sequence was obtained.
Genome extraction and purification.
One hundred milligrams of lung or PoT was ground using a Beadbeater apparatus (Mini-Beadbeater-96; BioSpec Products Inc., OK, USA) in phosphate-buffered saline (PBS) and then centrifuged at full speed for 1 min, and supernatant was used for viral extraction. Two hundred microliters of serum and OF was centrifuged for 5 min at 10,000 × g, and supernatant was used for viral extraction. Viral RNA was extracted using a quick-RNA viral kit (number R1035; Zymo Research, CA, USA) as described in the company's protocol. Thereafter, RNA was eluted using 50 μl of nuclease-free water (Corning, NY, USA). The total elution volume was used to isolate RNA with poly(A) tails with a magnetic bead purification method, the NEBNext poly(A) mRNA magnetic isolation module, as described by the company’s protocol, and poly(A) tail RNA was resuspended in 15 μl Tris buffer (number E7490; New England BioLabs, ON, Canada). First-strand cDNA then was synthesized using the nondirectional reaction step-up protocol of the NEBNext RNA first-strand synthesis module (number E7525) (New England BioLabs, ON, Canada), starting with 10 μl of isolated poly(A) tail RNA. Immediately after first-strand cDNA synthesis, the second DNA strand was synthesized using a NEBNext Ultra II nondirectional RNA second-strand synthesis module, as described by the manufacturer’s protocol, with a minor modification at the incubation step in the thermal cycler for 2 h (instead of 1.5 h) at 16°C (number E6111; New England BioLabs, ON, Canada). Double-stranded DNA (dsDNA) was then purified with AxyPrep Mag PCR clean-up kits (Axygen; Corning, NY, USA) using 1.8× beads and 70% ethanol. The purified dsDNA was diluted in 30 μl of 10 mM Tris-HCl at pH 8.0 and stored at –20°C for later use.
PRRSV WGS.
dsDNA was quantified using a Qubit dsDNA HS assay kit and a Qubit fluorometer (ThermoFisher Scientific, MA, USA). Sequencing libraries were prepared using a Nextera XT DNA library preparation kit (Illumina, CA, USA). Sequencing library construction was still performed even if the dsDNA quantification results were below the Qubit dsDNA HS assay kit threshold of detection (0.2 ng). Briefly, 0.2 to 0.3 ng of dsDNA was tagmented with 10 μl tagment DNA buffer (TD) and 5 μl Amplicon tagment mix (ATM) at 55°C for 5 min, using a Thermocycler TProfessional basic 96 (Biometra GmbH, Göttingen, Germany). The transposomes were inactivated with 5 μl of neutralizing tagment buffer (NT). Sequencing libraries were then amplified using an index adapter and Nextera PCR master mix (NMP) by following these PCR steps: 72°C for 3 min, 95°C for 30 s; 14 cycles of 95°C for 10 s, 55°C for 30 s, and 72°C for 30 s; a final elongation step at 72°C for 5 min; and a hold at 10°C until the next step. Sequencing libraries were then purified using AxyPrep Mag PCR clean-up kits (Axygen; Corning, NY, USA) as described by the Nextera XT protocol. Sequencing library quality was assessed using an Agilent high-sensitivity DNA kit with a Bioanalyzer (Agilent, CA, USA). Sequencing libraries were normalized using LNB1 beads (Nextera XT protocol) or the manual normalization protocol if the concentrations of the libraries were on the lower end of the Bioanalyzer curves. Sequencing libraries were sequenced in a v3 600-cycle cartridge (number MS-102-3003; Illumina) using an MiSeq Illumina benchtop sequencer (located at the Veterinary High-Throughput Sequencing Laboratory [VHTSL], at FMV), and PhiX was included at around 1% of the total sequencing libraries as a control to establish the sequencing run efficacy (Illumina, CA, USA).
Bioinformatic analyses.
At first, reads were trimmed for adaptors and quality by the MiSeq software during FastQ generation. Using CLC Genomic Workbench software (version 12.0.3; Qiagen, CA, USA), reads from a sample were assembled using the software’s built-in de novo assembly module, including another step of adaptor and quality trimming. Contigs obtained were checked against a database made using reference type 2 PRRSV genomes available online to identify the contigs that corresponded to full-length PRRSV sequences. These full-length genomes obtained by de novo assembly were then added to our PRRSV reference database. Second, reads from each sample were mapped against the same database. For each sample, the reference genome onto which the highest number of reads mapped and offered a uniform coverage was used as the reference genome for resequencing analysis using the software’s map-to-reference module. When more than one reference genome seemed to offer an appropriate depth and coverage, both were used separately for resequencing analysis, and the resulting sequences were compared to one another. Samples from which two PRRSV genomes could be assembled and possessed a nucleotide identity of <96% were considered to contain more than one PRRSV strain. The <96% nucleotide identity cutoff value was determined based on the sequencing error rate of <1% and the fact that we have ambiguity in less than 1% of the positions for each complete sequence. Thus, the worst-case scenario where both PRRSV coinfecting strains each could have a 1% error rate and 1% uncertainty could lead to identical strains with a nucleotide identity of ≥96%. Therefore, this nucleotide identity value was established as the cutoff value. PRRSV recombinant status was determined by comparing the location of a given strain’s individual ORFs on their respective phylogenetic trees. Bioinformatics and data analyses were performed using the CLC Genomic Workbench software (version 12.0.3; Qiagen, CA, USA). Phylogenetic trees were constructed using a maximum likelihood model with a type 1 PRRSV reference strain (Lelystad virus) used as an outgroup and with a bootstrap setting of 1,000. The trees were constructed with Geneious Prime software (Biomatter, version 2019).
Data availability.
All entire viral genomes of successfully sequenced PRRSV strains were submitted to GenBank with accession numbers MN865482 to MN865573 (see Table S1 in the supplemental material).
RESULTS
PRRSV WGS were obtained from a total of 88 swine clinical samples (an overall 66.67% PRRSV WGS success rate). In those clinical samples, the PRRSV WGS was successful in two OF, 13 PoT, 21 lung, and 52 serum samples. WGS made a difference over ORF5 sequencing in eight of those samples (Table 1). Ninety-two PRRSV entire genomes were obtained from those clinical samples (see Table S1 in the supplemental material). The additional 4 PRRSV genomes were the consequences of the identification of 4 PRRSV coinfections, samples in which two different PRRSV strains with a nucleotide identity below 96% were identified (listed in Table 1). Therefore, a total of 4.55% PRRSV coinfections were found among PRRSV WGS successful cases. The interstrain nucleotide identities of the 92 PRRSV entire viral genome sequences were found to vary between 79.57% and 99.94% (data not shown). At the viral gene level, the nsp2 and nsp7 nucleotide identities were found to be the least conserved between the 92 PRRSV Québec strains, with 66.04% and 64.38% nucleotide identities, respectively, indicating that ORF1a genetic diversity is a little higher than that of the other ORFs. It is worth noting that the overall genetic diversity was scattered throughout all the ORFs (Fig. 1 and Table 2). The highest nucleotide identities were found for the nsp4, E, N, and M gene-encoded proteins, with 92.41%, 86.94%, 86.13%, and 86.10%, respectively.
TABLE 1.
List of clinical samples containing more than one PRRSV strain (coinfection) and of strains that are classified differently by WGS than the ORF5 method
| Sample ID no. | Type of sample | Virus whole-genome sequencing characteristic | Effect on PRRSV classification |
|---|---|---|---|
| 1898026 | Serum | Coinfectiona | Both strains clustered in different lineages |
| 1952821 | Pool of tissues | Coinfection | Both strains clustered differently in the same lineage |
| 1968868 | Serum | Coinfection | Both strains clustered differently in the same lineage |
| 2153073 | Serum | Coinfection | Both strains clustered in different lineages |
| 1425619 | Lung | Recombinantb | Strain clustered differently in the same lineage, unlike ORF5 method |
| 1890826 #2c | Serum | Recombinant | Strain clustered in a different lineage, unlike ORF5 method |
| 1927781 | Serum | Recombinant | Strain clustered differently in the same lineage, unlike ORF5 method |
| 1952821 #1 | Pool of tissues | Recombinant | Strain clustered in a different lineage than that of ORF5 method |
| 2072533 | Serum | Recombinant | Strain clustered in a different lineage than that of ORF5 method |
| 2087409 | Lung | Recombinant | Strain clustered differently in the same lineage compared to ORF5 method |
At least two PRRSV different strains were identified in the same clinical sample and possessed a nucleotide identity of <96%.
PRRSV recombinant status was assigned when a strain clustered more than two nodes away in phylogenetic trees based on PRRSV whole-genome analysis compared to the ORF5 classification method.
The number after the sample identification (ID) number represents one of the two PRRSV strains that was identified within the same clinical sample.
FIG 1.
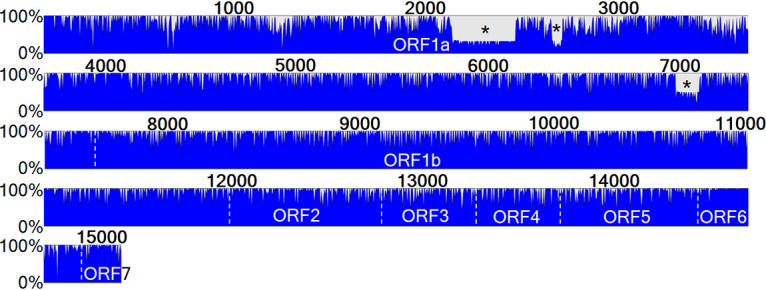
Nucleotide conservation status between Québec PRRSV strains. All 92 PRRSV sequenced strains are depicted here with the main ORFs shown. Three important and recurrent deletions in some of the strains are indicated with an asterisk. The scale to the left of the bar indicates the percentage of strains that shared identity at a given location, with a full blue bar signifying that all strains are identical at this position. The numbers on top of the blue bar indicate the nucleotide position.
TABLE 2.
Nucleotide conservation status of the main PRRSV-encoded proteinsa
| Corresponding protein | Lowest interstrain identity (%) | Corresponding protein | Lowest interstrain identity (%) |
|---|---|---|---|
| nsp1 | 76.15 | nsp11 | 84.01 |
| nsp2 | 66.04 | nsp12 | 82.47 |
| nsp3 | 80.09 | GP2 | 84.70 |
| nsp4 | 92.41 | E | 86.94 |
| nsp5 | 78.82 | GP3 | 82.35 |
| nsp6 | 77.08 | GP4 | 84.73 |
| nsp7 | 64.38 | GP5a | 82.05 |
| nsp8 | 82.61 | GP5 | 82.95 |
| nsp9 | 85.75 | M | 86.10 |
| nsp10 | 84.50 | N | 86.13 |
The highest nucleotide identities obtained between strains were ≥99.8% for all gene-encoded proteins. The lowest interstrain nucleotide identities between major PRRSV proteins are indicated.
Three deletions were identified by WGS in ORF1a of most wild-type strains (Fig. 1). The first one, located in the nsp2 coding region at nt 2159 to 2491, is 333 nt long. The second one, also in the nsp2 coding region, was found around nt 2717 to 2773 with a span of 57 nt. Both deletions occurred together and were found in 63 of the 92 PRRSV Québec sequenced strains. The third deletion was found in the nsp7 coding region, at positions 7106 to 7228, and was 123 nt long. The nucleotide positions of the deletions are given relative to the coding sequence of the MLV vaccine strain used as a reference. All strains that harbor the nsp7 deletion also have the nsp2 deletions, but some strains that do not possess the nsp7 deletion have the nsp2 deletions. Only 47 of the 92 sequenced strains have the nsp7 deletions.
Based on the ORF5 sequencing classification method, all 92 PRRSV strains were found to belong to lineages 1, 5, and 8 (Fig. 2B). Interestingly, 18 PRRSV strains were considered to be MLV vaccine-like strains (i.e., with nucleotide identities of >96%) if the tree was built using ORF5, when 17 strains were considered to be MLV vaccine-like strains when using the whole genome. It is worth noting that one discrepancy was found concerning MLV vaccine-like strain relatedness classification. In fact, PRRSV strain 2072533 was classified as an MLV vaccine-like strain based on the ORF5 method, whereas it was not genetically related to the MLV vaccine-like strain based on WGS results, with nucleotide identities of 98.51% and 84.01%, respectively (Fig. 2). Moreover, PRRSV strain 2072533 was identified to be a recombinant virus, which explains the discrepancy between the ORF5 and WGS classification methods (Table 1). No difference was found for the ATP vaccine-like and Fostera vaccine-like classification statuses (Fig. 2A compared to B). Consequently, misclassification by ORF5 sequencing of PRRSV vaccine-like strains was revealed by WGS in 3.23% of samples.
FIG 2.


PRRSV whole genome and ORF5 gene nucleotide phylogenetic tree comparison. (A and B) PRRSV WGS nucleotide phylogenetic tree (A) and PRRSV ORF5 nucleotide phylogenetic tree (B). The brackets indicate the PRRSV type 2 lineages. MLV, ATP, Fostera, PrimePac, and Prevacent correspond to commercially available live attenuated vaccines. Strains 97-7895, MN30100, VR2385, and MN184 are American reference strains. Strain PA8 is the first Canadian PRRSV whole-genome-sequenced strain, and strain IAF-Klop is a Québec reference strain. The branches for the Lelystad virus strain (type 1 PRRSV reference strain, which was used as an outgroup) should be three times longer but were shortened to fit the graph. PRRSV coinfecting strains, i.e., two different strains that were identified in the same clinical samples, are indicated with a number 1 or number 2. Colored arrows identify potential PRRSV recombinant strains that are clustering differently when WGS and ORF5 phylogenetic trees are compared. The scale bars represent a 5% difference in nucleotide identity.
As illustrated in Fig. 2, the PRRSV whole-genome sequence phylogenetic tree has allowed us to construct a more robust classification, based on the bootstrap values, compared to the ORF5 sequencing method. More interestingly, six PRRSV strains (identified by colored arrows in Fig. 2) were found to cluster differently within the WGS phylogenetic tree compared to the ORF5 phylogenetic tree. A summary of those classification differences is presented in Table 1. One of the reasons that could explain this difference is recombination events, at some point in time, between two PRRSV strains during coinfection in swine. As an example of this phenomenon, the 2072533 PRRSV strain nucleotide identity with a presumed parental PRRSV strain (the MLV vaccine strain) is shown in Fig. 3, in which the recombination position was identified around nt 12100. The nucleotide identity between these two PRRSV strains for the first 12,100 nt of the 5′ end (i.e., encompassing ORF1a, ORF1b, ORF2a, and ORF2) is 80.46%, whereas it is 98.63% (data not shown) for the 3′ end region of the genome (the last 3,000 nt of the genome). For strain 1952821 number 1, the first half of the genome (mainly ORF1a) shares 85.53% nucleotide identity with the MLV vaccine strain, whereas the second half (mostly ORF1b to ORF7) shares 96.55% nucleotide identity with the MLV vaccine strain (data not shown), suggesting again a recombination between a wild-type and vaccine-like strain. For the other possible recombinant strains, the specific recombination point could not be determined, either because the parental strains are unknown or because those strains may have undergone several recombination events over time. Overall, 6.52% possible recombinant viruses out of a total of 92 PRRSV strains were found among PRRSV WGS successful cases. It is worth noting that 2 of the 6 possible recombinant viruses were found in PRRSV coinfected samples. However, this is to be expected, given that coinfection is a prerequisite for recombination events to occur, and taking into account that more than one-third of produced virions during coinfections can be PRRSV recombinant viruses (24).
FIG 3.
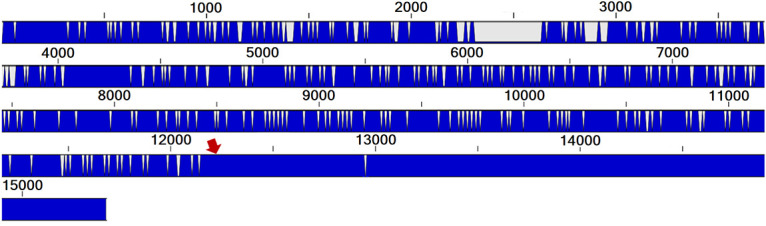
Comparison of the PRRSV recombinant strain 2072533 with one of its possible parental strains, the MLV vaccine strain. The red arrow points to the region of the genome where the recombination probably occurred. A full blue line indicates that both strains are identical in that given nucleotide position, and gray/white indicates that they differ at that position. The numbers on top of the blue bar indicate the nucleotide positions.
In four of the 88 successfully sequenced PRRSV-positive samples, two different PRRSV strains were found (Table 1), representing 4.55% coinfections among PRRSV WGS successful cases, as indicated above. The nucleotide identities of these four pairs of PRRSV were 88.8%, 92.36%, 91.57%, and 81.83% for samples 1890826, 1952821, 1968868, and 2153073, respectively (data not shown). A graph of the nucleotide conservation status of the PRRSV strains in sample 2153073 is shown in Fig. 4 as an example. Interestingly, the 2153073 number 2 strain possesses the nsp2 and nsp7 deletions, like the Québec wild-type strains, whereas its coinfecting strain, 2153073 number 1, is more related to ATP vaccine-like strains (sharing 99.02% nucleotide identity). All coinfections were identified from an individual animal (i.e., sample coming from a single animal and not from a pool of different animals). Interestingly, coinfections were identified in three serum samples (5.77% of WGS successful sera) and one PoT sample (7.14% of WGS successful PoT) (Table 1 and Table S1).
FIG 4.
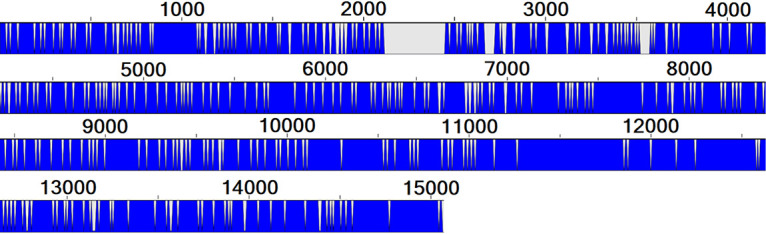
Nucleotide conservation status of two PRRSV coinfecting strains found in sample 2153073. Nucleotide identity between the two strains was found in sample 2153073. A full blue line indicates that both strains are identical in that given nucleotide position, and gray/white indicates that they differ at that position. The nucleotide identity between both PRRSV strains is 81.83%. The numbers on top of the blue bar indicate the nucleotide positions.
DISCUSSION
The first observation that can be easily made from our type 2 PRRSV complete genome analyses is that many of the PRRSV strains currently circulating in Québec appear to have three relatively large deletions (Fig. 1). The first two of these occur in the region coding for the nsp2 protein, and the third one is in the region coding for the nsp7 protein. While the exact implication of these deletions is unknown, several investigations have been conducted on type 2 PRRSV strains possessing various nsp2 deletions, especially with Asian PRRSV strains. The nsp2 protein is known to play a role in both immune system modulation and pathogenicity (29, 51). For example, it is believed that some amino acids in the nsp2 protein play a role in evading the ubiquitin-ISG15 system of the innate immune response (52, 53). There are also many reports of highly pathogenic or epidemic strains recognizable by nsp2 deletions (28, 30). The importance of nsp2 in the pathogenicity of PRRSV is what makes these deletions so interesting and why they warrant further investigations. It is worth noting that these specific nsp2 deletions can also be found in some PRRSV strains that were reported in the United States (i.e., GenBank accession numbers KT258000 and EF532803), meaning that they are not markers of a specific North American country but are probably specific to lineage 1 or at least to a subset of lineage 1 PRRSV strains. The involvement of the third deletion, in the region coding for the nsp7 protein into the pathogenicity of PRRSV, is much more elusive. PRRSV pathogenicity studies involving nsp7 deletions and mutation experiments are scarce and usually involve pinpoint mutations or very short deletions (25). However, at least one study has reported that nucleotide mutations at some nsp7-specific site could lead to changes in PRRSV replication efficiency in vitro. It is worth noting that to our knowledge, this specific nsp7 deletion does not seem to have been previously reported by other investigators. Therefore, this deletion certainly requires further investigation.
Overall, the whole-genome diversity of PRRSV type 2 strains in Québec is quite high, as expected based on ORF5 diversity, with up to 20.43% difference in nucleotide identities between strains. This diversity is higher in regions coding for proteins considered more or less accessory and lower for those considered essential, like the polymerase nsp9 or the helicase nsp10, as well as the structural proteins (54) (Table 2). Interestingly, the most conserved nucleotide encoded by the gene was found to be the serine protease nsp4 (Table 2), which is the main PRRSV protease responsible for cleaving most of the replicase polyprotein into its smaller functional parts (26).
Type 2 PRRSV WGS classification stays similar for most PRRSV strains compared to the ORF5 classification method, which includes 9 lineages, although few PRRSV strains appeared to be of an uncertain lineage when looking at the whole-genome sequences. This could be because WGS may have improved the depth of the genomic analyses compared to the ORF5 method (i.e., 15,000-nt-based analyses compared to 603-nt-based analyses, respectively). Two examples of this are the strains 2108698 and 2137004. While both clustered inside lineage 8 based on the ORF5 phylogenetic tree, they could just as easily be grouped with the reference strain MN30100 of lineage 9 based on whole-genome sequencing. Another example of this phenomenon is the reference strain IAF-klop. We know it belongs to lineage 1, but the whole genome seems to cluster more closely to lineages 5 to 9. Another possible explanation for this is that this specific strain has been passaged repeatedly in vitro in the MARC-145 cell line (55). Lastly, based on whole-genome sequencing, both strains from the 1890896 sample seem to belong to an uncertain lineage. The limited availability of PRRSV whole-genome sequence data and the relatively frequent occurrence of viral genome recombination events raise the need to come up with a genomic classification system that would take into account the higher variability of some nonstructural proteins and the incidence of viral genome recombination events. Such recombination events pose a real challenge to the current monophyletic lineage classification system.
A more adequate classification of the strains could be made using both the nsp2 and ORF5 gene sequences in a manner akin to influenza subtyping (27). The nsp2 coding region seems to routinely contain deletions that can serve as markers of the sublineage as well as being implicated in pathogenicity, making it a good candidate for typing of different strains (28–30). WGS remains, however, the method of choice compared to nsp2/ORF5 sequencing for a few reasons. First, the nsp2 coding region is more than 3,000 nt long and highly variable, making its amplification by PCR for Sanger sequencing quite difficult. Second, while adding a second gene from the 5′ end of the genome would greatly improve odds of catching recombinants, simply doing a second Sanger sequencing instead of whole-genome sequencing would not detect coinfections in swine samples.
The 4.55% occurrence of PRRSV coinfections among PRRSV WGS-positive cases was in line with what has been previously reported for swine influenza virus (SIV). In fact, more than one subtype (such as H1N1 and H3N2) of SIV is found in at least 2% of the tested samples, not accounting at all for coinfections with strains from the same subtype (31).
The six possible recombinant PRRSV strains (a prevalence of 6.52%) that cluster differently, based on WGS analyses, are also of high importance. In the field, veterinarian practitioners and swine producers are mostly interested in knowing if a strain within a PRRSV-positive sample is genetically related to a PRRSV vaccine-like or wild-type strain and if it is related to previous infections on that farm or to other known outbreaks. A PRRSV strain changing its clustering by WGS due to a possible recombination event can have an important effect on the clinical interpretation, as it can change its relatedness to a known high- or low-pathogenicity strain.
From a clinical point of view, the 2072533 strain would have been identified by veterinarian practitioners as an MLV vaccine-like strain based on the ORF5 gene method, whereas, in fact, it is not an MLV vaccine-like strain based on WGS. Overall, PRRSV WGS has changed the vaccine-like strain status of one PRRSV strain, i.e., 3.23% of the vaccine-like related cases (data not shown).
While these analyses have been conducted on Québec PRRSV strains, there is nothing to suggest that these findings could not extend to most, if not all, regions currently affected by PRRSV type 2 outbreaks, as both intra- and interlineage recombination events have been described and as coinfections can also occur with type 2 PRRSV strains from the same or different lineages. These types of recombination events are known to have occurred elsewhere (56, 57).
The main limit of our current study is that since Canada is exempt from type 1 PRRSV, we cannot say if these findings also extend to type 1 PRRSV strains. While type 2 PRRSV is the most frequent PRRSV found in both China and the United States, type 1 is also known to occur and the latest data are currently limited (32). It is also currently unknown if recombination between both viral species causing PRRS is possible in a clinical setting. Both species now becoming endemic to regions where the other one used to predominate could be something to investigate in the future. It should be noted, however, that the lack of type 1 PRRSV strains does not seem to have hindered the Québec diversity of type 2 PRRSV strains.
To our knowledge, this is the first time that a study has investigated the entire viral genome of several Canadian PRRSV type 2 strains. In addition to allowing the description of 3 substantial deletions in the ORF1a region of many contemporary lineage 1 strains, WGS appears to enable a better interpretation of clinical cases in about 9.10% of PRRSV cases due to either PRRSV coinfecting strains or the presence of PRRSV recombinant strains. Again, to our knowledge, this is the first time that both the proportion of misclassifications and the incidence of coinfections of type 2 PRRSV strains using whole-genome sequencing data have been quantified. In those cases, this additional information (that more than 1 strain was present in a sample or that the strain is not related to the same strains as those we would have thought with the ORF5 sequencing method) could lead directly to more appropriate preventive and palliative measures being applied by the veterinarians and swine producers, which could have a positive impact on the overall health of swine herds around the world.
Supplementary Material
ACKNOWLEDGMENTS
This research was financially supported by the Ministère de l’Agriculture, des Pêcheries et de l’Alimentation du Québec (MAPAQ) Innov’Action program. Carl A. Gagnon was financially supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) discovery grant and a Canadian Swine Research and Development Cluster (CSRDC) grant. Christian Lalonde was a recipient of a scholarship from the CRIPA, a research network financially supported by the Fonds de recherche du Québec–Nature et technologies (FRQNT).
We have no potential conflicts of interest regarding the research, authorship, and/or publication of this article to declare.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Holtkamp DJ, Kliebenstein JB, Neumann E, Zimmerman JJ, Rotto H, Yoder TK, Wang C, Yeske P, Mowrer CL, Haley CA. 2013. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod 21:72. doi: 10.2460/javma.2005.227.385. [DOI] [Google Scholar]
- 2.Mussel A. 2011. A risk, benefit, strength, weakness, opportunity and threat analysis for the control and possible eradication of porcine reproductive and respiratory syndrome (PRRS) virus within the Canadian swine herd. Canadian Swine Health Board, Ottowa, Canada. [Google Scholar]
- 3.Christianson WT. 1992. Stillbirths, mummies, abortions, and early embryonic death. Vet Clin North Am Food Anim Pract 8:623–639. doi: 10.1016/s0749-0720(15)30708-8. [DOI] [PubMed] [Google Scholar]
- 4.Pol JM, van Dijk JE, Wensvoort G, Terpstra C. 1991. Pathological, uhrastructural, and changes caused by Lelystad virus in experimentally induced infections of mystery swine disease (synonym porcine epidemic abortion and respiratory syndrome (PEARS)). Vet Q 13:137–143. doi: 10.1080/01652176.1991.9694298. [DOI] [PubMed] [Google Scholar]
- 5.Loula T. 1991. Mystery pig disease. Agripractice 12:23–34. [Google Scholar]
- 6.Bilodeau R, Dea S, Sauvageau R, Martineau G. 1991. Porcine reproductive and respiratory syndrome in Quebec. Vet Rec 129:102–103. doi: 10.1136/vr.129.5.102. [DOI] [PubMed] [Google Scholar]
- 7.Albina E. 1997. Porcine reproductive and respiratory syndrome: ten years of experience (1986-1996) with this undesirable viral infection. Vet Res 28:305–352. [PubMed] [Google Scholar]
- 8.Collins JE, Benfield DA, Christianson WT, Harris L, Hennings JC, Shaw DP, Goyal SM, McCullough S, Morrison RB, Joo HS. 1992. Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J Vet Diagn Investig 4:117–126. doi: 10.1177/104063879200400201. [DOI] [PubMed] [Google Scholar]
- 9.Halbur P, Paul P, Frey M, Landgraf J, Eernisse K, Meng X-J, Andrews J, Lum M, Rathje J. 1996. Comparison of the antigen distribution of two US porcine reproductive and respiratory syndrome virus isolates with that of the Lelystad virus. Vet Pathol 33:159–170. doi: 10.1177/030098589603300205. [DOI] [PubMed] [Google Scholar]
- 10.Rossow KD, Morrison RB, Goyal SM, Singh GS, Collins JE. 1994. Lymph node lesions in neonatal pigs congenitally exposed to porcine reproductive and respiratory syndrome virus. J Vet Diagn Investig 6:368–371. doi: 10.1177/104063879400600316. [DOI] [PubMed] [Google Scholar]
- 11.Harms P, Sorden S, Halbur P, Bolin S, Lager K, Morozov I, Paul P. 2001. Experimental reproduction of severe disease in CD/CD pigs coinfected with PRRSV and type 2 porcine circovirus. Vet Pathol 38:528–539. doi: 10.1354/vp.38-5-528. [DOI] [PubMed] [Google Scholar]
- 12.Galina L, Pijoan C, Sitjar M, Christianson W, Rossow K, Collins J. 1994. Interaction between Streptococcus suis serotype 2 and porcine reproductive and respiratory syndrome virus in specific pathogen-free piglets. Vet Rec 134:60–64. doi: 10.1136/vr.134.3.60. [DOI] [PubMed] [Google Scholar]
- 13.Cho JG, Dee SA, Deen J, Trincado C, Fano E, Jiang Y, Faaberg K, Murtaugh MP, Guedes A, Collins JE, Joo HS. 2006. The impact of animal age, bacterial coinfection, and isolate pathogenicity on the shedding of porcine reproductive and respiratory syndrome virus in aerosols from experimentally infected pigs. Can J Vet Res 70:297–301. [PMC free article] [PubMed] [Google Scholar]
- 14.Linhares DC, Johnson C, Morrison RB. 2015. Economic analysis of vaccination strategies for PRRS control. PLoS One 10:e0144265. doi: 10.1371/journal.pone.0144265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nathues H, Alarcon P, Rushton J, Jolie R, Fiebig K, Jimenez M, Geurts V, Nathues C. 2017. Cost of porcine reproductive and respiratory syndrome virus at individual farm level–an economic disease model. Prev Vet Med 142:16–29. doi: 10.1016/j.prevetmed.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Spilman MS, Welbon C, Nelson E, Dokland T. 2009. Cryo-electron tomography of porcine reproductive and respiratory syndrome virus: organization of the nucleocapsid. J Gen Virol 90:527–535. doi: 10.1099/vir.0.007674-0. [DOI] [PubMed] [Google Scholar]
- 17.Johnson CR, Griggs TF, Gnanandarajah J, Murtaugh MP. 2011. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J Gen Virol 92:1107–1116. doi: 10.1099/vir.0.030213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meulenberg JJ. 2000. PRRSV, the virus. Vet Res 31:11–21. doi: 10.1051/vetres:2000103. [DOI] [PubMed] [Google Scholar]
- 19.Cao QM, Subramaniam S, Ni Y-Y, Cao D, Meng X-J. 2016. The non-structural protein Nsp2TF of porcine reproductive and respiratory syndrome virus down-regulates the expression of swine leukocyte antigen class I. Virology 491:115–124. doi: 10.1016/j.virol.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Treffers EE, Napthine S, Tas A, Zhu L, Sun Z, Bell S, Mark BL, van Veelen PA, van Hemert MJ, Firth AE, Brierley I, Snijder EJ, Fang Y. 2014. Transactivation of programmed ribosomal frameshifting by a viral protein. Proc Natl Acad Sci U S A 111:E2172–E2181. doi: 10.1073/pnas.1321930111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelsen CJ, Murtaugh MP, Faaberg KS. 1999. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol 73:270–280. doi: 10.1128/JVI.73.1.270-280.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murtaugh MP, Elam M, Kakach L. 1995. Comparison of the structural protein coding sequences of the VR-2332 and Lelystad virus strains of the PRRS virus. Arch Virol 140:1451–1460. doi: 10.1007/BF01322671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Z, Collin E, Peddireddi L, Clement T, Gauger P, Hause BM. 2017. Genetic diversity in envelope genes of contemporary US porcine reproductive and respiratory syndrome virus strains influences viral antigenicity. Res Vet Sci 115:432–441. doi: 10.1016/j.rvsc.2017.07.027. [DOI] [PubMed] [Google Scholar]
- 24.Liu D, Zhou R, Zhang J, Zhou L, Jiang Q, Guo X, Ge X, Yang H. 2011. Recombination analyses between two strains of porcine reproductive and respiratory syndrome virus in vivo. Virus Res 155:473–486. doi: 10.1016/j.virusres.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M, Cao Z, Xie J, Zhu W, Zhou P, Gu H, Sun L, Su S, Zhang G. 2013. Mutagenesis analysis of porcine reproductive and respiratory syndrome virus nonstructural protein 7. Virus Genes 47:467–477. doi: 10.1007/s11262-013-0957-4. [DOI] [PubMed] [Google Scholar]
- 26.van Dinten LC, Rensen S, Gorbalenya AE, Snijder EJ. 1999. Proteolytic processing of the open reading frame 1b-encoded part of arterivirus replicase is mediated by nsp4 serine protease and is essential for virus replication. J Virol 73:2027–2037. doi: 10.1128/JVI.73.3.2027-2037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.World Health Organization. 1980. A revision of the system of nomenclature for influenza viruses: a WHO memorandum. Bull World Health Organization 58:585–591. [PMC free article] [PubMed] [Google Scholar]
- 28.Choi H-W, Nam E, Lee YJ, Noh Y-H, Lee S-C, Yoon I-J, Kim H-S, Kang S-Y, Choi Y-K, Lee C. 2014. Genomic analysis and pathogenic characteristics of type 2 porcine reproductive and respiratory syndrome virus nsp2 deletion strains isolated in Korea. Vet Microbiol 170:232–245. doi: 10.1016/j.vetmic.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Kim D-Y, Kaiser TJ, Horlen K, Keith ML, Taylor LP, Jolie R, Calvert JG, Rowland RR. 2009. Insertion and deletion in a non-essential region of the nonstructural protein 2 (nsp2) of porcine reproductive and respiratory syndrome (PRRS) virus: effects on virulence and immunogenicity. Virus Genes 38:118–128. doi: 10.1007/s11262-008-0303-4. [DOI] [PubMed] [Google Scholar]
- 30.Yu L-X, Wang X, Yu H, Jiang Y-F, Gao F, Tong W, Li L-W, Li H-C, Yang S, Chen P-F, Yang D-Q, Zhang W-C, Tong G-Z, Zhou Y-J. 2018. The emergence of a highly pathogenic porcine reproductive and respiratory syndrome virus with additional 120aa deletion in Nsp2 region in Jiangxi, China. Transbound Emerg Dis 65:1740–1748. doi: 10.1111/tbed.12947. [DOI] [PubMed] [Google Scholar]
- 31.Choi Y, Goyal S, Kang S, Farnham M, Joo H. 2002. Detection and subtyping of swine influenza H1N1, H1N2 and H3N2 viruses in clinical samples using two multiplex RT-PCR assays. J Virol Methods 102:53–59. doi: 10.1016/s0166-0934(01)00442-6. [DOI] [PubMed] [Google Scholar]
- 32.Wang A, Zhang J, Shen H, Zheng Y, Feng Q, Yim-Im W, Gauger PC, Harmon K, Zhu S, An T-Q, Li G. 2019. Genetic diversity of porcine reproductive and respiratory syndrome virus 1 in the United States of America from 2010 to 2018. Vet Microbiol 239:108486. doi: 10.1016/j.vetmic.2019.108486. [DOI] [PubMed] [Google Scholar]
- 33.Wu W-H, Fang Y, Rowland RR, Lawson SR, Christopher-Hennings J, Yoon K-J, Nelson EA. 2005. The 2b protein as a minor structural component of PRRSV. Virus Res 114:177–181. doi: 10.1016/j.virusres.2005.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allende R, Lewis T, Lu Z, Rock D, Kutish G, Ali A, Doster A, Osorio F. 1999. North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J Gen Virol 80:307–315. doi: 10.1099/0022-1317-80-2-307. [DOI] [PubMed] [Google Scholar]
- 35.Drew TW, Meulenberg JJ, Sands JJ, Paton DJ. 1995. Production, characterization and reactivity of monoclonal antibodies to porcine reproductive and respiratory syndrome virus. J Gen Virol 76:1361–1369. doi: 10.1099/0022-1317-76-6-1361. [DOI] [PubMed] [Google Scholar]
- 36.Mardassi H, Mounir S, Dea S. 1994. Identification of major differences in the nucleocapsid protein genes of a Quebec strain and European strains of porcine reproductive and respiratory syndrome virus. J Gen Virol 75:681–685. doi: 10.1099/0022-1317-75-3-681. [DOI] [PubMed] [Google Scholar]
- 37.Shi M, Lam TT-Y, Hon C-C, Murtaugh MP, Davies PR, Hui RK-H, Li J, Wong LT-W, Yip C-W, Jiang J-W, Leung FC-C. 2010. Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J Virol 84:8700–8711. doi: 10.1128/JVI.02551-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lambert M-È, Delisle B, Arsenault J, Poljak Z, D'Allaire S. 2019. Positioning Quebec ORF5 sequences of porcine reproductive and respiratory syndrome virus (PRRSV) within Canada and worldwide diversity. Infect Genet Evol 74:103999. doi: 10.1016/j.meegid.2019.103999. [DOI] [PubMed] [Google Scholar]
- 39.Shi M, Lemey P, Singh Brar M, Suchard MA, Murtaugh MP, Carman S, D'Allaire S, Delisle B, Lambert M-È, Gagnon CA, Ge L, Qu Y, Yoo D, Holmes EC, Chi-Ching Leung F. 2013. The spread of type 2 porcine reproductive and respiratory syndrome virus (PRRSV) in North America: a phylogeographic approach. Virology 447:146–154. doi: 10.1016/j.virol.2013.08.028. [DOI] [PubMed] [Google Scholar]
- 40.Dewey C, Charbonneau G, Carman S, Hamel A, Nayar G, Friendship R, Eernisse K, Swenson S. 2000. Lelystad-like strain of porcine reproductive and respiratory syndrome virus (PRRSV) identified in Canadian swine. Can Vet J 41:493–494. [PMC free article] [PubMed] [Google Scholar]
- 41.Murtaugh MP, Stadejek T, Abrahante JE, Lam TT, Leung FC-C. 2010. The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Res 154:18–30. doi: 10.1016/j.virusres.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 42.Petry D, Holl J, Weber J, Doster AR, Osorio FA, Johnson R. 2005. Biological responses to porcine respiratory and reproductive syndrome virus in pigs of two genetic populations. J An Sci 83:1494–1502. doi: 10.2527/2005.8371494x. [DOI] [PubMed] [Google Scholar]
- 43.Petry D, Lunney J, Boyd P, Kuhar D, Blankenship E, Johnson R. 2007. Differential immunity in pigs with high and low responses to porcine reproductive and respiratory syndrome virus infection. J Anim Sci 85:2075–2092. doi: 10.2527/jas.2006-721. [DOI] [PubMed] [Google Scholar]
- 44.Kim W-I, Lee D-S, Johnson W, Roof M, Cha S-H, Yoon K-J. 2007. Effect of genotypic and biotypic differences among PRRS viruses on the serologic assessment of pigs for virus infection. Vet Microbiol 123:1–14. doi: 10.1016/j.vetmic.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Pirzadeh B, Gagnon CA, Dea S. 1998. Genomic and antigenic variations of porcine reproductive and respiratory syndrome virus major envelope GP5 glycoprotein. Can J Vet Res 62:170–177. [PMC free article] [PubMed] [Google Scholar]
- 46.Pirzadeh B, Dea S. 1998. Immune response in pigs vaccinated with plasmid DNA encoding ORF5 of porcine reproductive and respiratory syndrome virus. J Gen Virol 79:989–999. doi: 10.1099/0022-1317-79-5-989. [DOI] [PubMed] [Google Scholar]
- 47.Gonin P, Pirzadeh B, Gagnon CA, Dea S. 1999. Seroneutralization of porcine reproductive and respiratory syndrome virus correlates with antibody response to the GP5 major envelope glycoprotein. J Vet Diagn Investig 11:20–26. doi: 10.1177/104063879901100103. [DOI] [PubMed] [Google Scholar]
- 48.Han J, Rutherford MS, Faaberg KS. 2009. The porcine reproductive and respiratory syndrome virus nsp2 cysteine protease domain possesses both trans-and cis-cleavage activities. J Virol 83:9449–9463. doi: 10.1128/JVI.00834-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Music N, Gagnon CA. 2010. The role of porcine reproductive and respiratory syndrome (PRRS) virus structural and non-structural proteins in virus pathogenesis. Anim Health Res Rev 11:135–163. doi: 10.1017/S1466252310000034. [DOI] [PubMed] [Google Scholar]
- 50.Yuan S, Nelsen CJ, Murtaugh MP, Schmitt BJ, Faaberg KS. 1999. Recombination between North American strains of porcine reproductive and respiratory syndrome virus. Virus Res 61:87–98. doi: 10.1016/S0168-1702(99)00029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou L, Zhang J, Zeng J, Yin S, Li Y, Zheng L, Guo X, Ge X, Yang H. 2009. The 30-amino-acid deletion in the Nsp2 of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China is not related to its virulence. J Virol 83:5156–5167. doi: 10.1128/JVI.02678-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frias-Staheli N, Giannakopoulos NV, Kikkert M, Taylor SL, Bridgen A, Paragas J, Richt JA, Rowland RR, Schmaljohn CS, Lenschow DJ, Snijder EJ, García-Sastre A, Virgin HW. 2007. Ovarian tumor domain-containing viral proteases evade ubiquitin-and ISG15-dependent innate immune responses. Cell Host Microbe 2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. 2012. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J Virol 86:3839–3850. doi: 10.1128/JVI.06466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang Y, Snijder EJ. 2010. The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res 154:61–76. doi: 10.1016/j.virusres.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reyes YH, Provost C, Traesel CK, Jacques M, Gagnon CA. 2018. Actinobacillus pleuropneumoniae culture supernatant antiviral effect against porcine reproductive and respiratory syndrome virus occurs prior to the viral genome replication and transcription through actin depolymerization. J Med Microbiol 67:249–264. doi: 10.1099/jmm.0.000659. [DOI] [PubMed] [Google Scholar]
- 56.Wang A, Chen Q, Wang L, Madson D, Harmon K, Gauger P, Zhang J, Li G. 2019. Recombination between vaccine and field strains of porcine reproductive and respiratory syndrome virus. Emerg Infect Dis 25:2335–2337. doi: 10.3201/eid2512.191111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, Li Y, Xiao S, Yang X, Chen X, Wu P, Song J, Ma Z, Cai Z, Jiang M, Zhang Y, Yang Y, Zhang Z, Zhou Z, Sheng J, Wang H. 2019. High-frequency mutation and recombination are responsible for the emergence of novel porcine reproductive and respiratory syndrome virus in northwest China. Arch Virol 164:2725–2733. doi: 10.1007/s00705-019-04373-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All entire viral genomes of successfully sequenced PRRSV strains were submitted to GenBank with accession numbers MN865482 to MN865573 (see Table S1 in the supplemental material).