

Abstract
Since its first emergence from China in late 2019, the SARS-CoV-2 virus has spread globally despite unprecedented containment efforts, resulting in a catastrophic worldwide pandemic. Successful identification and isolation of infected individuals can drastically curtail virus spread and limit outbreaks. However, during the early stages of global transmission, point-of-care diagnostics were largely unavailable and continue to remain difficult to procure, greatly inhibiting public health efforts to mitigate spread. Furthermore, the most prevalent testing kits rely on reagent-and time-intensive protocols to detect viral RNA, preventing rapid and cost-effective diagnosis. Therefore the development of an extensive toolkit for point-of-care diagnostics that is expeditiously adaptable to new emerging pathogens is of critical public health importance. Recently, a number of novel CRISPR-based diagnostics have been developed to detect COVID-19. Herein, we outline the development of a CRISPR-based nucleic acid molecular diagnostic utilizing a Cas13d ribonuclease derived from Ruminococcus flavefaciens (CasRx) to detect SARS-CoV-2, an approach we term SENSR (Sensitive Enzymatic Nucleic-acid Sequence Reporter). We demonstrate SENSR robustly detects SARS-CoV-2 sequences in both synthetic and patient-derived samples by lateral flow and fluorescence, thus expanding the available point-of-care diagnostics to combat current and future pandemics.
Introduction
Following emergence from China in late 20191–3, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1,4 has spread to almost every country despite unprecedented control efforts5. Compared to H1N1, Ebola, MERS, and SARS-CoV-1 outbreaks of recent decades, this novel coronavirus represents the first pandemic characterized by widespread global transmission coupled with significant mortality. Pre-symptomatic and asymptomatic carriers have been identified as major contributors to the prolific spread of SARS-CoV-24,6,7 however, in many cases these patients go unidentified and unisolated, exacerbating the spread of the disease. Thus, robust identification and isolation of all infected individuals are essential for controlling disease transmission. The SARS-CoV-2 pandemic thus presents an unparalleled global public health emergency, which spurred the urgent development of molecular diagnostics and therapeutics for timely patient identification, isolation and treatment. The economic, health, and societal damage wrought by SARS-CoV-2, highlights the importance of expanding and improving on current diagnostic technologies to identify and prevent future pandemics.
Early diagnostics detected SARS-CoV-2 infection through the amplification of viral RNA (vRNA) by real-time reverse transcription polymerase chain reaction (RT-PCR)8–10. These tests were time consuming11, limited by reagents12, required advanced equipment, and yielded significant false-negatives13–16 possibly exacerbated by genetic variation within the targeted viral genomic sequences17. Next generation sequencing-based diagnostics reduced false-negative rates, but still require specialized equipment and are slow (~12 hours)18. Therefore, developing alternative technologies with the potential to yield cost- and time-effective point-of-care diagnostics demands investment.
CRISPR-Cas nucleases can be easily programmed to target nucleic acids in a sequence-specific manner19–21, making them prime candidates for the detection and diagnosis of viral genetic material, and forming the CRISPR-based diagnostics (CRISPRDx) pipeline22–25. These systems rely on Type II Cas enzymes to physically bind target sequences26, or collateral cleavage by Type V or Type VI enzymes to detect DNA24,25,27 or RNA species, respectively22,23,28. Since pandemic onset, an array of innovative diagnostics and prophylactics relying on these technologies have been adapted to detect or target SARS-CoV-2 with unprecedented speed26,29–40, most notably represented by the DETECTR (DNA Endonuclease Targeted CRISPR Trans Reporter)24,25 and SHERLOCK (Specific High-Sensitivity Enzymatic Reporter unLOCKing)22,23 systems (Summarized in Fig. S1, Table S1).
The SHERLOCK system combines isothermal amplification of target sequences, followed by target recognition via Leptotrichia wadei Cas13a (LwaCas13a) and collateral cleavage of a bystander ssRNA probe to report the presence of a target22. This system has undergone significant optimization since its first development in 2017. This includes improvement of i) sensitivity, by the inclusion of an accessory protein to amplify signal or substitution of RPA with LAMP23,41,42, ii) specificity, by primer and guide optimization22,23, iii) throughput, by multiplexing detection using additional enzymes (including a cocktail of LwaCas13a, PsmCas13b (Prevotella sp. MA2016), CcaCas13b (Capnocytophaga canimorsus Cc5), and AsCas12a (Acidaminococcus sp. BV3L6))23, and iv) validation as a point-of-care diagnostic by using lateral flow and ultrafast RNA extraction methods23,31,33,43. Ideally, to maximize all the capabilities of SHERLOCK and expand the CRISPRDx toolkit, it is important to evaluate alternative Cas enzymes that can complement or supplement the system.
Similar to Cas ribonucleases used in other CRISPRDx systems, Cas13d enzymes such as RfxCas13d (CasRx), exclusively target RNA species that trigger subsequent collateral cleavage of bystander RNA44–46. Collateral cleavage is initiated following on-target ssRNA cleavage by the HEPN domain-based endoRNase heterodimer, which activates trans-cleavage of nonspecific bystander RNAs20,44,46,47. Furthermore, Cas13d enzymes are approximately 20% smaller than Cas13a-Cas13c effectors, and do not require a Protospacer Flanking Sequence (PFS)20,44,46,48, presenting an advantage for protein production and flexible targeting. While the genetic modulatory effects of CasRx have been thoroughly characterized in Drosophila, zebrafish, and human cells44,45,49, and its putative prophylactic properties against SARS-CoV-2 have been demonstrated40, its potential as a diagnostic system has not yet been explored.
In an effort to expand the CRISPRDx toolkit, herein, we report the first use of CasRx44 as a molecular diagnostic, developing a unique system we term SENSR (Sensitive Enzymatic Nucleic-acid Sequence Reporter) and demonstrating robust detection of SARS-CoV-2 viral sequences (Fig. 1A). Following on-target cleavage, CasRx exhibits collateral cleavage of off-target nucleic acids44,45, a feature shared by Cas nucleases used in other CRISPRDx systems22–25. We exploit the collateral cleavage activity of CasRx to detect SARS-CoV-2 in both synthetic templates as well as in patient-derived samples via fluorescence-based readout, and paper-based lateral flow assay. To maximize specificity, we performed an extensive bioinformatic analysis to identify novel conserved and specific viral targets to minimize false-negative and false-positive rates, respectively. We demonstrate that SENSR facilitates attomolar sensitivity in just under two hours total reaction time. The detection limit of SENSR is comparable to, though slightly less than, previously established CRISPRDx systems31,35,38 and shows promise for improvement.
Figure 1. Harnessing SENSR to detect evidence of SARS-CoV-2 viral transcripts.

[A] Overview of assay workflow. Following extraction of viral RNA, the detection protocol requires three distinct steps, the last of which differs based on desired output detection method. In the first reaction, specific target sequences within the viral RNA are reverse transcribed (RT) into cDNA and amplified by RPA at 42°C for 45m, while also adding T7 promoter sequences to the 5’ terminus (T7, purple). In the next reaction, in vitro transcription occurs simultaneously with CasRx collateral cleavage activation (pink) by recognition and cleavage of the target RNA sequence (purple) through the sequence-specific targeting activity of the gRNA (emerald). Addition of a probe conjugated to fluorescein and a quencher can facilitate readout by fluorescence following probe cleavage (top right). Addition of a probe conjugated to fluorescein and biotin facilitates readout by lateral flow assay (bottom right). [B] Schematic of the SARS-CoV-2 genome, RPA reagents, gRNA target sites, and synthetic amplicon position. The E (envelope) and N (nucleocapsid) genes are enlarged to depict the design schematic in more detail (purple and blue respectively). The schematic depicts the relative position of the RT-RPA primers and gRNAs used in the amplification step. The gRNA target map outlines all six gRNAs tested through the course of this work, with their relative positions. The E-gene encompases 227 total nt while the N-gene encompases 1,260 nt, and gRNA lengths and positions are shown to scale. Synthetic target amplicon denotes the length of the synthetic viral genome fragment used to test assay cleavage and its relative position within the gene coding sequence. [C] Preliminary characterization of CasRx fluorescence detection by N-targeting gRNAs (gRNA-Z, -AA, -AC) with the addition of 300 fM dsDNA template (N-gene template, 524bp) to an IVT-coupled cleavage reaction. Shown as Background Subtracted Units, A.U. over time (4 technical replicates each, Mean and SEM). gRNA-Z was selected for further downstream analysis. [D] Preliminary characterization of CasRx fluorescence detection by E-targeting gRNAs (gRNA-R, -T, -V) with the addition of 300 fM dsDNA template (E-gene template, 253 bp) to an IVT-coupled cleavage reaction. Plotted as Background Subtracted Units, A.U. over time (4 technical replicates each, Mean and SEM). gRNA-T was selected for further downstream analysis. [E] The in vitro cleavage properties of the E-targeting gRNA-T. In Lane 1 the absence of gRNA-T results in no cleavage of the E-gene fragment (228 nt). In Lane 2 the addition of gRNA-T results in cleavage of the E-gene template resulting in loss of the distinct 228 nt band and appearance of lower molecular weight cleavage species. In Lane 3, the off-target N-gene template (500 nt) remains uncleaved in the presence of CasRx and the E-targeting gRNA-T. In Lane 4, the absence of CasRx results in no cleavage of either E or N-gene templates. In Lane 5, the on-target cleavage of the E-gene template results in additional collateral cleavage of the N-gene template, resulting in the loss of both bands from the gel and the accumulation of lower molecular weight cleavage products. [F] The in vitro cleavage properties of the N-targeting gRNA-Z. In Lane 1, the absence of gRNA-Z results in no cleavage of the N-gene fragment (500 nt). In Lane 2, the addition of gRNA-Z results in cleavage of the N-gene template resulting in loss of the distinct 500 nt band and appearance of lower molecular weight cleavage species. In Lane 3, the off-target E-gene template (228 nt) remains uncleaved in the presence of CasRx and the N-targeting gRNA-Z. In Lane 4, the absence of CasRx results in no cleavage of either E or N-gene templates. In Lane 5, on-target cleavage of the N-gene template results in additional collateral cleavage of the E-gene template, resulting in the loss of both bands from the gel and the accumulation of lower molecular weight cleavage products.
Results
Development of the SENSR protocol
Derived from protocols originally developed for (Fig. S1, Table S1) SHERLOCK22,23,31,48, we designed a two-step protocol for the detection of nucleic acids using recombinant CasRx purified after expression in E. coli (Fig. S2). The protocol requires an initial 45-minute isothermal preamplification reaction combining reverse transcriptase and recombinase polymerase amplification (RT-RPA)50 producing a short dsDNA amplicon encompassing the CasRx target site and containing a T7 promoter sequence. This is followed by an in vitro transcription (IVT)-coupled cleavage reaction which converts the dsDNA amplicons into ssRNA, recognizable by CasRx for cleavage, resulting in collateral cleavage of a bystander ssRNA probe. Collateral cleavage of the probe is analyzed by either fluorescence or lateral flow readouts, thus indicating the initial presence or absence of SARS-CoV-2 genomic sequences (Fig. 1A, Fig S3).
Target selection and reagent validation
To ensure high analytical specificity, we identified conserved gRNA target sites using a bioinformatic pipeline to identify 30 nt target sequences conserved among the first 433 published SARS-CoV-2 genomes available at Genbank on April 7th, 2020, and without homology to other coronaviruses (ViPR, Virus Pathogen Resource, n = 3,164). This yielded a panel of gRNA target sites (n=8846) less likely to result in false positives or negatives due to sequence constraints (Fig. S4, Table S2, S3). Because RT-qPCR assays recommended by the WHO and CDC target the envelope (E) and nucleocapsid (N) genes within the SARS-CoV-2 genome, we selected these genes as the targets of SENSR for system validation (Fig. 1B)8,51. The bioinformatic analysis revealed multiple specific sequences within the N-gene (n = 150), to which we designed three gRNAs (gRNA-Z, -AA, -AC) (Table S2, Fig. S5A). However, the stringent bioinformatic search criteria did not identify targets specific to SARS-CoV-2 within the E-gene due to high sequence homology with the SARS-CoV-1 E-gene. We, therefore, relaxed the criteria to include target sites sharing homology with distantly related SARS-CoV-1 which is presently absent from the general population. From this, we selected three E-targeting guides (gRNA-R, -T, -V), with two displaying complete (gRNA-T (433/433), gRNA-V (433/433)) and one with nearly complete (gRNA-R (430/433)) conservation among the available SARS-CoV-2 genomes (Table S4, Fig. S5A). To validate these gRNAs, we generated synthetic SARS-CoV-2 targets encompassing the E- and N-genes (Table S5), and tested for in vitro cleavage with purified CasRx protein (Fig. S2). Initial characterization of on-target cleavage properties revealed significant degradation of target transcripts for all guides tested (Fig. S5B, C) motivating further assessment of all candidates. To determine the most effective gRNAs for use in SENSR, we monitored fluorescence accumulation over time in an IVT-coupled cleavage reaction for each gRNA. All gRNAs induced robust fluorescence within minutes, with the exception of gRNA-AA and -AC which produced no signal (Fig. 1C, D). gRNA-T (E-targeting) and gRNA-Z (N-targeting) were selected for downstream analysis due to their robust cleavage activity as well as sequence conservation among all publicly available SARS-CoV-2 patient sample isolates (Fig. 1B–D, Table S2, S3).
Fluorescence-based detection of SARS-CoV-2
We and others have recently demonstrated that on-target cleavage activates a secondary collateral cleavage property of CasRx44,45. We initially evaluated the in vitro collateral cleavage activity of CasRx with gRNA-T and gRNA-Z through gel electrophoresis. By incubating CasRx, gRNA-T or gRNA-Z, and varying the addition of synthetic templates, we found CasRx collateral cleavage was only activated when the synthetic template added complemented the gRNA target sequence (Fig. 1E, F). We sought to exploit this tandem, collateral cleavage activity to cleave a bystander fluorescent probe in trans, facilitating detection of SARS-CoV-2 by fluorimetry (Fig. 1A, Fig. S3). The quenched fluorescent reporter within RNaseAlert v2 has been used to report collateral cleavage of Leptotrichia wadeii (LwaCas13a), Capnocytophaga canimorsus Cc5 (CcaCas13b), and PsmCas13b (Prevotella sp. P5–125) in the SHERLOCK systems22,23,48, but our preliminary analysis of RNaseAlert v2 failed to yield fluorescence with CasRx in the presence of a synthetic target (Student’s t-test gRNA-T: p=0.4294, gRNA-Z: p=0.1510) (Fig. S6A). Prior work suggested CasRx preferentially cleaves targets containing poly-U stretches44, with complementary work demonstrating collateral cleavage of targets rich in both poly-A and poly-U stretches46. To develop a probe cleavable by CasRx, we therefore generated two custom 6 nucleotide ssRNA probes, poly-A and poly-U, each conjugated to a 5’ fluorescent molecule (6-FAM) and a 3’ fluorescence quencher (FQ), whose separation following cleavage results in detectable fluorescence accumulation (Fig. 1A). Following incubation of CasRx, with either gRNA-T or -Z, in the presence or absence of their respective synthetic targets (E- or N-gene), the poly-U probe yielded significant fluorescence for 10000 copies/μL compared to the no-template control (NTC) (Student’s t-test gRNA-T, gRNA-Z: p<0.0001), while the poly-A probe produced no detectable fluorescence signal (Student’s t-test gRNA-T: p=0.5953, gRNA-Z: p=0.7935), suggesting a preference for poly-U stretches by CasRx, and motivating its use in the remainder of experiments (Fig. S6A, Table S5).
Following probe selection, we moved to evaluate the collateral cleavage activity in the context of fluorescence. We set to determine if the gRNA incubated with the respective target sequence dictates the increase in fluorescence signal over time. To do so, we incubated CasRx, gRNA-T or gRNA-Z, the modified poly-U probe, and varied the addition of synthetic templates, while fluorescence data were acquired on a plate reader. We observed fluorescence signal only accumulated, and thus collateral cleavage activated, when the synthetic template added to the reaction complemented the gRNA target sequence (Fig. 2A, B). After optimizing preamplification reaction input volume using 100 copies/μL of synthetic RNA, and determining 50% preamplification reaction volume input to be optimal (Fig. S6B), we then determined the LOD by fluorescence for gRNA-T and gRNA-Z titrated at log scale from 10,000 to 0 copies/μL. We determined the LOD to be 100 copies/μL (Fig. 2C, D), indicating SENSR exhibits attomolar sensitivity comparable to other CRISPRDx systems22,24. These results demonstrate CasRx can robustly detect and report the presence of synthetic SARS-CoV-2 RNA via fluorescence readout.
Figure 2. SARS-CoV-2 detection by SENSR via Fluorescence and Lateral Flow assay.
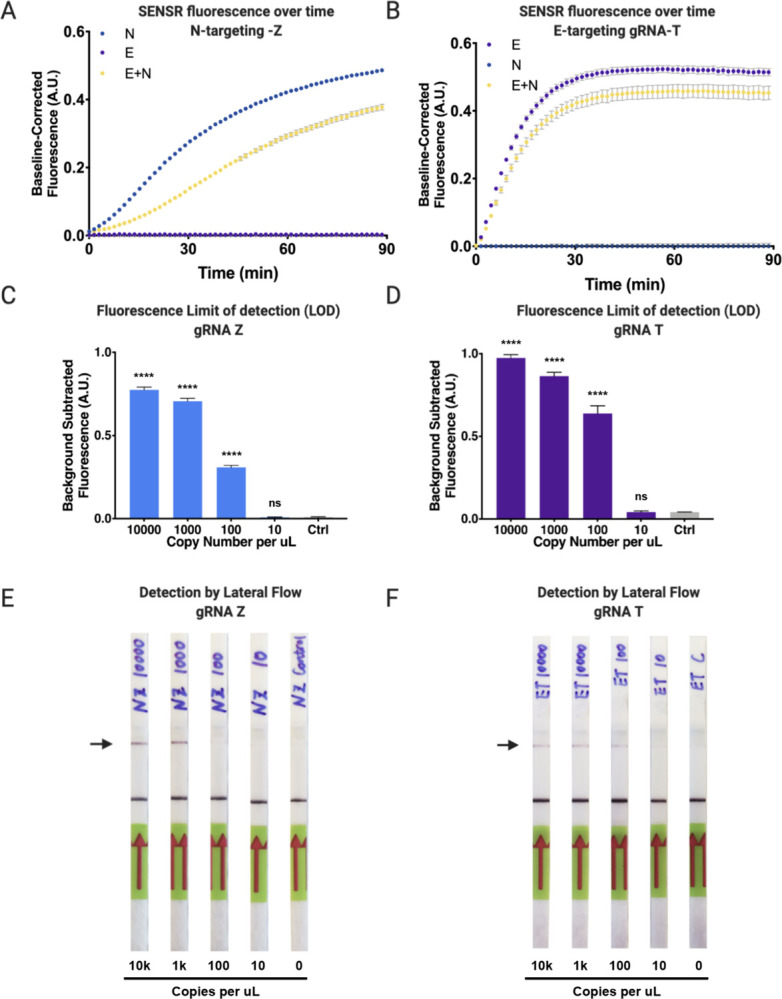
[A] Cleavage properties of E-targeting gRNA-T in fluorescence context. Fluorescence detection of E-gene in an IVT-coupled coupled context. gRNA-T incubated in three different contexts: with E-gene template, with N-gene template, or with both E- and N-gene template. Accumulation of fluorescence occurs when gRNA-T is incubated with the E-gene target template leading to cleavage of the ssRNA probe. Collateral cleavage is not observed when gRNA-T is incubated with the non-target N-gene template. [B] Cleavage properties of N-targeting gRNA-Z in fluorescence context. Fluorescence detection of N-gene in an IVT-coupled coupled context. gRNA-Z incubated in three different contexts: with N-gene template, with E-gene template, or with both N- and E-gene template. Accumulation of fluorescence occurs when gRNA-Z is incubated with the N-gene target template leading to cleavage of the ssRNA probe. Collateral cleavage is not observed when gRNA-Z is incubated with the non-target E-gene template. [C] CasRx nucleic acid limit of detection (LOD) for gRNA-Z detection via fluorescence following cleavage of N-gene target. Total copy number of 10000, 1000, 100, 10, or 0 of viral RNA template input into initial RT-RPA reaction, followed by CasRx detection by fluorescence. Performing a one-way ANOVA followed by a Dunnett’s test call copy numbers to the NTC significance was found for 10000, 1000, and 100 copies (p<0.0001) and no significance was found for 10 copies (p=0.9999). Results shown as background-subtracted fluorescence (A.U.) following 90 minute fluorescent readout (4 technical replicates each, Mean and SD). [D] CasRx nucleic acid LOD for gRNA-T detection via fluorescence following cleavage of E-gene target. Total copy number of 10000, 1000, 100, 10, or 0 of viral RNA template input into initial RT-RPA reaction, followed by CasRx detection by fluorescence. Performing a one-way ANOVA followed by a Dunnett’s test call copy numbers to the NTC significance was found for 10000, 1000, and 100 copies (p<0.0001) and no significance was found for 10 copies (p>0.9999). Results shown as background-subtracted fluorescence (A.U.) following 90 minute fluorescent readout (4 technical replicates each, Mean and SD). [E] LOD of the gRNA-Z recognition of N-gene target by lateral flow assay. CasRx detection reaction incubated for 1h prior to lateral flow assay. [F] LOD of the gRNA-T recognition of E-gene target by lateral flow assay. CasRx detection reaction incubated for 1h prior to lateral flow assay.
Lateral flow assay development
Collateral cleavage by CasRx can additionally be exploited to detect synthetic SARS-CoV-2 RNA by lateral flow assay, facilitating detection by simple paper test strip and eliminating the need for expensive laboratory equipment (Fig. 1A, Fig. S3). Similar to assays developed by others23,48, we developed a 6 nucleotide ssRNA probe conjugated with 5’ 6-FAM and 3’ biotin (Bio) compatible with Millenia HybriDetect lateral flow strips (Table S5). In brief, collateral cleavage results in separation of 6-FAM from biotin, detectable following capillary action down a paper dipstick imprinted with streptavidin or anti-FAM secondary antibodies at distant ends (Fig. 1A). The absence or presence of the upper band therefore indicates a negative or positive result, respectively (Summarized in Fig. 1A, Fig. S3). Using this protocol, we demonstrated that SENSR can be adaptable to readout by lateral flow, and determined the LOD of synthetic SARS-CoV-2 RNA to be as low as ~100 copies/μL, however with variability between guides (Fig. 2E, F). These results confirm that SENSR, like other CRISPRDx systems, can be adapted for read-out by lateral flow, indicating the potential for future application within point-of-care rapid diagnostic tests.
Specificity of SENSR against known possible off-targets
Diagnostic assays require stringent specificity parameters to limit false- negatives/positives. Because many Cas effectors tolerate some degree of mismatch52–54, unintended false-positives can occur as a result of cleaving closely related off-target sequences. In a health-care setting, SENSR is unlikely to be exposed to randomly generated high-identity sequences, and will more likely encounter closely related natural homologs. Therefore, we identified the four highest-identity natural homologous sequences to the gRNA-T and gRNA-Z target sites via BLAST. In each case, SARS-CoV-1 variants, Bat coronaviruses, and Pangolin coronaviruses were identified as the most closely related potential off-targets (OT), containing 2 or 3 mismatches, with gRNA-Z also targeting an additional unknown marine virus and a porcine genome sequence with 7 mismatches (Fig. S7A, B, Table S6). We synthesized these sequences as DNA templates containing a T7 promoter and assayed for off-target cleavage tolerance of gRNA-T and gRNA-Z in an IVT-coupled cleavage reaction analyzed via fluorescence detection. We found that gRNA-T was sufficiently specific to exclusively recognize the synthetic E-gene fragment (ANOVA, p<0.001), precluding recognition of any off-targets, which showed no significant differences in fluorescent signal with the NTC (ANOVA, Dunnett’s test OT1: p=0.9998, OT2: p=0.5242, OT3: p=0.6633, OT4: p=0.8475) (Fig. S7C). gRNA-Z demonstrated detection of the N-gene synthetic template and the most similar pangolin coronavirus off-target templates compared to the NTC (N, OT1, OT2: p<0.0001), thus presenting some degree of mismatch tolerance for gRNA-Z. Fluorescence signal for OT3 and OT4 did not differ from the NTC (ANOVA, Dunnett’s test OT3: p=0.8877, OT4: p>0.9999) (Fig. S7D).
CasRx-based detection of SARS-CoV-2 from patient isolates
We next sought to determine the capability of SENSR to detect SARS-CoV-2 from infected patient samples and directly compared these results to RT-qPCR-validated diagnostics. RT-qPCR analysis of patient samples was performed by targeting the N-, S-, and Orf1ab-genes (Table S7), and accordingly, we selected gRNA-Z to directly compare SENSR fluorescence detection to N-gene RT-qPCR Ct values. We performed fluorescence detection analysis on 42 RT-qPCR validated positive (n = 21) and negative (n = 21) patient samples. By fluorescence, SENSR yielded no false-positives among negative patient samples demonstrating 100% specificity (0/21), and obtained a conservative 57% concordance with confirmed positive samples (12/21) when the threshold for detection is set at S/N > 2 (Fig. 3A, B, Table S7). For RT-qPCR analysis of viral infections, a lower Ct value indicates a higher viral load within an isolated sample. SENSR demonstrated robust detection of infected patient samples with Ct values ≤ 20, detecting SARS-CoV-2 vRNA in (8/9) such samples (Sample ID: 1–9) (Fig. 3A, Table S7). SENSR could detect SARS-CoV-2 vRNA up to a maximum Ct value ≤ 28 with a moderate 25% (4/16) false-negative rate (Fig. 3A, B, Table S7). Next, we confirmed detection of SARS-CoV-2 can be achieved by lateral flow analysis. We performed lateral flow analysis on the 12 positive samples resulting in S/N > 2, and correlating to a Ct ≤ 28, to determine if SENSR can operate as a point-of-care diagnostic. Using lateral flow, we observed 92% (11/12) concordance with SENSR fluorescence analysis (Fig. 3C). These findings confirm SENSR can be successfully adapted for the rapid detection of SARS-CoV-2 from patient samples, however, further optimization will be required to improve sensitivity and consistency.
Figure 3. SENSR detection of positive SARS-CoV-2 validated patient samples.

[A] SENSR fluorescence analysis of RT-qPCR validated patient samples using gRNA-Z for detection. Samples 1–21 are patient samples positive for SARS-CoV-2 listed in order of ascending RT-qPCR Ct values for the N-gene where low Ct value is equal to a high viral load. The black and gray rings represent the Ct values for N, S, and Orf1ab genes, where black represents the N-gene Ct values and gray represents the S and Orf1ab Ct values. Samples N1–N5 are the five samples negative for SARS-CoV-2 with the highest recorded signalto-noise ratios (S/N) of all negative samples analyzed with the highest S/N = 1.8. The dashed line represents the S/N = 2 threshold used to determine a positive detection of SARS-CoV-2. The asterisks indicate a positive detection of SARS-CoV-2 in the patient sample. [B] Summary of fluorescence detection results for the RT-qPCR validated positive and negative patient samples. [C] Lateral flow based detection of the 12 samples from [A] that resulted in a positive detection of SARS-CoV-2 (S/N > 2). The top band represents the test band and the bottom the control band. An increase in saturation of the top band indicates a positive detection of SARS-CoV-2 in the sample. A positive result was also determined by comparing to the NTC, which served as a baseline for a negative result.
Discussion
With an increasingly interconnected world and expanding global population, future pandemics are inevitable. The COVID-19 pandemic spread prolifically in the early months of 2020, with containment elusive in part due to the scarcity of point-of-care diagnostics. The seemingly infinite adaptability of CRISPR has, or promises to, accelerate the development of everything from life-saving gene therapies55–57 and pig-to-human organ donations58; to disease-eradicating gene drives59–61 and possibly the re-animation of the Woolly Mammoth62,63 - with CRISPR-based diagnostics (CRISPRDx) being no exception. Though still nascent, CRISPRDx, like other CRISPR technologies, has proven fast to develop, highly flexible, capable of multiplexing, making it the ideal toolkit from which to develop expeditious future point-of-care diagnostics. The CRISPRDx technologies developed prior to the COVID-19 pandemic, such as SHERLOCK and DETECTR, may have helped halt disease transmission had they been deployed earlier and implemented more widely. Therefore, it is important to prepare now, well in advance of the next pandemic, by perfecting and expanding the CRISPRDx toolkit to the bounds of its capabilities.
Complementing the rapidly expanding CRISPRDx toolkit (Fig. S1, Table S1), here we outline the use of RfxCas13d (CasRx) to diagnose SARS-CoV-2 using both synthetic targets and patient derived samples. SENSR amplifies nucleic acid sequences through an isothermal alternative to PCR then detects the target sequences by exploiting the native collateral cleavage activity of CasRx (Fig. 1A), providing proof-of-principle that Cas13d can be adapted as a point-of-care diagnostic. We lend further evidence that CasRx cleavage results in activation of an off-target collateral cleavage property (Fig. 1 E, F)44,64, with a preference for poly-U stretches (Fig. S6A). This feature can be harnessed to detect viral sequences in a single reaction or possibly even in a multiplexed reaction combined with other Cas enzymes lacking a poly-U preference23,48. We identify highly conserved and specific targets to SARS-CoV-2 to prevent false-negatives (Fig. S4, Table S2, S3), and demonstrate detection with attomolar sensitivity by both fluorescence and lateral flow readouts comparable to other previously developed systems (Fig. 2C–F)31,38,39,65. We further demonstrate that SENSR has the potential to be so specific to SARS-CoV-2, and further prove it can be adapted to detect vRNA in infected patient isolates (Fig. 3A–C).
SENSR provides a robust proof-of-principle of viral detection by CasRx, and as such, it should be considered a candidate diagnostic system; however, it requires optimization in advance of deployment. Optimizing SENSR diagnostics can be pursued through a number of avenues. While some groups have improved specificity by selectively generating synthetic mismatches in guide sequences22, the gRNAs tested herein have moderate target specificity (Fig. S7). RT-RPA also exhibits nonspecific amplification66–68, and therefore, alternative isothermal amplification technologies, such as RT-LAMP41,69, could be implemented to improve specificity. Although RT-LAMP is likely to improve specificity as well as sensitivity, the requirement of two separate reactions remains problematic, increasing the likelihood of contamination due to sample transfers70. RT-LAMP and CasRx operate optimally at divergent temperatures (60–65°C and 37°C, respectively), which may be incompatible within a one-step molecular diagnostic, though advances have been made in this sphere33,71,72. Thus alterations to existing isothermal amplification technologies or discovery of novel thermostable Cas nucleases with collateral cleavage activity could facilitate a more highly-sensitive Cas-based molecular diagnostic that operates in one reaction at a single temperature.
Beyond amplification, improvement to gRNA design criteria could drastically improve gRNA selection for diagnosis and consequently the response time to future disease outbreaks. Currently, there remains no robust study attempting to characterize the in vitro collateral cleavage activity for varying Cas13 gRNA sequences, thus limiting efficient gRNA design and target selection for Cas13-based diagnostics. In this study, we observed significant variation in gRNA collateral cleavage activity, including two gRNAs (gRNA-AA and gRNA-AC) incapable of producing fluorescence signal (Fig. 2A) and kinetic variation exemplified by temporal differences in fluorescence signal accumulation for gRNA-T and gRNA-Z (Fig. 1E, F). We also observed mild off-target activity for gRNA-Z (Fig. S7D) and found variation in reporter saturation for lateral flow between gRNAs (Fig. 2E, F), which has been found in other systems31,38. Understanding gRNAspecific differences of Cas13 collateral cleavage will allow the development of functional gRNA target sequence libraries for use in future pandemics. Furthermore, robust exploration for CasRx gRNA truncations and permutations should be undertaken to generate gRNAs intolerant of target site polymorphisms, or to even distinguish between strains22, thus improving the specificity of SENSR. Optimizing workflow, deployment, and distribution, while taking steps to reduce the risk of contamination, is imperative to develop CasRx-based diagnostics to their full potential. Although optimization is required, we demonstrate that detection with CasRx is a promising advancement for detecting viral infections, and could be improved to become a powerful molecular diagnostic with numerous applications.
CasRx-based diagnostic systems may present a worthy advancement for CRISPRDx due to the fundamental characteristics of the Cas13d family. Like LwaCas13a, Cas13d is more flexible than most other Cas enzymes because it lacks a protospacer flanking sequence (PFS) requirement28,44,46, permitting targeting of any sequence without constraint. In addition, some native Cas13d systems include a WYL1-domain-containing accessory protein, which has been demonstrated to increase the on-target and collateral cleavage efficiency of the Cas13d effectors46,73, suggesting potential for future implementation. Furthermore, because they target RNA, next-generation Cas13-based systems may be capable of direct recognition of RNA, possibly at the single molecule level, without need for a prior reverse transcription (RT) and/or amplification step. This property could enable direct detection of many emerging viral threats including, but not limited to; bunyaviruses74, zoonotic viruses such as Ebola, hanta, and Lassa75; arboviruses such as dengue, chikungunya, and Zika23,76,77, and other coronaviruses such as MERS, SARS-CoV-1, as well as those yet undiscovered78,79. CasRx-based diagnostics systems could detect endemic pathogens capable of zoonotic transmission through livestock and wild animals such as influenza or other coronaviruses78,80,81 which may have been able to prevent past pandemics82, and avert mass herd culling resulting in billions of dollars of losses83,84. Beyond detection in patients and livestock, SENSR could be adapted to detect pathogens in insect disease vectors as well as infected individuals85, facilitating rapid one-pot field detection of mosquito-borne pathogens in areas lacking laboratory infrastructure86. However, SENSR is not limited to detection of RNA species, and could also be used to detect pathogen DNA (Fig. S8). By including an RNA polymerization step, this same technology could be harnessed to track evidence of insecticide resistance alleles87, released transgenic cargoes59,60, or the presence of Borrelia burgdorferi in a tick plucked from a hiker’s leg.
Pushing the boundaries of viral sequence recognition with CRISPR-Cas nucleases is not only of interest for genetic engineering and diagnostics, but also for therapeutics as well. The adaptability of CasRx RNA-targeting has recently been demonstrated to be a potentially powerful anti-COVID therapeutic40 as well as for other viruses88. Together with acute diagnostics, these technologies could promise a new mode of response to future viral outbreaks via a ‘plug-n-chug’ model, in which complementary diagnostics and therapeutics could be systematically rolled out almost immediately after completion of a viral genome sequence. Similar to LwaCas13a, CasRx could also be adapted to massively multiplexed arrays to facilitate identification of viral pathogens on a large scale37. Establishing these tools and frameworks now, could expedite response times and help prevent future outbreaks, avoiding the economic and health consequences which have resulted from poor preparedness to the current pandemic.
Materials and Methods
CasRx subcloning, protein expression and purification
To produce an expression plasmid for CasRx protein production, we cloned the human codon optimized CasRx coding sequence into the expression vector, pET-His6-MBP-TEV-yORF (Series 1-M) (purchased from QB3 MacroLab, Berkeley) using the Gibson assembly method (Gibson et al., 2009). In brief, the CasRx coding sequence was PCR amplified from plasmid OA-1050E (Addgene plasmid # 13241664) using primers 1136I.C1 and 1136I.C2 (Table S5). The fragment was purified and subcloned into the restriction enzyme cutting site EcoRI, downstream of the His-MBP recombinant protein in pET-His6-MBP-TEV-yORF, generating the final pET-6xHis-MBP-TEV-CasRx (OA-1136J; Addgene plasmid # 153023) plasmid.
Protein expression, culture, cell lysis, affinity and further downstream protein purification were performed as previously described (Fig. S2)44. In brief, to facilitate protein expression in liquid culture, pET-His6-MBP-TEV-CasRx was transformed into Rosetta2 (DE3) pLysS cells (Novagen, 71403). Starter cultures in LB were supplemented with kanamycin and chloramphenicol and incubated at 37° C overnight. 20 mL of starter culture were used to inoculate 1L of TB media supplemented with the same antibiotics. Cultures were allowed to grow until OD600 ~0.5, cooled on ice, induced with 0.2 mM IPTG, and then grown for 20 hours at 18° C. Cells were then pelleted, freeze-thawed, resuspended, lysed via sonication and clarified by centrifugation. Protein purification was performed by gravity Ni-NTA affinity chromatography (Thermo Scientific™ HisPur™ Ni-NTA Resin) followed by removal of the 6xHis-MBP tag by TEV protease concurrently with overnight dialysis. Further purification was achieved by cation exchange using a 5-mL HiTrap SP HP using a gradient of 125 mM to 2M NaCl in 50 mM Tris-HCl, 7.5% v/v glycerol, 1 mM DTT. The protein was finally purified by gel filtration chromatography in 50 mM Tris-HCl, 600 mM NaCl, 10% glycerol, 2 mM dithiothreitol on a Superdex® 200 16/600 column. Fractions were pooled, concentrated to ~ 2 mg/mL and stored at −80 C in the same buffer.
Production of target SARS-CoV-2 RNA and gRNAs
To detect viral genomic sequences, we designed two synthetic dsDNA gene fragments containing a T7 promoter sequence upstream of gene segments corresponding to the SARS-CoV-2 envelope (E) and nucleocapsid (N) protein coding regions (GeneBank Accession # MN908947). The 253bp SARS-CoV2 E-gene segment was ordered and synthesized as a custom gBlock® from Integrated DNA Technologies (IDT) and amplified using primers 1136Q-F and 1136Q-R (Table S5). A 500bp SARS-CoV2 N-gene segment was amplified from a plasmid 1136Y (Catalog # 10006625)65 using primers 1136X-F and 1136X-R (Table S5). These two SARS-CoV-2 gene targets were amplified using PCR and then purified using the MinElute PCR Purification Kit (QIAGEN #28004). We also designed eight synthetic dsDNA templates containing nucleotide variations from the native SARSCoV-2 E- and N-gene (4 synthetic targets each gene) that were used for gRNA off-target analysis and ordered as a gBlock® from IDT. Primers 1136-OFF-F and 1136-OFF-R1~1136-OFF-R5 were used to amplify these sequences (Table S5). The synthetic targets were chosen based on sequence homology identified using NCBI BLAST searches against gRNA-T and gRNA-Z. 40nt regions flanking the mismatch target sequences were included in the 5’ and 3’ ends of the 30 nt stretch in order to allow amplification analysis via RT-RPA.
We designed gRNAs targeting the synthetic vRNA gene segments using criteria previously outlined (Fig. S5)64 and generated these following a previously described templateless PCR protocol89. The primers used to amplify these gRNAs, as well as their final sequence are outlined in Table S5. We synthesized both the synthetic vRNA and gRNAs through in vitro transcription (IVT) using MEGAscript™ T7 Transcription Kit (Invitrogen™ #AM1334), followed by DNaseI digestion and purification using the MEGAclear™ Transcription Clean-Up Kit (Invitrogen™ #AM1908).
In vitro gRNA cleavage assays
To test the in vitro cleavage efficiency of gRNAs, we performed preliminary in vitro cleavage assays to test on-target cleavage, off-target cleavage, and collateral-cleavage properties. On-target cleavage assays were prepared with RNA templates for E-gene (1000 ng) or N-gene (1500 ng), followed by addition of CasRx (112 ng) and 10 ng of each gRNA in a 2:1 molar ratio. Reactions were prepared in 20 mM HEPES pH 7.2 and 9mM MgCl2, incubated at 37°C for one hour, denatured at 85°C for 10 min in 2X RNA loading dye (New England Biolabs, #B0363) and loaded on 2% 1X TBE agarose gel stained with SYBR™ gold nucleic acid staining (Invitrogen #S11494). Off-target cleavage assays were assembled similarly with the non-targeting synthetic vRNA template. Collateral-cleavage assays were prepared with both synthetic vRNA templates simultaneously and same quantities of gRNA and CasRx described above.
Bioinformatics of SARS-CoV-2 SENSR target sites.
433 SARS-CoV-2 genomes were downloaded from NCBI Virus (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide&VirusLineage_ss=SARS-CoV-2,%20taxid:2697049) and 3,164 non-SARS-CoV-2 Coronavirinae genomes were downloaded from Virus Pathogen Resource (https://www.viprbrc.org/brc/home.spg?decorator=corona_ncov) on April 7, 2020. To assess the specificity of our probes, all possible 30 nt sequences were extracted from the two genome sets using a Perl script (File S1) generating 52,712 and 8,338,305 unique fragments from SARS-CoV-2 and non-SARS-CoV-2 genomes, respectively. The probes designed to target E and N genes based on Corman et al. 2020 (https://www.eurosurveillance.org/content/10.2807/1560-7917.ES.2020.25.3.2000045) were cross-referenced against the extracted sequences to identify numbers of targeted genomes in each set. Four of six probes perfectly matched sequences in all 433 SARS-CoV-2 genomes. Two others, 1136R-E-Protein-gRNA1 and 1136S-N-Protein-gRNA1, matched 430 and 426 SARS-CoV-2 genomes, respectively. Of 3,164 non-SARS-CoV-2 viruses, the probes matched between 1 and 10 genomes, mostly from bat hosts (Summarized in Table S2). To identify a comprehensive set of possible targets that are specific to SARS-CoV-2 genomes, 16,645 30 nt sequences that perfectly matched all 433 SARS-CoV-2 genomes were filtered to remove the ones that were also found in any of the 3,164 non-SARS-CoV-2 genomes to produce a set of 8,846 SARS-CoV-2-specific sequences (Table S4). To check for possible cross-reactivity with human transcripts, the probes were mapped to the human transcriptome (GRCh38, ENSEMBL release 99, ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/) comprising both coding and non-coding RNAs using bowtie 1.2.3 allowing up to two mismatches (-v 2). None of the 8,846 sequences mapped to the human transcriptome to confirm their specificity to SARS-CoV-2. To visualize the distribution of the specific targets along the SARS-CoV-2 genome, probe density was calculated using a sliding window of 301 nt for each position of the reference SARS-CoV-2 genome NC_045512 (https://www.ncbi.nlm.nih.gov/nuccore/NC_045512) and plotted in R (Fig. S3). Viral genes that are affected by each probe were identified using the intersect function of bedtools (Table S3).
RT-RPA amplification of viral genomic sequences
Prior to all detection assays a pre-amplification step using RT-RPA was performed in order to amplify the SARS-CoV-2 target sequence. These protocols were initially developed and optimized on mock viral genome fragments and later validated against patient samples. To amplify the target sequences from the synthetic vRNA, we performed RT-RPA38 (protocol summarized in Fig. S3). In short, RT-RPA primers were designed to amplify 30 nt gRNA spacer regions flanked by 30 nt priming regions from the synthetic vRNA template while also incorporating a T7 promoter sequence into the 5’ end of the dsDNA gene fragment with +2 G’s thus increasing transcription efficiency (Fig. 1B)90. RT-RPA was performed at 42°C for 45 min by combining M-MuLV-RT (NEB #M0253L) with TwistAmp® Basic (TwistDx #TABAS03KIT). All RT-RPA primer sequences can be found in Table S1.
Fluorescence-based detection of SARS-CoV-2
For fluorescence-based detection, we developed a simple in vitro transcription-coupled cleavage assay with a fluorescence readout using 6-Carboxyfluorescein (6-FAM) as our fluorescent marker. To facilitate fluorescence detection, we developed a 6 nt poly-U probe conjugated to a 5’−6-FAM and a 3’-IABlkFQ (FRU, Table S5) and custom ordered from IDT. In total volumes of 15μL, the following reaction mix was prepared; 5.62μL water, 0.4μL HEPES, pH 7.2 (1M), 0.18μL MgCl2 (1M), 3.2μL rNTPs (25mM each), 2μL CasRx (55.4 ng/μL), 1μL RNase inhibitor (40U/μL), 0.6μL T7 Polymerase (50U/μL), 1 μL gRNA (10 ng/μL), and 1μL FRU probe (2uM). This was followed by the addition of 5μL (50% amplification vol) of the amplified target RNA from the RT-RPA preamplification mix (described above) or no-template control, which initiates the reaction following incubation at 37°C for 90 min. Experiments were immediately run on a LightCycler® 96 (Roche #05815916001) at 37°C under 5 sec acquisition followed by 5 sec incubation for the first 15 min, followed by 5 sec acquisition and 55 sec incubation for up to 75 min. Fluorescence readouts were analyzed over-time by normalization to templateless controls at each respective time point or through background subtracted fluorescence by subtracting the initial fluorescence value from the final value.
Lateral Flow-based detection of SARS-CoV-2
For lateral flow-based detection we modified the HybriDetect® system to detect the presence of SARS-CoV-2 sequences using SENSR38. In brief, we designed a ssRNA probe composed of a 6 nt poly-U probe conjugated on opposite ends with a 5’−6-FAM and a 3’-biotin which was custom ordered from IDT (LFRU, Table S5). Following incubation of 5.22μL water, 0.4μL HEPES, pH 7.2 (1M), 0.18μL MgCl2 (1M), 2μL CasRx (55.4ng/μL), 1μL gRNA (10 ng/μL), 5μL RT-RPA reaction mix, 1μL T7 polymerase (50 U/mL), 3.2μL rNTPs (25mM each), 1μL LFRU probe (20 uM), at 37°C for 60 min. 80μL of HybriDetect Assay buffer was added to each reaction and mixed thoroughly. Next, the lateral flow dipstick was placed into the reaction and allowed to flow upwards by capillary action for a maximum of 2 min. The presence or absence of upper or lower bands was analyzed to detect evidence of SARS-CoV-2 by collateral cleavage. The presence of a solitary upper band or both an upper and lower band indicates a positive result, a solitary lower band with a faint upper band was interpreted as a negative result.
Patient samples ethics statement
Human samples from patients were collected under University of California San Diego’s Human Research Protection Program protocol number 200470 for negatives (PI: Lauge Farnaes), and under a waiver of consent from clinical samples from San Diego County for positives (PI: Kristian Andersen), as part of the SEARCH Alliance activities. Samples were de-identified as required by these protocols prior to testing and analysis under University of California San Diego Biological Use Authorization protocols R1806 and 2401.
RNA extraction and processing of patient samples
Patient nasopharyngeal samples were collected and RNA was extracted using Omega Bio-Tek Mag-Bind Viral DNA/RNA 96 Kit (Omega Cat. No. M6246–03), following the manufacturer’s protocol for KingFisher Flex platform.
RT-qPCR validation of SARS-CoV-2 infection in patient samples
Patient samples were determined to be SARS-CoV-2 positive or negative TaqPath™ COVID-19 Combo Kit RT-qPCR assay as described in (https://www.fda.gov/media/136112/download), and reducing the RT-qPCR reaction volumes to 3μl and diluting the MS2 phage control to improve the limit of detection of the assay. The presence of SARS-CoV-2 viral RNA was analyzed using primers targeting the N, S, and Orf1ab genes with an MS2 control. All RT-qPCR assays were run using TaqPath™ 1-Step RT-qPCR Master Mix (ThermoFisher #A15299) and thermocycling conditions were run following the CDC recommended protocol (https://www.fda.gov/media/136112/download). Fluorescence data were acquired on a QuantStudio 5 qPCR machine (Applied Biosystems).
SENSR detection of patient samples
To detect the presence of SARS-CoV-2 in patient samples using SENSR, we tested this system against RT-qPCR validated samples. We obtained SARS-CoV-2 positive (N=21) and negative (N=21) samples and ran fluorescence analysis of these samples in triplicate. Samples were subject to preamplification using RT-RPA and incubated in an IVT-coupled cleavage reaction, as previously described. Data for analysis were acquired on LightCycler® 96 (Roche #05815916001) following the protocol previously described. We processed the data by generating background subtracted fluorescence data for each replicate by subtracting the final (90 min) fluorescence value from the initial (0 min) fluorescence value. Noise was set as the average of the three no-template control (NTC) background subtracted values. S/N was then calculated by dividing the background subtracted value for each replicate by the noise. The S/N for each sample was then determined by taking the average of the three independent S/N ratios in the triplicates. Samples were determined to be positive if S/N > 2 and negative if S/N < 2. Lateral flow analysis was run on samples that were determined as positives from the SENSR fluorescence analysis. The samples were assayed and analyzed following the previously described lateral flow methods and images were taken using a smartphone. Positives and negatives were determined in comparison to the NTC samples and using a positive control (synthetic template) as a standard.
Supplementary Material
Acknowledgements:
Funding: We thank the SEARCH (San Diego Epidemiology And Research for COVID-19 Health) Alliance for providing clinical samples. This work was supported in part by UCSD Seed Funds for Emergent COVID-19 Related Research, a Directors New Innovator award from NIH/NIAID (DP2 AI152071-01) and a DARPA Safe Genes Program Grant (HR0011-17-2-0047) awarded to O.S.A., and a Director’s Pioneer award from NCCIH (DP1 AT010885) to R.K., and the UCSD Return to Learn program via the EXCITE (EXpedited COVID-19 IdenTification Environment) lab. The views, opinions and/or findings expressed should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government.
Footnotes
Competing Interests: O.S.A. has a patent pending on this technology. All other authors declare no significant competing financial, professional, or personal interests that might have influenced the performance or presentation of the work described.
Data and Materials Availability: Bioinformatic scripts can be found in File S1. A limited quantity of CasRx enzyme will be made available upon request.
References
- 1.Zhu N. et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mallapaty S. Animal source of the coronavirus continues to elude scientists. Nature (2020) doi: 10.1038/d41586-020-01449-8 [DOI] [PubMed] [Google Scholar]
- 3.Andersen K. G., Rambaut A., Lipkin W. I., Holmes E. C. & Garry R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C., Horby P. W., Hayden F. G. & Gao G. F. A novel coronavirus outbreak of global health concern. The Lancet vol. 395 470–473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO. Coronavirus disease 2019 (COVID-19) Situation Report - 93. https://www.who.int/docs/default-source/coronaviruse/situationreports/20200422-sitrep-93-covid-19.pdf?sfvrsn=35cf80d7_4 (2020). [Google Scholar]
- 6.Bai Y. et al. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA (2020) doi: 10.1001/jama.2020.2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai C.-C. et al. Asymptomatic carrier state, acute respiratory disease, and pneumonia due to severe acute respiratory syndrome coronavirus 2 (SARSCoV-2): Facts and myths. J. Microbiol. Immunol. Infect. (2020) doi: 10.1016/j.jmii.2020.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corman V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 25, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Office of the Commissioner. Coronavirus (COVID-19) Update: FDA Authorizes First Test for Patient At-Home Sample Collection. https://www.fda.gov/news-events/press-announcements/coronavirus-covid19-update-fda-authorizes-first-test-patient-home-sample-collection (2020). [Google Scholar]
- 10.Shen M. et al. Recent advances and perspectives of nucleic acid detection for coronavirus. J Pharm Anal 10, 97–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.CDC. Coronavirus Disease 2019 (COVID-19). Centers for Disease Control and Prevention https://www.cdc.gov/coronavirus/2019ncov/lab/index.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Flab%2Frt-pcr-detection-instructions.html (2020). [Google Scholar]
- 12.Kan Alice, The Science Advisory Board contributing writer. Shortage of RNA extraction kits threatens coronavirus testing. Scienceboard.net https://www.scienceboard.net/index.aspx?sec=sup&sub=can&pag=dis&ItemID=564 (2020). [Google Scholar]
- 13.Li D. et al. False-Negative Results of Real-Time Reverse-Transcriptase Polymerase Chain Reaction for Severe Acute Respiratory Syndrome Coronavirus 2: Role of Deep-Learning-Based CT Diagnosis and Insights from Two Cases. Korean J. Radiol. 21, 505–508 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kucirka L., Lauer S., Laeyendecker O., Boon D. & Lessler J. Variation in False Negative Rate of RT-PCR Based SARS-CoV-2 Tests by Time Since Exposure. medRxiv 2020.04.07.20051474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y. et al. Evaluating the accuracy of different respiratory specimens in the laboratory diagnosis and monitoring the viral shedding of 2019-nCoV infections. MedRxiv (2020) doi: 10.1101/2020.02.11.20021493 [DOI] [Google Scholar]
- 16.Whitman J, Hiatt J, Mowery C, Shy B, Yu R, Yamamoto T, Rathore U, Goldgof F, Whitty C, Woo J, Gallman A, Miller T, Levine A, Nguyen D, Bapat S, Balcerek J, Bylsma S, Lyons A, mLi S, Wai-yi A, Gillis-Buck E, Steinhart Z, Lee Y, Apathy R, Lipke M, Smith J, Zheng T, Boothby I, Isaza E, Chan J, Acenas D, Lee J, Macrae T, Kyaw T, Wu D, Ng D, Gu W, York V, Eskandarian H, Callaway P, Warrier L, Moreno M, Levan J, Torres L, Farrington L, Loudermilk R, Koshal K, Zorn K, Garcia-Beltran W, Yang D, Astudillo M, Bernstein B, Gelfand J, Ryan E, Charles R, Iafrate J, Lennerz J, Miller S, Chiu C, Stramer S, Wilson M, Manglik A, Ye C, Krogan N, Anderson M, Cyster J, Ernst J, Wu A, Lynch K, Bern C, Hsu P, Marson A. Test performance evaluation of SARS-CoV-2 serological assays. Dropbox (2020). [Google Scholar]
- 17.Liu W. Sr. Non-specific Primers Reveal False-negative Risk in Detection of COVID-19 Infections. Public and Global Health (2020) doi: 10.1101/2020.04.07.20056804 [DOI] [Google Scholar]
- 18.Office of the Commissioner. Coronavirus (COVID-19) Update: FDA Authorizes First Next Generation Sequence Test for Diagnosing COVID-19. https://www.fda.gov/news-events/press-announcements/coronavirus-covid19-update-fda-authorizes-first-next-generation-sequence-test-diagnosingcovid-19 (2020).
- 19.Jinek M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abudayyeh O. O. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zetsche B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gootenberg J. S. et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science vol. 356 438–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gootenberg J. S. et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S.-Y. et al. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov 4, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azhar M. et al. Rapid, field-deployable nucleobase detection and identification using FnCas9. bioRxiv 2020.04.07.028167 (2020) doi: 10.1101/2020.04.07.028167 [DOI] [Google Scholar]
- 27.Harrington L. B. et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freije C. A. et al. Programmable Inhibition and Detection of RNA Viruses Using Cas13. Mol. Cell 76, 826–837.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukama O. et al. An ultrasensitive and specific point-of-care CRISPR/Cas12 based lateral flow biosensor for the rapid detection of nucleic acids. Biosensors and Bioelectronics 112143 (2020) doi: 10.1016/j.bios.2020.112143 [DOI] [PubMed] [Google Scholar]
- 30.Hajian R. et al. Detection of unamplified target genes via CRISPR-Cas9 immobilized on a graphene field-effect transistor. Nat Biomed Eng 3, 427–437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patchsung M. et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat Biomed Eng (2020) doi: 10.1038/s41551-020-00603-x [DOI] [PubMed] [Google Scholar]
- 32.Lucia C., Federico P.-B. & Alejandra G. C. An ultrasensitive, rapid, and portable coronavirus SARS-CoV-2 sequence detection method based on CRISPR-Cas12. bioRxiv (2020) doi: 10.1101/2020.02.29.971127 [DOI] [Google Scholar]
- 33.Joung J. et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. Infectious Diseases (except HIV/AIDS) (2020) doi: 10.1101/2020.05.04.20091231 [DOI] [Google Scholar]
- 34.Ding X., Yin K., Li Z. & Liu C. All-in-One Dual CRISPR-Cas12a (AIOD-CRISPR) Assay: A Case for Rapid, Ultrasensitive and Visual Detection of Novel Coronavirus SARS-CoV-2 and HIV virus. (2020) doi: 10.1101/2020.03.19.998724 [DOI] [PMC free article] [PubMed]
- 35.Broughton J. P. et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. (2020) doi: 10.1038/s41587-020-0513-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rauch J. N. et al. A Scalable, Easy-to-Deploy, Protocol for Cas13-Based Detection of SARS-CoV-2 Genetic Material. bioRxiv 2020.04.20.052159 (2020) doi: 10.1101/2020.04.20.052159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ackerman C. M. et al. Massively multiplexed nucleic acid detection using Cas13. Nature (2020) doi: 10.1038/s41586-020-2279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F., Abudayyeh O. O. & Jonathan S. G. A protocol for detection of COVID-19 using CRISPR diagnostics. (2020).
- 39.Metsky H. C., Freije C. A., Kosoko-Thoroddsen T.-S. F., Sabeti P. C. & Myhrvold C. CRISPR-based surveillance for COVID-19 using genomically-comprehensive machine learning design. bioRxiv 2020.02.26.967026 (2020) doi: 10.1101/2020.02.26.967026 [DOI] [Google Scholar]
- 40.Abbott T. R. et al. Development of CRISPR as a prophylactic strategy to combat novel coronavirus and influenza. bioRxiv 2020.03.13.991307 (2020) doi: 10.1101/2020.03.13.991307 [DOI] [Google Scholar]
- 41.Howson E. L. A. et al. Defining the relative performance of isothermal assays that can be used for rapid and sensitive detection of foot-and-mouth disease virus. J. Virol. Methods 249, 102–110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinton D. M. Sherlock CRISPR SARS-CoV-2 Kit. (2020).
- 43.Myhrvold C. et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konermann S. et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buchman A. B. et al. Programmable RNA Targeting Using CasRx in Flies. CRISPR J 3, 164–176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan W. X. et al. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol. Cell 70, 327–339.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang C. et al. Structural Basis for the RNA-Guided Ribonuclease Activity of CRISPR-Cas13d. Cell 175, 212–223.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kellner M. J., Koob J. G., Gootenberg J. S., Abudayyeh O. O. & Zhang F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kushawah G., del Prado J. A. N. & Martinez-Morales J. R. CRISPR-Cas13d induces efficient mRNA knock-down in animal embryos. bioRxiv (2020). [DOI] [PubMed] [Google Scholar]
- 50.Piepenburg O., Williams C. H., Stemple D. L. & Armes N. A. DNA detection using recombination proteins. PLoS Biol. 4, e204 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu X. et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 26, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tambe A., East-Seletsky A., Knott G. J., Doudna J. A. & O’Connell M. R. RNA Binding and HEPN-Nuclease Activation Are Decoupled in CRISPR-Cas13a. Cell Rep. 24, 1025–1036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng T. et al. Profiling single-guide RNA specificity reveals a mismatch sensitive core sequence. Sci. Rep. 7, 40638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teng F. et al. CDetection: CRISPR-Cas12b-based DNA detection with sub-attomolar sensitivity and single-base specificity. Genome Biol. 20, 132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu S. et al. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia. Blood 133, 2255–2262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maeder M. L. et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25, 229–233 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Inc., K. N. & Kernel Networks Inc. Single Ascending Dose Study in Participants With LCA10. Case Medical Research (2019) doi: 10.31525/ct1nct03872479 [DOI] [Google Scholar]
- 58.Niu D. et al. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science vol. 357 1303–1307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Esvelt K. M., Smidler A. L., Catteruccia F. & Church G. M. Emerging technology: concerning RNA-guided gene drives for the alteration of wild populations. Elife 3, e03401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li M. et al. Development of a confinable gene drive system in the human disease vector Aedes aegypti. eLife vol. 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Champer J., Buchman A. & Akbari O. S. Cheating evolution: engineering gene drives to manipulate the fate of wild populations. Nat. Rev. Genet. 17, 146–159 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Church G. Please reanimate. Sci. Am. 309, 12 (2013). [DOI] [PubMed] [Google Scholar]
- 63.The Woolly Mammoth Revival. https://reviverestore.org/projects/woolly-mammoth/.
- 64.Buchman A. et al. Programmable RNA Targeting using CasRx in Flies. bioRxiv 2020.04.03.023606 (2020) doi: 10.1101/2020.04.03.023606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Broughton J. P. et al. Rapid Detection of 2019 Novel Coronavirus SARSCoV-2 Using a CRISPR-based DETECTR Lateral Flow Assay. Infectious Diseases (except HIV/AIDS) (2020) doi: 10.1101/2020.03.06.20032334 [DOI] [Google Scholar]
- 66.Daher R. K., Stewart G., Boissinot M., Boudreau D. K. & Bergeron M. G. Influence of sequence mismatches on the specificity of recombinase polymerase amplification technology. Mol. Cell. Probes 29, 116–121 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Daher R. K., Stewart G., Boissinot M. & Bergeron M. G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 62, 947–958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luo G.-C., Yi T.-T., Jiang B., Guo X.-L. & Zhang G.-Y. Betaine-assisted recombinase polymerase assay with enhanced specificity. Anal. Biochem. 575, 36–39 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Notomi T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, E63 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sefers S. & Schmitz J. E. Molecular Contamination and Amplification Product Inactivation in Advanced Techniques in Diagnostic Microbiology: Volume 1: Techniques (eds. Tang Y.-W. & Stratton C. W.) 505–526 (Springer International Publishing, 2018). [Google Scholar]
- 71.Cai S., Jung C., Bhadra S. & Ellington A. D. Phosphorothioated Primers Lead to Loop-Mediated Isothermal Amplification at Low Temperatures. Anal. Chem. 90, 8290–8294 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Joung J. et al. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N. Engl. J. Med. (2020) doi: 10.1056/NEJMc2026172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang H., Dong C., Li L., Wasney G. A. & Min J. Structural insights into the modulatory role of the accessory protein WYL1 in the Type VI-D CRISPR-Cas system. Nucleic Acids Res. 47, 5420–5428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noronha L. E. & Wilson W. C. Comparison of two zoonotic viruses from the order Bunyavirales. Curr. Opin. Virol. 27, 36–41 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Wang L.-F. & Crameri G. Emerging zoonotic viral diseases. Rev. Sci. Tech. 33, 569–581 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Gould E., Pettersson J., Higgs S., Charrel R. & de Lamballerie X. Emerging arboviruses: Why today? One Health 4, 1–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Charrel R. N. et al. Emergence of Toscana virus in Europe. Emerg. Infect. Dis. 11, 1657–1663 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Guarner J. Three Emerging Coronaviruses in Two Decades. Am. J. Clin. Pathol. 153, 420–421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Torremorell M., Allerson M., Corzo C., Diaz A. & Gramer M. Transmission of influenza A virus in pigs. Transbound. Emerg. Dis. 59 Suppl 1, 68–84 (2012). [DOI] [PubMed] [Google Scholar]
- 81.Shi J. et al. H7N9 virulent mutants detected in chickens in China pose an increased threat to humans. Cell Res. 27, 1409–1421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mena I. et al. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacKenzie D. Many millions of pigs wiped out as African swine fever spreads. New Scientist vol. 244 6 (2019). [Google Scholar]
- 84.Parry J. Ten years of fighting bird flu. Bull. World Health Organ. 85, 3–4 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee R. A. et al. Ultrasensitive CRISPR-based diagnostic for field-applicable detection of Plasmodium species in symptomatic and asymptomatic malaria. Proceedings of the National Academy of Sciences 202010196 (2020) doi: 10.1073/pnas.2010196117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Choumet V. & Desprès P. Dengue and other flavivirus infections. Rev. Sci. Tech. 34, 473–8, 467–72 (2015). [PubMed] [Google Scholar]
- 87.Faucon F. et al. Identifying genomic changes associated with insecticide resistance in the dengue mosquito Aedes aegypti by deep targeted sequencing. Genome Res. 25, 1347–1359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blanchard E. L. et al. Treating Influenza and SARS-CoV-2 via mRNA-encoded Cas13a. bioRxiv 2020.04.24.060418 (2020) doi: 10.1101/2020.04.24.060418 [DOI] [PubMed] [Google Scholar]
- 89.Li M., Akbari O. S. & White B. J. Highly Efficient Site-Specific Mutagenesis in Malaria Mosquitoes Using CRISPR. G3 8, 653–658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brieba L. G., Padilla R. & Sousa R. Role of T7 RNA polymerase His784 in start site selection and initial transcription. Biochemistry 41, 5144–5149 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.