

Abstract
Sleep is a crucial physiological process for our survival and cognitive performance, yet the factors controlling human sleep regulation remain poorly understood. Here, we identified a missense mutation in a G protein-coupled neuropeptide S receptor 1 (NPSR1) that is associated with a natural short sleep phenotype in humans. Mice carrying the homologous mutation exhibited less sleep time despite increased sleep pressure. These animals were also resistant to contextual memory deficits associated with sleep deprivation. In vivo, the mutant receptors showed increased sensitivity to neuropeptide S exogenous activation. These results suggest that the NPS/NPSR1 pathway might play a critical role in regulating human sleep duration and in the link between sleep homeostasis and memory consolidation.
INTRODUCTION
Sleep remains a relatively understudied phenomenon, despite being essential in some form to most vertebrate life. Although humans spend about one-third of their lives in the sleep state, an understanding and recognition of its importance for our well-being are severely lacking. Sleep of sufficient duration, continuity, and depth is necessary to maintain high cognitive performance during wake and to prevent certain physiological changes that may predispose individuals to many adverse health outcomes (1–6). On average, people require about 8 to 8½ hours of sleep each day to function optimally (7). However, current surveys indicate that 35 to 40% of the adult U.S. population sleeps less than 7 hours on weekday nights (8), a duration known to lead to cumulative deficits in behavioral alertness and vigilant attention (9).
Sleep duration varies greatly (and appear to be normally distributed in the general population) among individuals and are heavily influenced by both genetic and environmental factors (10–15), making their investigation particularly challenging. The specific mechanisms underlying these differences are largely unknown, and until the recent identification of the first human gene/mutation linked to a short sleep duration trait, there was no knowledge regarding genetic contributions to short sleep in humans (16). People with this trait [familial natural short sleep (FNSS)] have a lifelong tendency to sleep only 4 to 6 hours per night while still feeling well rested (16). Anecdotally, these individuals also do not seem to bear the greater load of comorbid disorders traditionally associated with chronically restricted sleep. Identification of human FNSS genes presents an opportunity to study not only the genetics of human sleep dynamics but also the relationship between sleep homeostasis and health.
In this study, we identified another FNSS family and report a mutation in the NPSR1 gene causing a short sleep phenotype. Neuropeptide S receptor 1 (NPSR1) is a G protein-coupled receptor whose cognate ligand, neuropeptide S (NPS), has been reported to modulate arousal and sleep behaviors (17). Administration of NPS in mice increases wakefulness and hyperactivity (17). We recreated the putative FNSS mutation—NPSR1-Y206H—in mice and found that those carrying the homologous mutation showed a short sleep phenotype similar to human FNSS. Further, they appeared to be resistant to certain memory deficits associated with sleep deprivation (SD). Correspondingly, we showed the NPSR1-Y206H substitution changed downstream signaling dynamics and neuron behaviors in mouse brain in response to NPS treatment. These data suggest a causative role for the NPSR1-Y206H mutation in the short sleep phenotype and advance our understanding of the genetic players in human sleep variability with potential therapeutic implications.
RESULTS
The NPSR1-Y206H mutation was found in one FNSS family
In one of our identified FNSS families (K50226), the habitual total sleep time of two subjects was characterized (fig. S1, questionnaires) to be much shorter (5.5 and 4.3 hours) than the average optimal sleep duration in the general population (8 to 8 ½ hours) (Fig. 1A) (18). To pinpoint potential genetic drivers, we performed whole exome sequencing of DNA samples from these two individuals and identified a shared point mutation in the NPSR1 gene. This mutation converts a tyrosine into a histidine at position 206 (Y206H) in both of the two known isoforms (NPSR1A and B). The mutated residue is located in one of the highly conserved extracellular domains of NPSR1 (Fig. 1, B and C). NPSR1-Y206H is a rare mutation not found in the Exome Aggregation Consortium database and with a frequency of 4.06 × 10−6 in the Genome Aggregation database. NPS has been previously reported to promote arousal in rodents (17). Moreover, a homozygous NPSR1-N107I polymorphism (Fig. 1C) was reported to be associated with slightly reduced (~20 min) sleep duration in the human population by genetic association studies (19, 20). Together, these data support the possibility that this mutation may be causal for the FNSS trait in affected individuals.
Fig. 1. NPSR1-Y206H mutation was identified in a natural short sleep family.
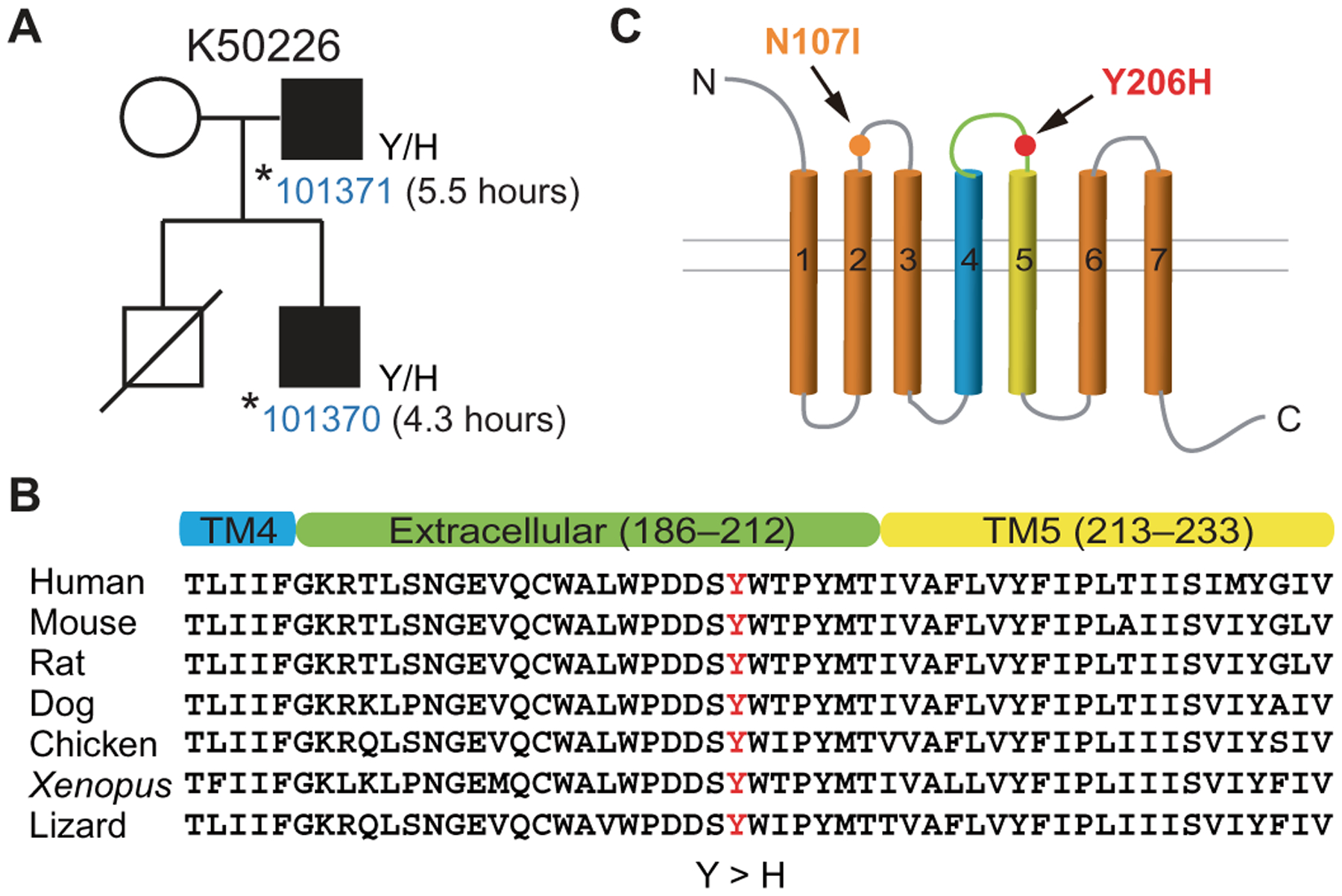
(A) Pedigree of the family (K50226) carrying the NPSR1 mutation. The self-reported total sleep time per 24-hour day of mutation carriers is indicated. (B) NPSR1-Y206H is localized to the extracellular domain between transmembrane domains 4 and 5 (TM4/5) and is highly conserved among vertebrate NPSR1 orthologs. (C) Schematic of the mutations (Y206H and N107I) in the extracellular loops of NPSR1.
Npsr1-Y206H mice show increased mobile time
Given the highly conserved protein sequence between mouse and human NPSR1 (fig. S2A), we generated an Npsr1-Y206H knock-in mouse model using CRISPR-Cas9. Endogenous Npsr1 mRNA expression was comparable between wild-type (WT; Npsr1+/+) and mutant (Npsr1+/m) mice (fig. S2B). We first monitored the mobile time of WT and Npsr1-Y206H mice using infrared video recording (ANY-maze) (21). Npsr1-Y206H mice demonstrated more mobile time and greater traveled distance than WT during both light and dark phases (Fig. 2A and fig. S3A). The mutant mice displayed significantly reduced mobile episodes (P = 0.0005; fig. S3B), suggesting that they had more consolidated mobile periods.
Fig. 2. Npsr1-Y206H mice demonstrate reduced sleep time.
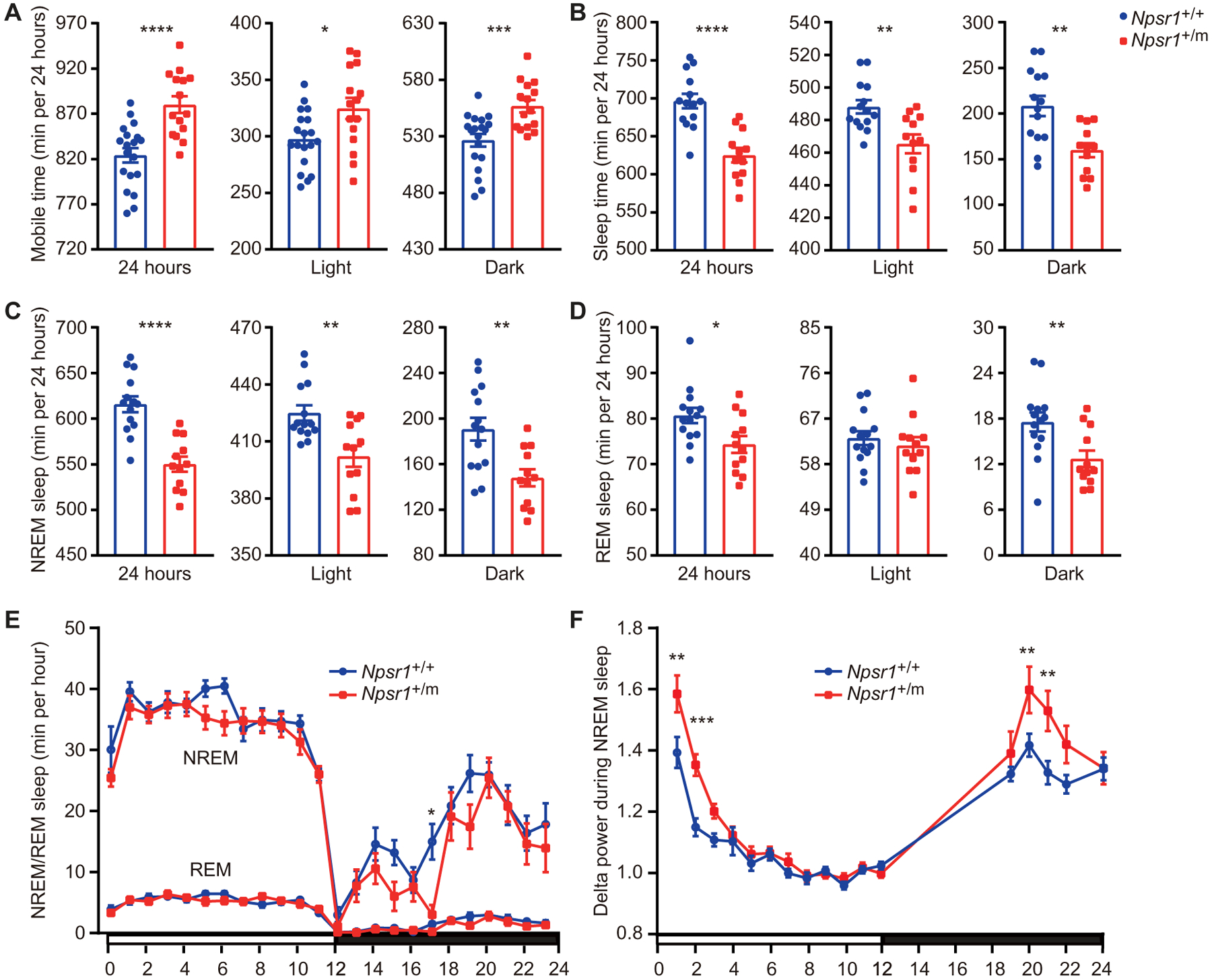
(A) Mouse movement was tracked by ANY-maze under LD 12:12. Total mobile time in 24 hours, light phase, and dark phase were calculated in Npsr1+/+ (n = 19) and Npsr1+/m (n = 15) mice. (B to D) Total sleep (B), NREM sleep (C), and REM sleep (D) time within 24 hours, light phase, and dark phase measured by EEG/EMG were calculated in Npsr1+/+ (n = 14) and Npsr1+/m (n = 12) mice. (E) NREM and REM sleep time were plotted hourly over 24 hours in Npsr1+/+ (n = 14) and Npsr1+/m (n = 12) mice. (F) NREM sleep delta power normalized to the average value during ZT9–12 was plotted hourly in Npsr1+/+ (n = 14) and Npsr1+/m (n = 12) mice over 24 hours. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, two-tailed Student’s t test (A to D); two-way repeated-measures (RM) ANOVA, post hoc Sidak’s multiple comparisons test (E and F). Data are mean ± SEM.
Npsr1-Y206H mice spend less time sleeping
To study sleep architecture, we next monitored WT and Npsr1-Y206H mice with electroencephalogram/electromyogram (EEG/EMG) recordings. Consistent with the ANY-maze results, Npsr1-Y206H mice exhibited a reduced total sleep time of 71 min compared to WT (P < 0.0001; Fig. 2B and fig. S3C). This difference remained statistically significant when assessed individually in both the light (P = 0.003) and dark phases (P = 0.0018; Fig. 2B). EEG data indicated that a reduction in non–rapid eye movement (NREM) sleep in mutants was the primary contributor to the short-sleep phenotype in both the light and dark phases, with the most pronounced change in the dark phase (Fig. 2, C and E). Npsr1-Y206H mice also showed significantly reduced rapid eye movement (REM) sleep during the dark phase (P = 0.0084; Fig. 2, D and E). We also examined the sleep phenotype of Npsr1 knockout (KO; Npsr1−/−) mice. No difference in mobile time or sleep duration was detected in WT versus KO littermates (fig. S3, D and E). Together, these results suggest that the NPSR1-Y206H mutation is likely the genetic cause of natural short sleep behavior in both mouse and human mutation carriers.
Consistent with the reduced mobile episodes in the ANY-maze assay, Npsr1-Y206H mice showed a significant reduction in wake bouts during the dark period (P = 0.0056) and an increase of episode duration during both the light (P = 0.0028) and dark phases (P = 0.0132) (fig. S4, A and B). Npsr1-Y206H mice also showed fewer episodes of NREM/REM sleep than WT (fig. S4, C and E), whereas the mean duration of each NREM/REM episode was unchanged (fig. S4, D and F). Together, these results suggest that the NPSR1-Y206H mutation contributes to shorter sleep duration through a combination of longer average wake-bout length with lower wake-bout number and reduced number of REM/NREM bouts with unchanged bout length.
Npsr1-Y206H mice sustain higher sleep pressure
Sleep pressure in mammals increases concomitantly with wake time. Spectral analysis of EEG showed an increase in the delta range (1 to 4 Hz) power during NREM sleep in Npsr1-Y206H mice (fig. S5A), a feature classically associated with increased sleep need. We next compared EEG delta power during NREM sleep across the light-dark cycle between mutant and control mice. During the dark phase, Npsr1-Y206H mice accumulated higher delta power than WT, possibly the result of increased wake time, which dissipates during the light phase (Fig. 2F). This result indicates that Npsr1-Y206H mice, although having reduced sleep time, can sustain higher sleep pressure.
Recently, quantitative phosphoproteomic analysis was performed in sleep-deprived WT mice and the Sleepy mutant mouse models. High sleep pressure was found to be associated with induction of cumulative phosphorylation of the brain proteome that dissipated during sleep (22). We measured expression of these proteins, called sleep-need-index phosphoproteins (SNIPPs), in mutant and WT NPSR1 mice as a correlate metric for sleep pressure. Phosphorylation of the SNIPPs EF2 and synapsin-1 was consistently increased in Npsr1-Y206H mice at ZT2 and ZT22 but not at ZT11 when sleep pressure is lowest (fig. S5, B and C). These molecular data are consistent with the Npsr1-Y206H mutant animals exhibiting increased sleep pressure in the early light and late dark phases (Fig. 2F).
Npsr1-Y206H mice have normal recovery sleep after SD
To further characterize these mice, we examined their sleep features under high sleep pressure by subjecting them to 6-hour SD (ZT0–6). As expected, Npsr1-Y206H mice displayed greater delta power than WT mice in the first hour after SD (Fig. 3A). Phosphorylation of EF2 and synapsin-1 was also consistently increased in Npsr1-Y206H mice after SD (fig. S6, A and B). Similar to what was observed without SD, Npsr1-Y206H mice displayed higher delta power than WT mice during the late dark phase. In addition, Npsr1-Y206H mice exhibited significantly reduced sleep (P = 0.0013) [REM (P = 0.0028) and NREM (P = 0.0016)] time accompanied with increased sleep pressure in the dark period immediately after SD (Fig. 3, B to D). The mutant mice showed comparable sleep gain with WT after SD (Fig. 3E), indicating that Npsr1-Y206H mice have a sleep rebound process that is similar to WT. Collectively, our results suggest that NPSR1-Y206H mutation promotes wakefulness in the presence of high sleep pressure.
Fig. 3. Npsr1-Y206H mice have normal recovery sleep after SD.
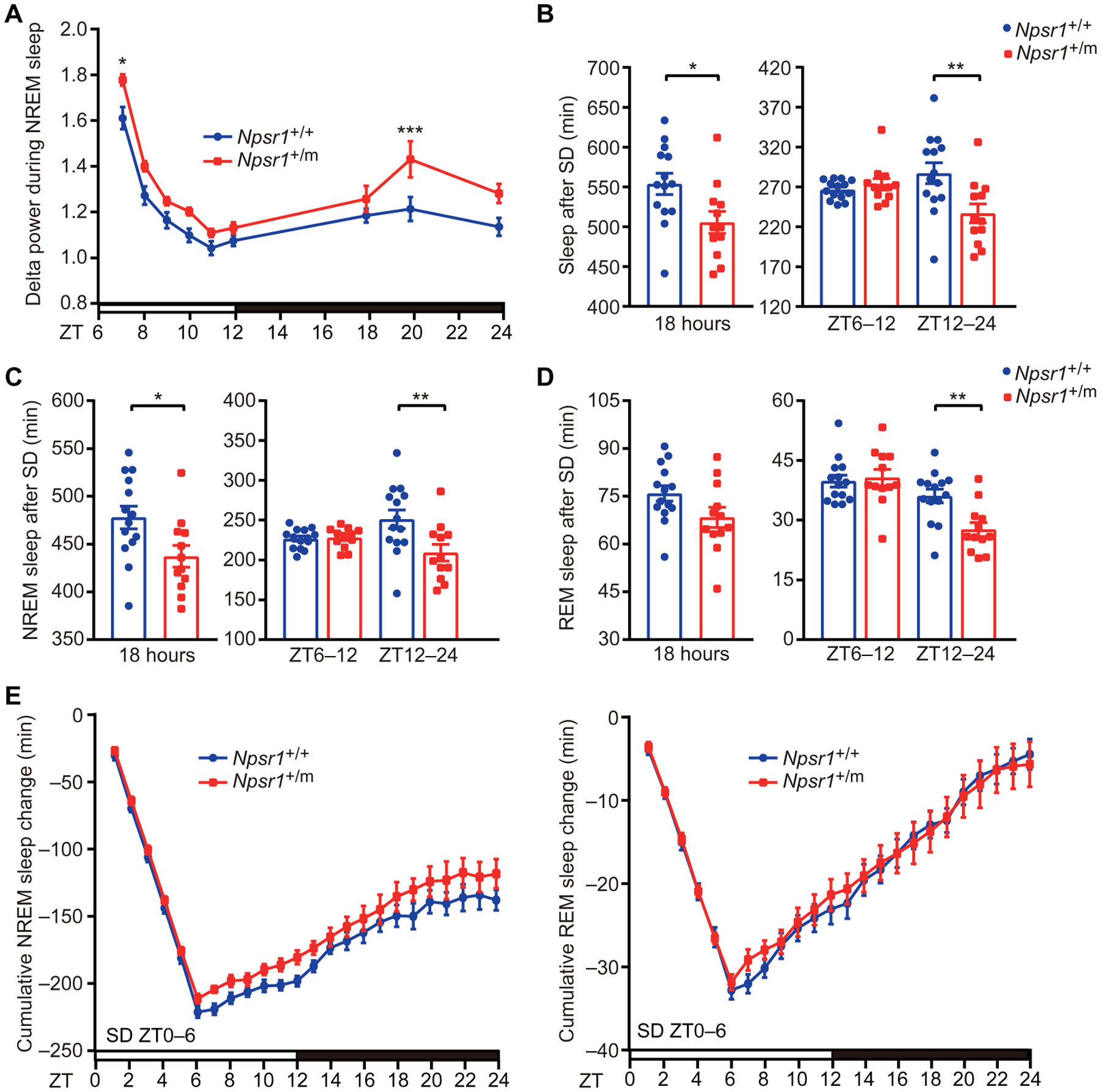
(A) NREM sleep delta power after SD (ZT0–6) normalized to the average NREM delta power during ZT9–12 of the baseline recording was plotted every hour in Npsr1+/+ (n = 14) and Npsr1+/m (n = 12) mice for 18 hours. (B to D) Total (B), NREM (C), and REM (D) sleep were calculated during indicated time periods for Npsr1+/+ (n = 14) and Npsr1+/m (n = 12) mice after 6 hours of SD (ZT0–6). (E) Cumulative NREM and REM sleep loss and gain compared with baseline conditions for the SD experiment in Npsr1+/+ (n = 14) and Npsr1+/m (n = 12) mice. *P < 0.05, **P < 0.01, and ***P < 0.001, two-tailed Student’s t test (B to D, left); two-way RM ANOVA, post hoc Sidak’s multiple comparisons test [A, B to D (right), and E]. Data are means ± SEM.
Npsr1-Y206H mice show increased phospho–cAMP response element–binding protein in the brain
NPSR1 is a receptor coupled to both Gs and Gq signaling and widely expressed in mammalian brain (23, 24). To determine whether the Y206H mutation alters the physiological function of NPSR1, we used brain lysates to quantify phospho–cAMP response element–binding protein (CREB)—a common downstream effector of both the Gs and Gq pathways (25). Increased phospho-CREB was observed in the cortex of Npsr1-Y206H mice at ZT2 and ZT22 (Fig. 4, A and B). To validate the putative activity of NPS on phospho-CREB, we then performed intracerebroventricular injection of NPS. Different concentrations of NPS were injected at ZT11, when phospho-CREB is comparable between WT and mutant mice. A dose-dependent increase of phospho-CREB was observed in WT mice after NPS injection, which was completely blocked by NPSR1 antagonist SHA 68 (Fig. 4, C to E) (26, 27). The increase of phospho-CREB was further enhanced in Npsr1-Y206H mice (Fig. 4, C and D). No effect was observed in Npsr1 KO mice (Fig. 4F), confirming NPS signaling is specific to NPSR1. These results revealed that NPS induces phospho-CREB in the mouse cortex, which was augmented in mutant mice, suggesting that mutant protein is likely to be more active in vivo.
Fig. 4. Mutant NPSR1 is more active in vivo.
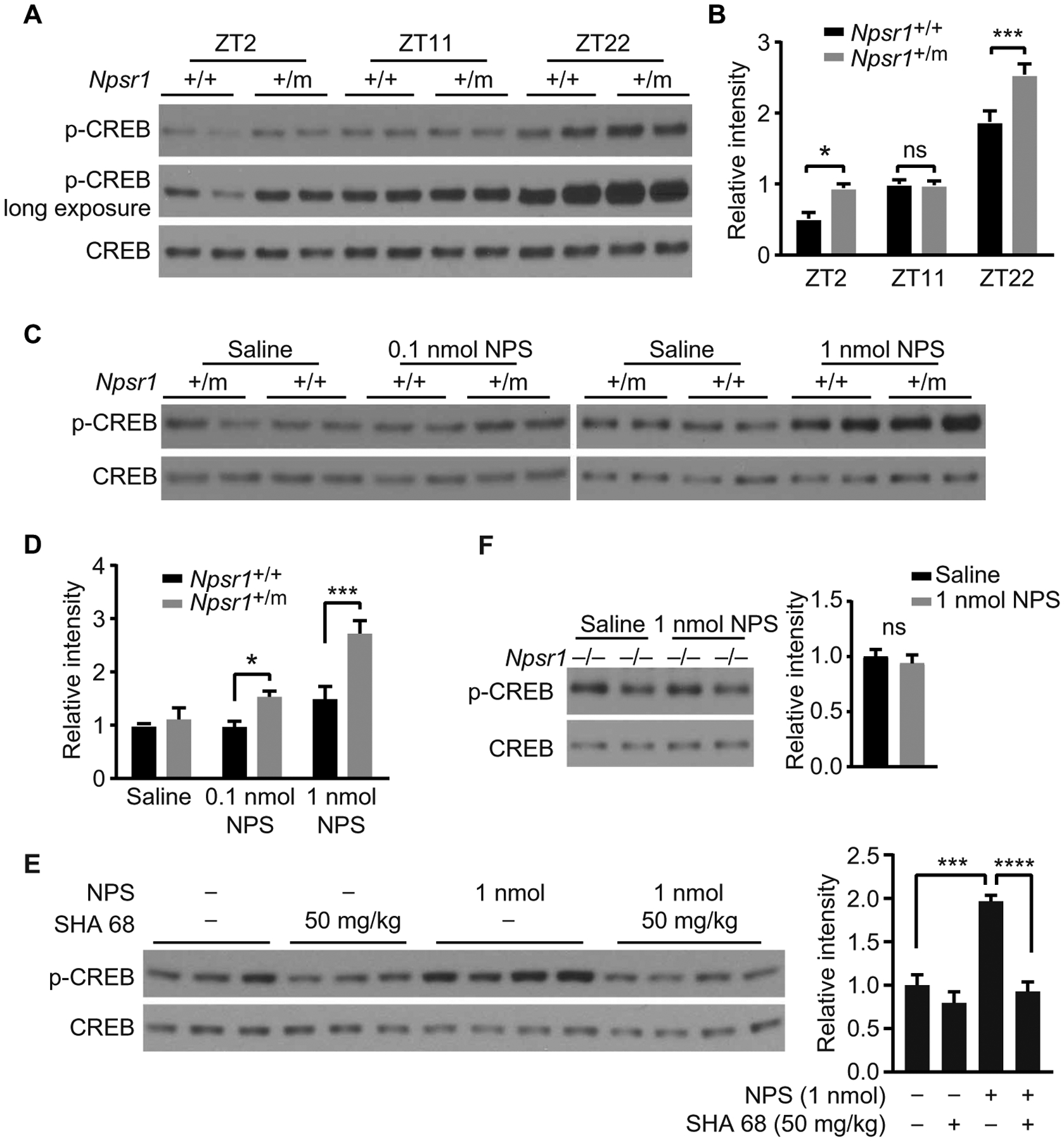
(A) p-CREB immunoblots for brain lysates collected at indicated time points from Npsr1+/+ and Npsr1+/m mice. (B) Quantified results of (A) from Npsr1+/+ (ZT2, n = 4; ZT11, n = 5; ZT22, n = 4) and Npsr1+/m mice (ZT2, n = 4; ZT11, n = 5; ZT22, n = 3). (C) Immunoblots for brain lysates collected at ZT11 from Npsr1 +/+ and Npsr1+/m mice after different doses of NPS. (D) Quantified results of (C) from Npsr1+/+ (saline, n = 4; 0.1 nmol NPS, n = 3; 1 nmol NPS, n = 4) and Npsr1+/m mice (saline, n = 4; 0.1 nmol NPS, n = 3; 1 nmol NPS, n = 3). (E) Western blot of p-CREB expression in WT brain lysates collected at ZT11 after SHA 68 (intraperitoneal) and/or NPS (intracerebroventricular) injection. Group sizes: Veh/SHA 68 (50 mg/kg) and saline, n = 3; Veh/SHA 68 (50 mg/kg) and NPS (1 nmol), n = 4. (F) Immunoblots for brain lysates collected at ZT11 from Npsr1−/− mice after saline (n = 4) or 1 nmol NPS injection (n = 4). Quantified results are shown in panels on the right. *P < 0.05, ***P < 0.001, and ****P < 0.0001, ns, not significant. Two-way ANOVA, Sidak’s multiple comparisons test (B and D). One-way ANOVA, Dunnett’s multiple comparisons test (E). Two-tailed Student’s t test (F). Data are means ± SEM.
Neurons from Npsr1-Y206H mice are hypersensitive to NPS
Because NPS/NPSR1 signaling was reported to trigger calcium mobilization in neurons (28), we performed single-cell calcium imaging on acutely isolated brain slices prepared from WT and mutant mice (Fig. 5A). Neurons in the centromedial thalamus (CMT) have been found to induce NREM-wake transitions (29), and Npsr1 mRNA is highly expressed in this area (24). We thus analyzed calcium signaling in this region (Fig. 5B), categorizing NPS responsive cells into four distinct groups based on their GCaMP signal response pattern after NPS treatment (Fig. 5C). The proportion of cells in all groups was lower in brain slices from Npsr1 KO mice (Fig. 5D), indicating that the GCaMP signals monitored here were primarily mediated by the NPS/NPSR1 pathway. In the mutant brain slices, there was a significantly (P < 0.0001) higher ratio of cells with a group 2 (fast and long lasting) type activation response to NPS (Fig. 5D). We also compared the calcium response of the neurons in the lateral hypothalamus (LH), which is another well-defined sleep-regulating center in the brain. There is no difference between WT and KO slices in all the groups, suggesting that NPS at this dose is not sufficient to initiate the NPSR1-dependent calcium response in the WT cells (fig. S7). This is probably due to the relatively lower expression of NPSR1 in this region compared with that of CMT (24). Nonetheless, the percentage of cells in group 2 is higher in the mutant slices, suggesting that the mutant cells have lower threshold for NPS response (fig. S7). Together, these results further support the conclusion that mutant receptors are more active in vivo.
Fig. 5. Calcium imaging of CMT neurons shows increased activity for one subtype in Npsr1-Y206H mice.
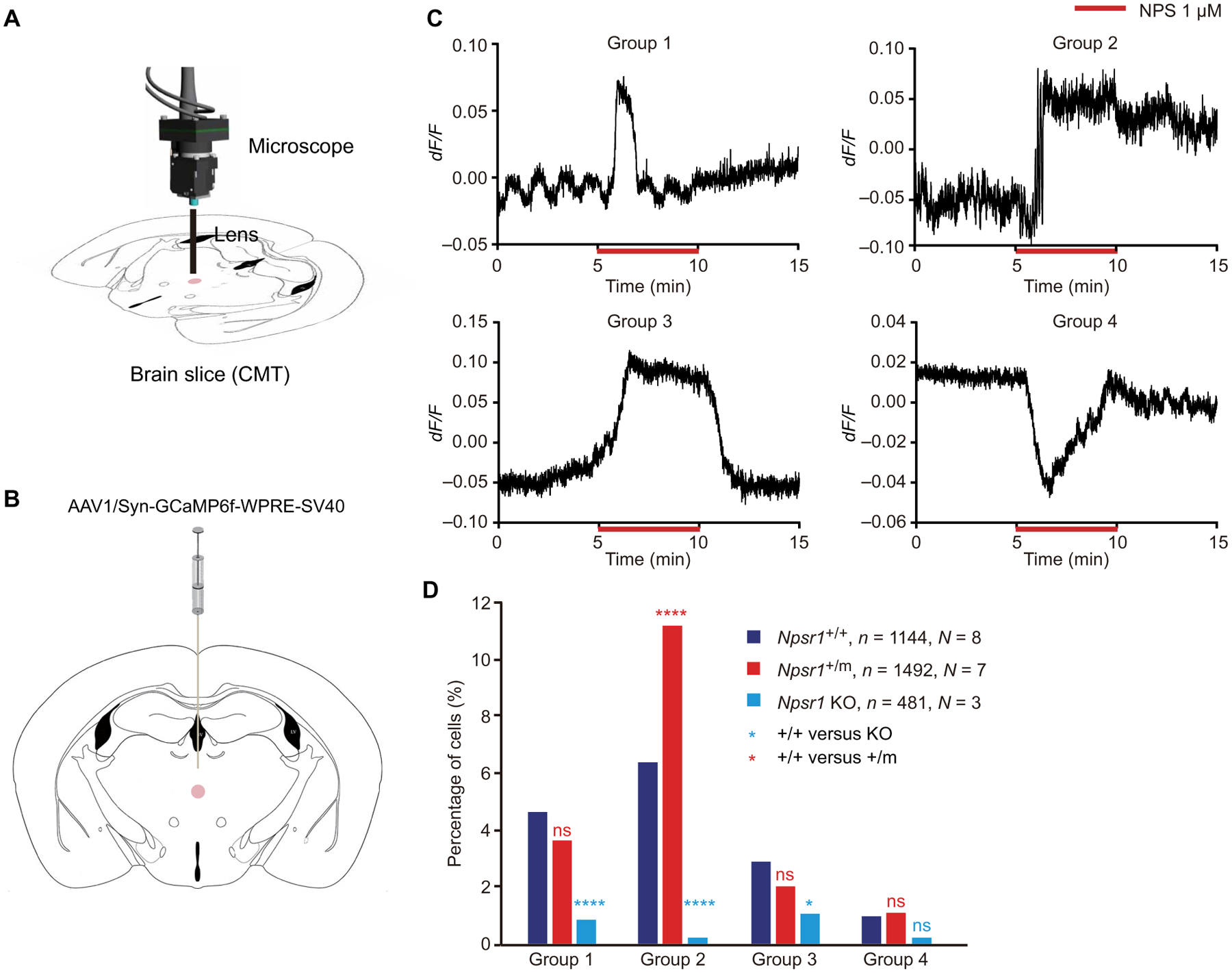
(A) Schematic of calcium imaging setup for recording the activity of CMT neurons in brain slices. (B) Schematic of anatomy for CMT. The injection/recorded area is marked with a pink circle. (C) Representative GCaMP fluorescence traces in different categories of cells that responded differentially to NPS treatment. Group 1, pulse activation; group 2, fast and long-lasting activation; group 3, fast activation and recovery; and group 4, inhibition. (D) Percentage of cells that show different types of response to NPS treatment in Npsr1+/+ (n = 8), Npsr1+/m (n = 7), and Npsr1−/− (n = 3) brain slices. *P < 0.05 and **** P < 0.0001; chi-square test (D). n, number of cells; N, number of animals.
Contextual memory of Npsr1-Y206H mice is more resilient to sleep loss
Accumulated sleep pressure caused by prolonged wakefulness can impair cognitive function (6, 30, 31). However, the cognitive performance of human FNSS subjects seemed unimpaired despite long-term reduced sleep duration (32). To see whether this phenomenon can be replicated in the Npsr1-Y206H mice, we subjected them to the contextual fear conditioning test, a memory-based assay known to be sensitive to sleep loss (33, 34). WT and Npsr1-Y206H mice were trained during either the early light phase (ZT3–4) (Fig. 6A) or the late dark phase (ZT23–24) (Fig. 6B) followed by testing at 24 and 48 hours after training. These time windows were chosen because SD needs to be carried out during the sleep (light) phase, and the changes of phospho-CREB and sleep pressure were most prominent at these two time windows. Npsr1-Y206H mice had similar performance as WT on both days 1 and 2 despite having less overall sleep time and a higher sleep pressure (Figs. 2, B and F, and 6). We subjected another group of mice to 6-hour SD immediately after training (Fig. 6, A and B), which has been shown to impair memory consolidation (33, 34). WT mice showed significantly reduced freezing time (P = 0.0179, ZT3; P = 0.0488, ZT23) on day 1, indicating impaired memory consolidation. Npsr1-Y206H mice exhibited no loss of freezing time upon testing on day 1 after SD, suggesting that contextual memory of mutant mice is more resistant to sleep loss. The mutant mice exhibited similar freezing times with WT mice on day 2, implying a preserved extinction process of contextual memory.
Fig. 6. Contextual memory of Npsr1-Y206H mice is resistant to sleep loss.
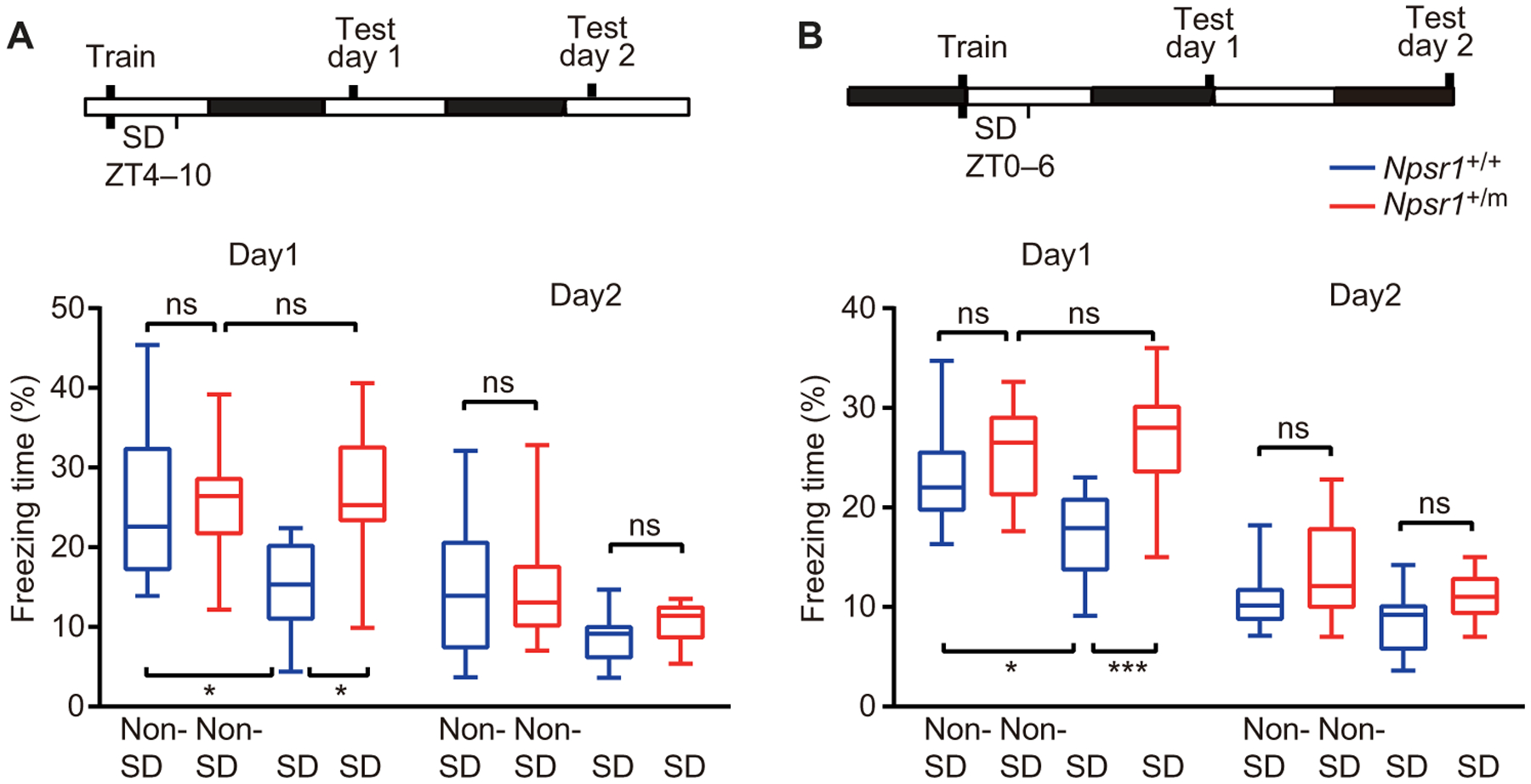
(A and B) Mice were trained in contextual fear conditioning at the beginning of the light phase (A) or at the end of the dark phase (B). Freezing response in the trained context was tested 24 (day 1) and 48 (day 2) hours after training. Percentage of time freezing during the 5 min of the test without SD [A, Npsr1+/+ (n = 17) and Npsr1+/m (n = 12); B, Npsr1+/+ (n = 12) and Npsr1+/m (n = 11)] or with SD [A, Npsr1+/+ (n = 10) and Npsr1+/m (n = 10); B, Npsr1+/+ (n = 10) and Npsr+/m (n = 11)] is shown in box and whisker plots. *P < 0.05 and *** P < 0.001. One-way ANOVA multiple comparisons followed by Tukey’s multiple comparisons test.
DISCUSSION
The physiological function of the NPS/NPSR1 pathway was first deciphered in 2004 (17). Although NPS/NPSR1 signaling was shown to have strong wake-promoting effects, most of the findings were derived from studies of rodents, and limited data are available about its effect on human sleep regulation. Initially found to be linked to an increased susceptibility for asthma (35), the homozygous NPSR1-N107I polymorphism in the human population was also reported to be associated with slightly reduced (~20 min) sleep duration by genetic association studies (19, 20), although there was no effort made to understand if/how this polymorphism was causative of the association (or just genetically linked to it). The fact that NPSR1-Y206H mutant mice have reduced sleep and that Npsr1 KO mice have no sleep phenotype supports NPSR1-Y206H as a gain-of-function mutation. Similarly, the NPSR1-N107I is also a hyperactive form (gain of function) in cultured cells (23). Both Y206 and N107 residues are located at the extracellular domains, implying that these mutations may increase ligand affinity or agonist efficacy rather than produce more constitutively active forms of receptor. Y206H differs from the N107I polymorphism in several ways. First, the incidence of Y206H (4.06 × 10−6) is much lower than that of N107I (~0.45, http://exac.broadinstitute.org) in the population. Second, heterozygous Y206H human carriers (>2 hours shorter sleep) have a much stronger sleep phenotype than homozygous N107I (~20 min shorter sleep) carriers (heterozygous N107I human carriers do not show sleep duration difference). Third, the Y206 residue of NPSR1 is well conserved in vertebrates, whereas the corresponding 107 residues in most vertebrates including mouse are Ile. All these features of the Y206H mutation support its causative role for FNSS and, thus, strengthen the important role of NPS/NPSR1 in modulating sleep duration. Npsr1-Y206H mice displayed more wakefulness in the presence of higher sleep pressure. Considering the wake-promoting function of NPS/NPSR1 (17) and our finding that Npsr1-Y206H is a gain-of-function mutation, we propose that the short sleep phenotype of human FNSS or Npsr1-Y206H mice results from strong induction of arousal. This hypothesis is consistent with the highly energetic behavioral traits observed in human FNSS, despite lifelong short sleep.
Npsr1 is widely expressed in the brain, and our results indicate hypersensitivity of mutant NPSR1 to NPS in two separate regions that were tested. Together, these results suggest that the wake-promoting phenotype in Npsr1-Y206H mice is likely mediated by the hyperactivity of mutant NPSR1 in sleep-wake-regulating nuclei including (but not limited to) CMT and LH. High expressions of both Npsr1 and NPS were detected in the paraventricular thalamus, which was recently shown to be critical for wakefulness (36). Moreover, moderate expression of Npsr1 was found in some nuclei of basal forebrain regions, another known sleep regulatory area. Further in-depth investigation is needed to determine all the loci participating in NPS/NPSR-mediated sleep regulation.
Chronic partial SD or restricted sleep often results in severe behavioral, physiological, psychiatric, and cognitive disorders, including fatigue, lapses in behavioral alertness, increased risk of obesity and diabetes, adverse cardiometabolic outcomes, depression, and deficits in cognitive performance (1, 37). Studies of healthy adults showed that restriction of sleep time to 4 or 6 hours per night over 14 consecutive days would result in cumulative, dose-dependent deficits in performance on all cognitive tasks (30). Humans with NPSR1-Y206H and the mutant mouse model have a lifelong reduction in daily sleep time (resembling restricted sleep for regular sleepers) accompanied by higher sleep pressure demonstrated in mice. However, this type of chronic SD seems to be “benign” and has no obvious detrimental effects to these FNSS subjects based on our assessments of the human subjects and the learning and memory assays done in the mice. In our growing clinical database of FNSS, we have observed that FNSS subjects are healthy, energetic, optimistic, with high pain threshold, and do not seem to suffer from adverse effects of chronic restricted sleep. It is possible that there is a mechanism in these FNSS human and mice compensating for the negative effects normally caused by sleep loss. Alternatively, these FNSS humans and mice are impervious to the negative effects caused by sleep loss. Further studies are needed to distinguish these possibilities. NPS has been reported to facilitate learning and memory (38, 39) and elicit anxiolytic (17) and antinociceptive effects (40, 41). It is possible that NPSR1-Y206H is also responsible for at least some of the seemingly protective traits (in addition to short sleep behavior) for these human mutation carriers. Whether these protective traits are secondary to short and efficient sleep or independently regulated by NPS/NPSR1 signals warrants further investigation.
Although a wake-promoting function of NPS/NPSR1 has been demonstrated by central administration of NPS (17), studies from KO mice were inconclusive as the KO mice have a minimal (or controversial) sleep phenotype under baseline conditions depending on the mouse background (42–45). The lack of a sleep phenotype in KO studies does not necessarily undermine the importance of NPSR1 in sleep regulation. It is not uncommon that KO mutations give no obvious phenotype, whereas gain-of-function dominant mutants provide critical mechanistic insight (46). Identification of the NPSR1-Y206H mutation in FNSS subjects presents an opportunity to reveal the mechanism of sleep-regulation function by NPSR1.
The main limitation of this study was the difference between the human and mouse sleep patterns. Despite the highly conserved genomic sequences, humans and mice display different features in sleep behaviors. Humans spend most sleep time at night and almost no sleep time during the day, whereas mice sleep both in the light and dark phases, with about 70% sleep time in the light phase and 30% in the dark phase. Moreover, mouse sleep is more fragmented than human sleep and does not occur in a consolidated bout as it does in humans. These differences probably result from varied sleep regulatory mechanisms between human and mice, which may contribute to differed phenotypes caused by the same genetic mutation. This could at least partly explain the difference in sleep phenotype observed in NPSR1 human mutation carriers (reduced by 2 to 4 hours during the rest phase) and Npsr1-Y206H mice (reduced sleep is mostly confined to the active phase for 1 hour).
The homology of NPSR1 across evolutionary time is less than that in many genes regulating critical biological processes such as cell cycle regulation or in cell excitability. The protein sequence identity of hNPSR1 to the following species is as follows: mouse (80%), dog (61%), Xenopus (51%), and zebrafish (20%). This fits with the differences mentioned above in sleep, even from human to mice. Thus, although there is much to learn from studies of sleep and sleep-like behavior in many model systems, it seems likely that there will also be interesting differences in sleep regulation in humans compared with other organisms. It will be important to better understand not only the similarities but also the differences in human sleep compared with other organisms.
In sum, we identified the NPSR1-Y206H mutation from the FNSS subjects. Mice carrying the mutation showed a similar phenotype to human FNSS. These data support a causal role of the NPSR1-Y206H mutation for the human short sleep phenotype. Thus, the NPS/NPSR1 pathway provides a potential therapeutic target to improve human sleep and treat sleep-related disorders.
MATERIALS AND METHODS
Study design
The objective of this study was to identify the genetic mutation that underlies the short sleep phenotype of FNSS individuals of kindred #50226. Exome sequencing was performed on DNA from two FNSS subjects. Contribution of the NPSR1-Y206H mutation to the short sleep phenotype was tested by generating Npsr1-Y206H knock-in mice using CRISPR-Cas9. Sleep phenotypes of mutant mice were examined with EEG and video recording. Molecular characterizations, including single-cell calcium imaging, were carried out to investigate the functional alterations of mutant NPSR1. To measure the effect of short sleep on the memory function of Npsr1 mutant mice, WT and mutant mice were subjected to contextual fear condition tests with or without SD. Similar numbers of Npsr1-Y206H mutant and WT littermates were assigned to each group. The sample size in animal studies was determined on the basis of previous experience with similar animal studies. For each experiment, the sample size indicated in the figure legend reflects the number of independent biological replicates. The experimenters were blind to the genotype of the animals during behavioral tests, calcium imaging, NPS/SHA injection, protein sample preparation, and EEG scoring. We excluded only mice with unreadable EEG signals from the data analysis. Raw data are reported in data file S1.
Nomenclature
For humans—gene (NPSR1), protein (NPSR1). For mouse—gene (Npsr1), protein (NPSR1). +/+ refers to WT animals or unaffected human subjects, and +/m refers to heterozygous mutant animals or affected human subjects.
Short sleeper characterization and identification of candidate gene
Human research subjects for this study were voluntary participants. All human participants signed a consent form approved by the Institutional Review Boards at the University of Utah and the University of California, San Francisco (10–03952). Self-reported habitual sleep-wake schedules were obtained during structured interviews by one of the authors (C.R.J. and L.J.P.). Blood sample collection and DNA preparation were performed as previously described (16).
Exome sequencing
Whole exome sequencing was performed by Beijing Genomics Institute using the Ion Torrent platform, and variants were called using the Torrent Unified Variant Caller. For each family, Variant Tools (v2.5.0) (47) was used to create a variant database, which was annotated with family members’ phenotypes, database for nonsynonymous SNPs’ functional predictions (dbNSFP) (48), SNP effect (SnpEff) (49), and allele frequencies from 1000 Genomes (50) and ExAC (51). Variants were selected as candidates using the following criteria: (i) They were heterozygous in affected family members and homozygous WT in unaffected family members; (ii) their allele frequency in both the 1000 Genomes and ExAC datasets was less than 0.001; (iii) they were potentially deleterious because they had a “high” predicted impact in SnpEff, or were called as “damaging” by Sorting Intolerant From Tolerant (SIFT) (52), or were categorized as either “probably damaging” or “possibly damaging” by HumDiv-trained PolyPhen-2 (53); and (iv) they did not belong to a gene with a high load of rare deleterious mutations. The latter criterion was evaluated on the basis of each gene’s number of variants in the ExAC and 1000 Genomes datasets with minor allele frequency <0.001 and “moderate” or “high” SnpEff impact. Genes in the top 10% by this metric were excluded from consideration. For each family, candidates were selected for follow-up Sanger sequencing from among the filtered variants based on neuronal gene expression (genecards.org) and known gene function related to neuronal signaling.
Generation of Npsr1-Y206H knock-in and KO mice
Npsr1-Y206H knock-in mice were generated using a CRISPR-Cas9–mediated approach. Briefly, DNA template for single guide RNA (sgRNA) was amplified with primers containing the T7 promoter and the sgRNA-targeting sequences. Primer sequences are as follows: forward, ′TTAATACGACTCACTATAGGAGCAATGAATA AGTGTGCAAGTTTTAGAGCTAGAAATAGC′; reverse, ′AAAAGCACCGACTCGGTGCC′. The sgRNA was then transcribed (MEGAshortscript T7 Kit, Life Technologies) and purified (MEGAclear Kit, Life Technologies) in ribonuclease-free water. The oligo DNA sequence for recombination is as follows: ′gtccagcccgtggcctctcacctgataattgccaagggaatgaagtacaccagaaaggcgac gatggtcatgtacggggtccagtgcgagtcatccggccacagtgcccagcactgcacctcaccattggaaagtgtccttttcccaaatatgatcagcgtgggaatggaga′.
Superovulated female C57BL/6J mice were mated to C57BL/6J stud males, and fertilized zygotes were collected from oviducts. Cas9 protein (50 ng/ml), sgRNA (20 ng/ml), and targeting oligo DNA (20 ng/ml) were mixed and injected into the pronucleus of fertilized zygotes. Injected zygotes were implanted into oviducts of pseudopregnant CD1 female mice. Founders were genotyped by polymerase chain reaction (PCR) and sequencing. Mice were then crossed with C57BL/6J mice for at least four generations to dilute out potential off-target affects. Two independent lines were chosen for experiments and gave similar results in all tests, demonstrating that the findings were not due to insertion affects. Npsr1 KO mice were obtained as a by-product when generating the knock-in mice. Two founders were found missing the whole exon encoding Y206, which is predicted to cause frameshift from Glu160 (371 amino acids total).
Animal studies
All experimental animals were singly housed on a light-dark (LD) 12:12 cycle and given ad libitum access to food and water. Male mice were used for all behavioral experiments including ANY-maze, EEG, and fear conditioning tests. Mice were at least 8 weeks old at the time of surgery. Littermates were used for studies comparing WT and mutant mice. We noticed that mouse sleep behaviors were affected by light intensity. For the experiments that compared the sleep time and locomotor activity between WT and mutant mice, the light intensity of the room was strictly controlled between 80 and 100 lux.
All experimental protocols were approved by the University of California, San Francisco Institutional Animal Care and Use Committee following the NIH’s Guide for the Care and Use of Laboratory Animals.
ANY-maze monitoring
Mice were kept in individual cages with free access to food and water. Mice were monitored by infrared camera and tracked by an automatic video tracking system (RRID SCR_014289, Stoelting). Mice were entrained to LD 12:12 for 1 week, and then locomotor activity was recorded for 3 to 4 days. Walking distance and mobile times were calculated using the ANY-maze software and data were averaged.
EEG/EMG implantation
Four guide holes were made using a 23-gauge surgical needle placed epidurally over the frontal cortical area (1 mm anterior to bregma and 1 mm lateral to the midline) and over the parietal area (3 mm posterior to bregma and 2.5 mm lateral to midline). One ground screw and three screws with leads were placed into the skull through the holes. The screws with leads were then soldered onto a six-pin connector EEG/EMG headset (Pinnacle Technologies). For EMG recordings, EMG leads from the headset were placed into the neck muscle. The headset was then covered with black dental cement to form a solid cap atop the mouse’s head. The incision was then closed with Vetbond (3M, Santa Cruz Biotech), and animals were given a subcutaneous injection of marcaine (0.05 mg/kg) before recovery on a heating pad. Behavioral experiments were conducted 3 weeks later to allow for sufficient recovery and for viral expression.
EEG/EMG recording and scoring
For EEG/EMG recording, mice were singly housed and habituated to the recording cable for 7 days in LD 12:12. Tethered preamplifiers were attached to the headset of the mice. The signals were relayed through commutators that allowed the animal to move freely. Data were acquired through the Sirenia software package (Pinnacle Technologies) (54).
Sleep was scored semiautomatically with the Sirenia Sleep Pro software in 10-s epochs for wakefulness, NREM, and REM sleep, and then subsequently hand scored by researchers blinded to genotype with the assistance of spectral analysis using fast Fourier transformation. In general, wakefulness was defined as desynchronized low-amplitude EEG and heightened tonic EMG activity with phasic bursts. NREM sleep was defined as synchronized, high-amplitude, low-frequency (0.5 to 4 Hz) EEG and substantially reduced EMG activity compared with wakefulness. REM sleep was defined as having a pronounced theta rhythm (4 to 9 Hz) with no EMG activity.
To examine sleep-wake behavior under baseline conditions, EEG/EMG signals were recorded and analyzed for the entire two consecutive days from the onset of the light phase. Sleep (NREM and REM sleep) time and power spectrum were averaged data from two consecutive days. For SD, mice were sleep deprived for 6 hours from the onset of the light phase by gently touching when they started to recline and lower their heads. Food and water were available. EEG/EMG signals were recorded and analyzed for the entire 18 hours after SD.
For spectral analysis, artifacts and state transition epochs were excluded. Relative NREM EEG power spectra were calculated at a 0.1-Hz resolution. Individual differences were normalized by expressing each frequency bin as a percentage of total EEG power over a 24-hour period for each mouse. As various behavioral states tend to have different EEG power—which could affect total power depending on each individual’s relative time spent in each state—the relative contribution to total power of each state was weighted by the respective time spent in that state (55). The time course of delta power (1.0 to 4.0 Hz) in NREM sleep was computed as previously described (22, 55–57). Change of NREM sleep delta power across the light-dark cycle was determined by the delta band of NREM sleep and normalized to the average NREM sleep delta power during ZT9–12 of the baseline recording day (58, 59). In the dark phase, especially the early dark phase, NREM sleep is absent in some time points. As a result, delta power is not presented for every hour. Intervals were chosen so that every mouse had NREM sleep for the time point shown in the figure.
Stereotaxic viral injection
Animals were anesthetized with 2% isoflurane and placed in a stereotaxic head frame on a heating pad. Opthalmic ointment was applied to the eyes to prevent drying. A midline incision was made down the scalp, and a craniotomy was made using a dental drill. A 10-μl NanoFil Hamilton syringe (WPI) with a pulled glass needle was used to infuse virus with a microsyringe pump (UMP3; WPI) and its controller (Micro4; WPI). Virus was infused at a rate of 50 nl/min. After infusion, the needle was kept at the injection site for 10 min and then slowly withdrawn at 0.01 mm/s. All stereotaxic coordinates were relative to bregma. AAV1/Syn-GCaMP6f.-WPRESV40 was injected into the CMT [−1.5 mm anteroposterior (AP), 0.0 mm mediolateral (ML), −3.7 mm dorsoventral (DV)] or LH (−1.5 mm AP, 1.0 mm ML, −5.0 mm DV) with a total of 300 nl of virus.
Intracerebroventricular injection
Before drug injections, mice were anesthetized with 2% isoflurane and placed in a stereotaxic head frame on a heating pad. For each mouse, the bregma was located without exposure of the skull (60). A guarded 23-gauge needle was used to punch a hole 0.2 mm posterior to the bregma and 1.0 mm lateral to the midline. The Nanofil syringe (WPI) was used to inject NPS (5857, Tocris) (0.1 or 1 nmol in 2 μl of saline) or vehicle (saline) into the right cerebral ventricle at a depth of 2.5 mm from the skull at ZT11. Mice were allowed to recover for 5 min and then placed back in the home cage. Vehicle or SHA 68 (SML1459–25MG, Sigma-Aldrich) [50 mg/kg in phosphate-buffered saline (PBS), 10% cremophor EL] were injected (intraperitoneally) 10 min before NPS. Brain tissues were collected 1 hour after the intracerebroventricular injections.
Calcium imaging in explants
Male Npsr1 mice (either WT or mutant, 8 to 12 weeks) were infused with AAV1/Syn-GCaMP6f.-WPRE-SV40 virus (300 nl) into the CMT or LH using the coordinates described above 2 to 4 weeks before slice preparation. Slices were prepared following the previously reported protocol (61). Briefly, animals were anesthetized under isoflurane and briefly perfused intracardially with 10 ml of ice-cold N-methyl-d-glucamine (NMDG) solution [92 mM NMDG, 30 mM NaHCO3, 25 mM glucose, 20 mM Hepes, 10 mM MgSO4, 5 mM sodium ascorbate, 3 mM sodium pyruvate, 2.5 mM KCl, 2 mM thiourea, 1.25 mM NaH2PO4, and 0.5 mM CaCl2 (pH 7.3, 300 mOsm, bubbled with 95% O2 and 5% CO2)]. The brains were then quickly removed and placed into additional ice-cold NMDG solution for slicing. Coronal slices were cut using a Leica VT1200S vibratome at 300-μm thickness and warmed to 36.5°C for 10 min. Slices were transferred to room temperature (22 to 24°C) Hepes holding solution containing 92 mM NaCl, 30 mM NaHCO3, 25 mM glucose, 20 mM Hepes, 5 mM sodium ascorbate, 3 mM sodium pyruvate, 2.5 mM KCl, 2 mM thiourea, 2 mM MgSO4, 2 mM CaCl2, and 1.25 mM NaH2PO4 (pH 7.3, 300 mOsm, bubbled with 95% O2 and 5% CO2) for 1 to 2 hours.
After incubation, slices were transferred to the recording chamber and constantly perfused with room temperature (22° to 25°C) recording solution containing 119 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 24 mM NaHCO3, 12.5 mM glucose, 2 mM CaCl2, and 2 mM MgSO4 (pH 7.3, 300 mOsm, bubbled with 95% O2 and 5% CO2) at a rate of 4 ml/min.
An integrated microscope (nVista HD, Inscopix) was used to image the GCaMP signal from the slice. We chose this small microscope because the lens that collects the light can go directly into the perfusion buffer from above to image the surface layer cells, which are usually healthier and more accessible to the drug. In addition, the recording and data processing software are commercially available and user-friendly. Although the resolution of the images might be lower than those gathered by some confocal microscopes, it is sufficient for our purposes here. We used the data acquisition software (nVista, Inscopix) to acquire the images [a range of 6 to 10% of LED (light-emitting diode) intensity, gain 2 to 3, 2.5 to 5 fps). During each recording, the recording solution containing NPS (1 μM) was turned on and off at the indicated time point.
The video was then analyzed by the Inscopix Data Processing (Inscopix) software. Each video was processed with spatial crop, spatial filter, motion correction, and dF/F calculation. Regions of interest (ROIs; considered as a single cell) were manually selected on the basis of the shape and dF/F changes throughout the recording. ROIs that exhibited short bursts of dF/F changes or fluctuations during the recording were analyzed. ROIs that showed steady decreases, increases, or maintained constant ΔF/F were excluded from further analysis. The dF/F changes were then aligned with the time window of NPS treatment, and the cells were categorized into four groups based on the alignment. Researchers were blinded to the genotype of the slice when processing and analyzing the data to ensure the same criteria applied to calculate both the WT and mutant cells.
Protein extraction and immunoblot analysis
Cortex and deep brain structures (striatum, thalamus, and hypothalamus) of WT and mutant brains were dissected in ice-cold PBS treated with protease and phosphatase inhibitors (#11697498001 and #5892970001, Roche). Protein was extracted by homogenizing the tissues with 2 ml of radioimmunoprecipitation assay buffer [10 mM tris-HCl (pH 7.4 to 7.6), 150 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate, 1 mM EDTA, 1% NP-40, proteinase inhibitor, and phosphatase inhibitor]. Western blotting was performed according to standard procedures using the corresponding antibodies. Antibodies were used at concentrations recommended by the manufacturer. Antibodies used in this study included anti-CREB (phospho S133) (#9198, Cell Signaling), anti-CREB (#9197, Cell Signaling), anti-EF2 (phospho T56/T58) (ab82981, Abcam), anti-EF2 (#2332, Cell Signaling), anti–synapsin-1 (phospho S605) (#88246, Cell Signaling), and anti–synapsin-1 (sc8295, Santa Cruz Biotech). Band intensities were determined using ImageJ software (NIH).
RNA isolation and real-time PCR
Total RNA was isolated from frozen tissues (brain) with TRIzol reagent (Thermo Fisher Scientific). A total of 5 μg of total RNA was reverse transcribed using the Superscript IV Kit (Thermo Fisher Scientific). cDNA was then quantified using SYBR Green real-time PCR analysis with the QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific). The real-time PCR data were normalized to Actb.
Contextual fear conditioning
Mice were handled gently for 2 min/day for five consecutive days before each experiment and were placed in the experiment room 1 hour before training or test to acclimate to the new environment. On the day of training, at either ZT3 or ZT23, mice were allowed to explore the conditioning chamber for 3 min before two (ZT3) or three (ZT23) 2-s 0.6-mA foot shocks with 1-min interval. Animals were left in the chamber for an additional 1 min after the shock and then returned to their home cage. Twenty-four hours after training, to test mice at the same time of the day, mice were returned to the training chamber for 5 min, and freezing responses were measured. Freezing responses were analyzed using automated tracking software and expressed as a percentage of total time spent in the testing chamber. In the SD group, mice were subjected to 6-hour SD after training.
Statistical analysis
The following methods were used to determine statistical significance: unpaired t test, one-way analysis of variance (ANOVA), two-way ANOVA, and chi-square test. Unless otherwise stated, all values are presented as means ± SEM. Original data are provided in data file S1. Data are judged to be statistically significant when P < 0.05. In figures, asterisks denote statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. All statistical analysis was performed using GraphPad Prism 7 software.
Supplementary Material
Fig. S1. Wake and sleep questionnaires for FNSS.
Fig. S2. Generation of Npsr1-Y206H mice.
Fig. S3. Sleep/wake measurements in Npsr1-Y206H and Npsr1 KO mice.
Fig. S4. EEG data analysis of sleep/wake behavior in Npsr1-Y206H mice.
Fig. S5. High sleep pressure was observed in Npsr1-Y206H mice.
Fig. S6. Hyperphosphorylated SNIPPs were observed in Npsr1-Y206H mice after SD.
Fig. S7. Calcium imaging of LH neurons shows increased activity for one subtype in Npsr1-Y206H mice.
Data file S1. Raw data.
Acknowledgments:
We are grateful to all of the FNSS families participating in our studies. We also thank the University of North Carolina Virus Core for supplying AAV, and all other members in the Ptáček and Fu laboratories for the helpful discussions.
Funding: This work was supported by NIH grant HG005946 to P.-Y.K., NS099333 to L.J.P., NS072360 and NS104782 to Y.-H.F., and by the William Bowes Neurogenetics Fund to L.J.P. and Y.-H.F. Y.M. was supported by NIH T32 HL007731. The generation of mouse models was also supported by NIH P30 DK063720 to the Diabetes Center at UCSF.
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Grandner MA, Chakravorty S, Perlis ML, Oliver L, Gurubhagavatula I, Habitual sleep duration associated with self-reported and objectively determined cardiometabolic risk factors. Sleep Med. 15, 42–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y, Wheaton AG, Chapman DP, Croft JB, Sleep duration and chronic diseases among US adults age 45 years and older: Evidence from the 2010 Behavioral Risk Factor Surveillance System. Sleep 36, 1421–1427 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manoharan S, Jothipriya A, Sleep duration and mortality—A systematic review. J. Pharm. Sci. Res 8, 867–868 (2016). [Google Scholar]
- 4.Schmid SM, Hallschmid M, Schultes B, The metabolic burden of sleep loss. Lancet Diabetes Endocrinol. 3, 52–62 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Curcio G, Ferrara M, De Gennaro L, Sleep loss, learning capacity and academic performance. Sleep Med. Rev 10, 323–337 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Banks S, Dinges DF, Behavioral and physiological consequences of sleep restriction. J. Clin. Sleep Med 3, 519–528 (2007). [PMC free article] [PubMed] [Google Scholar]
- 7.Consensus Conference Panel, Watson NF, Badr MS, Belenky G, Bliwise DL, Buxton OM, Buysse D, Dinges DF, Gangwisch J, Grandner MA, Kushida C, Malhotra RK, Martin JL, Patel SR, Quan SF, Tasali E, Twery M, Croft JB, Maher E, Barrett JA, Thomas SM, Heald JL, Joint Consensus Statement of the American Academy of Sleep Medicine and Sleep Research Society on the recommended amount of sleep for a healthy adult: Methodology and discussion. Sleep 38, 1161–1183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Wheaton AG, Chapman DP, Cunningham TJ, Lu H, Croft JB, Prevalence of healthy sleep duration among adults—United States, 2014. Morb. Mortal. Wkly. Rep 65, 137–141 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Belenky G, Wesensten NJ, Thorne DR, Thomas ML, Sing HC, Redmond DP, Russo MB, Balkin TJ, Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: A sleep dose-response study. J. Sleep Res 12, 1–12 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Sehgal A, Mignot E, Genetics of sleep and sleep disorders. Cell 146, 194–207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basner M, Fomberstein KM, Razavi FM, Banks S, William JH, Rosa RR, Dinges DF, American time use survey: Sleep time and its relationship to waking activities. Sleep 30, 1085–1095 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legates TA, Fernandez DC, Hattar S, Light as a central modulator of circadian rhythms, sleep and affect. Nat. Rev. Neurosci 15, 443–454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamoto-Mizuno K, Mizuno K, Effects of thermal environment on sleep and circadian rhythm. J. Physiol. Anthropol 31, 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Castro JM, The influence of heredity on self-reported sleep patterns in free-living humans. Physiol. Behav 76, 479–486 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Partinen M, Kaprio J, Koskenvuo M, Putkonen P, Langinvainio H, Genetic and environmental determination of human sleep. Sleep 6, 179–185 (1983). [DOI] [PubMed] [Google Scholar]
- 16.He Y, Jones CR, Fujiki N, Xu Y, Guo B, Holder JL, Rossner MJ, Nishino S, Fu YH, The transcriptional repressor DEC2 regulates sleep length in mammals. Science 325, 866–870 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, De Lecea L, Civelli O, Neuropeptide S: A neuropeptide promoting arousal and anxiolytic-like effects. Neuron 43, 487–497 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Hirshkowitz M, Whiton K, Albert SM, Alessi C, Bruni O, Don Carlos L, Hazen N, Herman J, Adams Hillard PJ, Katz ES, Kheirandish-Gozal L, Neubauer DN, O’Donnell AE, Ohayon M, Peever J, Rawding R, Sachdeva RC, Setters B, Vitiello MV, Ware JC, National Sleep Foundation’s updated sleep duration recommendations: Final report. Sleep Health 1, 233–243 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Spada J, Sander C, Burkhardt R, Häntzsch M, Mergl R, Scholz M, Hegerl U, Hensch T, Genetic association of objective sleep phenotypes with a functional polymorphism in the neuropeptide S receptor gene. PLOS ONE 9, e98789 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottlieb DJ, O’Connor GT, Wilk JB, Genome-wide association of sleep and circadian phenotypes. BMC Med. Genet 8, S9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher SP, Godinho SIH, Pothecary CA, Hankins MW, Foster RG, Peirson SN, Rapid assessment of sleep-wake behavior in mice. J. Biol. Rhythms 27, 48–58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Ma J, Miyoshi C, Li Y, Sato M, Ogawa Y, Lou T, Ma C, Gao X, Lee C, Fujiyama T, Yang X, Zhou S, Hotta-Hirashima N, Klewe-Nebenius D, Ikkyu A, Kakizaki M, Kanno S, Cao L, Takahashi S, Peng J, Yu Y, Funato H, Yanagisawa M, Liu Q, Quantitative phosphoproteomic analysis of the molecular substrates of sleep need. Nature 558, 435–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinscheid RK, Xu Y-L, Okamura N, Zeng J, Chung S, Pai R, Wang Z, Civelli O, Pharmacological characterization of human and murine neuropeptide s receptor variants. J. Pharmacol. Exp. Ther 315, 1338–1345 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Clark SD, Duangdao DM, Schulz S, Zhang L, Liu X, Xu YL, Reinscheid RK, Anatomical characterization of the neuropeptide S system in the mouse brain by in situ hybridization and immunohistochemistry. J. Comp. Neurol 519, 1867–1893 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Shaywitz AJ, Greenberg ME, CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem 68, 821–861 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Ruzza C, Rizzi A, Trapella C, Pela M’, Camarda V, Ruggieri V, Filaferro M, Cifani C, Reinscheid RK, Vitale G, Ciccocioppo R, Salvadori S, Guerrini R, Calo G’, Further studies on the pharmacological profile of the neuropeptide S receptor antagonist SHA 68. Peptides 31, 915–925 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Okamura N, Habay SA, Zeng J, Chamberlin AR, Reinscheid RK, Synthesis and pharmacological in vitro and in vivo profile of 3-oxo-1,1-diphenyl-tetrahydrooxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68), a selective antagonist of the neuropeptide S receptor. J. Pharmacol. Exp. Ther 325, 893–901 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erdmann F, Kügler S, Blaesse P, Lange MD, Skryabin BV, Pape HC, Jüngling K, Neuronal expression of the human neuropeptide S receptor NPSR1 identifies NPS-induced calcium signaling pathways. PLOS ONE 10, e0117319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gent TC, Bandarabadi M, Herrera CG, Adamantidis AR, Thalamic dual control of sleep and wakefulness. Nat. Neurosci 21, 974–984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Dongen HPA, Maislin G, Mullington JM, Dinges DF, The cumulative cost of additional wakefulness: Dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep 15, 117–126 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Meisel C, Bailey K, Achermann P, Plenz D, Decline of long-range temporal correlations in the human brain during sustained wakefulness. Sci. Rep 7, 11825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pellegrino R, Kavakli IH, Goel N, Cardinale CJ, Dinges DF, Kuna ST, Maislin G, Van Dongen H. P. a., Tufik S, Hogenesch JB, Hakonarson H, Pack AI, A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans. Sleep 37, 327–1336 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graves LA, Heller EA, Pack AI, Abel T, Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn. Mem 10, 168–176 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, Huang T, Brown KM, Li XY, Descalzi G, Kim SS, Chen T, Shang YZ, Zhuo M, Houslay MD, Abel T, Sleep deprivation impairs cAMP signalling in the hippocampus. Nature 461, 1122–1125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laitinen T, Polvi A, Rydman P, Vendelin J, Pulkkinen V, Salmikangas P, Mäkelä S, Rehn M, Pirskanen A, Rautanen A, Zucchelli M, Gullstén H, Leino M, Alenius H, Petäys T, Haahtela T, Laitinen A, Laprise C, Hudson TJ, Laitinen LA, Kere J, Characterization of a common susceptibility locus for asthma-related traits. Science 304, 300–304 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Ren S, Wang Y, Yue F, Cheng X, Dang R, Qiao Q, Sun X, Li X, Jiang Q, Yao J, Qin H, Wang G, Liao X, Gao D, Xia J, Zhang J, Hu B, Yan J, Wang Y, Xu M, Han Y, Tang X, Chen X, He C, Hu Z, The paraventricular thalamus is a critical thalamic area for wakefulness. Science 362, 429–434 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Goel N, Rao H, Durmer JS, Dinges DF, Neurocognitive consequences of sleep deprivation. Semin. Neurol 29, 320–339 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han RW, Yin XQ, Chang M, Peng YL, Li W, Wang R, Neuropeptide S facilitates spatial memory and mitigates spatial memory impairment induced by N-methyl-d-aspartate receptor antagonist in mice. Neurosci. Lett 455, 74–77 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Okamura N, Garau C, Duangdao DM, Clark SD, Jüngling K, Pape HC, Reinscheid RK, Neuropeptide S enhances memory during the consolidation phase and interacts with noradrenergic systems in the brain. Neuropsychopharmacology 36, 744–752 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng YL, Zhang JN, Chang M, Li W, Han RW, Wang R, Effects of central neuropeptide S in the mouse formalin test. Peptides 31, 1878–1883 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Li W, Chang M, Peng YL, Gao YH, Neuropeptide S produces antinociceptive effects at the supraspinal level in mice. Regul. Pept 156, 90–95 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Ruzza C, Pulga A, Rizzi A, Marzola G, Guerrini R, Calo G ’, Behavioural phenotypic characterization of CD-1 mice lacking the neuropeptide S receptor. Neuropharmacology 62, 1999–2009 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Duangdao DM, Clark SD, Okamura N, Reinscheid RK, Behavioral phenotyping of Neuropeptide S receptor knockout mice. Behav. Brain Res 205, 1–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fendt M, Buchi M, Bürki H, Imobersteg S, Ricoux B, Suply T, Sailer AW, Neuropeptide S receptor deficiency modulates spontaneous locomotor activity and the acoustic startle response. Behav. Brain Res 217, 1–9 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Zhu H, Mingler MK, McBride ML, Murphy AJ, Valenzuela DM, Yancopoulos GD, Williams MT, Vorhees CV, Rothenberg ME, Abnormal response to stress and impaired NPS-induced hyperlocomotion, anxiolytic effect and corticosterone increase in mice lacking NPSR1. Psychoneuroendocrinology 35, 1119–1132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Y, Toh KL, Jones CR, Shin JY, Fu YH, Ptáček LJ, Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell 128, 59–70 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.San lucas FA, Wang G, Scheet P, Peng B, Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics 28, 421–422 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Jian X, Boerwinkle E, dbNSFP v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat 34, E2393–E2402 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM, A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG; Exome Aggregation Consotrium, Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ng PC, Henikoff S, SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31, 3812–3814 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR, A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J, Fuller PM, The GABAergic parafacial zone is a medullary slow wave sleep-promoting center. Nat. Neurosci 17, 1217–1224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franken P, Dudley CA, Estill SJ, Barakat M, Thomason R, O’Hara BF, McKnight SL, NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: Genotype and sex interactions. Proc. Natl. Acad. Sci 103, 7118–7123 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cirelli C, Locus ceruleus control of slow-wave homeostasis. J. Neurosci 25, 4503–4511 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vassalli A, Franken P, Hypocretin (orexin) is critical in sustaining theta/gamma-rich waking behaviors that drive sleep need. Proc. Natl. Acad. Sci 114, E5464–E5473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Franken P, Chollet D, Tafti M, The homeostatic regulation of sleep need is under genetic control. J. Neurosci 21, 2610–2621 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mang GM, Franken P, Sleep and EEG phenotyping in mice. Curr. Protoc. Mouse Biol 2, 55–74 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Kim HY, Lee DK, Chung B-R, Kim HV, Kim Y, Intracerebroventricular injection of amyloid-β peptides in normal mice to acutely induce alzheimer-like cognitive deficits. J. Vis. Exp 10.3791/53308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ting JT, Daigle TL, Chen Q, Feng G, Acute brain slice methods for adult and aging animals: Application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Wake and sleep questionnaires for FNSS.
Fig. S2. Generation of Npsr1-Y206H mice.
Fig. S3. Sleep/wake measurements in Npsr1-Y206H and Npsr1 KO mice.
Fig. S4. EEG data analysis of sleep/wake behavior in Npsr1-Y206H mice.
Fig. S5. High sleep pressure was observed in Npsr1-Y206H mice.
Fig. S6. Hyperphosphorylated SNIPPs were observed in Npsr1-Y206H mice after SD.
Fig. S7. Calcium imaging of LH neurons shows increased activity for one subtype in Npsr1-Y206H mice.
Data file S1. Raw data.