

Plasmodium (malaria) and helminth parasite coinfections are frequent, and both infections can be affected by the host gut microbiota. However, the relationship between coinfection and the gut microbiota is unclear. By performing comprehensive analyses on blood/stool samples from 130 individuals in Colombia, we found that the gut microbiota may have a stronger relationship with the number of P. vivax (malaria) parasites than with the number of helminth parasites infecting a host. Microbiota analysis identified more predictors of the P. vivax parasite burden, whereas analysis of blood samples identified predictors of the helminth parasite burden. These results were unexpected, because we expected each parasite to be associated with greater differences in its biological niche (blood for P. vivax and the intestine for helminths). Instead, we find that bacterial taxa were the strongest predictors of P. vivax parasitemia levels, while circulating TGF-β levels were the strongest predictor of helminth parasite burdens.
KEYWORDS: Colombia, Plasmodium vivax, STH, soil-transmitted helminths, Trichuris trichiura, malaria, microbiota
ABSTRACT
The role of the gut microbiota during coinfection with soil-transmitted helminths (STH) and Plasmodium spp. is poorly understood. We examined peripheral blood and fecal samples from 130 individuals who were either infected with Plasmodium vivax only, coinfected with P. vivax and STH, infected with STH alone, or not infected with either P. vivax or STH. In addition to a complete blood count (CBC) with differential, transcriptional profiling of peripheral blood samples was performed by transcriptome sequencing (RNA-Seq), fecal microbial communities were determined by 16S rRNA gene sequencing, and circulating cytokine levels were measured by bead-based immunoassays. Differences in blood cell counts, including an increased percentage of neutrophils, associated with a transcriptional signature of neutrophil activation, were driven primarily by P. vivax infection. P. vivax infection was also associated with increased levels of interleukin 6 (IL-6), IL-8, and IL-10; these cytokine levels were not affected by STH coinfection. Surprisingly, P. vivax infection was more strongly associated with differences in the microbiota than STH infection. Children infected with only P. vivax exhibited elevated Bacteroides and reduced Prevotella and Clostridiaceae levels, but these differences were not observed in individuals coinfected with STH. We also observed that P. vivax parasitemia was higher in the STH-infected population. When we used machine learning to identify the most important predictors of the P. vivax parasite burden (among P. vivax-infected individuals), bacterial taxa were the strongest predictors of parasitemia. In contrast, circulating transforming growth factor β (TGF-β) was the strongest predictor of the Trichuris trichiura egg burden. This study provides unexpected evidence that the gut microbiota may have a stronger link with P. vivax than with STH infection.
INTRODUCTION
Coinfection with soil-transmitted helminths (STH) and a malaria parasite is a common occurrence due to the geographical overlap of these infections (1). Both parasites can manipulate the host immune response to allow for their own persistence. Helminths can regulate the host immune system to prevent their elimination, which simultaneously protects the host against excessive inflammation (2). Although this may be beneficial for the worm and the host in many cases, it also allows other foreign antigens to remain hidden, thus possibly affecting the immune response to other pathogens (3). The effects of helminth coinfection are therefore complex, and epidemiological studies have often resulted in contradictory conclusions. One potential reason for contradictory results in the literature is that STH infection may render the host more susceptible to coinfection with other pathogens but may also reduce the severity of morbidity resulting from inflammation in response to these other infections. For example, in response to malaria, the host produces high levels of proinflammatory cytokines that help control the parasite but also contribute to pathology (4). In areas of endemicity, repeated infections lead to the development of partially protective immunity to malaria, which frequently results in persistent infections with low levels of parasites (5).
Several studies have found an association between malaria severity and the presence of STH. However, the direction of the association is dependent on the study and the species of STH in the coinfection. One study from Senegal showed that children who tested positive for Ascaris, Ancylostoma, or Trichuris were more susceptible to Plasmodium falciparum infection than their uninfected peers (6). However, another study, also in Senegal, showed that Schistosoma haematobium infection was associated with lower P. falciparum densities (7). When it comes to pathogenesis, a study in Thailand found that Ascaris infection was protective against cerebral malaria (as opposed to uncomplicated malaria cases), suggesting that Ascaris in particular might protect against morbidity resulting from malaria infection (8). It is hypothesized that people infected with helminths are more susceptible to P. falciparum infection but experience severe morbidity and mortality from these infections less frequently (9, 10). In mouse models of coinfection, there is similar confusion over the contrasting effects of helminths, depending on the nature of the rodent malaria model (11). Variation in the gut microbiota between individual mammalian hosts may further confound some of these complex interactions.
There is now evidence that the composition of the gut microbiota in the mammalian host can affect disease severity and protect against infection with malaria parasites (12). In mice, Plasmodium infection can affect the composition of the gut microbiota, changing their susceptibility to other infections or to malaria-induced pathology (13, 14). Different gut bacterial communities of mice from different vendors were responsible for differences in parasite burden and mortality after infection with Plasmodium (15). These studies in mouse models indicate that malaria can induce changes in the gut microbiome of the host but also that the composition of the host microbiome can modulate the development of pathology induced by malaria. Another study in mice found that Escherichia coli among the microbiota expressing α-gal conferred protection against Plasmodium sp. infection (16). Among human subjects, minimal differences in the composition of the gut microbiome were found in children before and after malaria followed by artesunate treatment (17), indicating important differences from mouse models of infection in this respect. However, as with mice, a human-subject study from Mali suggested that the gut microbiome may affect susceptibility to P. falciparum infection; individuals who were at a lower risk of being infected with P. falciparum had significantly higher proportions of Bifidobacterium and Streptococcus in their gut microbiomes (18).
The effects of helminths on the gut microbiota have also been increasingly documented (19). By characterizing the microbiota of a group of indigenous Malaysians known as the Orang Asli using 16S rRNA gene sequencing, we found previously that STH-infected individuals have greater microbial diversity than uninfected individuals (20). Deworming treatment reduces microbial diversity and shifts the community balance by increasing the abundance of Bacteroides and reducing that of Clostridiaceae in this population (21). Furthermore, the parasite burden of Trichuris infection has one of the largest effect sizes on microbial variation (22). Nonetheless, every study population has unique characteristics, and at other study sites, such trends have not been observed (23, 24). Large-intestine-dwelling helminths, such as Trichuris trichiura, may have different effects on the microbiota from small-intestine- and tissue-dwelling helminths (19). Additionally, the effects of anthelmintic drugs on the microbiota are still not definitively understood. Finally, how helminths, malaria, and the microbiota interact to influence the immune response remains unclear.
Here, we examine the association of STH and Plasmodium vivax coinfection with the gut microbiota and immune response through a cross-sectional study based in Colombia. We identified the most important predictors of both T. trichiura and P. vivax parasitemia levels by using machine learning models to integrate the various multiomic measurements. Surprisingly, the gut microbiota may have a stronger relationship with P. vivax infection than with STH infection, since multiple bacterial taxa were identified as the strongest predictors of parasitemia levels in P. vivax-infected individuals.
RESULTS
To study the effects of P. vivax and STH coinfection, we collected blood and stool samples from 130 children aged 4 to 16 years, living in Tierralta, Córdoba, Colombia. Healthy children infected (n = 39) or not (n = 31) with STH were confirmed to be PCR negative for P. vivax in the blood. Children with acute P. vivax malaria (n = 60) were split into those who were infected with any STH (n = 27) and those who were not (n = 33). These two groups had comparable proportions of female and indigenous individuals, as well as similar age distributions (Table 1). Notably, children coinfected with STH had higher P. vivax parasitemia measurements (Table 1; Fig. 1) (P, 0.04 by the Wilcoxon signed-rank test). When STH types were examined separately (see Fig. S1A in the supplemental material), the median P. vivax parasitemia level of individuals in any STH infection group was higher than that for individuals with no STH infection, but the number of individuals in each category is small, and there are no statistically significant differences in P. vivax parasitemia between each pair of groups (Fig. S1B).
TABLE 1.
Characteristics of the study populationa
| Characteristic | Value for group |
Differences between groupsb | |||
|---|---|---|---|---|---|
| Uninfected | P. vivax infected | STH infected | Coinfected | ||
| No. of participants | 31 | 33 | 39 | 27 | |
| No. female | 16 | 18 | 13 | 8 | No sig. diffs. by two-proportion z-test |
| No. indigenous | 0 | 11 | 0 | 7 | Hospital-collected populations similar (two-proportion z-test) |
| Age (yr) | 9.1 ± 2.9 | 12.8 ± 3.2 | 10.6 ± 2.2 | 12.0 ± 2.98 | Differences by ANOVA; sig. diffs. between all pairs except P. vivax and coinfection |
| P. vivax parasitemia (no. of parasites/μl) | 0 | 2,554 ± 2,346 | 0 | 3,901 ± 2,660 | Differences by ANOVA (P = 0.04) between P. vivax and coinfection |
| No. infected with T. trichiura | 0 | 0 | 36 | 25 | No sig. diffs. by two-proportion z-test |
| T. trichiura egg count/g | N/A | N/A | 38 ± 44 | 50 ± 88 | No sig. diffs. by Mann-Whitney test |
| No. infected with A. lumbricoides | 0 | 0 | 11 | 4 | No sig. diffs. by two-proportion z-test |
| A. lumbricoides egg count/g | N/A | N/A | 2,115 ± 3,825 | 347 ± 659 | No sig. diffs. by Mann-Whitney test |
| No. infected with hookworm | 0 | 0 | 3 | 11 | Sig. diff. by two-proportion z-test (P = 0.001) |
| Hookworm egg count/g | N/A | N/A | 295 ± 502 | 37 ± 65 | No sig. diffs. by Mann-Whitney test |
| Location of collection | School | Hospital | School | Hospital | |
The study population included four groups of children: those infected with P. vivax only, those infected with STH only, those infected with both P. vivax and STH, and those not infected with either P. vivax or STH. Children infected with P. vivax (with or without STH infection) were enrolled in the hospital before receiving treatment. Children who were not infected with P. vivax (with or without STH infection) were enrolled from a school within the hospital’s catchment area.
sig. diffs., significant differences.
FIG 1.
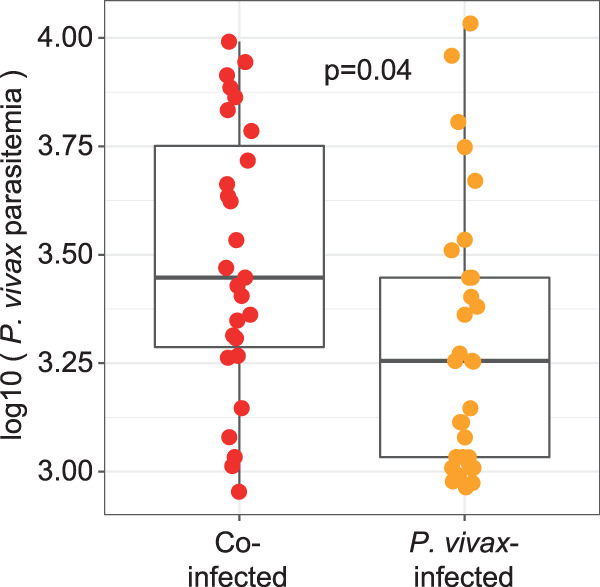
P. vivax parasitemia is higher in STH-coinfected individuals. Box plots show the P. vivax parasitemia levels (expressed as the number of parasites per microliter) of children with both P. vivax and STH infections (red circles) and children with P. vivax infections only (orange circles). P. vivax parasitemia is significantly higher in individuals who are coinfected with STH (P, 0.04 by the Wilcoxon signed-rank test). These box plots (and all other box plots shown) represent the median, interquartile range (box), and 95% confidence interval (whiskers).
(A) Among the 66 individuals infected with STH in this study, T. trichiura was the most common STH infection. However, Ascaris sp. and hookworm infections were present as well. (B) P. vivax parasitemia for each individual in the P. vivax-only and STH-plus-P. vivax groups. In this box plot, samples are grouped by the species or combination of species of STH infecting their host. None of these groups are statistically significant (ANOVA P value, 0.265). Download FIG S1, EPS file, 0.05 MB (52.1KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The frequencies of infection with Trichuris trichiura and Ascaris lumbricoides were similar for children who were coinfected with P. vivax and those who were not. Although more individuals had hookworm infections in the P. vivax coinfected group (n = 11) than in the P. vivax-negative group (n = 3), hookworm prevalence was relatively low (21%), compared to T. trichiura prevalence (92%). There were no significant differences in the egg counts for each of these three STH between children infected with STH only and those coinfected with STH and P. vivax (Table 1). The complete breakdown of the different STH infections is shown in Fig. S1A. While each STH type may have a different relationship with P. vivax, due to the limited number of individuals infected with Ascaris and hookworm, subset analysis was not possible. Because of the preponderance of T. trichiura and the limited frequency of coinfection with other STHs, the observations in this study are primarily for T. trichiura infection.
A complete blood count with differential (CBC w/diff) analysis of the blood of the 130 children in this study provided data on cellular composition and other blood parameters. A principal-component analysis (PCA) plot based on all the measurements from the CBC w/diff shows that children infected with P. vivax only or with STH coinfection largely clustered separately from those not infected with P. vivax (Fig. 2A). Hence, infection with P. vivax, but not with STH, significantly alters multiple blood parameters. Individuals with P. vivax infection diverged from individuals without P. vivax infection along the PC1 axis (Fig. 2A). This indicates that PC1 can explain an important component of the variance that separates P. vivax-infected and uninfected individuals. Identification of factors with negative loading on PC1 (Fig. 2B) indicates that there is a negative association between P. vivax infection and platelets/hematocrit (Fig. 2B and C), which is consistent with the known consequences of P. vivax infection (25, 26). Additionally, identification of factors with positive loading on PC1 (Fig. 2B) indicates that there is a positive association between P. vivax infection and the percentage of neutrophils/monocytes in the blood (Fig. 2B). Indeed, platelets and hematocrit were significantly lower in individuals infected with P. vivax, regardless of STH infection (Fig. 2C), and the percentages of monocytes and neutrophils were elevated in individuals with P. vivax infection (Fig. 2C). Box plots for the remaining 12 variables can be found in Fig. S2. Notably, we did not include asymptomatic P. vivax-infected individuals in this study, and we specifically screened our control groups to be negative for P. vivax infection. Hence, all P. vivax-infected individuals in this study were symptomatic and were recruited in the hospital setting, while all other individuals (uninfected or infected with STH only) were recruited from the community.
FIG 2.

CBC w/diff analysis can distinguish individuals with P. vivax infection but not those with STH infection. (A) Principal-component analysis based on results from CBC w/diff analysis. Each point represents the result for one child and is color-coded by infection status, as throughout the figures. Data are shown in red for children coinfected with P. vivax and STH, in orange for children infected with P. vivax only, in dark blue for children with STH alone, and in light blue for children who were not infected with either parasite. Ellipses show the areas covering 90% of the samples from each group. (B) The factors loading for PC1 reflect the amounts of variance shared by these parameters (either negatively [in blue] or positively [in red]) with the PC1 values. MCV, mean corpuscular volume. (C) Box plots of the two variables most negatively associated with PC1 (platelets and hematocrit) as well as the two variables most positively associated with PC1 (the percentages of monocytes and neutrophils) are shown with the same color scheme. Box plots for the remaining 12 clinical variables can be found in Fig. S2.
Additional box plots of clinical data. Download FIG S2, EPS file, 0.7 MB (772.8KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To examine the relationship between specific cytokines and infection status, we measured the circulating levels of 13 cytokines in plasma samples. A PCA plot based on these variables shows that most individuals had similar levels of these cytokines, except for some individuals with P. vivax infection either with (red dots) or without (orange dots) STH coinfection (Fig. 3A). Interleukin 6 (IL-6), IL-8, IL-10, and IL-4 are the variables that contributed the most to the positive loading along the PC1 axis (Fig. 3B). These four cytokines were elevated in individuals infected with P. vivax, with or without STH coinfection (Fig. 3C).
FIG 3.

Elevation of plasma cytokine levels in some individuals infected with P. vivax. (A) Principal-component analysis of levels of 13 different circulating cytokines measured by bead-based immunoassays. Each point represents the result for one child and is color-coded by infection status, including children coinfected with P. vivax and STH (red), children infected with P. vivax only (orange), children with STH alone (dark blue), and children who were not infected with either parasite (light blue). Ellipses show the areas covering 90% of the samples from each group. (B) The factors positively loading for PC1 reflect the amounts of variance shared by these parameters with the PC1 values. (C) Box plots of the four variables (IL-6, IL-8, IL-10, and IL-4) most positively associated with PC1.
To identify associations between the gut microbial communities and infection status, we performed 16S rRNA gene sequencing on stool samples collected from all children in the study. Shannon diversity did not differ significantly between comparison groups (Fig. S3; P, 0.244 by analysis of variance [ANOVA]). A nonmetric multidimensional scaling (NMDS) plot was constructed to map each sample onto 2-dimensional space, using Jaccard distances (Fig. 4A). Most individual samples clustered together, though some samples from children with P. vivax infections only (without STH coinfection; shown in orange) clustered separately along the first axis. Results were similar based on Bray-Curtis and unweighted UniFrac distances, as shown in Fig. S4. When additional information on STH species (including hookworm and Ascaris) was added into Fig. 4A, we saw no obvious clustering based on STH species (Fig. S5). When the microbiota compositions of children with P. vivax infection only (orange) were compared with those of children with STH coinfection (red), Bacteroides was elevated in individuals without STH coinfection. Prevotella copri, a Prevotella sp., and Clostridiaceae were elevated in coinfected children (Fig. 4B and C). Prevotella copri, a Prevotella sp., and Clostridiaceae were less abundant, and Bacteroides was more abundant, in samples from the P. vivax-only group than in any of the other groups. These results indicate that differences in gut microbial composition are associated with P. vivax infection in some individuals, but none of the P. vivax-infected individuals who were coinfected with STH showed similar alterations in microbial composition.
FIG 4.

Effects of STH coinfection on the microbiota of P. vivax-infected patients. (A) NMDS plot based on Jaccard distances between stool microbiota samples (based on 16S rRNA gene sequencing), colored according to STH-P. vivax infection status. Some individuals with P. vivax infections only (orange) had a distinct microbiota and clustered separately along the first axis. (B) LDA effect sizes calculated using LEFSe (58) are shown, comparing samples from individuals infected with P. vivax alone with samples from those coinfected with P. vivax and STH. Microbes that were significantly (P < 0.01) elevated in individuals infected with P. vivax only are shown in orange, and microbes that were significantly elevated in individuals coinfected with P. vivax and STH are shown in red. This comparison was made based on the visual differentiation of these groups in panel A and the comparability of these two hospital-based groups. (C) Box plots for the three top microbes enriched in samples from coinfected individuals and the top microbe enriched in samples from P. vivax-infected individuals.
Shannon diversity is plotted for each sample analyzed by 16S rRNA gene sequencing, grouped by infection status. Download FIG S3, EPS file, 0.1 MB (70.7KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
(A) An NMDS plot based on Bray-Curtis distance between stool microbiota samples (based on 16S rRNA gene sequencing), colored according to STH and P. vivax infection status. (B) An NMDS plot based on unweighted UniFrac distance between stool microbiota samples (based on 16S rRNA gene sequencing), colored according to STH and P. vivax infection status. As in Fig. 4A, some individuals with P. vivax infections only (orange) had a distinct microbiota and clustered separately along the first axis. Download FIG S4, EPS file, 0.3 MB (268.2KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
An NMDS plot based on Jaccard distance between stool microbiota samples (based on 16S rRNA gene sequencing), colored according to STH (species coded by color) and P. vivax (triangles represent samples from infected individuals) infection status. Some individuals with P. vivax infections only (purple circles) had a distinct microbiota and clustered separately along the first axis. This plot was generated to determine whether the broad definition of STH was masking any major differences in microbiota composition associated with the specific STH species. Download FIG S5, EPS file, 0.1 MB (57.9KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We next performed transcriptome sequencing (RNA-Seq) of the total blood samples that were collected into PAXgene tubes from enrolled children. A PCA plot based on the total RNA-Seq data set shows that most samples had similar transcriptional profiles. There were some individual outliers, but these samples came from all four groups of children (Fig. 5A). Volcano plots comparing gene expression across all six pairwise comparisons between the four groups are shown in Fig. S6. Genes that have a log2 fold change greater than 1 and meet the adjusted P value cutoff of 0.01 are highlighted. DESeq analysis identified 30 genes that were significantly upregulated in children with P. vivax infection only (Fig. 5B). Functional enrichment analysis of these 30 genes using PANTHER (27) indicated that many of these genes are involved in neutrophil function (Fig. 5C). Hence, P. vivax infection appears to be associated with an increase in neutrophil activation in the peripheral blood. As noted above, CBC w/diff analysis had indicated that the percentage of neutrophils was elevated in individuals with P. vivax infection, regardless of STH infection status (Fig. 2C). However, the absolute numbers of neutrophils were similar across all groups (Fig. S2B). Hence, this neutrophil signature may reflect an increased activation state of blood neutrophils and not just an increase in cell number. DESeq analysis comparing STH-infected individuals with uninfected individuals (Fig. S6D), and comparing individuals coinfected with P. vivax and STH with those infected with P. vivax only (Fig. S6E), did not identify any genes that were significantly different, indicating that the transcriptional effects of helminth infection on the blood cells are relatively minor. In other comparisons where one comparison group was infected with malaria and the other was not (such as uninfected individuals versus coinfected individuals [Fig. S6F]), there were significant differences in gene expression between the groups. These are shown in Fig. S6 by marking the P. vivax-infected comparison group with a red M. Finally, DESeq analysis of all P. vivax-infected individuals using raw P. vivax parasitemia as the outcome variable did not identify any genes significantly associated (adjusted P value, >0.05) with parasitemia.
FIG 5.

RNA-Seq of the peripheral blood identifies genes upregulated by P. vivax infection but not by STH infection. (A) PCA plot based on the 50% most variable genes with at least 400 reads across all samples. Ellipses show the areas covering 90% of the samples from each group, revealing the almost complete overlap between groups. (B) Differential abundance analysis by DESeq identifies 30 genes that are upregulated in individuals infected with P. vivax only compared to uninfected individuals (with an adjusted P value of <0.05 and a log2 fold change of ≥1). (C) Biological processes overrepresented in these 30 genes relative to all genes in the Homo sapiens Gene Ontology database (accessed 8 October 2019), using Fisher’s exact test in PANTHER (27). The top 10 specific subclasses (from hierarchically sorted output) are shown, based on the lowest FDR.
Volcano plots comparing gene expression across all six pairwise comparisons between the four groups. Genes that have a log2 fold change greater than 1 and meet the adjusted P value cutoff of 0.01 are highlighted. The comparison group that is infected with P. vivax is shown with an M for malaria. Download FIG S6, EPS file, 0.3 MB (279.4KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Gene set enrichment analysis (GSEA) using a blood transcriptional module (BTM) database (28, 29) identified pathways enriched in monocytes, Toll-like receptor (TLR) and inflammatory signaling, and immune activation in individuals infected with P. vivax only relative to uninfected individuals (Fig. S7). Similar results were found in the comparison of coinfected individuals with uninfected individuals (Fig. S7).
GSEA based on BTM database. mirroring Fig. S6. (A) Coinfection versus no infection; (B) P. vivax-only versus STH infection; (C) coinfection versus STH infection; (D) STH infection versus no infection (no significant differences); (E) coinfection versus P. vivax-only infection; (F) P. vivax-only infection versus no infection. Download FIG S7, EPS file, 0.1 MB (70.9KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Having assembled a heterogeneous set of blood and stool measurements from this study, we wanted to integrate the data and determine which variables were the most important predictors of the four separate groups by feeding all the variables into a random forest model. Since we were most interested in the effects of coinfection, we focused on the differences between individuals with P. vivax infection only and those with P. vivax and STH coinfection. When the variables were sorted by importance to the predictive model (the mean decrease in Gini when the variable was omitted from the model), 8/10 of the most important variables were microbes (Fig. 6A). As expected, some of these microbes overlapped with those identified by linear discriminant analysis (LDA) (Fig. 4): [Prevotella], Prevotella, Bacteroides, Clostridiaceae, and Cetobacterium. (Greengenes taxonomy includes operational taxonomic units [OTUs]) with contested classifications in square brackets.) Other important variables include one gene (NDUFA6, encoding NADH:ubiquinone oxidoreductase subunit A6; not identified as differentially expressed by DESeq2) and P. vivax parasitemia. As noted above, STH coinfection was associated with higher P. vivax parasitemia (Table 1 and Fig. 1). However, it should be noted that the out-of-box error rate for this model was 38%, and a random model would have an error rate of approximately 50%. Hence, the large number of variables we measured provided only a small amount of information that could differentiate between STH-coinfected and P. vivax-only-infected individuals. However, this heterogeneous model selects some of the same microbes identified by LDA (Fig. 4), providing additional support for the conclusion that the most distinct differences between P. vivax-infected individuals with STH infection and those without STH infection were microbial.
FIG 6.

Integrative analysis of heterogeneous data sets using random forest models to identify the strongest predictors of coinfection and parasite burden. (A) The random forest model selected eight microbes (brown) in the top 10 predictors of whether a P. vivax-infected child is also infected with STH. Bars represent the mean decrease in Gini when a variable is removed from the model; a larger decrease means that the variable is more different between individuals infected with P. vivax only and those with both P. vivax and STH infections. Bars are color-coded based on the type of variable: microbes measured by 16S rRNA gene sequencing are shown in brown, genes measured by RNA-Seq in red, cytokines in yellow, and demographic variables from a questionnaire in purple. The model included 4,046 variables: 38 microbial genera, 3,907 genes, 85 measurements from blood tests, including CBC w/diff and cytokine levels, and 16 variables from the demographic questionnaire. Of the 10 variables shown, all were higher in the coinfected group, except for NDUFA6 and Bacteroides, which were higher in the P. vivax-only group. However, the strongest predictors of whether an individual is infected with P. vivax or not (see Fig. S8A) are found in the data from clinical bloodwork and cytokine panels. (B) The random forest model selected seven microbes in the top 10 predictors of P. vivax parasitemia. In this continuous-outcome model, the increase in node purity represents the importance of the variable to the model; higher numbers mean that the variable is more important for predicting P. vivax parasitemia. The model included the same variables as those in panel A, except that the group (the response variable in panel A) was removed and P. vivax parasitemia was made into a response rather than a predictor. (C) To examine one of these important variables, we created a scatter plot, which shows that Prevotella is correlated with P. vivax parasitemia (r2 = 0.13; P = 0.005 [among those infected with P. vivax]). Results for samples from children infected with P. vivax only are shown in orange, and those from children coinfected with STH are shown in red. (D) The random forest model selected TGF-β as the top predictor of the T. trichiura egg count. Other variables that are predictive of egg burden include several genes (from RNA-Seq results), the child’s height, and one microbe. The model included the same variables as those in panel A, except that the T. trichiura egg count was made into a response rather than a predictor. (E) TGF-β is correlated with the T. trichiura egg count (r2 = 0.16; P = 0.002 [among those infected with T. trichiura]). Results for samples from children infected with STH only are shown in blue, and those from children coinfected with P. vivax are shown in red.
(A) Random forest model selection of the top 10 predictors of whether a study subject is in the P. vivax-only group or the uninfected group. Each bar represents the mean decrease in Gini when a variable is removed from the model; a larger decrease means that the variable is more different between the two comparison groups. The model includes microbial, transcriptomic, clinical, and demographic variables, as in Fig. 6. (B) Random forest model selection of the top 10 predictors of whether a study subject is in the STH-only group or the uninfected group. (C) Random forest model selection of the top 10 predictors of whether a study subject is in the P. vivax-only group or the STH-only group. Download FIG S8, EPS file, 0.02 MB (20.3KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
When the same integrative analysis was done to compare P. vivax-only-infected individuals with uninfected individuals, we found that IL-10, IL-6, platelets, and the number of lymphocytes were the most important predictors (Fig. S8A) of P. vivax infection. This was consistent with the results shown in Fig. 2 and 3. When we compared STH-only-infected and uninfected individuals, we found that the monocyte percentages and numbers, as well as MCP.1 and TGF.B1, were the most important predictors of STH infection (Fig. S8B). This is consistent with the results presented in Fig. 2C, showing that the percentage of monocytes is lower in uninfected individuals than in all other groups. The association with transforming growth factor β (TGF-β) is explored further in Fig. 6D and E.
Next, we wanted to determine what variables are important predictors of parasite burden for P. vivax and Trichuris trichiura. At 92% prevalence, T. trichiura was by far the most common STH found in this community. First, a random forest model was run with the continuous outcome of P. vivax parasitemia (Fig. 6B). Strikingly, some of the same microbes identified in the categorical classification above (Fig. 6A) were also identified as important predictors of P. vivax parasitemia levels. Of the 10 most important predictors of parasitemia, 7 were bacterial taxa, highlighting the potential importance of gut bacterial communities in P. vivax infection. Microbes that were predictive both of whether a person was coinfected with STH and of P. vivax parasitemia included [Prevotella], Prevotella, and Slackia. The number of neutrophils was also predictive of P. vivax parasitemia, which is consistent with the other results (Fig. 2 and 5), indicating a role for neutrophils in individuals infected with P. vivax. When we plotted Prevotella read counts against P. vivax parasitemia (Fig. 6C), the coefficient of correlation was low (r2 = 0.128), but the P value for this correlation was significant at 0.005, suggesting a small but significant relationship between these two variables.
When we ran the random forest model to predict the T. trichiura egg count, the strongest predictor was TGF-β (Fig. 6D). When we plotted TGF-β levels against the T. trichiura egg count (Fig. 6E), the coefficient of correlation was low (r2 = 0.158), but the P value for this correlation was significant at 0.002 (Fig. 6E). Notably, although the parameters measured by CBC w/diff (Fig. 2) and plasma cytokine measurements (Fig. 3) appeared to be better at distinguishing individuals from separate groups, these parameters were mostly poor predictors of parasite burdens and coinfection status (with the exception of TGF-β and neutrophil numbers). Of the 10 most important predictors for the T. trichiura egg count, 7 parameters were transcript levels from the RNA-Seq analysis of whole blood. In order to examine the relationship between the T. trichiura egg count and blood RNA-Seq analysis directly, we performed DESeq with the T. trichiura egg count as the outcome. This identified nine genes with adjusted P values below 0.05, shown in Table S1. The top gene identified by the random forest model (RNF10) and the top gene identified by DESeq (DEFA1B) are plotted against the T. trichiura egg count in Fig. S9. Hence, treating the T. trichiura egg count as a continuous variable identified a more significant relationship between egg burden and the expression of specific transcripts in the blood than when STH-infected individuals were compared to uninfected individuals as categorical variables (Fig. S6D and E). In summary, we found that stool sample analysis for bacterial communities identified more predictors of P. vivax parasitemia, whereas blood sample analysis identified more predictors of the T. trichiura egg count, which is the opposite of where the respective parasites reside.
Raw gene read counts for RNF10 (above) and DEFA1B (below) are plotted against the T. trichiura egg count. Download FIG S9, EPS file, 0.1 MB (62.3KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DESeq output with the T. trichiura egg count as the outcome. Download Table S1, DOCX file, 0.01 MB (14.1KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
In this study, we took a multiomic approach toward investigating the effects of coinfection with soil-transmitted helminths and P. vivax. To our surprise, we found that the gut microbiota communities had a stronger association with malaria infection than with STH infections. We had previously reported, in both cross-sectional (20) and longitudinal (21) studies on the Orang Asli in Malaysia, that T. trichiura infection impacted microbial diversity and the composition of the microbiota in infected individuals. In contrast, the current study found that the microbiota of uninfected and STH-infected individuals are not significantly different (Fig. 4A; see also Fig. S5 in the supplemental material). This suggests that the environment in which the microbiota and STH exist (all the other cofactors, such as the diet, age, lifestyle, and other previous and current infections of the human host) may determine whether or not STH affect the gut microbiota. This could explain why the effects of STH on the gut microbiome have not been consistent between studies, even when investigators examine the same STH species (23, 24, 30, 31). Thus, we conclude that the effects of STH infections on the microbiota are dependent on the context of infection.
Here, we observed an association of acute P. vivax infections with microbiome composition; however, the direction of the causal relationship between malaria and the microbiota is still not clear. It could be interpreted as differences in the intestinal microbiota communities being induced by infection with P. vivax or as the result of individuals with particular microbiome characteristics being more susceptible to P. vivax infections. Previous longitudinal studies observed that there are minimal effects of P. falciparum infection on microbiome composition (17) but that the intestinal microbiota composition is associated with the prospective risk of P. falciparum infection (18). Taken together, these studies suggest that the preexisting microbiota composition may affect susceptibility to malaria.
We cannot rule out the possibility that the microbiota samples were impacted by antimalarial treatment, since the fecal samples from P. vivax-infected participants were collected 1 day after the start of antimalarial treatment in the hospital. However, STH-coinfected individuals were also sampled at the same time point after antimalarial treatment, so any effects on coinfected and P. vivax-only-infected individuals should be equivalent.
The effect of STH infections on malaria development and pathogenesis is a highly relevant clinical issue because of the major overlap between these two parasites worldwide (32). We found here that STH-infected individuals have higher P. vivax parasitemia (Fig. 1). In the absence of longitudinal data, it is unclear why individuals with STH infections have higher P. vivax parasitemia. Perhaps some individuals are more susceptible to both infections, or perhaps STH infection induces an immunological environment that is more permissive to P. vivax parasites. Previous studies in Southeast Asia (33) and Africa (34–37) also reported that helminth coinfection had a positive association with the occurrence of malaria caused by P. falciparum or P. vivax infections. While STH may render coinfected people more susceptible, it does not appear to cause increased severity of symptoms (38, 39). In fact, there are also reports that helminth coinfection may protect against certain aspects of pathogenesis, such as P. vivax-induced anemia (40). The relationship between STH infection and P. vivax parasitemia could be mediated by immune modulation induced by helminth infections, which resulted in higher tolerance to disease (41). However, no indication of immune modulation by helminth infection is reflected in the blood transcriptomes of these study participants (Fig. S6 and S7), although DESeq results do suggest that some genes are associated with T. trichiura infection (Table S1).
We also observed an increase in the percentage of neutrophils among white blood cells and in the expression of neutrophil-specific genes in P. vivax infections regardless of STH coinfection, confirming previous evidence that neutrophil activation is an important response to malaria (42). Indeed, a strong neutrophil response with the formation of extracellular traps is observed in acute-phase P. falciparum patients (43). However, a previous study (44) described a decrease in neutrophils and neutrophil-specific genes in P. vivax infections, but this was observed only in first-time infections, possibly accounting for the differences from our results.
In this study, we found that of all the multiomic measurements that we performed, the level of circulating TGF-β was the variable most predictive of the T. trichiura egg burden. Just based on this association, it is not clear whether TGF-β is important for worm expulsion or colonization. However, the relationship between TGF-β and Trichuris infection has been established in the murine model of Trichuris muris infection (45). In this model, TGF-β was shown to act on CD4+ T cells to promote chronic infection instead of worm expulsion. Antibody-mediated blockade of TGF-β activity could protect mice from infection, indicating that increased TGF-β levels may be beneficial for the colonization of the host by the parasite. One possible mechanism may be the induction of Foxp3+ CD4+ regulatory T cells (Tregs) that promote chronic infection by suppressing protective immunity. A previous study on African children found that both Trichuris trichiura and Ascaris lumbricoides infection burdens were associated with IL-10 and TGF-β production from peripheral blood cells (46). Notably, this study also showed that there was a negative relationship between IL-10 and TGF-β production and the ability of these cells to produce TH1 or TH2 cytokines against worm and bacterial antigens. In a different study, a healthy volunteer who ingested eggs from the porcine parasite Trichuris suis also expressed higher levels of TGF-β in the ascending and transverse colon after infection, indicating a direct link between infection and TGF-β expression in humans (47). Finally, there is also evidence from human genetic association studies that TGF-B1 could be important in T. trichiura infection. Polymorphisms in the TGF-B1 gene that affect allergy and asthma are also associated with increased susceptibility to Trichuris trichiura infection, as well as increased IL-10 production (48). Overall, our study provides additional evidence that the link between TGF-β and Trichuris infection may be one of the most important immunological relationships between the host and this parasite.
An important caveat of our analysis is that for the random forest regression model we used, the variances explained were −10% (for P. vivax parasitemia [Fig. 6C]) and −23% (for T. trichiura egg burden [Fig. 6D]), suggesting severely limited power to predict parasitemia from other types of data. For predicting coinfection membership, the model correctly classified 26/33 samples as P. vivax infected but incorrectly classified 16/27 coinfected samples as only P. vivax infected (Fig. 6A). This indicated that the large set of multiomic parameters we measured provided only a small amount of information that could be used to differentiate between samples from these two groups. These models were more useful for identifying specific parameters that were interesting in the data sets and for generating hypotheses than for any substantive predictive value. Much larger samples would have to be collected to improve upon the current studies.
While the gut microbiota may play a role in Plasmodium infections and contribute to the complexities of coinfection with STH, additional interventional studies are needed to determine causality and the directionality (i.e., cause or consequence) of the relationship of the gut bacterial communities with the parasites. Surprisingly, we found that the gut microbiota may have a stronger association with P. vivax infection than with STH infection. One possible explanation is that STH coinfection could be affecting bacterial communities in a way that makes the host more permissive to P. vivax infection. Alternatively, P. vivax could be affecting the gut microbiota in a way that is prevented by STH infection. Longitudinal interventional studies that include deworming and antimalarial treatment could provide greater causal evidence for these linkages.
MATERIALS AND METHODS
Ethics statement.
Written informed consent was obtained from parents or guardians of all study participants. Ethical approval was granted by the Committee on Human Ethics of the Health Sciences Department of the University of Córdoba, Monteria, Colombia, in Acta 007 on 22 May 2017.
Study design and sample collection.
Sample collection was performed between February and June 2019 in the municipality of Tierralta, Colombia. Tierralta is situated in the southwest of the department of Córdoba (8°10′N, –76°04′E).
Children with P. vivax malaria were recruited at Hospital San José of Tierralta. P. vivax infection was diagnosed by a thick blood smear, performed in the morning. Children returned in the afternoon for diagnosis and, if positive, for treatment with chloroquine (10 mg/kg of body weight, followed by 7.5 mg/kg at 24 and 48 h) and primaquine (0.25 mg/kg for 14 days), according to the National Health Institute of Colombia guidelines. None of the children were hospitalized. Blood and stool samples were collected on the same day or the next day after P. vivax malaria diagnosis. In the hospital, children were asked whether they had used antimalarials in the past 15 days before sampling for the diagnosis of malaria, and all answered no. The malaria diagnosis was done in 3 to 4 h, whereupon the children received a prescription for the antimalarial treatment, so they were started on antimalarials on the same day. Therefore, both P. vivax-only and P. vivax- and STH-coinfected participants started the antimalarial treatment 1 day before the sampling for feces.
The groups of control children and STH-infected children were recruited at a local school, Institución Educativa Santa Fe de Ralito, within the municipality of Tierralta. The catchment area of the school is within the larger catchment area of the hospital. An informational meeting with the parents of children recruited for the study was held to explain the procedures and to distribute screw-cap containers. Study participants returned the stool samples on the next day; at that time, study coordinators also collected blood samples. All study participants received anthelmintic treatment (400 mg albendazole) after Kato-Katz results were obtained, on average 3 to 5 days after sample collection. Thus, all stool and blood samples were collected prior to treatment administration.
Collection and processing of stool samples.
Samples were transported on the same morning to the Universidad de Córdoba, where they were divided into two portions: one portion was processed on the same day by the Kato-Katz method, and the other portion was stored at –20°C until DNA was extracted. All study participants received anthelmintic treatment (400 mg albendazole) after Kato Katz results were obtained—on average 3 to 5 days after sample collection. Thus, all stool and blood samples were collected prior to treatment administration. Although researchers did not ask about recent antibiotic use, it is unlikely that any of the study participants had taken antibiotics in the previous 2 weeks, since none of the participants reported diarrhea or other symptoms that might have prompted them to take antibiotics.
The species diagnosis and intensity of infection were determined by the Kato-Katz method, and results were recorded as eggs per gram, following WHO guidelines (https://apps.who.int/iris/handle/10665/63821). The numbers of eggs per gram were recorded for Trichuris trichiura, Ascaris lumbricoides, and hookworm. DNA was extracted using the DNeasy PowerSoil kit (Qiagen) according to the manufacturer’s instructions.
Microbiome sequencing and analysis.
The 16S rRNA gene was amplified at the V4 hypervariable region and was sequenced according to a multiplexing protocol described elsewhere (49, 50) on the Illumina MiSeq system by paired-end sequencing for 2 × 150 bp reads. Automated 16S rRNA amplicon PCR libraries using GTC F/R primers were added to each sample; samples were then normalized and pooled. Microbiome bioinformatics were performed with QIIME 2 2019.4 (51). Raw sequence data were demultiplexed and quality filtered using the q2-demux plugin and denoising with DADA2 (52). Amplicon sequence variants were aligned with mafft (53) and were used to construct a phylogeny with fasttree (54). Alpha and beta diversity metrics were estimated using q2-diversity after samples were rarefied (subsampled without replacement) to 25,000 sequences per sample. Taxonomy was assigned to amplicon sequence variants (ASVs) using the q2‐feature‐classifier (55) classify‐sklearn, a naive Bayes taxonomy classifier, against the Greengenes (version 13.8) 99% OTU reference sequences (56).
QIIME2 output files were imported into R using Phyloseq (57), and Jaccard distances between samples were calculated using nonmetric multidimensional scaling (NMDS). Principal-component analysis was done based on this dissimilarity matrix. Ellipses were superimposed, representing a 95% confidence level.
To examine the relative abundances of microbial taxa between comparison groups, we performed linear discriminant analysis (LDA) effect size (LEfSe) tests (58) with a P value cutoff of 0.01, grouping samples by genus.
Collection and processing of blood samples.
Peripheral blood (5 ml in EDTA) was collected from each subject and processed on the same day by centrifugation at 1,372 × g for 5 min. Plasma was aliquoted and frozen at –80°C. Aliquots from all samples were shipped in dry ice to the New York University School of Medicine. Cytokines were determined by using LEGENDplex inflammation panel I from BioLegend (San Diego, CA) for IL-1β, IL-2, IL-4, IL-6, IL-10, IL-8, IL-12p70, IL-17A, IP10, tumor necrosis factor alpha (TNF-α), monocyte chemoattractant protein 1 (MCP1), gamma interferon (IFN-γ), and TGF-β1 on a FACSCalibur system (Becton, Dickinson, Franklin Lakes, NJ).
Peripheral blood was also collected in PAXgene Blood RNA tubes (PreAnalytiX; Qiagen, Valencia, CA) and was stored at –80°C until RNA isolation. RNA isolation was performed using the PAXgene blood RNA kit (PreAnalytiX; Qiagen, Valencia, CA) according to the manufacturer’s instructions. The celseq2 analysis method (https://yanailab.github.io/celseq2/) for processing CEL-Seq2 single-cell RNA-Seq data was performed for two lanes of a paired-end 50-cycle Illumina NovaSeq 6000 run conducted by the NYU School of Medicine Genome Technology Core. Per-read, per-lane FASTQ files were generated using the bcl2fastq2 Conversion software (version 2.2.0) to convert per-cycle binary base call (BCL) files outputted by the sequencing instrument into the FASTQ format. For generating the feature-cell unique molecular identifier (UMI) count matrix from the multiplexed FASTQ files as input, a global configuration in YAML format was first specified according to the associated list of CEL-Seq bar codes (6 bp in length, located 7 bases from the start of Read1) as well as applicable transcriptome annotation information for read alignment to the human reference hg19 and/or the mouse reference mm10 using the alignment program Bowtie2 (version 2.3.4.1); an experiment table containing relevant metadata (SAMPLE_NAME, CELL_BARCODES_INDEX, R1, R2) was defined; and the celseq2 software’s “COUNT_MATRIX” automated pipeline (version 0.5.3.3) was run in order to successively demultiplex the 6-bp CEL-Seq barcodes from the multiplexed data, map reads of human samples to the associated species reference, and finally generate the matrix of UMI counts for annotated genomic features.
RNA-Seq data analyses.
To perform PCA on the RNA-Seq data, we further filtered the above count matrix (where genes differential by sequencing batch were removed) and kept only genes with >10 counts in at least 10% of samples. We next performed a variance stabilizing transformation (vst) on the filtered count data matrix, generated as described above, and removed genes with low variance (bottom half) using the varFilter function in the R/Bioconductor genefilter package (59). These filtering steps resulted in 6,076 genes, on which we performed PCA using the vst-transformed count values.
We used DESeq2 (60) to analyze differences between gene expression in comparison groups. Significance limits for upregulated genes were set at a P value of <0.05, and the log2 fold change cutoff was set as ≥1. The genes identified by DESeq2 were then supplied to PANTHER; biological processes overrepresented in these genes relative to all genes in the Homo sapiens Gene Ontology (GO) database (accessed 8 October 2019) were enumerated, using Fisher’s exact test for significance in PANTHER (27). The top 10 specific subclasses (from hierarchically sorted output) were selected, based on the lowest false discovery rate (FDR). Gene set enrichment analysis (GSEA) was done using the blood transcription modules (BTMs) (28, 29).
Random forest analyses.
Metadata and clinical and cytokine test results were combined with 16S rRNA gene sequencing read counts (grouped by genus) and RNA-Seq read counts (filtering out all genes with <400 reads across all samples). Response variables used in separate analyses were as follows: group (comparing one infection category to another), P. vivax parasitemia (continuous outcome), and T. trichiura egg count (continuous outcome). Variables that provided redundant information were removed from the analyses (for example, the T. trichiura egg count was removed as a predictor when the response variable was the P. vivax-only-infected group versus the group coinfected with STH, since the observation of eggs in the stool was a determining characteristic for inclusion in the latter group). Analyses were done using randomForest in R (61). For the group analysis, a classification tree was used with 10,000 trees, and 63 variables were tried at each split. When the response variable was P. vivax parasitemia, the random forest regression model based on 10,000 trees used 1,346 variables at each split. The random forest regression model to predict the T. trichiura egg count was based on 10,000 trees and used 1,345 variables at each split. Individual scatter plots were made based on variables of interest identified by these analyses. In order to add a best-fit line and r2 to the plot, a linear model was run.
Data availability.
Raw data of microbial 16S rRNA sequencing have been deposited in the European Nucleotide Archive under accession number ERP119792. Raw RNA-Seq data have been deposited in NCBI’s Gene Expression Omnibus data repository under accession number GSE144792.
ACKNOWLEDGMENTS
This work was supported in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH; by NIH grants AI133977 and AI130945 to P.L.; and by Universidad de Córdoba FCS 01-19 resolución 1875 to Maria Fernanda Yasnot and Mayra Raciny-Aleman.
We acknowledge the helpful suggestions of Joseph Cooper Devlin, Kelly V. Ruggles, and Pratik Worah. We also acknowledge the Tierralta children who participated in the study, their families, and the Santa Fe de Ralito school in Tierralta, Colombia. Finally, thanks to Maria Camila Velasco, Ana Karina Nisperuza, and Jorge Arrieta for help with sample collection and laboratory analysis.
Footnotes
Citation Easton AV, Raciny-Aleman M, Liu V, Ruan E, Marier C, Heguy A, Yasnot MF, Rodriguez A, Loke P. 2020. Immune response and microbiota profiles during coinfection with Plasmodium vivax and soil-transmitted helminths. mBio 11:e01705-20. https://doi.org/10.1128/mBio.01705-20.
Contributor Information
David Serre, University of Maryland, Baltimore.
Jacques Ravel, University of Maryland School of Medicine.
REFERENCES
- 1.Mwangi TW, Bethony JM, Brooker S. 2006. Malaria and helminth interactions in humans: an epidemiological viewpoint. Ann Trop Med Parasitol 100:551–570. doi: 10.1179/136485906X118468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maizels RM, Yazdanbakhsh M. 2003. Immune regulation by helminth parasites: cellular and molecular mechanisms. Nat Rev Immunol 3:733–744. doi: 10.1038/nri1183. [DOI] [PubMed] [Google Scholar]
- 3.Hartgers FC, Yazdanbakhsh M. 2006. Co-infection of helminths and malaria: modulation of the immune responses to malaria. Parasite Immunol 28:497–506. doi: 10.1111/j.1365-3024.2006.00901.x. [DOI] [PubMed] [Google Scholar]
- 4.Moxon CA, Gibbins MP, McGuinness D, Milner DA, Marti M. 2020. New insights into malaria pathogenesis. Annu Rev Pathol 15:315–343. doi: 10.1146/annurev-pathmechdis-012419-032640. [DOI] [PubMed] [Google Scholar]
- 5.Bousema T, Okell L, Felger I, Drakeley C. 2014. Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat Rev Microbiol 12:833–840. doi: 10.1038/nrmicro3364. [DOI] [PubMed] [Google Scholar]
- 6.Spiegel A, Tall A, Raphenon G, Trape JF, Druilhe P. 2003. Increased frequency of malaria attacks in subjects co-infected by intestinal worms and Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg 97:198–199. doi: 10.1016/s0035-9203(03)90117-9. [DOI] [PubMed] [Google Scholar]
- 7.Briand V, Watier L, Le Hesran J-Y, Garcia A, Cot M. 2005. Coinfection with Plasmodium falciparum and Schistosoma haematobium: protective effect of schistosomiasis on malaria in Senegalese children? Am J Trop Med Hyg 72:702–707. doi: 10.4269/ajtmh.2005.72.702. [DOI] [PubMed] [Google Scholar]
- 8.Nacher M, Gay F, Singhasivanon P, Krudsood S, Treeprasertsuk S, Mazier D, Vouldoukis I, Looareesuwan S. 2000. Ascaris lumbricoides infection is associated with protection from cerebral malaria. Parasite Immunol 22:107–113. doi: 10.1046/j.1365-3024.2000.00284.x. [DOI] [PubMed] [Google Scholar]
- 9.Druilhe P, Tall A, Sokhna C. 2005. Worms can worsen malaria: towards a new means to roll back malaria? Trends Parasitol 21:359–362. doi: 10.1016/j.pt.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Degarege A, Veledar E, Degarege D, Erko B, Nacher M, Madhivanan P. 2016. Plasmodium falciparum and soil-transmitted helminth co-infections among children in sub-Saharan Africa: a systematic review and meta-analysis. Parasit Vectors 9:344. doi: 10.1186/s13071-016-1594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knowles SCL. 2011. The effect of helminth co-infection on malaria in mice: a meta-analysis. Int J Parasitol 41:1041–1051. doi: 10.1016/j.ijpara.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Ippolito MM, Denny JE, Langelier C, Sears CL, Schmidt NW. 2018. Malaria and the microbiome: a systematic review. Clin Infect Dis 67:1831–1839. doi: 10.1093/cid/ciy374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniguchi T, Miyauchi E, Nakamura S, Hirai M, Suzue K, Imai T, Nomura T, Handa T, Okada H, Shimokawa C, Onishi R, Olia A, Hirata J, Tomita H, Ohno H, Horii T, Hisaeda H. 2015. Plasmodium berghei ANKA causes intestinal malaria associated with dysbiosis. Sci Rep 5:15699. doi: 10.1038/srep15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mooney JP, Lokken KL, Byndloss MX, George MD, Velazquez EM, Faber F, Butler BP, Walker GT, Ali MM, Potts R, Tiffany C, Ahmer BMM, Luckhart S, Tsolis RM. 2015. Inflammation-associated alterations to the intestinal microbiota reduce colonization resistance against non-typhoidal Salmonella during concurrent malaria parasite infection. Sci Rep 5:14603. doi: 10.1038/srep14603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villarino NF, LeCleir GR, Denny JE, Dearth SP, Harding CL, Sloan SS, Gribble JL, Campagna SR, Wilhelm SW, Schmidt NW. 2016. Composition of the gut microbiota modulates the severity of malaria. Proc Natl Acad Sci U S A 113:2235–2240. doi: 10.1073/pnas.1504887113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yilmaz B, Portugal S, Tran TM, Gozzelino R, Ramos S, Gomes J, Regalado A, Cowan PJ, d’Apice AJF, Chong AS, Doumbo OK, Traore B, Crompton PD, Silveira H, Soares MP. 2014. Gut microbiota elicits a protective immune response against malaria transmission. Cell 159:1277–1289. doi: 10.1016/j.cell.2014.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandal RK, Crane RJ, Berkley JA, Gumbi W, Wambua J, Ngoi JM, Ndungu FM, Schmidt NW. 2019. Longitudinal analysis of infant stool bacteria communities before and after acute febrile malaria and artemether-lumefantrine treatment. J Infect Dis 220:687–698. doi: 10.1093/infdis/jiy740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yooseph S, Kirkness EF, Tran TM, Harkins DM, Jones MB, Torralba MG, O’Connell E, Nutman TB, Doumbo S, Doumbo OK, Traore B, Crompton PD, Nelson KE. 2015. Stool microbiota composition is associated with the prospective risk of Plasmodium falciparum infection. BMC Genomics 16:631. doi: 10.1186/s12864-015-1819-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cortés A, Peachey L, Scotti R, Jenkins TP, Cantacessi C. 2019. Helminth-microbiota cross-talk—a journey through the vertebrate digestive system. Mol Biochem Parasitol 233:111222. doi: 10.1016/j.molbiopara.2019.111222. [DOI] [PubMed] [Google Scholar]
- 20.Lee SC, Tang MS, Lim YAL, Choy SH, Kurtz ZD, Cox LM, Gundra UM, Cho I, Bonneau R, Blaser MJ, Chua KH, Loke P. 2014. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis 8:e2880. doi: 10.1371/journal.pntd.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramanan D, Bowcutt R, Lee SC, Tang MS, Kurtz ZD, Ding Y, Honda K, Gause WC, Blaser MJ, Bonneau RA, Lim YAL, Loke P, Cadwell K. 2016. Helminth infection promotes colonization resistance via type 2 immunity. Science 352:608–612. doi: 10.1126/science.aaf3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SC, Tang MS, Easton AV, Devlin JC, Chua LL, Cho I, Moy FM, Khang TF, Lim YAL, Loke P. 2019. Linking the effects of helminth infection, diet and the gut microbiota with human whole-blood signatures. PLoS Pathog 15:e1008066. doi: 10.1371/journal.ppat.1008066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin I, Djuardi Y, Sartono E, Rosa BA, Supali T, Mitreva M, Houwing-Duistermaat JJ, Yazdanbakhsh M. 2018. Dynamic changes in human-gut microbiome in relation to a placebo-controlled anthelminthic trial in Indonesia. PLoS Negl Trop Dis 12:e0006620. doi: 10.1371/journal.pntd.0006620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper P, Walker AW, Reyes J, Chico M, Salter SJ, Vaca M, Parkhill J. 2013. Patent human infections with the whipworm, Trichuris trichiura, are not associated with alterations in the faecal microbiota. PLoS One 8:e76573. doi: 10.1371/journal.pone.0076573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coelho HCC, Lopes SCP, Pimentel JPD, Nogueira PA, Costa FTM, Siqueira AM, Melo GC, Monteiro WM, Malheiro A, Lacerda MVG. 2013. Thrombocytopenia in Plasmodium vivax malaria is related to platelets phagocytosis. PLoS One 8:e63410. doi: 10.1371/journal.pone.0063410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leal-Santos FA, Silva SBR, Crepaldi NP, Nery AF, Martin TOG, Alves-Junior ER, Fontes CJF. 2013. Altered platelet indices as potential markers of severe and complicated malaria caused by Plasmodium vivax: a cross-sectional descriptive study. Malar J 12:462. doi: 10.1186/1475-2875-12-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. 2019. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47:D419–D426. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, Schmidt DS, Johnson SE, Milton A, Rajam G, Kasturi S, Carlone GM, Quinn C, Chaussabel D, Palucka AK, Mulligan MJ, Ahmed R, Stephens DS, Nakaya HI, Pulendran B. 2014. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol 15:195–204. doi: 10.1038/ni.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kazmin D, Nakaya HI, Lee EK, Johnson MJ, van der Most R, van den Berg RA, Ballou WR, Jongert E, Wille-Reece U, Ockenhouse C, Aderem A, Zak DE, Sadoff J, Hendriks J, Wrammert J, Ahmed R, Pulendran B. 2017. Systems analysis of protective immune responses to RTS,S malaria vaccination in humans. Proc Natl Acad Sci U S A 114:2425–2430. doi: 10.1073/pnas.1621489114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Easton AV, Quiñones M, Vujkovic-Cvijin I, Oliveira RG, Kepha S, Odiere MR, Anderson RM, Belkaid Y, Nutman TB. 2019. The impact of anthelmintic treatment on human gut microbiota based on cross-sectional and pre- and postdeworming comparisons in Western Kenya. mBio 10:e00519-19. doi: 10.1128/mBio.00519-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosa BA, Supali T, Gankpala L, Djuardi Y, Sartono E, Zhou Y, Fischer K, Martin J, Tyagi R, Bolay FK, Fischer PU, Yazdanbakhsh M, Mitreva M. 2018. Differential human gut microbiome assemblages during soil-transmitted helminth infections in Indonesia and Liberia. Microbiome 6:33. doi: 10.1186/s40168-018-0416-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brooker S, Akhwale W, Pullan R, Estambale B, Clarke SE, Snow RW, Hotez PJ. 2007. Epidemiology of plasmodium-helminth co-infection in Africa: populations at risk, potential impact on anemia, and prospects for combining control. Am J Trop Med Hyg 77(6 Suppl):88–98. doi: 10.4269/ajtmh.2007.77.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burdam FH, Hakimi M, Thio F, Kenangalem E, Indrawanti R, Noviyanti R, Trianty L, Marfurt J, Handayuni I, Soenarto Y, Douglas NM, Anstey NM, Price RN, Poespoprodjo JR. 2016. Asymptomatic vivax and falciparum parasitaemia with helminth co-infection: major risk factors for anaemia in early life. PLoS One 11:e0160917. doi: 10.1371/journal.pone.0160917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuasha N, Hailemeskel E, Erko B, Petros B. 2019. Comorbidity of intestinal helminthiases among malaria outpatients of Wondo Genet health centers, southern Ethiopia: implications for integrated control. BMC Infect Dis 19:659. doi: 10.1186/s12879-019-4290-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Degarege A, Legesse M, Medhin G, Animut A, Erko B. 2012. Malaria and related outcomes in patients with intestinal helminths: a cross-sectional study. BMC Infect Dis 12:291. doi: 10.1186/1471-2334-12-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babamale OA, Ugbomoiko US, Heukelbach J. 2018. High prevalence of Plasmodium falciparum and soil-transmitted helminth co-infections in a periurban community in Kwara State, Nigeria. J Infect Public Health 11:48–53. doi: 10.1016/j.jiph.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Efunshile AM, Olawale T, Stensvold CR, Kurtzhals JAL, König B. 2015. Epidemiological study of the association between malaria and helminth infections in Nigeria. Am J Trop Med Hyg 92:578–582. doi: 10.4269/ajtmh.14-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Degarege A, Animut A, Legesse M, Erko B. 2009. Malaria severity status in patients with soil-transmitted helminth infections. Acta Trop 112:8–11. doi: 10.1016/j.actatropica.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 39.Hürlimann E, Houngbedji CA, Yapi RB, N’Dri PB, Silué KD, Ouattara M, Utzinger J, N'Goran EK, Raso G. 2019. Antagonistic effects of Plasmodium-helminth co-infections on malaria pathology in different population groups in Côte d’Ivoire. PLoS Negl Trop Dis 13:e0007086. doi: 10.1371/journal.pntd.0007086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melo GC, Reyes-Lecca RC, Vitor-Silva S, Monteiro WM, Martins M, Benzecry SG, Alecrim MDGC, Lacerda MVG. 2010. Concurrent helminthic infection protects schoolchildren with Plasmodium vivax from anemia. PLoS One 5:e11206. doi: 10.1371/journal.pone.0011206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frosch AEP, John CC. 2012. Immunomodulation in Plasmodium falciparum malaria: experiments in nature and their conflicting implications for potential therapeutic agents. Expert Rev Anti Infect Ther 10:1343–1356. doi: 10.1586/eri.12.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aitken EH, Alemu A, Rogerson SJ. 2018. Neutrophils and malaria. Front Immunol 9:3005. doi: 10.3389/fimmu.2018.03005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knackstedt SL, Georgiadou A, Apel F, Abu-Abed U, Moxon CA, Cunnington AJ, Raupach B, Cunningham D, Langhorne J, Krüger R, Barrera V, Harding SP, Berg A, Patel S, Otterdal K, Mordmüller B, Schwarzer E, Brinkmann V, Zychlinsky A, Amulic B. 2019. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci Immunol 4:eaaw0336. doi: 10.1126/sciimmunol.aaw0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vallejo AF, Read RC, Arevalo-Herrera M, Herrera S, Elliott T, Polak ME. 2018. Malaria systems immunology: Plasmodium vivax induces tolerance during primary infection through dysregulation of neutrophils and dendritic cells. J Infect 77:440–447. doi: 10.1016/j.jinf.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Worthington JJ, Klementowicz JE, Rahman S, Czajkowska BI, Smedley C, Waldmann H, Sparwasser T, Grencis RK, Travis MA. 2013. Loss of the TGFβ-activating integrin αvβ8 on dendritic cells protects mice from chronic intestinal parasitic infection via control of type 2 immunity. PLoS Pathog 9:e1003675. doi: 10.1371/journal.ppat.1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turner JD, Jackson JA, Faulkner H, Behnke J, Else KJ, Kamgno J, Boussinesq M, Bradley JE. 2008. Intensity of intestinal infection with multiple worm species is related to regulatory cytokine output and immune hyporesponsiveness. J Infect Dis 197:1204–1212. doi: 10.1086/586717. [DOI] [PubMed] [Google Scholar]
- 47.Williams AR, Dige A, Rasmussen TK, Hvas CL, Dahlerup JF, Iversen L, Stensvold CR, Agnholt J, Nejsum P. 2017. Immune responses and parasitological observations induced during probiotic treatment with medicinal Trichuris suis ova in a healthy volunteer. Immunol Lett 188:32–37. doi: 10.1016/j.imlet.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Costa RDS, Figueiredo CA, Barreto ML, Alcantara-Neves NM, Rodrigues LC, Cruz AA, Vergara C, Rafaels N, Foster C, Potee J, Campbell M, Mathias RA, Barnes KC. 2017. Effect of polymorphisms on TGFB1 on allergic asthma and helminth infection in an African admixed population. Ann Allergy Asthma Immunol 118:483–488.e1. doi: 10.1016/j.anai.2017.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Caporaso JG. 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Segaa N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gentleman R, Carey V, Huber W, Hahne F. 2019. genefilter: methods for filtering genes from high-throughput experiments. R package, version 1.70.0. [Google Scholar]
- 60.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liaw A, Wiener M. December 2002. Classification and regression by randomForest. R News 2/3:18–22. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Among the 66 individuals infected with STH in this study, T. trichiura was the most common STH infection. However, Ascaris sp. and hookworm infections were present as well. (B) P. vivax parasitemia for each individual in the P. vivax-only and STH-plus-P. vivax groups. In this box plot, samples are grouped by the species or combination of species of STH infecting their host. None of these groups are statistically significant (ANOVA P value, 0.265). Download FIG S1, EPS file, 0.05 MB (52.1KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Additional box plots of clinical data. Download FIG S2, EPS file, 0.7 MB (772.8KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Shannon diversity is plotted for each sample analyzed by 16S rRNA gene sequencing, grouped by infection status. Download FIG S3, EPS file, 0.1 MB (70.7KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
(A) An NMDS plot based on Bray-Curtis distance between stool microbiota samples (based on 16S rRNA gene sequencing), colored according to STH and P. vivax infection status. (B) An NMDS plot based on unweighted UniFrac distance between stool microbiota samples (based on 16S rRNA gene sequencing), colored according to STH and P. vivax infection status. As in Fig. 4A, some individuals with P. vivax infections only (orange) had a distinct microbiota and clustered separately along the first axis. Download FIG S4, EPS file, 0.3 MB (268.2KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
An NMDS plot based on Jaccard distance between stool microbiota samples (based on 16S rRNA gene sequencing), colored according to STH (species coded by color) and P. vivax (triangles represent samples from infected individuals) infection status. Some individuals with P. vivax infections only (purple circles) had a distinct microbiota and clustered separately along the first axis. This plot was generated to determine whether the broad definition of STH was masking any major differences in microbiota composition associated with the specific STH species. Download FIG S5, EPS file, 0.1 MB (57.9KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Volcano plots comparing gene expression across all six pairwise comparisons between the four groups. Genes that have a log2 fold change greater than 1 and meet the adjusted P value cutoff of 0.01 are highlighted. The comparison group that is infected with P. vivax is shown with an M for malaria. Download FIG S6, EPS file, 0.3 MB (279.4KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
GSEA based on BTM database. mirroring Fig. S6. (A) Coinfection versus no infection; (B) P. vivax-only versus STH infection; (C) coinfection versus STH infection; (D) STH infection versus no infection (no significant differences); (E) coinfection versus P. vivax-only infection; (F) P. vivax-only infection versus no infection. Download FIG S7, EPS file, 0.1 MB (70.9KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
(A) Random forest model selection of the top 10 predictors of whether a study subject is in the P. vivax-only group or the uninfected group. Each bar represents the mean decrease in Gini when a variable is removed from the model; a larger decrease means that the variable is more different between the two comparison groups. The model includes microbial, transcriptomic, clinical, and demographic variables, as in Fig. 6. (B) Random forest model selection of the top 10 predictors of whether a study subject is in the STH-only group or the uninfected group. (C) Random forest model selection of the top 10 predictors of whether a study subject is in the P. vivax-only group or the STH-only group. Download FIG S8, EPS file, 0.02 MB (20.3KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Raw gene read counts for RNF10 (above) and DEFA1B (below) are plotted against the T. trichiura egg count. Download FIG S9, EPS file, 0.1 MB (62.3KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DESeq output with the T. trichiura egg count as the outcome. Download Table S1, DOCX file, 0.01 MB (14.1KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
Raw data of microbial 16S rRNA sequencing have been deposited in the European Nucleotide Archive under accession number ERP119792. Raw RNA-Seq data have been deposited in NCBI’s Gene Expression Omnibus data repository under accession number GSE144792.