

Abstract
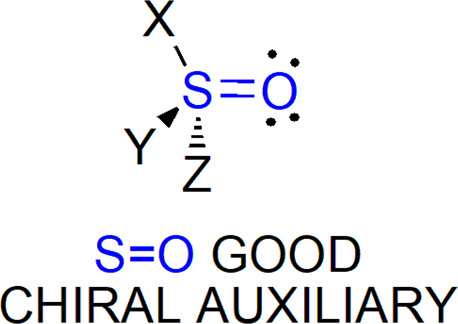
Chiral sulfinyl compounds, sulfoxides, sulfoximines, sulfinamides, and other derivatives, play an important role in asymmetric synthesis as versatile auxiliaries, ligands, and catalysts. They are also recognized as pharmacophores found in already marketed and well-sold drugs (e.g., esomeprazole) and used in drug design. This review is devoted to the modern methods of preparation of sulfinyl derivatives in enantiopure or enantiomerically enriched form. Selected new approaches leading to racemic products for which the asymmetric variant can be developed in the future are mentioned as well.
1. Introduction
In this review, we would like to focus on the recent achievements in the synthesis of chiral sulfinyl compounds. Their common feature, a stereogenic sulfur atom is connected to electronegative oxygen atom by a polar, partially dative bond, although in all structures in this review it is consistently shown as S=O moiety (Figure 1). We limit our discussion to organosulfur derivatives, i.e., compounds with at least one S–C bond. Consequently, sulfilimines (lacking S=O fragment), and sulfites or sulfates (no C–S bond present) will not be covered. Also sulfenyl compounds and sulfonyl derivatives will be only marginally mentioned as achiral starting materials or side products in the preparation of their sulfinyl counterparts.
Figure 1.
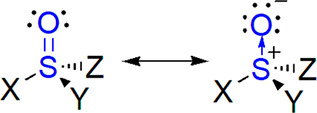
General formula of sulfinyl compounds showing resonance structures of sulfinyl group.
We dare to undertake this topic encouraged by the good reception of our review published in the year 2010 on the enantioselective preparation of chiral sulfoxides.1 Since then, hundreds of articles have appeared devoted to this group of compounds. Although chiral, enantiopure, or enantioenriched sulfoxides have been mainly prepared using previously developed methods, also new approaches to them have been described. This situation calls for a review which would include novel protocols and new interesting examples. The present paper is, however, not a simple update of our previous article as we have decided to extend our interest to other important groups of chiral sulfinyl compounds, which are also prepared in a stereoselective manner, e.g., sulfinamides, sulfinimines, sulfinates and thiosulfinates, sulfoximines, sulfonimidamides, and sulfonimidates.
These derivatives deserve attention due to their importance in modern synthetic, medicinal, and agricultural chemistry. The sulfinyl group is considered as a valuable chiral auxiliary, used in the stereoselective preparation of variety of useful chiral compounds. It is relatively easy to introduce and remove, configurationally stable, and capable of high asymmetric induction. As an example, tert-butane sulfinamide introduced by Ellman and co-workers finds numerous applications in the synthesis of enantiomerically pure amines and their derivatives, in many cases exhibiting significant biological activity.2 Recent reviews show the utility of various sulfur derivatives in organic synthesis.3,4 Nonracemic sulfinyl compounds are also used in catalytic stereoselective reactions, both as ligands for transition metals and as chiral organocatalysts. Three important reviews published in the last five years deal with this emerging application of chiral sulfoxides and their analogues in asymmetric catalysis.5−7
Chiral organosulfur derivatives are found in natural systems such as methionine sulfoxide, S-adenosyl methionine, allicin, or leinamycin 1 (Figure 2).8 Synthetic sulfinyl compounds often exhibit a significant biological activity. As a consequence, pharmaceutical and agrochemical industry manifest an increasing interest in these chemicals, part of which have been marketed as drugs, like esomeprazole 2 and its various derivatives used as antiulcer drugs or modafinil 3 (Figure 2). Recently, the better synthetic availability of sulfoximines (as exemplified by roniciclib 4), sulfonimidamides, and sulfonimidates has led to the increased interest in their possible application in drug design.9 These analogues of sulfones and sulfonamides, well recognized pharmacophores, offer additional possibilities of structural modifications which may affect their interactions within natural systems. In this context, the development of efficient methods of stereoselective preparation of sulfinyl compounds is of special importance.
Figure 2.
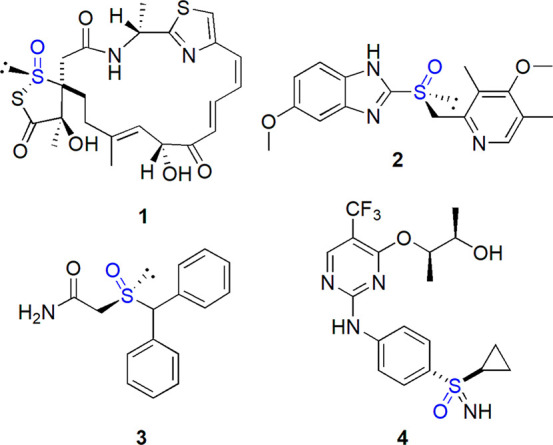
Examples of enantiomerically pure sulfinyl compounds exhibiting biological activity: leinamycin (1), esomeprazole (2), modafinil (3), roniciclib (4).
Articles describing methods of preparation of sulfoxides, but also of other sulfinyl compounds, are collected in several books. Among the most recent, the first one, entitled “Chiral Sulfur Reagents: Applications in Asymmetric and Stereoselective Synthesis”, written by Marian Mikołajczyk, Józef Drabowicz, and Piotr Kiełbasiński, was printed in 1997 and contains chapters devoted to various types of sulfoxides,10 sulfinic acid derivatives,11 and sulfoximines.12 Eleven years later, a book edited by Takeshi Toru and Carsten Bolm was issued. From the point of view of the present review, chapters on synthesis and application of chiral sulfinates,13 sulfinamides,14 and sulfoximines15 are particularly important.
Important review articles and highlights underscoring the significance of particular groups of sulfinyl compounds will be introduced in respective sections. As we do not intend to repeat the material covered by them, only the most representative examples and the latest papers published up to the year 2019 will be discussed.
2. Chiral Organic Compounds with a Tetrahedral Sulfur Stereocenter
The study of stereochemistry of organic sulfur compounds has a long and rich history.16 We shall limit our discussion to tetrahedral sulfur, which can become a stereogenic center when three (a lone pair of electrons takes the part of fourth substituent) or four different groups are attached to it. If we take into account only the chirality arising from the presence of the sulfinyl group, among trisubstituted derivatives sulfoxides are chiral if two carbon groups are different. A whole family of chiral compounds can be regarded as derivatives of sulfinic acids, which themselves are achiral due to proton exchange (however, chirality can result from isotopic substitution17): sulfinic halides, sulfinamides and sulfinimines (N-sulfinylimines), sulfinates, and thiosulfinates (also thiosulfinic acids and their salts are chiral, although they are prone to slow decomposition and rarely prepared17,18). Among their counterparts in which the place of lone electron pair is taken by a =NR′ fragment, sulfoximines require two different carbon groups for chirality, while derivatives of sulfonimidic acids are chiral by definition (Figure 3). Salts with three organic groups attached to sulfur should be also mentioned.
Figure 3.
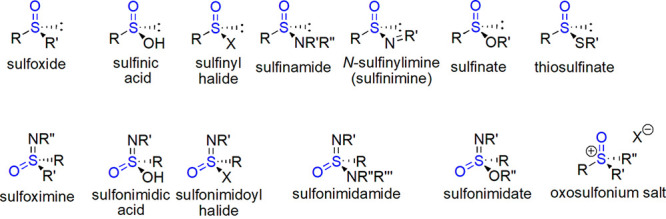
Main classes of chiral sulfinyl compounds. R,R′, R′′,R′′′ = alkyl or aryl, but when attached to N then also H; X = F, Cl, Br, I.
Certainly, these derivatives differ in terms of chemical and configurational stability. Sulfinyl chlorides have been mainly prepared as reactive intermediates without bothering about enantiomeric purity, and their storage is problematic (a contact with moisture evolves gaseous HCl).19−21 Sulfinic acids are prone to disproportionation, yielding diaryl thiosulfinates and sulfonic acids.22 Stability of sulfinyl derivatives was extensively studied by Kice and co-workers. A mechanism of racemization of thiosulfinates caused by nucleophiles or catalyzed by acids was analyzed as well as kinetics of alkaline hydrolysis of aryl thiosulfinates and thiosulfonates.23−25 It was shown that the presence of a bulky substituent at sulfur increases the stability of thiosulfinates, and most work was done for tert-butyl derivative (see section 5). Various factors affecting racemization were summarized in a chapter written by Mikołajczyk and Drabowicz.26
Several types of methods to prepare chiral sulfinyl compounds can be distinguished, taking into account which bond is formed in the course of reaction. Stereoselective oxidation of prochiral sulfur atom in sulfenyl compounds has been recognized as an efficient method of synthesis of nonracemic sulfoxides but is also of importance for other classes of organosulfur derivatives. Alternatively, one sulfinyl derivative can be converted into another, with concomitant formation of either sulfur–carbon or sulfur–heteroatom (N, O, S) bond.
In general, we can also consider possible transformations involving disubstituted (sulfides, disulfides, thiols, sulfenyl halides, sulfenic acid derivatives: sulfenates, sulfenamides), trisubstituted, and tetrasubstituted organosulfur compounds (Scheme 1). In conversions involving the first group to the members of the second and third one, chiral reactant or catalyst is necessary to yield optically active product (stereoselective formation of two new bonds constitutes a considerable challenge). Chirality transfer from another stereogenic center already present in the molecule is also possible in diastereoselective reactions.
Scheme 1. Possible Interconversions of Sulfinyl Derivatives.
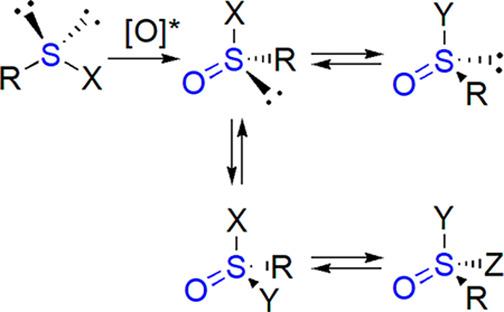
Other transformations engaging chiral tri- or tetrasubstituted derivatives can be stereospecific, proceeding with either retention or inversion of configuration of sulfur atom. As proven by early experiments, substitution reactions, which typically make use of nucleophilic reagents, are in most cases associated with inversion, while conversions connected with change of coordination number keep the arrangement of substituents (the fourth one takes place of the lone pair or vice versa, see also section 9.1).27−29
Kinetic resolution can be engaged in certain cases to increase the optical purity of desired products or to resolve racemic mixtures. Certain interconversions, like sulfonamide into sulfinylimine and vice versa, do not proceed at a stereogenic sulfur atom (this is also true for modifications within substituents, although certain reaction conditions may favor racemization). One should not forget about desymmetrization of achiral (e.g., meso) compounds, also resulting from modification of substituents.
Not all possible conversions can be treated as useful preparative methods. Yield and stereoselectivity are the main criteria, but economic and environmental aspects should be also taken into account. Procedures should be possibly general, operationally simple, and with the number of steps minimized. Last but not least, they should start from cheap and easily available materials. Therefore, deamidation of sulfoximines, which proceeds with retention of configuration, is not regarded as a practical synthetic route to enantiomerically pure sulfoxides.30 Similarly, reduction of sulfones has never emerged as a convenient route to sulfoxides.31,32
The subsequent sections will be devoted to the synthesis of particular groups of sulfinyl compounds, mainly in enantiomerically pure or enriched form. However, certain procedures leading to racemic mixtures will be also presented as at least part of them can be adapted to give single stereoisomeric products.
3. Preparation of Chiral, Nonracemic Sulfoxides
Our review on stereoselective preparation of chiral sulfoxides covered mainly literature from the years 2000–2008 (although referred also to classical papers).1 In recent years, several other review articles devoted to this group of organosulfur compounds have been published, differing in their scope, size, and general message as well as topicality. Three important papers concerning application of chiral sulfoxides in asymmetric catalysis have already been mentioned in the Introduction.5−7 A general review by Maguire and co-workers on the synthesis of enantioenriched sulfoxides, with a special attention paid to kinetic resolution, was published in 2011.33 Several articles were devoted to asymmetric oxidations, including sulfoxidations, with a focus on environmentally benign methods.34−36 A first part of a paper in which Franklin Davis described his “adventures in sulfur–nitrogen chemistry” (using his own words) described the preparation and use of chiral oxaziridine derivatives (known as Davis reagents) in enantioselective oxidation of sulfenyl compounds: sulfides, disulfides, and sulfenylimines.37 In 2018, a review by Han et al. was published, presenting the developments in the preparation of optically active sulfoxides in the years 2011–2016.38 An important issue of correct determination of stereochemical outcome of synthetic reactions has been discussed in detail. The problem is connected with self-disproportionation of enantiomers (SDE), the effect of change of enantiomeric composition during the purification process.
We do not intend to present once more the details of all methods used for preparation of chiral, enantiomerically pure, or enantiomerically enriched sulfoxides. Their description can be found in previous reviews. In this section, we shall focus on the main tendencies that could be observed in field in the past decade.
3.1. Chemical Methods
3.1.1. S=O Bond Formation
Enantioselective sulfoxidation remains the principal method of preparation of chiral, nonracemic sulfoxides. From the economical point of view, catalytic oxidation systems are preferred, although Davis’ oxaziridines are also used.39 Attempts with new chiral stoichiometric oxidants (N-chloramine derivatives) were not very encouraging (up to 23% ee).40 Consequently, transition metal catalysts (in particular, titanium- and vanadium-based) are commonly used.
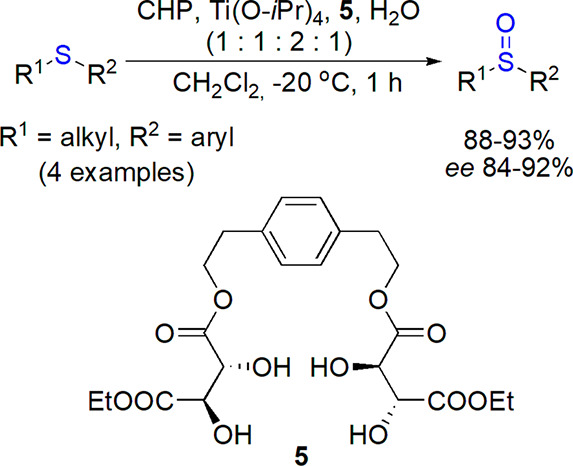
The long puzzling origin of the Kagan/Modena oxidation system based on Ti(IV) complexes of diethyl tartrate and tert-butyl hydroperoxide (TBHP) or cumene hydroperoxide (CHP) was studied by Corey’s group.41 DFT calculations were applied to compare geometries and energies of possible isomeric helical complexes formed in the course of reaction: the more stable P diastereomer was shown to preferentially yield one of the enantiomers of sulfoxide. This analysis led to design and synthesis of a chiral tetraol containing two tartrate moieties 5, which was found superior in the enantioselective oxidation of alkyl aryl sulfides in comparison with the original Kagan’s system (Scheme 2).
Scheme 2. Enantioselective Oxidation of Sulfides Catalyzed by Ti(IV) Complex with Chiral Tetraol 5(41).
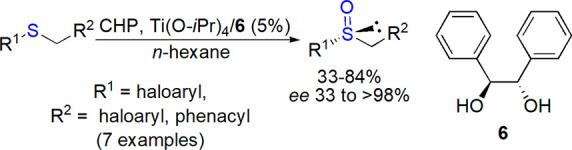
In a series of publications, Cardelliccio and co-workers thoroughly studied oxidation of sulfides by titanium/(S,S)- or (R,R)-hydrobenzoin (HB, 6) catalytic system.42−47 Through a combination of experimental and theoretical work, they found that benzyl aryl sulfides were particularly efficiently oxidized by TBHP (yields up to 92%, ee 84 to >98%), which was rationalized by different interactions in diastereomeric octahedral adducts formed by [Ti(HB)2]4+ with substrate and TBHP.42−44 However, they found that for aryl benzyl or aryl phenacyl sulfides containing fluorinated substituents, tert-butyl hydroperoxide was relatively inefficient but could be replaced with CHP, which resulted in satisfactory yields and much improved enantioselectivity (Scheme 3).45−48 Again, this observation was explained by the calculated degeneracy of the octahedral diastereomers formed when TBHP was applied, while in the case of CHP, additional interactions stabilized mainly one of complexes leading preferentially to one enantiomer.
Scheme 3. Enantioselective Oxidation Catalyzed by Titanium/(S,S)-Hydrobenzoin48.
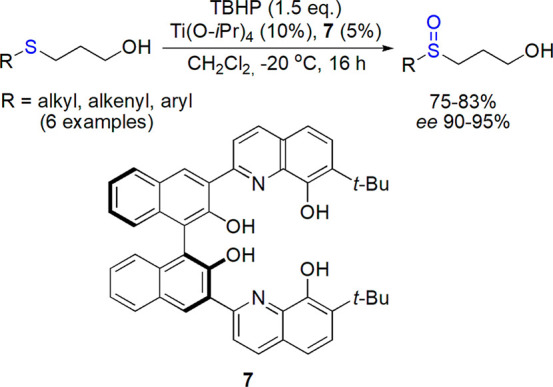
Talsi and Bryliakov explored titanium complexes with salan derivatives; oxidation of pyridylmethylthiobenzimidazoles led to enantiopure proton pump inhibitors.49−52 Also, other groups prepared various enantiomerically enriched sulfoxides using chiral tetradendate and tridendate Ti(IV) complexes as catalysts.53−56 An interesting dinuclear system based on 8-quinolinol-derived ligand 7, which was found efficient in epoxidation of a variety of homoallylic alcohols, was checked in sulfoxidation by Bhadra et al. (Scheme 4).57 γ-Hydroxypropyl sulfides were converted to the corresponding sulfoxides (difficult to prepare by other routes) in high yield and with ee values up to 95%; the presence and placement of hydroxyl group was found crucial for the observed stereochemical outcome.
Scheme 4. Oxidation of Hydroxypropyl Sulfides with a Catalytic System Based on Dinuclear Titanium Complex with Chiral Ligand 7 Described by Bhadra et al.57.
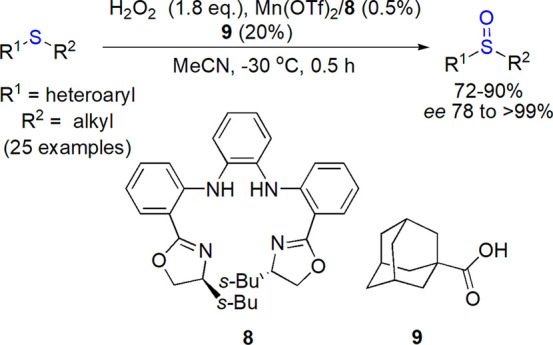
Modified Schiff bases were introduced by several research groups as chiral tridendate ligands for vanadium(IV), typically applied as VO(acac)2 in a two-phase system with hydrogen peroxide as oxidant.58−64 Manganese was used in combination of salen65,66 or another tetradendate, N4 ligand 8; the latter, with carboxylic acid 9 as additive and H2O2 as oxidant, allowed highly enantioselective, gram-scale preparation of various sulfoxides also in the flow system (Scheme 5).67−69 Chiral salen-type Fe(III) complexes were also used in successful sulfoxidations with peroxides.70,71 Nishiguchi et al. applied iron(III) complex with a Schiff base 11 in preparation of proton pump inhibitors and received esomeprazole 2 in 87% yield (12% of sulfone was formed) and >99% ee (Scheme 6).72 Optimized reaction conditions included slow addition of aqueous hydrogen peroxide and the use of lithium 4-dimethylaminobenzoate additive 12 and ethyl acetate as a solvent. Both ligand and carboxylate could be prepared in situ, and the reaction was conducted on a kilogram scale (up to 4.66 kg of sulfide 10). Other derivatives (lansoprazole, pantoprazole, rabeprazole) were obtained in 75–87% yield and 83–98% ee.
Scheme 5. Oxidation of Sulfides Catalyzed by Manganese Complex69.
Scheme 6. Synthesis of Esomeprazole 2 Catalyzed by Fe(III)-Chiral Schiff Base Complex72.
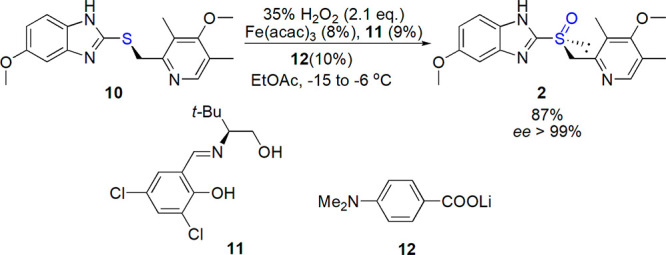
A new concept of asymmetric counteranion-directed catalysis (ACDC)73 was tested by Liao and List; the catalyst 13 consisted of an achiral iron(III) cationic complex and an enantiopure phosphate counteranion.74 High yields and enantioselectivities were noted, setting the new record for salen-Fe systems (Scheme 7).
Scheme 7. Chiral Anion as the Source of Asymmetric Induction in Sulfoxidation Catalyzed by Achiral Cationic Fe(III) Complex74.
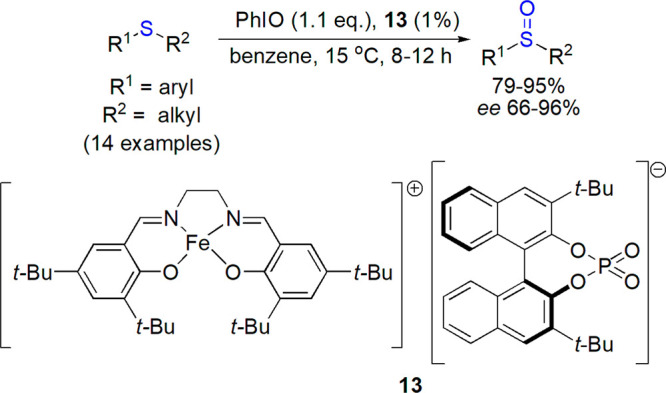
The best effects of asymmetric induction (up to 97% ee) were also achieved in the category of copper-Schiff base catalysts described by Maguire and co-workers.75 The first use of nucleic acid-bound copper(II) complex with bipyridine in sulfoxidation of thioanisole by aqueous hydrogen peroxide was reported by Cheng et al.76,77 A transfer of chirality for G-quadruplex DNA to the product was observed with the enantiomeric excess up to 56% (99% conversion). CD and fluorescence quenching measurements showed that the limited stereoselectivity may result from relatively weak, nonspecific interactions of both thioanisole and Cu complexes with the quadruplex.
Molybdenum(VI) complexes of chiral Schiff bases78,79 or imidazolium-based zwitterionic dicarboxylic acid derived from l-valine80,81 were also used in asymmetric oxidation of sulfides, albeit with moderate or low enantioselectivity, which could be improved by kinetic resolution.80
Chirality of coordination compounds can result not only from the presence of appropriate ligands but also from a stereogenic center located on the central ion. In possible catalytic conversions, this core is usually directly engaged in the catalytic action, and it is not surprising that chiral-at-metal complexes have already found application in various asymmetric transformations.82,83 Ye and co-workers explored the strategy based on the use of such octahedral ruthenium(II) or iridium(III) complexes with 2,2′-bipyridyl or 2-phenylpyridyl ligands, capable of catalyzing enantioselective oxidation of sulfides by m-CPBA but also of preferential coordination of one enantiomer of sulfoxide (dynamic thermodynamic resolution).84−87 Enantiomeric excess of up to 99% was reached for various derivatives, including modafinil acid and its analogues. Photooxidation of hydroxysulfides catalyzed by iridium complex of 2-phenylquinoline connected with coordination-based resolution of enantiomers was reported by Li and Ye in 2019 (Scheme 8).88
Scheme 8. One-Pot Photooxidation of Sulfide 14 Catalyzed by Iridium Complex 15 Coupled with Resolution of Sulfoxide 16 by Chiral-at-Metal complex Δ-17 Described by Li and Ye88.

Construction of supramolecular assemblies and metal–organic frameworks capable of selective oxidation of sulfides to sulfoxides was reported by several groups.89−93 Certain systems containing chiral subunits led to high enantioselectivities.91
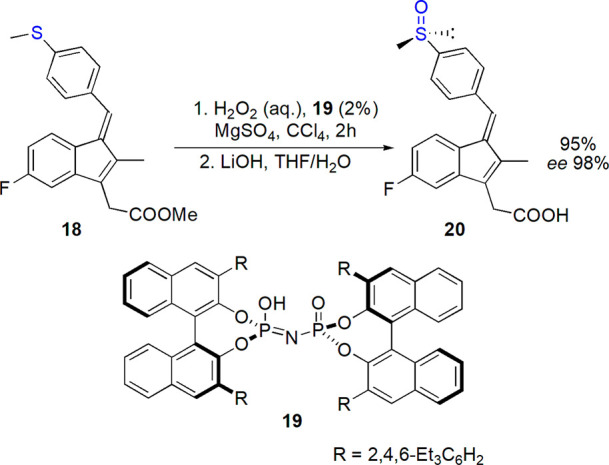
In our previous review on stereoselective synthesis of sulfoxides, we expressed our expectation that organocatalytic sulfoxidation would attract growing attention.1 Since 2010, the actual development has been not so impressive.94−96 However, promising results were obtained with flavin-derived catalysts97,98 and BINOL-based phosphoric acid derivatives.99−101 The latter allowed an enantioselective preparation of (R)-sulindac 20 (95% yield, 98% ee, Scheme 9). However, (R)-modafinil was identified as a more difficult goal, and organocatalyzed oxidation of its parent sulfide with hydrogen peroxide solution was performed with up to 26% ee.102
Scheme 9. Enantioselective Preparation of R-Sulindac with a Chiral Confined Brønsted Acid99.
3.1.2. S–C Bond Formation
Preparation of sulfoxides in Friedel–Crafts-type reactions dates back to the 1974 paper of Olah and Nishimura.103 Among recent applications, sulfinylation of indoles through electrophilic substitution with arylsulfinic acids described by Miao et al. is worth mentioning, as it was performed in water at room temperature without any additives.104 Another green Friedel–Crafts sulfinylation of aromatic compounds with sulfinic esters was carried out in ionic liquid and accelerated by ultrasound irradiation.105 Sulfinamides were applied in a similar, visible light-accelerated transformation of heteroaromatic substrates.106 A lack of source of chiral induction in all above reactions resulted in formation of racemic products.
In recent years, a considerable progress in the enantioselective synthesis of sulfoxides has been connected mainly with the development of methods based on the reactions of sulfenate anions with electrophiles. In 2004, structure, methods of generation, and reactions of sulfenic acid anions were reviewed by O’Donnell and Schwan.107 In the following years, efficient routes to these underexplored intermediates were introduced and their subsequent arylations catalyzed by transition metal complexes were described. Perrio and co-workers identified β-sulfinyl esters as versatile precursors of a sulfenate anion.108 Various bases (NaHMDS, KHMDS, NaH, LDA, n-BuLi, t-BuOK) were efficient in its generation, and reaction with benzyl or alkyl halides led to expected sulfoxides in up to 95% yield. To obtain enantioenriched products, the authors added enantiopure sparteine to the system and observed a weak asymmetric induction (ee up to 29%). The same method of generation of sulfenate anion from β-sulfinyl esters was used by Madec, Poli, and co-workers, who connected it with palladium-catalyzed allylic alkylation109 and arylation.110 Enantioenriched aryl–aryl and aryl–benzyl sulfoxides were prepared by this group by applying palladium catalyst with a chiral ligand.111 Josiphos-type derivatives appeared most efficient in this role (Scheme 10). A pseudodomino process of palladium-catalyzed arylation of anions obtained from allylic sulfoxides was also described.112 The achievements by Poli, Madec, and co-workers in the field were summarized in a short review published in 2010.113
Scheme 10. Stereoselective Preparation of Sulfoxides via Palladium-Catalyzed Arylation of Sulfenate Anion with Josiphos-Type Ligand111.
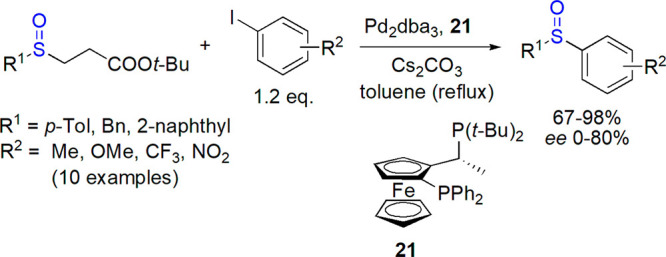
Perrio’s group focused their attention on a diverse approach involving the use of phase-transfer reagents serving both as catalysts and chirality source.114,115Cinchona alkaloid-based ammonium salt was found optimal for this task; it was applied in a two-phase system (aqueous NaOH/toluene–dichloromethane mixture), sulfinyl sulfone served as a starting material, and methyl iodide as an electrophile. The yields were high (up to 96%), but the enantioselectivity was moderate (up to 58% ee, Scheme 11).
Scheme 11. Chiral Phase-Transfer Catalyst 22 in Synthesis of Aryl Methyl Sulfoxides114.
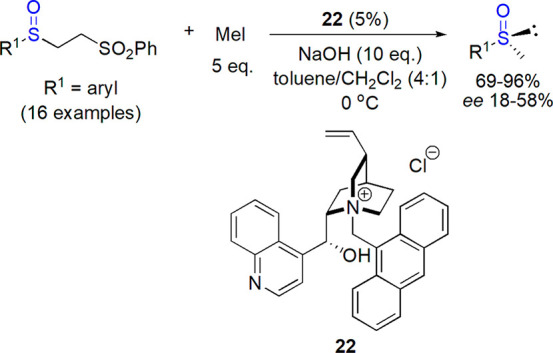
An improvement of methodology based on chiral phase-transfer catalyst was introduced by Zong et al., who described a highly enantioselective alkylation of sulfenate anions bearing heteroaryl substituents in the presence of novel halogenated chiral pentanidium salts (Scheme 12).116 The reaction was performed in cyclopentyl methyl ether or diethyl ether and concentrated aqueous cesium hydroxide; yields and enantioselectivities, particularly for benzyl derivatives, were high (up to 99% ee).
Scheme 12. Use of Iodinated Pentanidium Salt 23 as Phase-Transfer Agent for Preparation of Optically Active Benzyl Heteroaryl Sulfoxides116.
Another report from Perrio’s group explored a possibility of sulfenate anion generation by pyrolysis of tert-butyl sulfoxides followed by the action of K3PO4 as a base and palladium-catalyzed coupling with aryl halides or triflates.117 Induction of chirality in this process was only studied with a planar-chiral racemic substrate, for which 50% de was observed.
Also other research teams contributed to the development of chiral sulfinate methodology. Schwan and co-workers investigated mainly diastereoselective alkylations.118,119 Nolan and co-workers published their improvement to the existing methodology, which suffered from the limited availability of activated sulfoxides required for anion generation and the need of using aryl iodides.120 To overcome these drawbacks, they applied palladium complexed with N-heterocyclic carbene (NHC) as a precatalyst and observed a direct S-arylation of nonactivated aryl sulfoxides by aryl chlorides and bromides. The reaction was not performed in the asymmetric variant.
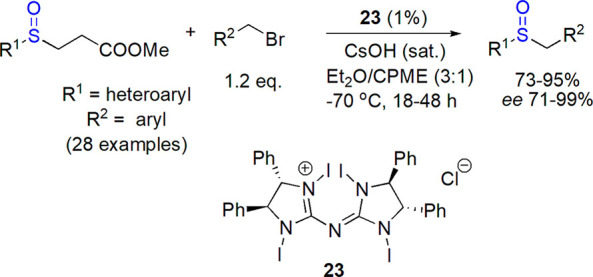
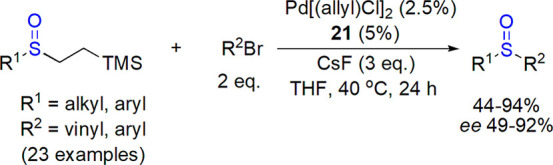
Walsh and co-workers studied palladium-catalyzed arylations of benzylic sulfoxides.121,122 Later on, they concentrated their efforts on finding mild conditions for generation of sulfenate.123,124 In their preparation of enantioenriched aryl–aryl sulfoxides, sodium tert-butoxide was chosen as a base, and the chiral catalyst was formed in situ from Pd(dba)2 and enantiopure ferrocene-based bis-phosphine 21.125 Nineteen sulfoxides were prepared, including these bearing heteroaryl groups, in 73–98% yield and 70–95% ee. An elegant route to two enantiomers of 3-quinolino 6-quinolino sulfoxide was proposed using the same catalytic system and regioisomeric starting materials. As an extension of their studies, Walsh and co-workers described the use of CsF to generate sulfenate anions and their Pd-catalyzed reaction with alkenyl and aryl bromides (Scheme 13).126 Ferrocene-based chiral phosphine ligand 21 and [Pd(allyl)Cl]2 as a palladium source were efficient in the process affording various enantioenriched (49–92% ee) sulfoxides in 44–94% yield.
Scheme 13. Enantioselective Reaction of Sulfenate Anions with Aryl and Alkenyl Bromides126.
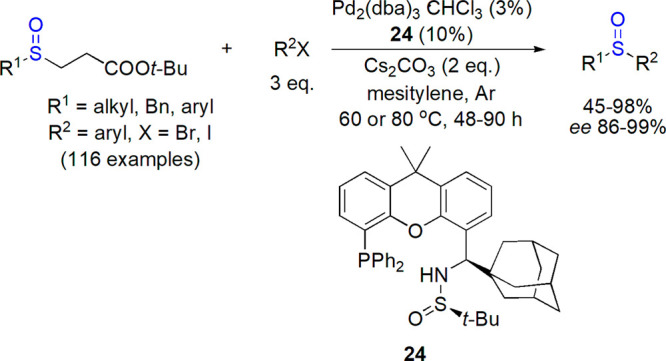
Aryl–aryl, but also alkyl– and benzyl–aryl sulfoxides were enantioselectively prepared by Zhang and co-workers via arylation of sulfenate anions catalyzed by palladium complexes.127 Among various chiral phosphine ligands tested, optimum results were obtained for a new xanthene-based phosphine derivative (R,RS)-24 bearing 1-adamantyl and sulfinamide moieties. Cs2CO3 was used as a base, Pd2(dba)3·CHCl3 as a palladium source, and mesitylene as a solvent (Scheme 14). The versatility of the system was demonstrated by the synthesis of over 100 different sulfoxides, including sulindac, in most cases with enantiomeric excess higher than 90%. A possibility of scaling-up of the protocol was also shown.
Scheme 14. Palladium-Catalyzed Preparation of Sulfoxides Reported by Zhang and Co-workers127.
Although palladium-catalyzed reactions of sulfenate anions were commonly performed, protocols in which this noble metal was not used were described as well. A CuBr2-catalyzed reaction of sulfenate anions generated from sulfoxide ester by cesium carbonate with benzyl radicals formed from arenes by tert-butyl perbenzoate was developed by Bolm and co-workers.128 Transition metal-free arylations were also reported by this group; they obtained racemic aryl–aryl and alkyl–aryl sulfoxides from β-sulfinyl ester and diphenyliodonium triflate with KOH as a base in a water–toluene biphasic system at room temperature.129
In a search for the cheaper methodology for sulfoxide synthesis under milder conditions, Zhang’s group developed a transition metal-free protocol of arylation of sulfenate anions.130 Aryl or alkyl sulfenate anions were generated at room temperature and reacted with diaryliodonium salts. A variety of aryl–aryl and alkyl–aryl sulfoxides were prepared in 62–91% yield. A possibility of scaling-up the reaction was demonstrated; however, it was not applied in the enantioselective preparation of sulfoxides. Racemic alkynyl sulfoxides were prepared by Waser and co-workers in an efficient metal-free method using ethynyl benziodoxolone (EBX) reagents to trap sulfenate anions.131
Synthetic methods based on reactions of sulfenate anions allow for the preparation of a variety of chiral sulfoxides, and in many cases yields and enantioselectivities are excellent. Despite of certain drawbacks (alkyl–alkyl sulfoxides cannot be prepared directly), these routes should be treated as an important alternative way of C–S bond formation for Andersen’s method based on nucleophilic substitution.
3.2. Biological Preparations
Biotechnological methods of preparation of chiral sulfoxides provide an interesting alternative to purely chemical systems. In most cases, oxidation with enzymes results in high enantioselectivity and turnover number. Possible drawbacks are connected mainly with sensitivity of biocatalysts and, sometimes, their wide substrate scope. A longer discussion can be found in our previous review;1 in 2018, sulfoxidations with biological systems were also summarized.132
Two main approaches to enzymatic synthesis of optically active sulfoxides can be distinguished. Whole-cell preparations are operationally simpler and do not require addition of cofactors, although the nature of active catalytic species is not certain. Not surprisingly, the majority of recent reports in this area concern the use of bacteria or fungi for sulfoxidation.133−140 However, the use of isolated enzymes for enantioselective oxidation of sulfides in the past decade was also described by several groups.141−144 Examples include monooxygenases, dioxygenase, and peroxidase.
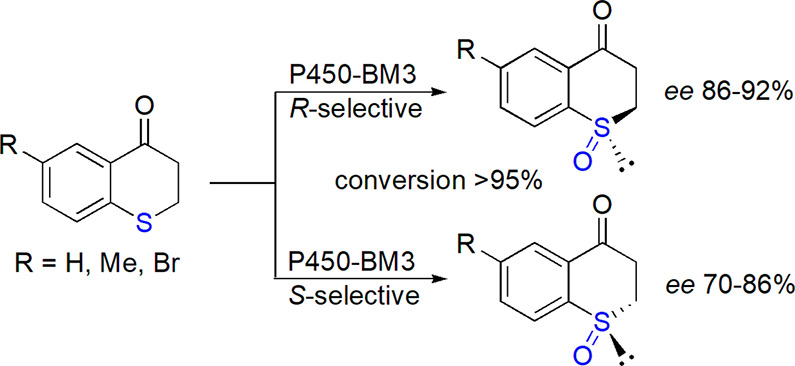
Several research groups reported on the enantioselective oxidation of sulfides catalyzed by appropriately modified enzymes into which metal cofactors were introduced. Such artificial metalloenzymes were based on normally redox-innocent transport protein145 or bovine serum albumin.146,147 In another approach, appropriately designed mutations in the heme pocket of dye-decolorizing peroxidase resulted in introduction of its ability to catalyze oxidation of sulfides with high conversion and enantioselectivity (both up to 99%).148 Among the latest biocatalytic stereoselective preparations of sulfoxides, asymmetric sulfoxidation of 1-thiochroman-4-one derivatives by cytochrome P450-BM3 monooxygenase was described by Reetz and co-workers (Scheme 15).149 Directed evolution allowed increasing of the stereoselectivity (ee increased from 50% for the wild-type enzyme to 86–93%) and even to reversing of the stereochemical preference.
Scheme 15. Sulfoxidation of Thiochromanone Derivatives by Cytochrome P450-BM3 Variants Obtained through Directed Evolution149.
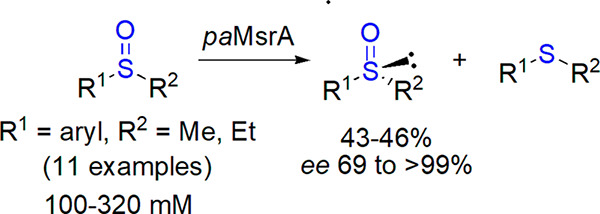
Enzymes can be used also for the reductive resolution of racemic sulfoxides. Electrochemical regeneration of DMSO reductase applied in such a process was described by Chen et al.150 Methionine reductase was also exploited by Chen and co-workers; newly identified homologues tolerated various substrates at high concentration and led to ca. 50% yield (maximum value for resolution) and >99% ee (Scheme 16).151−153
Scheme 16. Reductive Resolution of Alkyl Aryl Sulfoxides by Methionine Reductase paMsrA153.
4. Stereoselective Synthesis of Sulfinates
Esters of sulfinic acid RS(O)OR′ have been treated mainly as valuable synthetic intermediates for other sulfinyl derivatives. A relatively easy preparation of these configurationally stable derivatives bearing a chiral R′ fragment followed by separation of diastereomers allows their transformation into enantiomerically pure sulfoxides and other derivatives. Methods of preparation and major applications of sulfinates were reviewed in book chapters.11,13 Consequently, in this section, only a short overview of older contributions will be presented, with the stress on the possible diverse synthetic routes.
4.1. S=O Bond Formation
Stereoselective sulfoxidation as the method of preparation of optically active sulfinates has not gained popularity in comparison to other approaches, which make use of more available starting materials, are operationally simpler and more efficient. The attempts of enantioselective reactions of sulfenates with chiral oxidants (e.g., peroxocamphoric acid154) or catalytic oxidation systems (Kagan’s method155) were not particularly encouraging (maximum ee of 36% was reached).
Diastereoselective oxidation of benzenesulfenates bearing a phosphonate group at the ortho-position leading to the corresponding sulfinates was investigated by Vazeaux, Drabowicz, and their co-workers.156 Starting sulfenates 27 were obtained in the reaction of chiral alcohols with sulfenyl chloride 26 (in turn prepared from thiol 25, Scheme 17). Menthyl derivatives were oxidized with tested oxidants with high yield and variable diastereoselectivity (up to 76% de for NBS), and the direction of asymmetric induction could be changed if the appropriate enantiomer of chiral oxaziridine was applied. Among other chiral alcohols, 8-phenylmenthol 30 and trans-2-phenylcyclohexanol 31 led to reasonable results (also for (R)-Greene’s alcohol 34, and one of enantiomers of Davis’ oxaziridine 35 86% de was noted, while for the other enantiomer ent-35de = 9% and a reversed stereochemical preference was found).
Scheme 17. Diastereoselective Oxidation of Sulfenates Bearing a Phosphonate Group156.

4.2. S–O Bond Formation
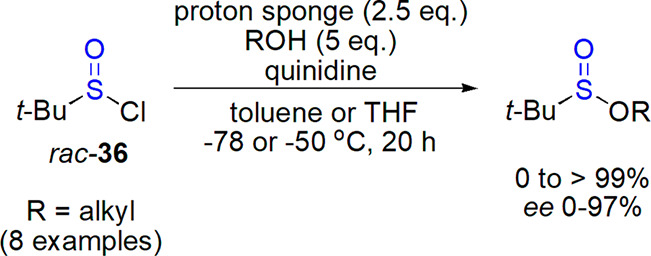
The first successful preparation of optically active sulfinates was achieved by Phillips in 1925, who obtained alkyl p-tolylsulfinates by transesterification.157 Since that time, numerous protocols have been described with chirality transfer from reactants (diastereoselective preparations) or auxiliaries used (enantioselective synthesis). The latter methods often utilized chiral tertiary amines, which were used in the reaction of racemic sulfinyl chlorides and achiral alcohols. As an example, preparation of sulfinates through enantioselective dynamic kinetic resolution of racemic tert-butanesulfinyl chloride was achieved by Ellman and co-workers.158 This compound was subjected to the reaction with benzyl alcohol in the presence of proton sponge and a chiral base (N-methyl imidazole-containing octapeptide) led to the best results (99% yield, 80% ee, for only 0.5% of the catalyst). This catalyst was later replaced by relatively cheap and available Cinchona alkaloids, and the method extended over other (mainly benzyl) alcohols (Scheme 18).159 Simultaneously, highly enantioselective preparation of arenesulfinates from racemic sulfinyl chlorides and achiral alcohols assisted by Cinchona alkaloids was reported by Shibata, Toru, and co-workers (up to 99% ee).160,161
Scheme 18. Enantioselective Preparation of Sulfinates from Racemic Sulfinyl Chlorides in the Presence of Quinidine159.
A variety of chiral alcohols have already found use in diastereoselective preparation of sulfinates. Menthol 29 has often been applied;162 Andersen’s method of stereoselective synthesis of sulfoxides is based on the crystalline (1R,2S,5R,SS)-(−) menthyl p-toluenesulfinate.163 Diacetone-d-glucose (DAG), a sugar-derived alcohol 33, was also identified as a particularly useful auxiliary (both are commercially available).164 Cholesterol, ephedrine, and various sugar alcohols including cyclodextrin should be also mentioned.
Mikołajczyk, Drabowicz, and co-workers thoroughly analyzed the stereospecifity of substitution at a stereogenic sulfur atom. Conversion of enantiomerically enriched sulfinamides to sulfinates in the presence of strong acids was shown to proceed with a predominant inversion of configuration.165 (S)-N,N-Diethyl p-toluene sulfinamide was first prepared from the corresponding menthyl sulfinate and Et2NMgBr. Its reactions with alcohols were found to proceed with medium to excellent stereospecifity, dependent mainly on the structure of alcohols; a complete inversion was observed for primary ones, and the steric hindrance exerted by secondary and tertiary alcohols resulted in partial racemization. Also, transesterification of sulfinates with 2-propanol in the presence of strong acids was primarily shown to proceed with a predominant inversion of configuration, with stereochemical outcome dependent on both sulfinate and acid used (up to 40% ee).166,167
The stereochemistry of conversion of sulfinamides to sulfinate esters was later found to be more complex.168 Bujnicki et al. investigated the effect of substituents of sulfinamide and the structure of alcohol and found that in certain cases a predominant retention of configuration was observed.169 It was associated with a combination of sterically hindered alcohol and a bulky leaving amine fragment. Moreover, the stereochemical preference could be changed by addition of inorganic salt to the system while solvent change did not affect much the stereoselectivity. Analysis of reaction kinetics allowed the authors to propose a mechanism of the reaction involving addition, leading to sulfurane intermediates capable of pseudorotation (forced by steric factors).
A novel approach to the preparation of sulfinate esters was introduced by Jacobsen et al.170 A mixed anhydride was prepared from sodium sulfinate and trimethylacetyl chloride, and reacted with various primary and secondary alcohols in the presence of trimethylamine. Esters were formed in 57–82% yield. Sulfination of nonactivated alcohols with the use of sulfonyl isocyanides was achieved by Ji et al.171 A double activation was provided by Lewis acid (bismuth(III) triflate) and Brønsted acid (CCl3COOH). Asymmetric variant of these two protocols was not proposed.
4.3. S–C Bond Formation
Reactions of sulfites and chlorosulfites with organometallic reagents also provided sulfinates with a variable yield and stereoselectivity. Optically active tert-butyl sulfinates were prepared from symmetrical dialkyl sulfites and tert-butylmagnesium chloride in the presence of chiral amines (the optimal results, ee up to 74%, were obtained for Cinchona alkaloids, bearing also a hydroxyl group; Scheme 19).172 An appropriate choice of the auxiliary allowed to prepare both optical antipodes of the product. An alternative method based on a kinetic resolution upon reaction of racemic thiosulfinates with Grignard reagents and quinine was also developed; it allowed preparation of nonracemic sulfinates with nonbulky alkyl or aryl substituents with ee up to 33%.
Scheme 19. Preparation of Optically Active Sulfinates from Dialkyl Sulfites in the Presence of Quinine172.
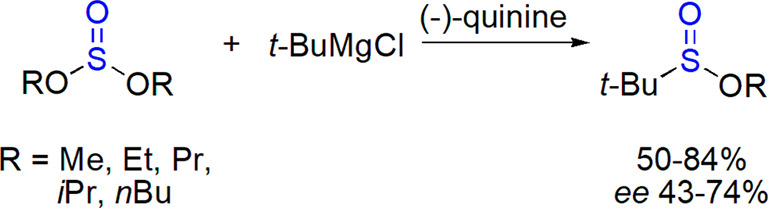
The use of chiral sulfites leading to diastereomeric mixtures of sulfinate products was also described.173,174 Kagan’s group converted the obtained, purified esters into enantiopure sulfoxides.173
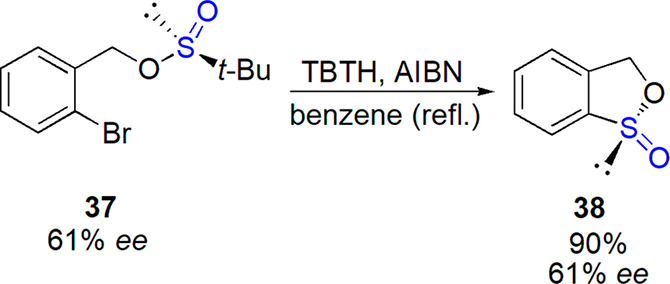
An interesting homolytic substitution process in which certain sulfinates under radical conditions (Bu3SnH, AIBN, heating in benzene) underwent an intramolecular cyclization to give cyclic esters (sultines) was reported by Coulomb et al.175 An optically enriched starting compound led to the product with a complete inversion of configuration (Scheme 20); the similar conclusion was drawn for the cyclization of chiral sulfinamides. When a new stereogenic center was created, a preference for trans configuration was observed and two sultines were formed with a complete diastereoselectivity.
Scheme 20. Stereospecific Radical Cyclization of Enantiopure Sulfinamide175.
4.4. Other Methods
Formation of two bonds in one process was also reported. Sulfinates (but only racemic) were prepared using aerobic, copper-catalyzed reaction of thiols and alcohols.176 Shyam et al. showed that also disulfides and thiosulfinates could be reacted with benzyl alcohol to give the desired ester, although in lower yield.
The racemic O-alkyl alkylsulfinates were resolved using formation of inclusion complexes with β-cyclodextrin.177 The highest optical purity of 70% was achieved for complexed O-isopropyl methylsulfinate.
5. Synthesis of Optically Active Thiosulfinates
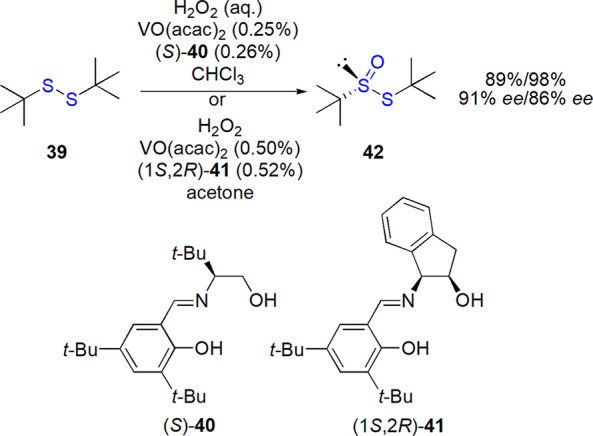
Thioesters of thiosulfinic acid (or disulfide S-oxides) belong to chiral sulfinyl compounds exhibiting limited chemical and configurational stability (see section 2). As the increase of steric hindrance exerted by groups connected to sulfur atoms results in the stabilization, majority of studies focused on tert-butyl tert-butane thiosulfinate 42 (Scheme 21). High-yielding and enantioselective methods have been developed for its preparation from a cheap and easily available tert-butyl disulfide 39, and thus this thiosulfinate is regarded as a valuable starting material for the synthesis of other enantiopure sulfinyl compounds: sulfinamides, sulfoxides, sulfinates, and others.27,178
Scheme 21. Preparation of tert-Butane tert-Butyl Thiosulfinate Developed by Ellman and Co-workers178,184−187.
Asymmetric sulfoxidation procedures are of particular importance, and they include chemical and enzymatic procedures. Other methods, although less significant and with limited practical use, should not be completely neglected.
5.1. S=O Bond Formation
5.1.1. Chemical Methods
The study on the synthesis of nonracemic disulfide monoxides started in 1960s with the works of Savige and Fava.154,179,180 In 1965, Kice and co-workers described an asymmetric oxidation of aryl disulfides to thiosulfinates.181 The use of chiral peroxycamphoric acid led to the optically active product in ca. 60% yield and very low enantioselectivity (ca. 3% ee based on the given specific rotation). Davis’ 2-sulfonyloxaziridines were slightly more efficient (14% ee for oxidation of tert-butyl disulfide, but only 2% ee for p-tolyl derivative),182 and Kagan’s system (t-BuOOH/Ti(O-iPr)4/DET) allowed to increase the enantiomeric excess to 52%.155
Further improvement was possible after introduction of vanadium Schiff base catalysts. In 1997, Ellman and co-workers developed an enantioselective method of preparation of tert-butane tert-butyl thiosulfinate 42 by oxidation of the corresponding disulfide 39 with hydrogen peroxide in a biphasic system in the presence of chiral vanadium complex (Schiff base 40 introduced by Bolm and Bienewald183 was used, Scheme 21).178,184 In subsequent studies, it was proven that slow addition of 30% aqueous H2O2 and the use of cosolvent with low miscibility with water (CHCl3 was found optimal) were of importance for the observed stereoselectivity (up to 91% ee was reached), which is otherwise limited by a nonenantioselective oxidation.185 Later, change of chiral ligand to the one derived from cis-1-aminoindan-2-ol and 3,5-di-tert-butylsalicylaldehyde (41) allowed to exchange the two-phase solvent system to acetone, and to apply the protocol on a kilogram scale (99% conversion, 85–86% ee).186,187 The product could be purified by a simple crystallization and obtained in both enantiomeric forms, depending on configuration of the Schiff base.187
An enantiopure analogue of tert-butylsulfinamide attached to a polystyrene support was also prepared in Ellman’s group using a dynamic resolution methodology.188 Its utility in the enantioselective synthesis of chiral amines and alkaloids was demonstrated.
Other chiral Schiff bases were tested by Ma et al. in vanadium-catalyzed enantioselective oxidation of disulfides by hydrogen peroxide.189tert-Butane tert-butyl thiosulfinate was obtained in 30–93% yield and 4–88% ee when ligands derived form 3,5-di-tert-butylsalicylaldehyde and aminoalcohols were applied. Also para-tolyl disulfide was oxidized by H2O2 under these conditions, albeit in lower yield and enantioselectivity (ee < 40%). Both esterified and reduced derivatives of Schiff bases were found to be inefficient chiral inducers.
5.1.2. Enzymatic Methods
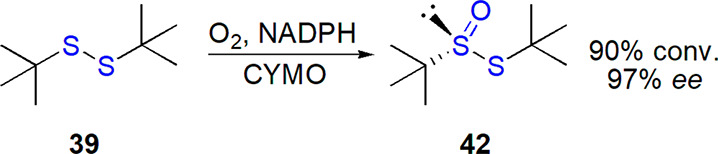
Cyclohexanone monooxygenase was applied by Colonna and co-workers in the asymmetric oxidation of disulfides.190 An excellent enantioselectivity (97% ee and 90% conversion) was observed only for tert-butyl disulfide 37 (Scheme 22), while three other thiosulfinates were obtained in low yield and ee up to 70%, which was attributed to their lower stability and ease of racemization. A regioselective oxidation of sulfur atom connected to tert-butyl substituent was noted for the unsymmetrical substrate, p-tolyl tert-butyl disulfide. Bovine serum albumin was tested as a chiral auxiliary in oxidation of disulfides; desired products were formed, but stereoselectivity was unsatisfactory.191 Instead, an oxidation system based on chiral fructose-derived dioxirane was applied, which led to high conversion and ee up to 75%.
Scheme 22. Oxidation of tert-Butyl Disulfide by Cyclohexanone Monooxygenase190.
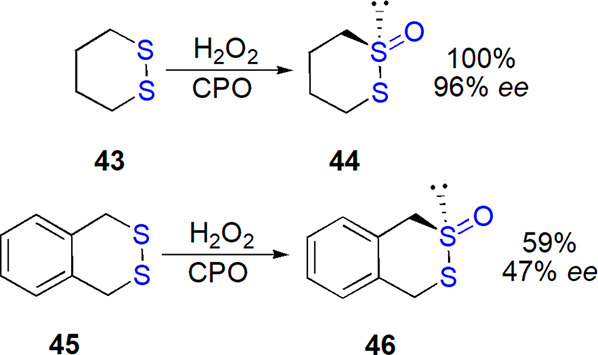
Also, Boyd et al. described oxidation of 1,2-disulfides using enzymatic methods.192−194 While acyclic substrates failed to give expected thiosulfinates under conditions used, 1,2-dithiane was converted into S-oxide quantitatively and with 96% ee by chloroperoxidase (CPO) and H2O2 (Scheme 23). Other tested systems: cyclohexanone monooxygenase and whole cells expressing arene dioxygenases were inefficient in this transformation. CPO catalyzed oxidation of a bicyclic substrate in 59% yield and 47% ee (Scheme 23); in both cases, configuration of the main isomer was established as S. A possibility of stereoselective deoxygenation of racemic dithiane-derived thiosulfinate by dimethyl sulfoxide reductase (DMSOR) to yield enantioenriched residual substrate (95% ee, S isomer) was demonstrated as well.192,193
Scheme 23. Oxidation of Cyclic Disulfides by Chloroperoxidase194.
5.2. S–S Bond Formation and Other Preparations
Drabowicz and Mikołajczyk explored various possibilities for preparation of nonracemic thiosulfinates from other sulfinyl derivatives. Thermal decomposition of di-tert-butyl sulfoxide in the presence of equimolar amount of enantiopure chiral amines led to the formation of nonracemic t-butyl t-butanethiosulfinate (Scheme 24).195 Although enantiomeric excess was low (1–26%, the latter value for quinine as inducer of chirality), both enantiomers of thiosulfinate could be prepared, and amines could be recovered after the reaction. Other thiosulfinates could not be prepared with this method, which was attributed to their instability under the conditions used.
Scheme 24. Preparation of Optically Active Thiosulfinate by Decomposition of Corresponding Sulfoxide195.
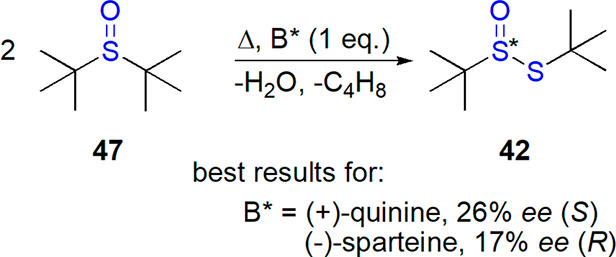
Chiral amines were also applied as chiral inducers in the condensation of sulfinyl chlorides with thiols.27 (−)-N,N-Dimethylamphetamine and (+)-N,N-dimethylfenchylamine allowed preparation of six enantioenriched (S)-thiosulfinates (Scheme 25). A reaction of tert-butyl thiol with arylsulfinic acids in the presence of chiral carbodiimide led to the corresponding thiosulfinate in up to 2% ee.196
Scheme 25. Synthesis of Enantioenriched Thiosulfinates from Sulfinyl Chlorides and Thiols27.
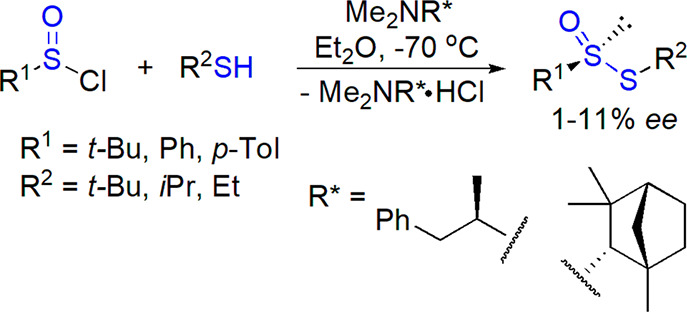
Nonracemic thiosulfinates were also prepared by Drabowicz and Mikołajczyk by treatment of enantiomerically enriched sulfinamides with thiols in the presence of trifluoroacetic acid.197 The reaction took place with predominant inversion of configuration, with stereospecifity in the range of 30 to >80%, dependent mainly on the structure of thiols.
tert-Butane tert-butyl thiosulfinate was obtained by Liao et al. by resolution of racemate using inclusion crystallization with (R)-binol, yielding both enantiomers with high enantiomeric purity (ee > 99%).198
6. Stereoselective Preparation of Sulfinamides and Sulfinimines
Trisubstituted sulfinyl compounds of the general formula R1S(O)NR2R3 (sulfinamides) or R1S(O)N=CR2R3 (sulfinimines/N-sulfinylimines) belong to most important chiral auxiliaries in asymmetric synthesis, used in preparation of a great variety of optically active heteroorganic compounds containing a nitrogen atom. Primary (R2 = R3 = H), secondary (R2 = alkyl, aryl, R3 = H), and tertiary amides can be prepared; the appropriate choice of R1–R3 groups results in modification of their properties, e.g., basicity and nucleophilicity of the nitrogen atom, which may influence the course of asymmetric reactions.
A chapter of “Chiral Sulfur Reagents” by Mikołajczyk, Drabowicz, and Kiełbasiński presents methods of stereoselective preparation of sulfinamides and N-sulfinylimines (named N-alkylidenesulfinamides throughout the chapter) and their synthetic applications published up to 1996.11 In their chapter from the book, edited by Toru and Bolm, Senanayake and co-workers covered the literature up to 2007, with a special focus of a general preparative route introduced in their laboratory.15 Ellman and co-workers published two review papers in which tert-butanesulfinamide was presented as a useful synthetic intermediate in asymmetric reactions.199,200N-Sulfinylimines, the family of compounds of great synthetic importance, were the subject of the second part of article by Davis, who briefly described the synthesis of enantiomerically pure compounds from his group, more widely showing their great potential as chiral auxiliaries and reactants.37 In their papers published in 2002 and 2009, respectively, Ellman’s group and Ferreira and co-workers narrowed their topic to tert-butanesulfinimines: their enantioselective preparation, postsynthetic modification, and use in asymmetric transformations, mainly in the synthesis of chiral amines.201,202
Typically, optically active sulfinamides are prepared from enantiomerically pure sulfinates, thiosulfinates, or sulfinyl chlorides (the latter generated in situ) by a nucleophilic substitution. However, other synthetic routes to these compounds have been reported as well. Sulfinamides and N-sulfinylimines can be mutually interconverted without the change of configuration of sulfur; importantly, such conversions can be stereoselective if a new stereogenic center or double bond is formed.
6.1. S=O Bond Formation
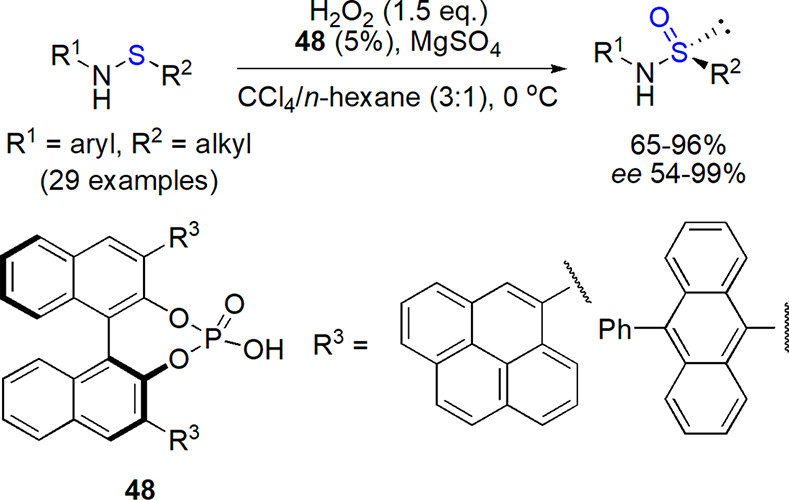
Only a few examples of preparation of sulfinamides by oxidation of corresponding sulfenamides were reported. This approach is hindered by a limited scope of available substrates, although they can be prepared, e.g., from thiols, disulfides or sulfenyl halides.203 Among recent examples, a nonstereoselective oxidation of various sulfenamides by KF/m-CPBA system in acetonitrile–water at 0 °C in 82–94% yield was performed by Datta et al.204 The reaction was fast (5–20 min), and overoxidation products were not formed. A recent report by Tang and co-workers on enantioselective oxidation of sulfenamides with hydrogen peroxide mediated by a chiral binaphthyl-based phosphoric acid derivative showed the possibility of preparation of nonracemic sulfinamides via S=O bond formation (Scheme 26).205 Under optimized reaction conditions (dichloromethane/35% aqueous H2O2/5% of catalyst/MgSO4 additive/0 °C), overoxidation to sulfonamide was not observed, and various derivatives were isolated in 65–96% yield and with high enantioselectivity (in most cases 90–99% ee). A proposed mechanism involved activation of reactants by a catalyst through hydrogen bond formation, and the observed stereochemical outcome was substantiated with DFT calculations of transition states. The prepared sulfinamides were further converted into other compounds from this class (vide infra) and sulfoxides.205
Scheme 26. Preparation of Optically Active Sulfinamides by Oxidation of Sulfenamides Catalyzed by Chiral Phosphoric Acid Derivative205.
Other attempts involving oxidation of sulfenyl derivatives formed from thiols or disulfides are worth mentioning. N-tert-Butanesulfinylphthalimide was prepared by treatment of the corresponding sulfenyl derivative (obtained from N-bromophthalimide and tert-butyl disulfide) with peroxyacetic acid.206 Ring opening under Lewis acid catalysis (samarium triflate was used) followed by the addition of ammonia yielded tert-butane sulfinamide. Unfortunately, all attempts to perform the process in a stereoselective manner were unsuccessful. Copper-catalyzed oxidative coupling of thiols or disulfides with amines was reported by Taniguchi.207 Reactions were carried out under air, and the presence of dioxygen was found necessary for the reasonable yield. Only achiral reactants and catalysts were tested.
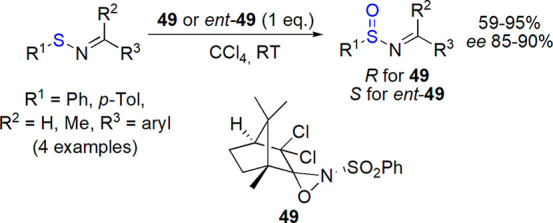
N-Sulfinylimines were also prepared by enantioselective oxidation of sulfenylimines. In 1997, Davis and co-workers described the use of enantiopure oxaziridines 49 for this purpose and obtained the desired products in 59–95% yield and enantiomeric excess up to 90% (improved to >97% by crystallization, Scheme 27).208 Kagan’s method was less efficient (80% yield, 13% ee). As preparation methods based on nucleophilic substitution were found more promising, this approach was abandoned.
Scheme 27. Enantioselective Oxidation of Sulfinylimines with Oxaziridines208.
6.2. S–N Bond Formation
Sulfinamides are conveniently obtained from other trisubstituted sulfinyl derivatives. Nonracemic sulfinates have been used in such conversions for over half of a century. In 1968, Colonna, Giovini, and Montanari prepared optically active p-toluenesulfinamides from the corresponding menthyl sulfinate and dialkylaminomagnesium bromides.209 The reaction proceeded with an inversion of configuration. Later on, this auxiliary introduced by Andersen,163 now commercially available, was used in the synthesis of other sulfinamides, mainly with the use of lithium amides. Davis and co-workers reacting this sulfinate with lithium amide followed by addition of aldehyde or ketone obtained the corresponding N-sulfinylimine with high enantioselectivity (Scheme 28).208,210,211
Scheme 28. Use of Menthyl Sulfinate in Stereoselective Preparation of Sulfinamides and Sulfinylimines37.
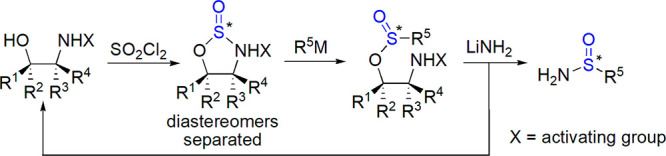
In 2002, Senanayake’s group introduced a versatile and stereoselective method for the synthesis of sulfinyl compounds with the use of N-activated oxathiazolidine oxides (Scheme 29).212,213 Their subsequent reactions with organometallic compounds and lithium amide led to a variety of tertiary alkyl and aryl sulfinamides.15 Reaction of N-acylated 1,2,3-oxathiazolidine-2-oxide with Grignard compounds was also used to prepare chiral sulfinamides by Qin et al.214
Scheme 29. Senanayake’s Approach to Chiral Sulfinamides15.
Nucleophilic substitution of enantioenriched tert-butane tert-butyl thiosulfinate (see Section 5.1) was established by Ellman and co-workers as a very convenient route to tert-butyl sulfoxides and, in particular, tert-butanesulfinamide (lithium amide was used, Scheme 30).178,184,186,187 No racemization during this step was observed, and the product could be obtained in multigram amounts. Compound 52 was identified as a valuable precursor for a range of enantiomerically pure tert-butanesulfinyl derivatives and, after removal of sulfinyl auxiliary, chiral nitrogen-containing compounds. As an example, reaction with aldehydes provided N-sulfinylimines; Grignard addition and cleavage with acid yielded optically active branched amines.178
Scheme 30. Preparation of Enantiomerically Pure tert-Butanesulfinamide and Its Transformations178,186,187.

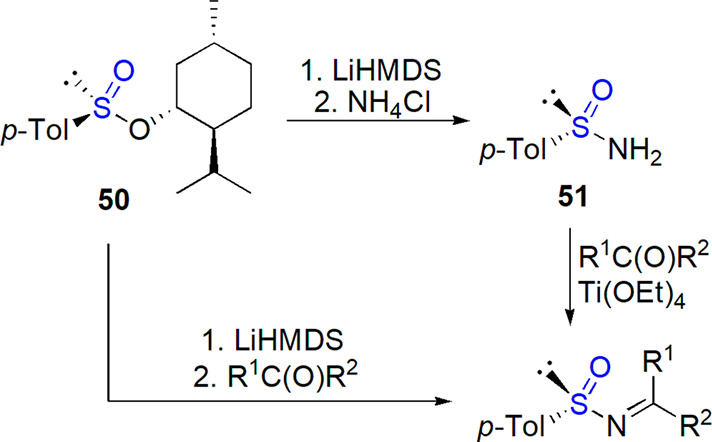
Various enantiomerically pure aldimines were prepared in a one-step condensation of sulfinamide 52 with aldehydes in the presence of dehydrating agents.215 Also, ketimines were formed with a significant E/Z preference from respective ketones when Ti(OEt)4 was applied as a Lewis acid (Scheme 30).215,216 The procedure was later optimized, and tetrahydrofuran and cyclopentyl methyl ether (preferred in the case of more challenging ketones) were identified as optimal solvents.217 The possibility of recovering the chiral auxiliary was investigated as well.218 After the cleavage of imine with HCl, fast racemizing sulfinyl chloride was separated from chiral amine hydrochloride. The dynamic chiral resolution of this sulfinyl derivative treated with ethanol in the presence of quinine and proton sponge (both could be reused as well) in cyclopentyl methyl ether led to tert-butanesulfinamide in high yield and enantiomeric purity (up to 87% ee).
Alternatively, reduction of ketimines with NaBH4 could be performed to give sulfinamides (66–86% yield, 80–94 de).219 Both enantiomers of amine were obtained with high ee, with configuration dependent on the choice of reducing agent (NaBH4 vs l-selectride).220 Addition of alkyl, aryl, alkenyl, and allyl carbanions to aldimines and ketimines was performed, and the resulting sulfinamides were cleaved by HCl in methanol, yielding a variety of enantioenriched branched amines.221 Other examples of the use of tert-butylsulfinyl auxiliary in asymmetric synthesis by Ellman’s group included preparation of (among others) amino acids,222−224 amino alcohols,225−227 cyclic amines,228,229 unsaturated amines,2,230,231 nitriles,232 aminophosphonates,233 aminoboronates,234 and total synthesis of natural products.235,236
Novel urea and thiourea organocatalysts bearing N-sulfinyl fragment as a chirality source and an acidifying agent were developed in Ellman’s laboratory.237 They were obtained in condensation of isocyanates or isothiocyanates with tert-butanesulfinamide and used in asymmetric aza-Henry reaction. High stereoselectivity was observed for derivatives also containing aminoindanol moiety. Other sulfinylureas were found efficient in enantioselective additions to nitroalkanes.238−242 Also enantiopure N-sulfinyl prolinamide was prepared via deprotonation of tert-butanesulfinamide and reaction with (S)-proline methyl ester, and its high efficiency in aldol reaction was demonstrated.243 Ellman’s group applied enantiopure tert-butanesulfinamide 52 to the preparation of chiral ligands. C2-Symmetric bis(N-sulfinylimine) derivatives and bis(sulfinyl)imidoamidine (SIAM) were tested in copper- and zinc-catalyzed asymmetric Diels–Alder reaction (up to 98% de and up to 98% ee),244 while N-sulfinylimine ligands bearing phosphine moiety were used in palladium-catalyzed allylic alkylation (yielding up to 96% ee),245 and iridium-catalyzed hydrogenation of stilbenes (up to 94% ee).246
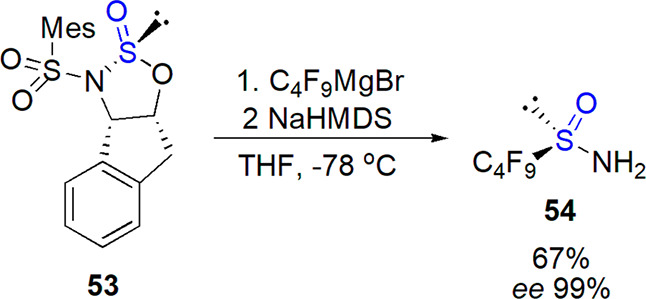
Later, Ellman and co-workers developed an improved synthesis of more electrophilic perfluorobutanesulfinamide 54 accomplished with the use of Senanayake’s 2-aminoindanol-derived sulfinyl transfer reagent213 and a suitable Grignard reagent prepared from EtMgBr and C4F9I.247,248 The sulfinate intermediate was treated with amine nucleophiles; NaHMDS led to the highest yield. The sulfinamide could be prepared on a gram scale in 67% yield and 99% ee using this two-step sequence (Scheme 31). Its condensation with ethyl glyoxylate provided N-sulfinyl imine ester, which was used in asymmetric transformations (Rh(III)-catalyzed C–H bond functionalization, aza-Diels–Alder reaction).247,249
Scheme 31. Preparation of Perfuorobutanesulfinamide248.
An alternative for tert-butane sulfinamide for the application in asymmetric synthesis was introduced by Li and co-workers.250 The novel auxiliary combined properties of para-tolyl- (aromatic chromophore) and tert-butyl-substituted (stability) derivatives. (R)-2-Phenyl-2-propyl sulfinamide 58 was prepared in four steps starting from the respective thiol, with H2O2/VO(acac)2/chiral Schiff base oxidation of disulfide as a key step for chiral induction (85% ee was further improved to >99% by washing with n-hexane, Scheme 32). This time, amidation of thiosulfinate was not straightforward and required the use of tert-butyldimethylsilyl amine (TBDMS-NH2) followed by deprotection. Reaction of aldimines and ketimine derived from this sulfinamide with allylmagnesium bromide proceeded with high diastereoselectivity, additionally increased by simple (group-assisted) purification.
Scheme 32. Preparation of Sulfinamide 58 by Li and Co-workers250.

Sulfinyl chlorides are less convenient in the stereoselective synthesis of sulfinamides due to their instability and configurational lability. However, several enantiopure arylsulfinamides were obtained by Zhu and Shi by a spontaneous crystallization of products of reaction of sulfinyl chlorides with (R)-N-benzyl-1-phenylethanamine.251 (R,SS)-Diastereomers were isolated from the reaction mixture as solids in ca. 30% yield, while mother liquor was enriched in (R,RS) isomers (50–58% de) and could be separated by chromatography. Enantiopure sulfinates were prepared from the corresponding sulfinamides using methanol and boron trifluoride etherate and later converted to primary (SS)-sulfinamides (which could not be obtained directly form tertiary counterparts).
A protocol for synthesis of sulfinamides from amines and sulfonyl chlorides reduced in situ with phosphine was developed by Harmata and co-workers.252 The reaction showed broad substrate scope, although the use of chiral amines did not result in diastereoselection. We observed modest diastereoselectivity (up to 17% de) when we reacted tosyl and nosyl chlorides with enantiomerically pure amines (both commercially available and bicyclic derivatives prepared in our laboratory) under modified Harmata’s conditions (Scheme 33).253 Products were isolated as single stereoisomers by chromatographic separation.
Scheme 33. Preparation of Chiral Epimeric Secondary Sulfinamides from Sulfonyl Chlorides and Enantiopure Amines253.
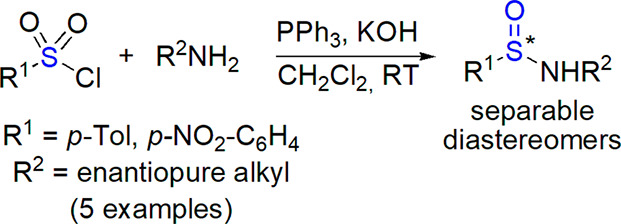
Tang and co-workers converted N-phenyl tert-butane sulfinamide prepared by enantioselective oxidation of its sulfenyl counterpart (see section 6.1) into other derivatives (for which direct oxidation was not successful) via N-protection and nucleophilic substitution with primary amines.205 While yields were moderate (55–70%), almost complete inversion of configuration was observed (94–97% ee). Copper-catalyzed transamidations of primary sulfinamides with O-benzoylhydroxy-substituted secondary and primary amines were performed by Bolm and co-workers in 61–83% and 24–79% yield, respectively.254 The use of enantiomerically pure amines led to 1:1 mixtures of diastereomers, which were separated by column chromatography to yield enantiopure sulfinamides.
N-Bromosuccinimide was used by Wei and Sun to activate tert-butylsulfinyl group in sulfoxides.255 Reaction with nitrogen, oxygen, or carbon nucleophiles led to sulfinamides, sulfinates, and other sulfoxides, respectively, generally in high yield. Only racemic compounds were prepared.
6.3. Dealkylation/Dearylation of Sulfoximines
Enantiopure sulfinamides were also obtained by dealkylation of sulfoximines with retention of configuration. First examples of this reactivity were reported in 1971. Schroeck and Johnson performed reduction with aluminum amalgam,256 Tsujihara et al.257 conducted pyrolysis of alkyl-substituted derivatives, and Williams et al. carried out tosylation.258 Schroeck and Johnson also observed a similar conversion (reductive elimination of N(CH3)2 or OPh group, respectively) of sulfonimidamides and sulfonimidates.256 More examples of stereoselective transformations of sulfoximines to sulfinamides caused by various triggers can be found in a recent review by Wiezorek et al. devoted to degradation of sulfoximines.30 These reactions, though interesting for the study of stability and stereochemistry of involved derivatives, cannot be regarded as useful preparative routes for optically active sulfinamides.
6.4. Other Methods
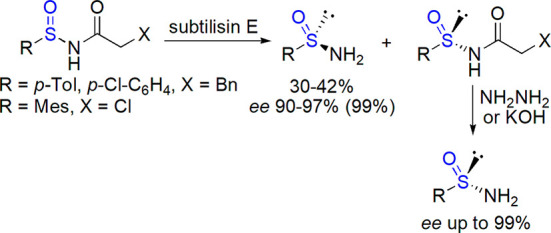
Various methods based on transformation of substituents or involving rarely used sulfur reagents were described. A biocatalytic route to optically active sulfinamides was developed by Kazlauskas and co-workers.259 Hydrolysis of racemic N-chloroacetyl and N-dihydrocinnamoyl arylsulfinamides were catalyzed by subtilisin E overexpressed in Bacillus subtilis, providing mainly (R) primary sulfinamides (Scheme 34). Their (S) counterparts could be formed after nonenzymatic hydrolysis of unreacted acylated sulfinamides. Yields were moderate, but an excellent enantioselectivity was observed in most cases. Gram-scale resolutions were also performed, and synthetically useful auxiliaries were obtained with 95–99% ee (after recrystallization). Enantioselective N-arylation of racemic primary sulfinamides with aryl iodides catalyzed by chiral Cu(I) complexes was described by Liu et al.260 Under optimized conditions, substituted sulfinamides were obtained in variable yield (37–93%) and medium stereoselectivity (5–60% ee).
Scheme 34. Biocatalytic Deracemization of N-Acyl Sulfinamides259.
Inspired by the rapid development in the sulfenate anion application in the preparation of sulfoxides (see section 3.1.2), Dai and Zhang decided to study electrophilic amidation of these anions generated from β-sulfinyl esters.261 Optimization of reaction conditions revealed CuI/bypy as the best catalytic system, lithium tert-butoxide as the most efficient base, with toluene as solvent. A variety of aliphatic and aromatic sulfinamides were prepared in 44–96% yield. The reaction could be performed in a gram scale; its asymmetric variant, however, was not described.
Wang et al. reported on a cross-coupling of arylboronic derivatives with aminosulfur trifluorides (DAST reagents) to afford diverse aromatic sulfinamides in yields up to 92%.262 The use of enantiopure tocopherol-derived boronic acid resulted in the 1:1 mixture of epimers; all other compounds were racemic.
7. Stereoselective Synthesis of Sulfoximines
About 60 years ago, Bentley and co-workers identified a novel sulfinyl compound from the treatment of protein with nitrogen trichloride.263−266 They were able to separate diastereomers of methionine sulfoximine and prepared other sulfoximines from sulfoxides using HN3.267,268 Since then, these stable monoaza analogues of sulfones have been recognized as valuable auxiliaries, ligands, and catalysts in asymmetric synthesis, building blocks in pseudopeptides and, in the recent years, also as drug candidates.
Sulfoximines share many properties with sulfoxides: they are chemically and (if chiral), also configurationally stable. They bear sulfinyl group capable of high induction of chirality. However, the presence of a nucleophilic imine nitrogen atom which can participate in hydrogen bonding or metal coordination makes them special.
A recent growing interest in the synthesis and utilization of nonracemic sulfoximines prompted several research groups to write reviews, general, or focused on specific types, transformations, and applications. Bolm and co-workers prepared a chapter in the book on organosulfur chemistry.14 Articles by this group dealt with properties and applications of fluorinated derivatives,269 methods of sulfur atom imidation of sulfides and sulfoxides,270 and routes of degradation of sulfoximines.30 The importance of these compounds in the area of drug discovery is highlighted in articles by Bolm’s group271 and Lücking et al.272,273 Harmata and Hong reviewed the chemistry of 2,1-benzothiazines, cyclic sulfoximines studied thoroughly by Harmata’s group.274 Bull, Degennaro, and Luisi decided to highlight the progress made in preparation of N-unsubstituted derivatives.275 Two reviews written in Chinese on synthesis and applications of sulfoximines can be added to the list.276,277
In this section we shall focus on showing diverse synthetic strategies and efficient routes to variously substituted derivatives. In their review published in 2000, Reggelin and Zur stated that the most chemistry of optically active sulfoximines emanated from several “key intermediates”, mainly S-methyl-S-phenyl-substituted compounds resolved through formation of diastereomeric salts with 10-camphorsulfonic acid.278,279 Though resolutions (mainly kinetic) still remain important (see section 7.5), enantiomerically pure or enriched compounds can be now prepared using a variety of pathways developed in the recent years.
7.1. S=O Bond Formation
Typical preparations of sulfoximines start from prochiral sulfide. Oxidation and imidation steps can be performed in different sequence (Scheme 35). However, introduction of NH or NR fragment into a sulfoxide was recognized as more attractive strategy than the oxidative transformation of sulfilimines (sulfilimides). They are relatively stable toward oxidation, and access to sulfoximines requires strong oxidizing agents, such as potassium permanganate, alkaline hydrogen peroxide, etc.280 As far as we know, chiral oxidants have not been used for this transformation. However, chirality transfer is possible as sulfilimines can be obtained as single enantiomers.281 Stereochemistry of oxidation of N-sulfonylsulfimines with alkaline solution of KMnO4 was studied by Kresze and Wustrow, who observed retention of configuration.282 Long reaction times are sometimes required for this oxidant, and yields can be small (for instance, only 10% for oxidation of thiane-derived sulfilimine).283 However, permanganates are still chosen for certain preparations. For example, sulfilimines bearing perfluoroalkyl substituents were prepared from appropriate sulfoxides by treatment with nitriles; oxidation with KMnO4 led to the corresponding racemic sulfoximines, either bearing N-acyl or NH fragment (depending on the environment).284
Scheme 35. Possible Pathways for Preparation of Sulfoximines from Sulfides.
An improvement was introduced to the method of preparation of the crop protection agent sulfoxaflor (known also under trade name Isoclast active).285 The N-CN sulfilimine was prepared from pyridine sulfide treated with iodobenzene diacetate and H2NCN in acetonitrile and then oxidized. Various oxidation methods were tested, including RuCl3/NaIO4 combination, but for practical and economic reasons, 40% aqueous NaMnO4 was chosen. As the insecticide is used as a mixture of isomers (two stereogenic centers are present in the molecule), stereoselectivity of the reaction was not studied.
Various sulfoximines were also prepared by oxidation of sulfilimines with ruthenium tetroxide, generated in situ from NaIO4 and substoichiometric amounts of RuO2.286,287
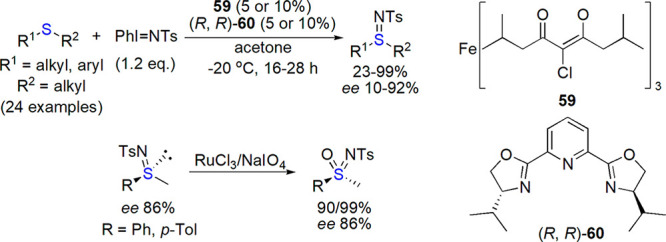
Racemic N-cyanosulfilimines, prepared by Bolm and co-workers from sulfides and cyanogen amine, were easily oxidized with m-CPBA into corresponding sulfoximines, and the cyano group was cleaved by treatment with trifluoroacetic acid and methanolysis.288 Later on, the enantioselective sulfimidation of sulfides with PhI=NTs catalyzed by transition-metal chiral complexes and bis-oxazoline ligands was studied (Scheme 36).289 The best results (yield reaching 99%, up to 82% ee) were noted for iron(III) acetylacetonates used as catalyst precursors. Oxidation of the obtained N-tosylsufilimines with NaIO4/RuCl3 or m-CPBA led to optically active sulfoximines with high enantioselectivity, from which a protecting group could be easily removed.
Scheme 36. Enantioselective Imination of Sulfides and Enantiospecific Oxidation of Sulfilimines289.
A chiral N-mesyloxycarbamate caused a stereoselective amination of thioethers in the presence of a chiral dirhodium(II) carboxylate catalyst to yield the corresponding sulfilimines.290,291 The possibility of preparation of sulfoximine through oxidation of sulfilimine with NaIO4 with RuCl3 as a catalyst was demonstrated; removal of chiral auxiliary with zinc in acetic acid yielded NH-sulfilimine as a single enantiomer (94%). Lebel and co-workers also applied iron catalyst in a similar preparation.292 Increase of yield was possible thanks to application of continuous flow technology. 1-Butylimidazole was used as a base to form 1-butylimidazolium mesylate as an ionic liquid byproduct.
Alternatively, chlorination can be applied to increase oxidation state of sulfur atom. Furukawa, Oae, and co-workers found that alkaline hydrolysis of N-chlorinated sulfilimines resulted in formation of NH-sulfoximines in high yields (up to 95%).293,294N-Chlorosuccinimide and sodium hypochlorite were used as chlorinating agents for NH-sulfilimine; both reaction steps were shown to proceed with a retention of configuration.
7.2. S=N Bond Formation
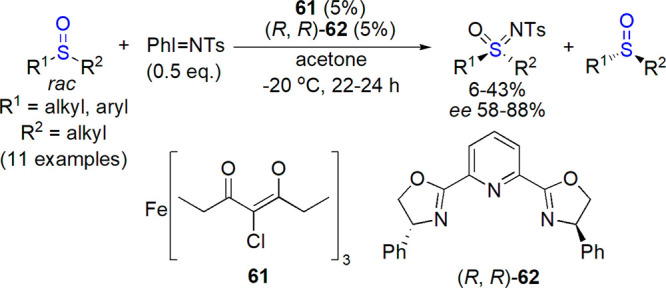
In a 2015 article by Bizet, Hendriks, and Bolm, methods for preparation of sulfoximines by imidation of sulfoxides (and sulfimides from sulfides) were comprehensively reviewed.270 Efficiency of various imidating agents: N-haloamides and similar compounds (chloramine T, MSH), aziridine derivatives, dioxazolones, azides, and iminoiodinanes was compared. Uncatalyzed reactions typically gave lower yields in comparison with protocols engaging metal complexes: copper, silver, rhodium, ruthenium, and iron. Most imidations of sulfoxides were performed with racemic reactants. However, several research groups checked the stereospecifity of the reaction using enantiopure or enantiomerically enriched sulfoxide as a starting material. In each case, a complete retention of configuration was observed. An interesting example of kinetic resolution from Bolm’s laboratory is noteworthy: racemic sulfoxides were treated with PhI=NTs in the presence of Fe(acacCl)361 and a chiral bis-oxazoline ligand 62, yielding sulfoximines with up to 88% ee (and up to 43% yield, Scheme 37).295
Scheme 37. Preparation of Optically Active Sulfoximines through Kinetic Resolution of Racemic Sulfoxides295.
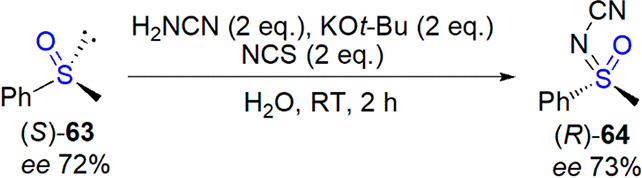
Among recent contributions from Bolm’s laboratory, an efficient and stereospecific preparation of N-cyanosulfoximines from sulfoxides was described by Dannenberg et al.296 Optimized reaction conditions included the use of cyanamide (2 equiv), potassium tert-butoxide (2 equiv) as base, and N-chlorosuccinimide (2 equiv) as oxidant, the set of reactants that were efficient for the synthesis of N-cyanosulfilimines from sulfides.288 Reaction was performed in water at room temperature, with yields varying from 23 to 98%. Retention of configuration was observed for imination of a representative, enantioenriched sulfoxide (Scheme 38).
Scheme 38. Stereospecific Formation of N-Cyanosulfoximine from Sulfoxide296.
NH-Sulfoximines were synthesized in the reaction of sulfoxides and triflic acid salts of hydroxylamine catalyzed by Fe(II) complex.297 No attempt was made to perform the reaction with enantioenriched sulfoxide or chiral iron catalyst.
The significant increase of yield (up to 91%) of imination of sulfoxides with NaN3 or TMSN3 was observed by Gutmann et al. when concentrated sulfuric acid was replaced by a fuming one.298 The two-phase (H2O/CH2Cl2) reaction could be performed under continuous flow conditions. However, the original optical purity of the reactant was not retained.
In 2015, Bull and co-workers described a rhodium-catalyzed transfer of carbamates to sulfoxides, yielding N-protected sulfoximines in good to excellent yields (54–98% under optimized conditions).299 BocNH2 and PhI(OAc)2 were used to generate BocN=IPh species in situ, Rh2(OAc)4 acted as a catalyst, and MgO served as a base. Carbamate protection allowed performing good-yielding Suzuki coupling with S-aryl substituents; on the other hand, Boc and Cbz groups were easily removed to afford NH-sulfoximines. As shown in one example, both N-transfer and deprotection proceeded with a complete retention of configuration (Scheme 39); other reactions were performed with racemic samples.
Scheme 39. Stereospecific Formation of Sulfoximine from Sulfoxide-Direct NH Transfer and Rh-Catalyzed Carbamate Transfer299,300.
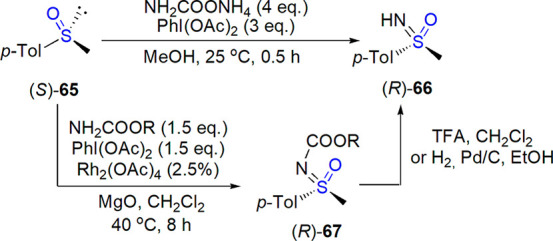
In the search of a convenient electrophilic nitrene source, Bull, Luisi, and co-workers tested ammonium salts for a direct NH transfer under metal-free conditions.300 The use of ammonium carbamate (4 equiv) and diacetoxyiodobenzene (3 equiv) in methanol at 25 °C resulted in fast formation of sulfoximines from the corresponding sulfoxides. The reaction showed a wide substrate scope. Stereochemical outcome of the process was checked for three enantioenriched sulfoxides, for which a complete retention of configuration was observed (an example is shown in Scheme 39).
Later, the same group reported the use of similar set of reactants for the one-pot preparation of sulfoximines from sulfides301 and conversion of thiols to sulfones and sulfonimidates (vide infra).302 The possibility of performing the synthesis in a flow reactor was demonstrated as well.303
7.3. S–C Bond Formation
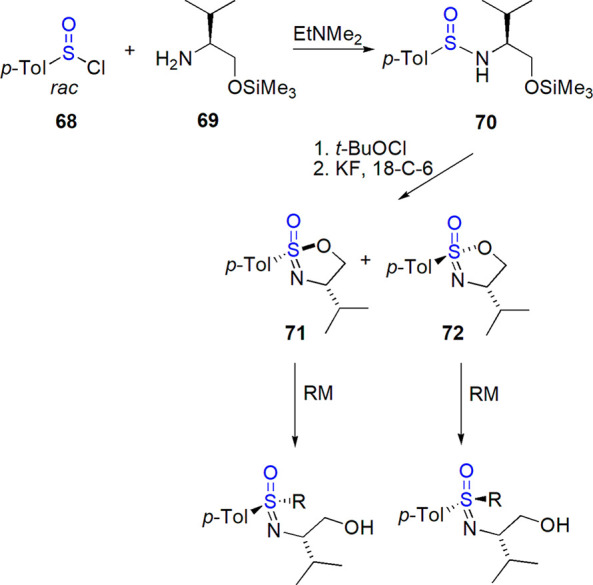
Sulfoximines can be obtained from other tetrasubstituted sulfinyl derivatives: sulfonimidoyl halides, sulfonimidamidates, and sulfonimidamides. Reggelin and co-workers developed a strategy based on preparation of two epimers of cyclic sulfonimidamidate (71 and 72, see also section 9.1) and their reaction with organometallic reagents, which proceeded with an inversion of configuration (Scheme 40).304 This synthetic precursor was also applied in the preparation of the first bis(NH-sulfoximine) in a diastereomerically pure (but racemic) form.305 Epimers of the same sulfinimidamidate were used in a one-pot synthesis of enantiopure cyclic oxathiazine S-oxides306 and other enantiopure sulfoximines.278
Scheme 40. Sulfoximines Obtained from Epimeric Sulfonimidamidates304.
A synthesis based on the use of sulfonimidates was also reported in 2018.307 They were shown to react with a variety of organometallic (mainly Grignard) compounds in THF. Yields were moderate to high; as only racemic reactants were used, stereochemical aspects of the transformation were not discussed.
Harmata and co-workers concentrated their attention on the preparation of derivatives of cyclic sulfoximine, 2,1-benzothiazine, a compound which found numerous applications in asymmetric synthesis.274 Reaction of sulfonimidoyl chloride 73 with alkynes under Lewis acid catalysis afforded 2,1-benzothiazines in 46–75% yield (Scheme 38).308 These cyclic sulfoximines were also obtained when alkenes were used instead of alkynes, and good yield was in certain cases (for example, cyclohexene addition) accompanied by high diastereoselectivity (Scheme 41).309 Reaction of sulfonimidoyl chloride with mono-, di-, and trisubstituted olefins in the presence of AlCl3 provided benzothiazines bearing two stereogenic centers with variable diastereoselectivity.310 The effect of alkene substitution on the reaction outcome was studied.
Scheme 41. Preparation of 2,1-Benzothiazines from Compound 73 and Alkynes or Alkenes308,309.
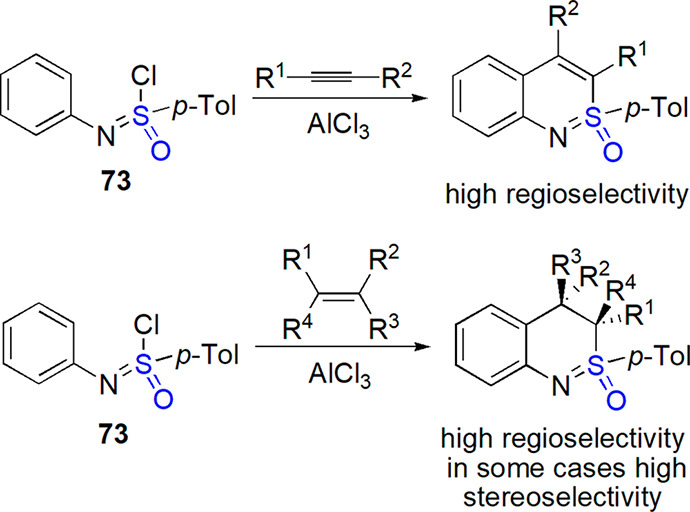
Also noncyclic sulfoximines were prepared from sulfonimidoyl chlorides or fluorides, as exemplified by the reaction of the latter with TMSCF3, leading to trifluoromethylated product.311 Diarylsulfoximines were prepared by Takata and co-workers from sulfonimidoyl chlorides (in turn obtained by treatment of sulfinyl chlorides with chloramine-T) and aromatic hydrocarbons catalyzed by Lewis acids.312
Synthesis of sulfoximines can start from trisubstituted derivatives, sulfinamides, or imines derived therefrom. Enantiopure N-tert-butylsulfinylimines bearing fluorinated alkyl substituents were applied in a [3+2]-cycloaddition reaction with arynes, yielding cyclic sulfoximines with excellent diastereo- and enantioselectivity (the configuration of sulfur stereocenter was retained, Scheme 42).313 A reverse process of conversion of cyclic sulfoximines into sulfonamides was investigated as well.
Scheme 42. [3+2]-Cycloaddition Leading to Cyclic Sulfoximines313.
Stockman and co-workers prepared a series of cyclic sulfoximines from nonracemic mesityl- or tert-butyl sulfinylimines via an aza-Darzens-type aziridination with 2-bromobut-2-enoic acid methyl ester; the formed vinyl aziridines spontaneously rearranged into diastereomerically pure products.314 The reaction could be performed either with isolation of the aziridine intermediate or in a one-pot fashion, typically in higher yield.
In very recent reports by Aota et al., a stereospecific synthesis of sulfoximines by a S-selective alkylation or arylation of sulfinamides was described.315,316 Protection of nitrogen atom with a pivaloyl group and the proper choice of reaction conditions (2 equiv of alkyl iodide or bromide, 1.1–1.2 equiv of NaH, 1.1–1.2 equiv of 15-crown-5 in dioxane, 11–168 h) allowed preparation of sulfoximines in 31–96% (but in most cases >80%) yield with a retention of configuration (Scheme 43).315 The protecting group could be removed by LiAlH4 treatment. Interestingly, starting with tert-butyl sulfinamide opened the route to both enantiomers of various derivatives with two similar alkyl chains via de-tert-butylation of sulfoximine obtained in the first step with TFA followed by the second alkylation. The reaction was conducted on a gram scale as well. A similar strategy was used for CuI-catalyzed arylation of sulfinamides with aryliodonium salts and N,N-dicyclohexylmethylamine in DMSO: conversion of N-pivaloyl tert-butane sulfinamide proceeded in 53–90% yield.316 De-tert-butylation with potassium tert-butoxide (85%) and second arylation (68–91%) yielded the desired enantiopure diaryl sulfoximines. The same group described also a stereoselective preparation of five-membered cyclic sulfoximines by cyclization of optically pure N-propargylsulfinamides.317
Scheme 43. Alkylation of Pivaloyl-Protected Sulfinamides315.
7.4. Formation of S=O and S=N Bonds
In 2017, three groups independently reported a convenient one-pot protocol for the synthesis of NH-sulfoximines from sulfides. Reactants which were found efficient for the conversion of sulfoxides to sulfoximines, diacetoxyiodobenzene (phenyliodine diacetate, PIDA), and ammonium carbamate were successfully applied by Bull, Luisi, and co-workers in nitrogen and oxygen transfer to sulfides (Scheme 44).301 Preparation of NH-sulfoximines from sulfides treated with PIDA and H2NCOONH4 was also described by Reboul and co-workers,318 and modified reaction conditions allowed extension of the methodology for derivatives bearing perfluoroalkyl substituents (Scheme 44).319 A similar procedure, but with ammonium carbonate ((NH4)2CO3) as the source of ammonia, was developed by Li and co-workers (Scheme 44).320 An asymmetric variant of this versatile, operationally simple method offering a broad substrate scope and performed under mild conditions (at room temperature under air) is of high demand, but it has not been announced yet.
Scheme 44. Preparation of Sulfoximine from Sulfides301,319,320.
The sulfoximination protocol was applied by Reboul’s group in the last step of the synthesis of drug candidate atuveciclib.321 An attempted stereoselective synthesis via enantiomerically enriched sulfoxide resulted in (S)-enantiomer with only 20% ee. The authors attributed this unsatisfactory result to the low enantioselectivity of sulfoxidation step for which Kagan’s conditions were applied.
7.5. Desymmetrization of Achiral or Racemic Sulfoximines. Kinetic Resolution
Desymmetrization of N-trialkylsilyl dimethyl sulfoximines was performed by their treatment with chiral bases and reacting the resulting anion with electrophiles, thus yielding enantioenriched derivatives difficult to obtain by other methods.322 Under optimized conditions, with chiral lithium amide prepared in situ, several electrophiles were tested with variable yield (6–86%) and enantioselectivity (18–70% ee).
The desymmetrization strategy was later used for the preparation of S,S-dialkyl sulfoximine 77, an optically active myristic acid analogue.323 However, a simple generation of anion from dimethyl derivative 75 followed by addition of dodecyl iodide resulted in a racemic product and a strategy was modified, finally yielding the desired sulfoximine with 58% ee (Scheme 45). The target compound was prepared in high enantiomeric purity using enantioselective oxidation of 4-bromophenyl methyl sulfide (Kagan’s method was applied), followed by a displacement of aryl group by Grignard reagent, iron-catalyzed imination, and deprotection of nitrogen atom.
Scheme 45. Desymmetrization of Sulfoximine 75 Yielding Myristic Acid Derivative323.
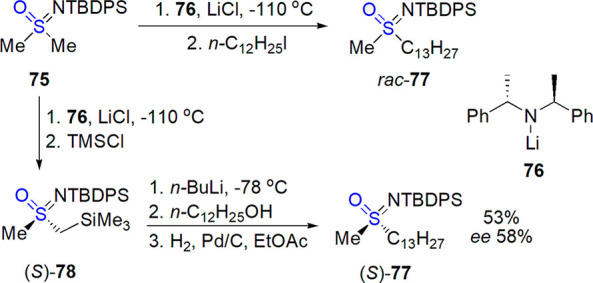
Kinetic resolution of NH-sulfoximines utilizing their amidation with enals catalyzed by chiral N-heterocyclic carbene catalysts was described by Dong et al.324 A broad variety of aryl-substituted sulfoximines afforded amides with reasonable yield and excellent stereoselectivity (up to 59% yield, up to 96% ee), and unreacted sulfoximines in up to 56% yield and up to 99% ee; both enantiomeric forms of each product could be obtained). The reaction could be performed on a gram scale, which was demonstrated by the efficient preparation of sulfoximine further transformed into optically active Betrixaban analogue, an inhibitor of human Factor Xa.
A stereodifferentiation of variety of sulfoximines upon annulation with diazo compounds catalyzed by rhodium(III) complex bearing a chiral Cpx ligand was independently described by the groups of Li325 and Cramer.326,327 A possible inversion of stereoselectivity caused by the change of solvent or suitable additives was noticed.
7.6. Modifications of Sulfoximines
Typical modifications of optically active sulfoximines concern substitution of imine nitrogen atom (however, they were also introduced into pseudopeptides by Bolm and co-workers using reactions of S-methyl substituent328−330). New attempts are usually performed with racemic substrates, but in many cases tests with enantiomerically pure compounds prove a retention of configuration of stereocenters. For example, in N-arylation of various sulfoximines with aryl bromides catalyzed by palladium salts, Bolm and co-workers also used a chiral (S)-bromide and (R)-sulfoximine and obtained a single diastereomer, strongly supporting the hypothesis of complete retention of configuration of both stereogenic centers (Scheme 46).331,332
Scheme 46. Reaction between (S)-Aryl Bromide and (R)-Sulfoximine332.
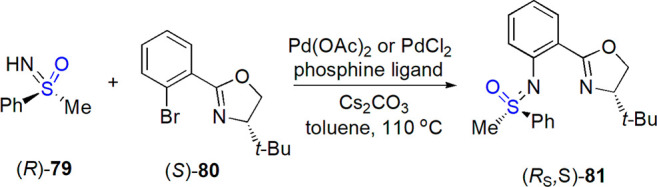
Palladium- and copper-catalyzed arylations of NH- or N-benzylsulfoximines developed in Bolm’s laboratory allowed preparation of chiral ligands for asymmetric synthesis.333,334 This group obtained also chiral thiourea organocatalysts from (S)-S-methyl-S-phenylsulfoximine by a direct reaction with isothiocyanate.335 Protocols for preparation of N-alkyl (acylation and reduction with complexed boranes)336 and N-alkynyl derivatives (through the reaction with methoxy(tosyloxy)iodobenzene) were described as well.337,338
To avoid using stereochemically labile sulfonimidoyl chlorides in the synthesis of enantiomerically pure benzothiazines, Harmata and Pavri adapted the N-arylation protocol developed by Bolm.331,339 The reaction between enantiopure NH-sulfoximine and ortho-bromobenzoates or analogous cinnamate derivative resulted in N-arylation; ring closure upon addition of a base such as sodium or potassium hydride yielded substituted 2,1-benzothiazines as single isomers (Scheme 47). The reaction could be performed in a one-pot version when o-bromobenzaldehydes were used as reactants. Benzothiazines were also obtained in the reaction of methyl phenylsulfoximine with o-bromocinnamates followed by base-induced cyclization.340,341 Products were formed as single stereoisomers in a highly stereospecific process.
Scheme 47. Synthesis of 2,1-Benzothiazines from Enantiopure Sulfoximine (R)-66 Described by Harmata and Pavri339.
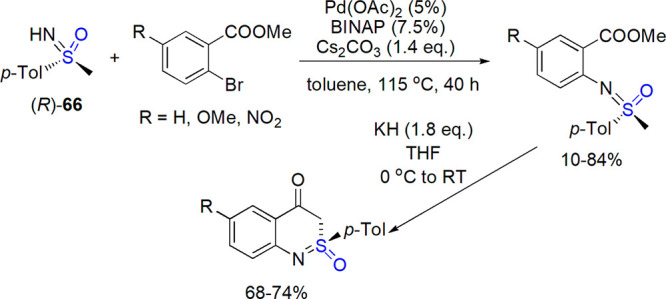
Harmata and co-workers described a palladium-catalyzed microwave-assisted reaction of aryl chlorides with NH-sulfoximine, which allowed preparation of a series of arylated sulfoximines and benzothiazines in good to excellent yields in a relatively short time.342 When aryl dichlorides were used, under various reaction conditions, only products of monosubstitution were observed.343
8. Sulfonimidoyl Halides
The title compounds, which can be regarded as halides of sulfonimidic acid, are relatively unstable (at least chlorides and bromides), moisture-sensitive compounds prepared as intermediates in the synthesis of other tetrasubstituted sulfinyl derivatives. Their chemistry was reviewed by Levchenko and co-workers in 2000.344 Here, we present several papers devoted to the preparation of these compounds, but they will also appear in other sections, especially in the one concerning sulfonimidamides.
Most frequently, sulfonimidoyl chlorides are obtained from sulfinyl chlorides, sulfinamides, but also from other tetrasubstituted sulfinyl derivatives. Their first successful preparation was reported by Levchenko and Kirsanov in 1960.345 Johnson and co-workers developed a new method of preparation of these compounds by oxidation of sulfinamides with chlorine or N-chlorobenzotriazole 82,346,347 later replaced by tert-butyl hypochlorite (Scheme 48).348 Retention of configuration during this transformation was established (due to the priority rules, this means that when chlorine substituent takes place of lone pair of electrons, (S)-sulfinamide was converted to (R)-sulfonimidoyl chloride).349 Stereospecific conversions of sulfonimidoyl chloride to sulfinamidate and sulfonimidamides was associated with inversion of configuration.
Scheme 48. Preparation of Sulfonimidoyl Chlorides from Sulfinyl Chlorides or Sulfinamides346−349.
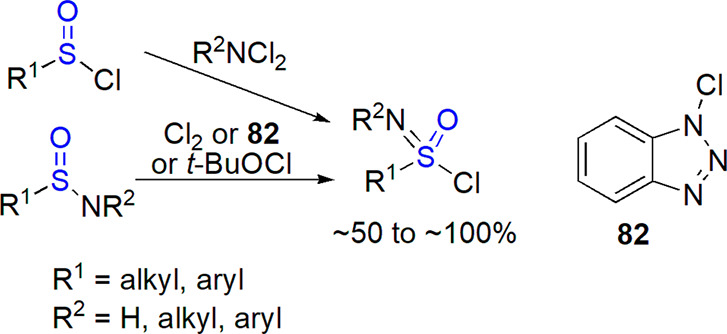
Jones and Cram reported on the preparation of epimeric N-carbomenthoxy-p-toluenesulfonimidoyl chlorides using two methods and proved that also imination of sulfinyl chloride proceeds with retention of configuration.28 A cycle of stereospecific transformations involving several sulfinyl derivatives was proposed.
Roy investigated the possibility of preparation of sulfonimidoyl chloride from other tetrasubstituted sulfinyl compounds.350 He reacted N-silylated sulfonamides with phosphorus chlorides and observed formation of the corresponding chlorides under appropriate conditions. He also obtained the first detectable sulfonimidoyl bromide. Correlation of configuration was not the part of the study.
Sulfonimidoyl fluorides have been recognized as more stable analogues of their chloride counterparts, which makes them more convenient as reaction intermediates that can be isolated without the need of immediate use. Unfortunately, the stereochemical course of the reactions in which they were prepared was not studied. First, sulfonimidoyl fluorides were prepared by Johnson and co-workers from the respective chlorides and diverse fluoride sources (NaF, KF, TBAF).351 These derivatives were used in the synthesis of sulfoximines. Novel sulfonimidoyl fluorides were prepared by van Leusen et al. by treatment of the corresponding chlorides with KF or KHF2.352N-Tosyl derivatives exhibited shelf stability; partial resolution to enantiomers by crystallization was achieved for one derivative. Fluorides were converted to the corresponding (racemic) isocyanides as well.
Selectfluor was found useful as a fluoride source in conversion of cyclic sulfoximine to the cyclic fluorinated 1-oxo-2H-1,2,4-benzothiadizine derivative, which was characterized by X-ray study.353 Quite recently, Sharpless and co-workers applied thionyl tetrafluoride 83 in a two-step reaction; in the first step, primary amines were treated with SOF4, which resulted in difluoride intermediate. Its reaction with alkyllithium yielded the desired products (Scheme 49).354
Scheme 49. Synthesis of Sulfonimidoyl Fluorides from SOF4 Reported by Sharpless and Co-workers354.
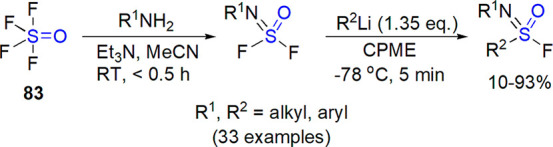
9. Preparation of Optically Active Sulfonimidates
Similarly to sulfoximines and sulfonimidamides, sulfonimidates have been known for over half a century and recently rediscovered, mainly as intermediates in the synthesis of sulfoximines. To fulfill these roles, they are not always required in enantiomerically pure form, and thus certain preparative methods rather neglect the fate of stereogenic center. Possible applications of these compounds are limited by their observed slow conversion to corresponding sulfonamides at elevated temperature.355
9.1. S–O Bond Formation
Typical preparations of sulfonimidates involve oxidative introduction of alkoxy group into sulfinamides. It is often, but not necessarily, realized in two steps. Already, in the 1970s, Johnson and co-workers prepared enantiopure sulfonimidates in a two-step procedure involving reaction of (S)-N-methylbenzenesulfinamide 84 with chlorine followed by treatment of obtained sulfonimidoyl chloride 85 with sodium phenoxide (Scheme 50).29 (S)-Sulfonimidate 87 was formed as a result of retention of configuration in the first step and inversion in the second one. Scheme 50 shows other possible conversions involving also sulfonimidamide 86 and sulfoximine 88, confirming that typically interconversions of tetrasubstituted derivatives are associated with the change of configuration, in contrast to those between three- and four-substituted chiral sulfinyl compounds.
Scheme 50. Transformations of Sulfinamide (S)-84 through Chloride (R)-85 to Sulfonimidamide (R)-86, Sulfonimidate (S)-87, and Sulfoximine (S)-8829.
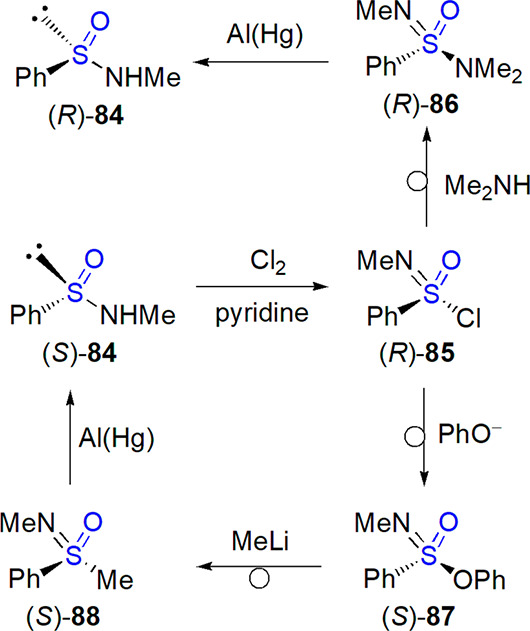
In 1993, Roy reported a conversion of racemic N-silylated sulfonimidoyl chlorides and bromides to the corresponding sulfonimidates in yields up to 78%.350 A quantitative desilylation with methanol resulted in NH-sulfonimidates.356 Thermally induced, Lewis acid-catalyzed polycondensation of these compounds as well as of certain silylated derivatives was observed, yielding poly(oxothiazene) polymers with an N=S backbone.
Sulfinamides bearing hydroxyl group in the N-substituent were shown to undergo cyclization to sulfonimidates. Reggelin and co-workers developed an efficient stereoselective synthesis of enantiomers of cyclic sulfonimidate that could be used as precursors for enantiopure sulfoximines (Scheme 40).304,357,358 The synthesis started from (S)-O-trimethylsilylvalinol and sodium salt of p-toluenesulfinic acid. The most efficient route included separation of epimeric sulfinamides by crystallization prior to the conversion of sulfonimidoyl chlorides with t-BuOCl; a fast, DBU-induced cyclization appeared at −78 °C, which precluded the loss of enantiomeric purity.357
Sulfonimidates were prepared by Malacria and co-workers through the reaction of sulfinamides with alcohols and iodosylbenzene (Scheme 51).359 The reaction proceeded smoothly for primary alcohols serving also as solvents (71–94% yield after 1 h in room temperature). When the reaction was conducted in acetonitrile with 3 equiv of alcohols, yields were slightly lower (39–95%) due to the competitive oxidation to sulfonamide. Also, secondary sulfinamide was efficiently converted to the corresponding sulfonimidate in 52% yield. Stereospecifity of the transformation was checked with two enantiomerically pure sulfinamides, which yielded the corresponding products with 62 and 72% ee, mainly with retention of configuration.360 This observation was confirmed by the conversion of enantiopure sulfinamides bearing an additional stereogenic center: diastereomeric products were formed with 51–76 de. X-ray structure of the major isomer of resulting sulfonimidate proved that configuration of sulfur stereocenter was retained.
Scheme 51. Preparation of Sulfonimidamidates from Sulfinamides by Malacria and Co-workers359,360.
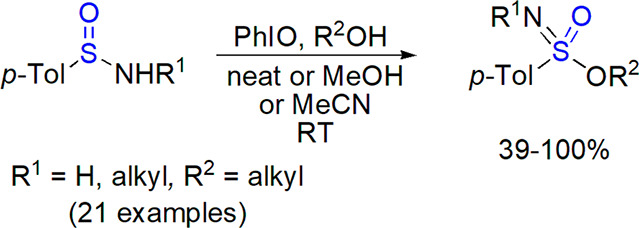
The above method was limited to N-alkyl derivatives. In 2006, Felim et al. reported the modified protocol which allowed preparation of N-aryl sulfonimidates, and better results were obtained when PhIO was replaced by more stable PhI(OAc)2 (or its phenyl-substituted derivatives) with MgO as a base.361
Similarly, a series of 27 sulfonimidates were prepared by oxidation of sulfinamides with PhIO in the presence of alcohols (15 min at room temperature, yields up to 93%).307 The resulting (racemic) compounds were further converted to sulfoximines (see section 7.4).
9.2. Other Methods
In a recent report of Luisi, Bull, and co-workers, thiols were reacted with ammonium carbamate and diacetoxyiodobenzene in methanol, yielding the corresponding sulfonamides or sulfonimidates, depending on reaction conditions (Scheme 52; shorter reaction time and an increase of the amount of ammonium carbamate resulted in higher yields of sulfonimidate).302 In this convenient one-pot protocol, a direct formation of both sulfur–oxygen and sulfur–nitrogen bonds was observed. Factors which would induce stereoselectivity of the process were not checked; all reactants, including a variety of thiols, were achiral, and chiral additives were not tested. A possibility of transformation of sulfonimidate to sulfoximine was demonstrated by the reaction of electron-rich 4-methoxyphenyl sulfonimidate with morpholine (80% yield, racemic).
Scheme 52. Preparation of Sulfonimidates from Thiols302.
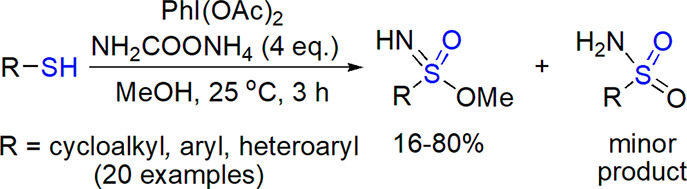
10. Stereoselective Preparation of Sulfonimidamides
Sulfonimidamides belong to the group of intrinsically chiral sulfinyl compounds. Their story is very similar to other tetrasubstituted derivatives, sulfoximines and sulfonimidates; first prepared in the 1960s by Levchenko and co-workers, long forgotten, in the 21st century they have experienced an impressive revival.362 It is connected with the discovery of their potential as biologically active substances, with possible pharmaceutical and agricultural applications363 but also as chiral auxiliaries for asymmetric synthesis. The progress in the synthesis of sulfonimidamides was reviewed in a 2018 article by Nandi and Arvidsson.362 Since then, several new publications have been issued. They will be presented in this section, together with representative examples of important contributions to the field.
Enantiopure sulfonimidamides were obtained either from other optically active sulfinyl derivatives or by separation of obtained isomers by HPLC. As the access to the starting material with two sulfur–nitrogen bonds is limited, one of these connections is formed in most preparations. The majority of described transformations involve sulfinamides and sulfonamides, and other tetrasubstituted derivatives (sulfonimidoyl halides or sulfonimidates) may serve as intermediates. Various approaches can differ by the substitution of imine and amine nitrogen atoms in the final product; postsynthetic modifications can change this pattern without altering the configuration of stereogenic sulfur atom.
10.1. Preparation of Sulfonimidamides from Sulfinamides
The first synthesis of optically active sulfonimidamide was reported by Johnson and co-workers.349 Chlorination of sulfinamide (S)-84 followed by reaction with dimethylamine resulted in product (R)-86 in 56% overall yield with inversion of configuration (Scheme 50).
More recently, conversion of sulfinamides to sulfonimidamides was studied by Bolm’s group. García Mancheño and Bolm investigated chlorination of p-tolylsulfinamides connected with immediate transformation of transient sulfonimidoyl chlorides by the amine nucleophile into the respective racemic sulfonimidamides in 50–97%.364 Two enantiopure N-benzoyl- and N-tert-butyloxycarbonyl-protected sulfonimidamides were efficiently prepared in several steps starting from the commercially available sodium 4-toluenesulfinate 89 (Scheme 53).365 Its conversion to enantiopure sulfinamide (S)-51 was achieved using menthol auxiliary. Oxidative chlorination of this sulfinamide with tert-butyl hypochlorite followed by reaction of transient (R)-sulfonimidoyl chlorides 92 and 93 with aqueous ammonia resulted in formation of enantiopure N-substituted (S)-sulfonimidamides 94 and 95 in 78% and 97% yield, respectively. Their configuration was confirmed by X-ray diffraction measurement. Amino-functionalized sulfonimidamides were prepared by oxidative chlorination of sulfinamides with t-BuOCl followed by the reaction with amine or bisamine.366 Alternatively, a reaction of N-benzoyl-protected sulfonimidamide with aziridinium salts obtained from aminoalcohols was performed. In the first case, enantiopure reactants were used and products were obtained as single stereoisomers, while the second method afforded separable mixtures of diastereomers. Applied in copper-catalyzed asymmetric Henry reaction, they led to product formation in reasonable yields and up to 95% ee.
Scheme 53. Preparation of Enantiopure Sulfonimidamides from Sodium Sulfinate365.
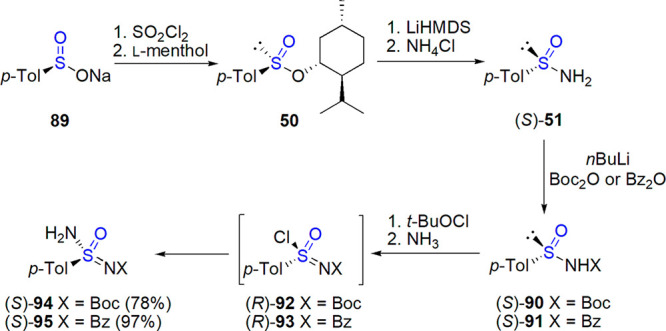
A direct amination of N-arylsulfinamides with N-benzoyl derivatives of secondary amines (piperidine, piperazine, or morpholine) catalyzed by CuBr was also reported by Bolm and co-workers.254 Chiral amines were not tested, and racemic products were obtained in 35–96% yield.
Oehlrich and co-workers described a synthesis of sulfonimidamides bearing a CF3 substituent from the corresponding sulfinamides. Their oxidative fluorination by the action of NCS and TBAF led to sulfonimidoyl fluoride intermediates, more stable as compared to their chloride counterparts; a subsequent reaction with amines led to the desired sulfinyl derivatives.367 Trifluoromethylated sulfonimidamides were also prepared by the same group from a bench-stable solid sulfoximine precursor. In both cases, the process could be conducted in a flow reactor. Unfortunately, stereoselectivity of the substitution was not the issue that deserved attention.368
Cathers and Schloss reported on the synthesis of dipeptides containing sulfonimidamides as mimics for transition states for metalloproteases and aspartic acid.369 Sulfinyl phthalimide prepared by oxidation of the respective sulfenyl derivative with m-CPBA was first treated with l-phenylalanine methyl ester to afford a separable mixture of diastereomers. Sulfinamide was then reacted with chlorine, followed by substitution of chloride with ammonia, which yielded a sulfonimidamide with a free NH2 group. Two more derivatives contained glycine benzyl ester and leucine methyl ester, the latter converted upon the crystallization attempt into a cyclic sulfonimidamide. The configuration of sulfur stereocenter in these compounds was, however, not studied.
Inspired by the work of Bull, Luisi, and co-workers,300 Lücking’s group applied ammonium carbamate combined with PhI(OAc)2 for NH transfer to tertiary sulfinamides.370NH-sulfonimidamides were formed in good yields (Scheme 54) and, as suggested by one experiment with enantiomerically enriched substrate, also in a stereospecific manner (though configuration of the product was not established).
Scheme 54. Synthesis of Sulfonimidamides from Tertiary Sulfinamides370.
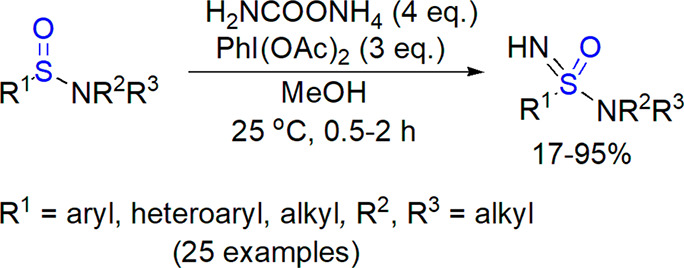
10.2. Preparation of Sulfonimidamides from Sulfonamides
Sulfonamides can be also converted to sulfonimidamides by a formal replacement of =O by a =NR fragment. Typically, sulfonimidoyl chlorides serve as intermediates of such preparations, as exemplified by the synthesis described by Chen and Gibson (Scheme 55).371 Yields varied from 20 to 89%; enantiomerically pure derivatives were obtained only by chiral HPLC separation of a racemic sample. Sulfonimidamide derivatives of amino acids were prepared by chlorination of sulfonamides with Ph3PCl2, followed by the reaction with enantiopure amino acid esters.372 Diastereomers formed in ca. 2:1 ratio were separated by column chromatography. Alternatively, a removal of N-tert-butyldiphenylsilyl (TBDPS) protecting group was performed yielding five-membered cyclic sulfonimidamides; preparative HPLC led to individual stereoisomers, and configuration of one of them was established by X-ray study.
Scheme 55. Preparation of Sulfonimidamides from Sulfonamides371.
A novel approach to the synthesis of sulfonimidamides was recently proposed by Grygorenko and co-workers who prepared a stable and versatile imidosulfuric diamide from the corresponding sulfonamide and used it in reactions with amines.373 A variety of sulfonimidamides were obtained in 83–94% yield, but only as racemates because no attempt was directed toward enantioselective variant of the reaction.
10.3. Other Preparations
Toth et al. prepared a series of sulfonimidamide–urea conjugates and evaluated their biological activity.374 These compounds were obtained as racemates from sulfides, disulfides, thiols, or sodium sulfinates, which were converted into the corresponding N-sulfinyl thioureas; their oxidative chlorination with t-butyl hypochlorite followed by amination afforded the target compounds. The use of (1S,2R)-norephedrine in the last step yielded a mixture of epimers which were separated to give enantiopure derivatives.
Sulfonimidamides were also obtained from unprotected sulfoximines in a copper-catalyzed dealkylation/amination process in which S-methyl or S-benzyl substituent was replaced by amines under O2 atmosphere.375 Yields were in certain cases high, however, the use of optically active sulfoximine resulted in racemic product.
Willis and co-workers developed a one-pot protocol using sulfinylamines, in particular a stable sulfinyltritylamine, as a starting point for sulfonimidamide synthesis.376 Three steps: treatment with organometallic compound, chlorination, and the reaction with amine led to the protected sulfonimidamides in 49–80% yield. The use of chiral amine led to separable mixture of diastereomers; other stereochemical aspects were not considered.
Enantiopure imidazolyl and 1,2,4-triazolyl sulfonimidoyl derivatives were prepared from sulfonimidoyl chlorides as reported by Kluge et al.377 Also, 1,2,3-triazole ring was introduced as a substituent of sulfur atom via click reaction of the corresponding azide by Nandi and Jesin.378
To overcome the drawbacks of previous methods (e.g., limited functional group tolerance), Bolm’s group described a new one-pot approach, involving sulfonimidate formation from N-tritylsulfinylamine, 4-fluorophenyldiazonium tetrafluoroborate, and 1-hydroxybenzotriazole in the presence of a base (N-methylpiperidine was found optimal) in dimethyl carbonate chosen as solvent (Scheme 56).379 Relatively stable sulfonimidates were converted to sulfonimidamides by the action of primary and secondary aliphatic amines. The method was found versatile, with high tolerance for various functional groups, although without any control of stereochemistry.
Scheme 56. Formation of Sulfonimidamidates from N-Tritylsulfinylamine379.
Bull, Luisi, and co-workers studied reaction of ammonium carbamate and iodosylbenzene with sulfenamides, a rarely used starting material, prepared from disulfides and amines.380 A one-pot formation of S=O and S–N bonds proceeded smoothly (45–95% yield after 1 h reaction in 2-propanol at 25 °C, with 2.5 equiv of PhIO as oxidant, 2.0 equiv of NH2COONH4, and 1.0 equiv of acetic acid additive, Scheme 57). The obtained racemic products could be further functionalized.
Scheme 57. One-Pot Preparation of Sulfonimidamides from Sulfenamides380.
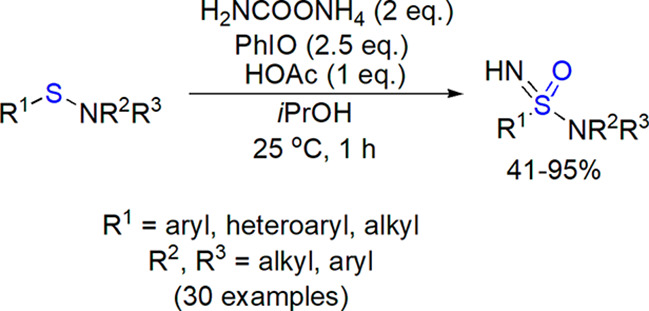
10.4. Modifications of Sulfonimidamides
Numerous modifications involving either imine or amine nitrogen atoms of prepared sulfonimidamide have been reported by several research groups. It is assumed that such reactions do not affect the sulfur stereocenter, and usually this has not been checked or simply racemic mixtures have been used in these transformations. Apparently enantiopure final products are not necessary for certain applications. Several recent examples will be briefly described as the protocols can be easily adapted for synthesis of optically active compounds.
Arvidsson’s group focused their attention on preparation of NH sulfonimidamides.381 Three methods were developed for deprotection of morpholine-substituted and imine-protected sulfonimidamidates under acidic, nucleophilic, and basic conditions. An efficient palladium-catalyzed arylation of imine nitrogen with aryl halides was also described. Arvidsson and co-workers reported also copper-catalyzed arylation using aryl boronic acids382 (a similar reaction was independently described by Battula et al.383), and N-acylation with aryl or heteroaryl halides and carbon monoxide released in situ from Mo(CO)6 in the presence of palladium catalyst, DBU, and triethylamine.384 Recently, N-trifluoromethylthiolated sulfonimidamides were prepared and their cytotoxic activity studied.385
Microwave-assisted, copper-catalyzed N-arylation of sulfonimidamides was also described by Malacria and co-workers.386 Even though hydroxyproline was used as ligand, the obtained products were racemic. The same team converted sulfonimidamides bearing NH2 group into nitrenes by the reaction with PhIO; these nitrogen-transfer agents were trapped by sulfides, sulfoxides, or olefins in the presence of Cu(II) salt.387 Reaction with enantiopure sulfoxide gave a mixture of separable diastereomers; otherwise diastereoselectivity was low in the case of sulfimide formation, while up to 50% de was noted for aziridination.
In 2018, 47 N-functionalized tertiary sulfonimidamides were prepared by Lücking and co-workers by arylation, alkylation, trifluoromethylation, cyanation, sulfonylation, alkoxycarbonylation, and aminocarbonylation of the =NH group.388 Their hydrolytic and metabolic stability were checked, indicating the potential of application in biological systems.
Modifications of sulfonimidamides were also introduced by Bolm and co-workers.389,390 Enantiopure derivatives were tested in asymmetric reactions. Novel organocatalysts were obtained in good yield from protected (S)-proline and two enantiomers of N-tosylsulfonimidamide.391 High diastereo- and enantioselectivity was observed when these compounds were applied in solvent-free asymmetric aldol reaction. N-Phosphorylation of single enantiomers of sulfonimidamides resulted in novel phosphoramidites (SIAPhos), which were used to prepare neutral rhodium and iridium complexes, the latter found efficient in asymmetric hydrogenation of enamides (70–92% ee).392
Kinetic resolution of racemic sulfonimidamides was described by Liang et al., who applied these compounds for amination of benzylic C–H bonds catalyzed by a chiral Rh(II) complex.393 A strong matched effect was observed between (S)-enantiomers of sulfinyl compound and the catalyst. The reaction of various benzylic substrates with racemic sulfonimidamide and PhI(OCOt-Bu)2 in the presence of (S)-form of rhodium complex resulted in formation of (1R,SS) product in 56–77% yield, 88–99% de, and 97–99% ee,, accompanied by recovery of unreacted substrate with (Rs) configuration.
11. Summary
Undoubtly, chiral sulfinyl compounds have proved their usefulness and versatility in modern stereoselective synthesis, serving as precursors, catalysts, and ligands in various preparations of important target molecules, mainly for biomedical applications. These derivatives, despite a common structural feature, constitute an omnifarious family. Consequently, certain methods of their preparation in enantiomerically pure form are similar, while part of synthetic routes remain applicable only for particular representatives. A possibility of mutual interconversions between various sulfinyl compounds is therefore worth emphasizing, because one precursor can be quite often transformed into other derivatives in a stereospecific manner without significant loss of optical purity (which can be in many cases improved by crystallization). More and more such chiral sulfinyl transfer agents have become easily available (in a short synthesis or from commercial sources), in both enantiomeric forms, and virtually any substitution pattern can be achieved. On the other hand, preparative methods based on catalytic enantioselective oxidation or imination of achiral organosulfur compounds involve chirality multiplication and are beneficial from the economic point of view. The possible disadvantages and difficulties (e.g., contamination by traces of toxic metals which can preclude biomedical applications) can be overcome if chiral organic molecules are applied as catalysts.
The development of synthetic methods is often inspired by the increased interest in a given family of compounds resulting from possible applications (both synthetic and biomedical). Quite often, a series of independent reports are published in a short period of time, followed by subsequent papers describing modifications and improvements. In the case of sulfinyl compounds, it can be observed that new approaches have been introduced mainly without much stress on stereochemical issues, which are included on the later stage. This raises hopes on further progress in the field, as asymmetric variants of newly introduced methods are needed. One recent example can be added to those cited in the previous sections. A one-pot synthesis of unsymmetrical sulfoxides was developed based on subsequent reactions of two organometallic reagents with DABSO (DABCO-bis(sulfur dioxide)) used as a source of sulfinyl group.394 A potential of this methodology and other protocols in which chiral sulfinyl compounds are obtained as racemic mixtures should serve as a driving force in future research in the field.
Glossary
Abbreviations Used
- 15-C-5
15-crown-5, 1,4,7,10,13-pentaoxacyclopentadecane
- 18-C-6
18-crown-6, 1,4,7,10,13,16-hexaoxacyclooctadecane
- ACDC
asymmetric counteranion-directed catalysis
- AIBN
azobis(isobutyronitrile)
- BINOL
1,1′-bi-2-naphthol
- Boc
tert-butoxycarbonyl
- Bz
benzoyl
- CHP
cumene hydroperoxide
- CPME
cyclopentyl methyl ether
- CPO
chloroperoxidase
- Cpx
chiral cyclopentadienyl ligand
- CYMO
cyclohexanone monooxygenase
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DABSO
DABCO-bis(sulfur dioxide)
- DAG
diacetone-d-glucose
- DAST
diethylaminosulfur trifluoride
- dba
dibenzylideneacetone
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DET
diethyl tartrate
- DMC
dimethyl carbonate
- DMSOR
dimethyl sulfoxide reductase
- EBX
ethynyl benziodoxolone
- HB
hydrobenzoin
- HOBt
1-hydroxybenzotriazole
- Josiphos
chiral diphosphine, ferrocene-based ligand
- KHMDS
potassium bis(trimethylsilyl)amide
- LDA
lithium diisopropylamide
- LiHMDS
lithium bis(trimethylsilyl)amide
- m-CPBA
meta-chloroperbenzoic acid
- MSH
O-(mesitylsulfonyl)hydroxylamine
- NaMDS
sodium bis(trimethylsilyl)amide
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- NHC
N-heterocyclic carbene
- paMsrA
methionine sulfoxide reductase A obtained from Pseudomonas alcaliphila
- PIDA
phenyliodine diacetate, PhI(OAc)2
- SIAM
bis(sulfinyl)imidoamidine
- SIAPhos
phosphorylated sulfonimidamides
- TBAF
tetrabutylammonium fluoride
- TBDMS-NH2
tert-butyldimethylsilyl amine
- TBDPS
N-tert-butyldiphenylsilyl
- TBHP
tert-butyl hydroperoxide
- TBTH
tributyltin hydride
- TFAA
trifluoroacetic acid anhydride
- TMS
trimethylsilyl
- Trt
trityl, triphenylmethyl
Biographies
Elżbieta Wojaczyńska received her M.Sc. Eng. degree in organic chemistry from the Wrocław University of Science and Technology in 1997. Her doctoral thesis on the enantioselective synthesis and application of chiral sulfoxides was completed under the supervision of Prof. Jacek Skarżewski in 2001. Her research at the Wrocław University of Science and Technology concentrates on the synthesis of new chiral building blocks and novel chiral ligands and catalysts for asymmetric synthesis and biomedical applications.
Jacek Wojaczyński graduated in chemistry from the Faculty of Mathematics, Physics and Chemistry of the University of Wrocław in 1993. Five years later, he defended his Ph.D. thesis on metalloporphyrin modifications at the Faculty of Chemistry of the same university (supervisor: Prof. L. Latos-Grażyński). He is a member of the Metalloporphyrin Chemistry Group at this faculty, and his research field includes oligoporphyrins, heme proteins, and degradation of oligopyrrolic macrocycles.
The authors declare no competing financial interest.
References
- Wojaczyńska E.; Wojaczyński J. Enantioselective Synthesis of Sulfoxides: 2000–2009. Chem. Rev. 2010, 110, 4303–4356. 10.1021/cr900147h. [DOI] [PubMed] [Google Scholar]
- Xu H. C.; Chowdhury S.; Ellman J. A. Asymmetric Synthesis of Amines Using tert-Butanesulfinamide. Nat. Protoc. 2013, 8, 2271–2280. 10.1038/nprot.2013.134. [DOI] [PubMed] [Google Scholar]
- Kaiser D.; Klose I.; Oost R.; Neuhaus J.; Maulide N. Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev. 2019, 119, 8701–8780. 10.1021/acs.chemrev.9b00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer I.; Velado M.; Fernández de la Pradilla R.; Viso A. From Allylic Sulfoxides to Allylic Sulfenates: Fifty Years of a Never-Ending [2,3]-Sigmatropic Rearrangement. Chem. Rev. 2017, 117, 14201–14243. 10.1021/acs.chemrev.7b00428. [DOI] [PubMed] [Google Scholar]
- Sipos G.; Drinkel E. E.; Dorta R. The Emergence of Sulfoxides as Efficient Ligands in Transition Metal Catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. 10.1039/C4CS00524D. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Rao M. Development of Chiral Sulfoxide Ligands for Asymmetric Catalysis. Angew. Chem., Int. Ed. 2015, 54, 5026–5043. 10.1002/anie.201411073. [DOI] [PubMed] [Google Scholar]
- Otocka S.; Kwiatkowska M.; Madalińska L.; Kiełbasiński P. Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis. Chem. Rev. 2017, 117, 4147–4181. 10.1021/acs.chemrev.6b00517. [DOI] [PubMed] [Google Scholar]
- Bentley R. Role of Sulfur Chirality in the Chemical Processes of Biology. Chem. Soc. Rev. 2005, 34, 609–624. 10.1039/b418284g. [DOI] [PubMed] [Google Scholar]
- Lücking U. Neglected Sulfur(VI) Pharmacophores in Drug Discovery: Exploration of Novel Chemical Space by the Interplay of Drug Design and Method Development. Org. Chem. Front. 2019, 6, 1319–1324. 10.1039/C8QO01233D. [DOI] [Google Scholar]
- Chiral Sulfoxides. In Chiral Sulfur Reagents: Applications in Asymmetric and Stereoselective Synthesis; Mikołajczyk M., Drabowicz J., Kiełbasiński P., Eds.; CRC Press: Boca Raton, FL, 1997; Chapter 3, pp 47–194. [Google Scholar]
- Chiral Sulfinic Acid Derivatives. In Chiral Sulfur Reagents: Applications in Asymmetric and Stereoselective Synthesis; Mikołajczyk M., Drabowicz J., Kiełbasiński P., Eds.; CRC Press: Boca Raton, FL, 1997; Chapter 2, pp 7–45. [Google Scholar]
- Chiral Sulfinimines and Sulfoximines. In Chiral Sulfur Reagents: Applications in Asymmetric and Stereoselective Synthesis; Mikołajczyk M., Drabowicz J., Kiełbasiński P., Eds.; CRC Press: Boca Raton, FL, 1997; Chapter 4, pp 195–240. [Google Scholar]
- Drabowicz J.; Kiełbasiński P.; Krasowska D.; Mikołajczyk M.. Asymmetric Synthesis of Optically Active Sulfinic Acid Esters. In Organosulfur Chemistry in Asymmetric Synthesis; Toru T., Bolm C., Eds.; Wiley-VCH: Wienheim, 2008; Chepter 2, pp 31–54. [Google Scholar]
- Worch C.; Mayer A. C.; Bolm C.. Synthesis and Use of Chiral Sulfoximines. In Organosulfur Chemistry in Asymmetric Synthesis; Toru T., Bolm C., Eds.; Wiley-VCH: Weinheim, 2008; Chapter 6, pp 209–232. [Google Scholar]
- Senanayake C. H.; Han Z.; Krishnamurthy D.. Synthesis and Use of Chiral Sulfinamides. In Organosulfur Chemistry in Asymmetric Synthesis; Toru T., Bolm C., Eds.; Wiley-VCH: Weinheim, 2008; Chapter 7, pp 233–264. [Google Scholar]
- Drabowicz J. Stereochemistry of Organic Sulfur Compounds: More than 100 Years of History, Current State and Further Challenges. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 145–148. 10.1080/10426507.2016.1252366. [DOI] [Google Scholar]
- Mikołajczyk M.; Łyżwa P.; Drabowicz J.; Wieczorek M.; Bujacz G. Thiosulfinic Acids: a New Class of Chiral Organosulfur Compounds. Angew. Chem., Int. Ed. Engl. 1989, 28, 97–98. 10.1002/anie.198900971. [DOI] [Google Scholar]
- Drabowicz J.; Łyżwa P.; Bujnicki B.; Mikołajczyk M. Thio- and Oxo-acids of Tricoordinated Sulfur: Synthetic and Stereochemical Aspects. Phosphorus, Sulfur Silicon Relat. Elem. 1994, 95, 293–312. 10.1080/10426509408034214. [DOI] [Google Scholar]
- Douglass I. B.; Koop D. A. On the Instability of Methanesulfinyl Chloride. J. Org. Chem. 1964, 29, 951–952. 10.1021/jo01027a511. [DOI] [Google Scholar]
- Schwan A. L.; Strickler R. R. Synthesis and Reactions of Sulfinyl Chlorides. An Update. Org. Prep. Proced. Int. 1999, 31, 579–615. 10.1080/00304949909355347. [DOI] [Google Scholar]
- Herak J. J. Chiral Sulfinyl Chlorides as Intermediates in Interconversion of 4-Oxoazetidine-2-sulfinic Acid Derivatives. Croat. Chem. Acta 1995, 68, 393–398. [Google Scholar]
- Kice J. L.; Bowers K. W. The Mechanism of the Disproportionation of Sulfinic Acids. J. Am. Chem. Soc. 1962, 84, 605–610. corrigendum J. Am. Chem. Soc.1962, 84, 5003. 10.1021/ja00863a021. [DOI] [Google Scholar]
- Kice J. L.; Large G. B. The Relative Nucleophilicity of Some Common Nucleophiles toward Sulfenyl Sulfur. The Nucleophile- and Acid-Catalyzed Racemization of Optically Active Phenyl Benzenethiolsulfinate. J. Am. Chem. Soc. 1968, 90, 4069–4076. 10.1021/ja01017a026. [DOI] [Google Scholar]
- Kice J.; Guaraldi G. The Relative Nucleophilicity of Some Common Nucleophiles toward Sulfinyl Sulfur. The Nucleophile-Catalyzed Hydrolysis of Aryl Sulfinyl Sulfones. J. Am. Chem. Soc. 1968, 90, 4076–4081. 10.1021/ja01017a027. [DOI] [Google Scholar]
- Kice J. L.; Rogers T. E. Mechanism of the Alkaline Hydrolysis of Aryl Thiolsulfinates and Thiolsulfonates. J. Am. Chem. Soc. 1974, 96, 8009–8015. 10.1021/ja00833a028. [DOI] [Google Scholar]
- Mikołajczyk M.; Drabowicz J.. Chiral Organosulfur Compounds. In Topics of Stereochemistry; Allinger N. L., Eliel E. L., Wilen S. H., Eds.; Wiley, 1982; Vol. 13, pp 333–470. [Google Scholar]
- Mikołajczyk M.; Drabowicz J. Optically Stable Thiosulphinate S-Esters: Asymmetric Synthesis and Nucleophilic Substitution at Sulphinyl Sulphur. J. Chem. Soc., Chem. Commun. 1976, 220–221. 10.1039/C39760000220. [DOI] [Google Scholar]
- Jones M. R.; Cram D. J. Stereochemistry of Sulfur Compounds. VII. Course of Substitution at Sulfur Attached to Four Different Ligands. J. Am. Chem. Soc. 1974, 96, 2183–2190. 10.1021/ja00814a031. [DOI] [Google Scholar]
- Johnson C. R.; Jonsson E. U.; Wambsgans A. Nucleophilic Substitution at Sulfur in Sulfonimidoyl Compounds: Synthesis of Sulfoximines. J. Org. Chem. 1979, 44, 2061–2065. 10.1021/jo01327a002. [DOI] [Google Scholar]
- Wiezorek S.; Lamers P.; Bolm C. Conversion and Degradation Pathways of Sulfoximines. Chem. Soc. Rev. 2019, 48, 5408–5423. 10.1039/C9CS00483A. [DOI] [PubMed] [Google Scholar]
- Still I. W. J.; Szilagyi S. A Convenient Procedure for the Reduction of Sulfones to Sulfoxides. Synth. Commun. 1979, 9, 923–930. 10.1080/00397917908064214. [DOI] [Google Scholar]
- Still I. W. J.; Ablenas F. J. Conversion of Sulfones to Sulfoxides via Hydride Reduction of (Aryloxy)sulfoxonium Salt Intermediates. J. Org. Chem. 1983, 48, 1617–1620. 10.1021/jo00158a009. [DOI] [Google Scholar]
- O’Mahony G. E.; Kelly P.; Lawrence S. E.; Maguire A. R. Synthesis of Enantioenriched Sulfoxides. Arkivoc 2011, i, 1–110. [Google Scholar]
- Srour H.; Le Maux P.; Chevance S.; Simonneaux G. Metal-Catalyzed Asymmetric Sulfoxidation, Epoxidation and Hydroxylation by Hydrogen Peroxide. Coord. Chem. Rev. 2013, 257, 3030–3050. 10.1016/j.ccr.2013.05.010. [DOI] [Google Scholar]
- Bryliakov K. P. Catalytic Asymmetric Oxygenations with the Environmentally Benign Oxidants H2O2 and O2. Chem. Rev. 2017, 117, 11406–11459. 10.1021/acs.chemrev.7b00167. [DOI] [PubMed] [Google Scholar]
- Volcho K.Recent Developments in the Catalytic Asymmetric Sulfoxidation Reactions. In Frontiers of Green Catalytic Selective Oxidations. Green Chemistry and Sustainable Technology; Bryliakov K., Ed.; Springer: Singapore, 2019. [Google Scholar]
- Davis F. A. Adventures in Sulfur-Nitrogen Chemistry. J. Org. Chem. 2006, 71, 8993–9003. 10.1021/jo061027p. [DOI] [PubMed] [Google Scholar]
- Han J.; Soloshonok V. A.; Klika K. D.; Drabowicz J.; Wzorek A. Chiral Sulfoxides: Advances in Asymmetric Synthesis and Problems with the Accurate Determination of the Stereochemical Outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. 10.1039/C6CS00703A. [DOI] [PubMed] [Google Scholar]
- Ławecka J.; Karczmarzyk Z.; Wolinska E.; Olender E.; Branowska D.; Rykowski A. A Novel Approach to Conformationally Strained 2,2′-Bipyridine Thiacrown Ethers and Their Chiral Sulfoxides by Sequential Homo-Coupling/DA–rDA Reaction of 5,5′-Bi-1,2,4-triazine-Containing Thiamacrocycles. Tetrahedron 2011, 67, 3098–3104. 10.1016/j.tet.2011.02.081. [DOI] [Google Scholar]
- İpek H.; Akdag A. Enantioselective Oxidation of Thioanisole to Methyl Phenyl Sulfoxide by Chiral Compounds Bearing N-Cl Bond. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1285–1293. 10.1080/10426507.2015.1012203. [DOI] [Google Scholar]
- Newhouse T. R.; Li X.; Blewett M. M.; Whitehead C. M. C.; Corey E. J. A Tetradentate Ligand for the Enantioselective Ti(IV)-Promoted Oxidation of Sulfides to Sulfoxides: Origin of Enantioselectivity. J. Am. Chem. Soc. 2012, 134, 17354–17357. 10.1021/ja305991k. [DOI] [PubMed] [Google Scholar]
- Naso F.; Capozzi M. A. M.; Bottoni A.; Calvaresi M.; Bertolasi V.; Capitelli F.; Cardellicchio C. A Combined Theoretical and Experimental Investigation on the Enantioselective Oxidation of Aryl Benzyl Sulfides in the Presence of a Chiral Titanium Catalyst. Chem. - Eur. J. 2009, 15, 13417–13426. 10.1002/chem.200902110. [DOI] [PubMed] [Google Scholar]
- Cardellicchio C.; Capozzi M. A. M.; Centrone C.; Naso F. Highly enantioselective oxidation of aryl benzyl sulfides. Phosphorus, Sulfur Silicon Relat. Elem. 2011, 186, 1193–1195. 10.1080/10426507.2010.529856. [DOI] [Google Scholar]
- Capozzi M. A. M.; Centrone C.; Fracchiolla G.; Naso F.; Cardellicchio C. Study of Factors Affecting Enantioselectivity in the Oxidation of Aryl Benzyl Sulfides in the Presence of Chiral Titanium Catalysts. Eur. J. Org. Chem. 2011, 2011, 4327–4334. 10.1002/ejoc.201100310. [DOI] [Google Scholar]
- Capozzi M. A. M.; Terraneo G.; Cavallo G.; Cardellicchio C. The search for exceptions in the highly enantioselective titanium catalysed oxidation of aryl benzyl sulfides. Tetrahedron 2015, 71, 4810–4816. 10.1016/j.tet.2015.05.036. [DOI] [Google Scholar]
- Capozzi M. A. M.; Capitelli F.; Bottoni A.; Calvaresi M.; Cardellicchio C. The Effect of the Fluorine Substitution on the Enantioselective Oxidation of Sulfides with Chiral Titanium Catalysts: A Combined Computational and Experimental Investigation. ChemCatChem 2013, 5, 210–219. 10.1002/cctc.201200456. [DOI] [Google Scholar]
- Capozzi M. A. M.; Bottoni A.; Calvaresi M.; Cardellicchio C. A Dichotomy in the Enantioselective Oxidation of Aryl Benzyl Sulfides: A Combined Experimental and Computational Work. Tetrahedron 2018, 74, 2041–2047. 10.1016/j.tet.2018.03.005. [DOI] [Google Scholar]
- Capozzi M. A. M.; Frascaro V.; Pescitelli G.; Cardellicchio C. New insights into the titanium-mediated enantioselective oxidation of fluorinated aryl benzyl sulfides and aryl phenacyl sulfides. Tetrahedron 2019, 75, 2406–2412. 10.1016/j.tet.2019.03.016. [DOI] [Google Scholar]
- Talsi E. P.; Bryliakov K. P. Catalytic Enantioselective Oxidation of Bulky Alkyl Aryl Thioethers with H2O2 over Titanium–Salan Catalysts. Eur. J. Org. Chem. 2011, 2011, 4693–4698. 10.1002/ejoc.201100557. [DOI] [Google Scholar]
- Talsi E. P.; Bryliakov K. P. Titanium–Salan-Catalyzed Asymmetric Sulfoxidations with H2O2: Design of More Versatile Catalysts. Appl. Organomet. Chem. 2013, 27, 239–244. 10.1002/aoc.2968. [DOI] [Google Scholar]
- Talsi E. P.; Rybalova T. V.; Bryliakov K. P. Isoinversion Behavior in the Enantioselective Oxidations of Pyridylmethylthiobenzimidazoles to Chiral Proton Pump Inhibitors on Titanium Salalen Complexes. ACS Catal. 2015, 5, 4673–4679. 10.1021/acscatal.5b01212. [DOI] [Google Scholar]
- Talsi E. P.; Bryliakov K. P. Ti-Salan Catalyzed Asymmetric Sulfoxidation of Pyridylmethylthiobenzimidazoles to Optically Pure Proton Pump Inhibitors. Catal. Today 2017, 279, 84–89. 10.1016/j.cattod.2016.03.006. [DOI] [Google Scholar]
- Rodygin K. S.; Rubtsova S. A.; Kutchin A. V.; Slepukhin P. A. One-Pot Synthesis and Asymmetric Oxidation of 2-Nitro-4-(trifluoromethyl)benzene Containing Sulfides. Phosphorus, Sulfur Silicon Relat. Elem. 2011, 186, 1885–1894. 10.1080/10426507.2010.547893. [DOI] [Google Scholar]
- Wang Y.; Wang M.; Wang L.; Wang Y.; Wang X.; Sun L. Asymmetric Oxidation of Sulfides with H2O2 Catalyzed by Titanium Complexes of Schiff Bases Bearing a Dicumenyl Salicylidenyl Unit. Appl. Organomet. Chem. 2011, 25, 325–330. 10.1002/aoc.1762. [DOI] [Google Scholar]
- Bühler S.; Goettert M.; Schollmeyer D.; Albrecht W.; Laufer S. A. Chiral Sulfoxides as Metabolites of 2-Thioimidazole-Based p38r Mitogen-Activated Protein Kinase Inhibitors: Enantioselective Synthesis and Biological Evaluation. J. Med. Chem. 2011, 54, 3283–3297. 10.1021/jm101623p. [DOI] [PubMed] [Google Scholar]
- Zhao G.; Tan R.; Zhang Y.; Luo X.; Xing C.; Yin D. Cooperative Chiral Salen TiIV Catalysts with Built-In Phase-Transfer Capability Accelerate Asymmetric Sulfoxidation in Water. RSC Adv. 2016, 6, 24704–24711. 10.1039/C6RA01130F. [DOI] [Google Scholar]
- Bhadra S.; Akakura M.; Yamamoto H. Design of a New Bimetallic Catalyst for Asymmetric Epoxidation and Sulfoxidation. J. Am. Chem. Soc. 2015, 137, 15612–15615. 10.1021/jacs.5b11429. [DOI] [PubMed] [Google Scholar]
- Zeng Q.; Weng W.; Xue X. Sulfide Oxidation Catalyzed by Vanadyl Complexes of N-Salicylidene α-Amino Acids at Low Catalyst Loading. Inorg. Chim. Acta 2012, 388, 11–15. 10.1016/j.ica.2012.03.024. [DOI] [Google Scholar]
- Il’ina I. V.; Koneva E. A.; Korchagina D. V.; Sal’nikov G. E.; Genaev A. M.; Volcho K. P.; Salakhutdinov N. F. Chiral Schiff Bases Synthesized from Terpenes of Pinane Series in Asymmetric Metal Complex Oxidation of Sulfides. Russ. J. Org. Chem. 2012, 48, 214–220. 10.1134/S1070428012020108. [DOI] [Google Scholar]
- Stingl K. A.; Weiß K. M.; Tsogoeva S. B. Asymmetric Vanadium- and Iron-Catalyzed Oxidations: New Mild (R)-Modafinil Synthesis and Formation of Epoxides Using Aqueous H2O2 as a Terminal Oxidant. Tetrahedron 2012, 68, 8493–8501. 10.1016/j.tet.2012.07.052. [DOI] [Google Scholar]
- Romanowski G.; Kira J. Oxidovanadium(V) Complexes with Chiral Tridentate Schiff Bases Derived from R(−)-Phenylglycinol: Synthesis, Spectroscopic Characterization and Catalytic Activity in the Oxidation of Sulfides and Styrene. Polyhedron 2013, 53, 172–178. 10.1016/j.poly.2012.12.038. [DOI] [Google Scholar]
- Romanowski G. Synthesis, Characterization and Catalytic Activity in the Oxidation of Sulfides and Styrene of Vanadium(V) Complexes with Tridentate Schiff Base Ligands. J. Mol. Catal. A: Chem. 2013, 368–369, 137–144. 10.1016/j.molcata.2012.11.023. [DOI] [Google Scholar]
- Aydin A. E. Synthesis of Novel β-Amino Alcohols and Their Application in the Catalytic Asymmetric Sulfoxidation of Sulfides. Tetrahedron: Asymmetry 2013, 24, 444–448. 10.1016/j.tetasy.2013.03.011. [DOI] [Google Scholar]
- Chuo T. H.; Boobalan R.; Chen C. Camphor-Based Schiff Base Of 3-Endo-Aminoborneol (SBAB): Novel Ligand for Vanadium-Catalyzed Asymmetric Sulfoxidation and Subsequent Kinetic Resolution. ChemistrySelect 2016, 1, 2174–2180. 10.1002/slct.201600379. [DOI] [Google Scholar]
- Voss F.; Herdtweck E.; Bach T. Hydrogen Bond Induced Enantioselectivity in Mn(Salen)-Catalysed Sulfoxidaton Reactions. Chem. Commun. 2011, 47, 2137–2139. 10.1039/c0cc04636a. [DOI] [PubMed] [Google Scholar]
- Wang C.; Kurahashi T.; Fujii H. Structure and Reactivity of an Iodosylarene Adduct of a Manganese(IV)–Salen Complex. Angew. Chem., Int. Ed. 2012, 51, 7809–7811. 10.1002/anie.201202835. [DOI] [PubMed] [Google Scholar]
- Dai W.; Li G.; Wang L.; Chen B.; Shang S.; Lv Y.; Gao S. Enantioselective Oxidation of Sulfides with H2O2 Catalyzed by a Pre-Formed Manganese Complex. RSC Adv. 2014, 4, 46545–46554. 10.1039/C4RA09832C. [DOI] [Google Scholar]
- Dai W.; Mi Y.; Lv Y.; Chen B.; Li G.; Chen G.; Gao S. Development of a Continuous-Flow Microreactor for Asymmetric Sulfoxidation Using a Biomimetic Manganese Catalyst. Adv. Synth. Catal. 2016, 358, 667–671. 10.1002/adsc.201501023. [DOI] [Google Scholar]
- Dai W.; Shang S.; Lv Y.; Li G.; Li C.; Gao S. Highly Chemoselective and Enantioselective Catalytic Oxidation of Heteroaromatic Sulfides via High-Valent Manganese(IV)–Oxo Cation Radical Oxidizing Intermediates. ACS Catal. 2017, 7, 4890–4895. 10.1021/acscatal.7b00968. [DOI] [Google Scholar]
- Chakraborty D.; Malik P.; Goda V. K. A New Methodology for the Oxidation of Sulfides with Fe(III) Catalysts. Appl. Organomet. Chem. 2012, 26, 21–26. 10.1002/aoc.1859. [DOI] [Google Scholar]
- Bera P. K.; Kumari P.; Abdi S. H. R.; Khan N. H.; Kureshy R. I.; Subramanian P. S.; Bajaj H. C. In Situ-Generated Chiral Iron Complex as Efficient Catalyst for Enantioselective Sulfoxidation Using Aqueous H2O2 as Oxidant. RSC Adv. 2014, 4, 61550–61556. 10.1039/C4RA09237F. [DOI] [Google Scholar]
- Nishiguchi S.; Izumi T.; Kouno T.; Sukegawa J.; Ilies L.; Nakamura E. Synthesis of Esomeprazole and Related Proton Pump Inhibitors through Iron-Catalyzed Enantioselective Sulfoxidation. ACS Catal. 2018, 8, 9738–9743. 10.1021/acscatal.8b02610. [DOI] [Google Scholar]
- Mahlau M.; List B. Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem., Int. Ed. 2013, 52, 518–533. 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]
- Liao S.; List B. Asymmetric Counteranion-Directed Iron Catalysis: A Highly Enantioselective Sulfoxidation. Adv. Synth. Catal. 2012, 354, 2363–2367. 10.1002/adsc.201200251. [DOI] [Google Scholar]
- O’Mahony G. E.; Eccles K. S.; Morrison R. E.; Ford A.; Lawrence S. E.; Maguire A. R. Investigation of Steric and Electronic Effects in the Copper-Catalysed Asymmetric Oxidation of Sulfides. Tetrahedron 2013, 69, 10168–10184. 10.1016/j.tet.2013.08.063. [DOI] [Google Scholar]
- Cheng M.; Li Y.; Zhou J.; Jia G.; Lu S.-M.; Yang Y.; Li C. Enantioselective Sulfoxidation Reaction Catalyzed by a G-Quadruplex DNA Metalloenzyme. Chem. Commun. 2016, 52, 9644–9647. 10.1039/C6CC03016E. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Cheng M.; Hao J.; Jia G.; Li C. Fluorescence Spectroscopic Insight into the Supramolecular Interactions in DNA-Based Enantioselective Sulfoxidation. ChemBioChem 2018, 19, 2233–2240. 10.1002/cbic.201800393. [DOI] [PubMed] [Google Scholar]
- Chakravarthy R. D.; Suresh K.; Ramkumar V.; Chand D. K. New Chiral Molybdenum Complex Catalyzed Sulfide Oxidation with Hydrogen Peroxide. Inorg. Chim. Acta 2011, 376, 57–63. 10.1016/j.ica.2011.05.033. [DOI] [Google Scholar]
- Mohammadnezhad G.; Debel R.; Plass W. Molybdenum(VI)-Promoted Sulfoxidation in the Presence of Tridentate Sugar-Based Schiff-Base Ligands. J. Mol. Catal. A: Chem. 2015, 410, 160–167. 10.1016/j.molcata.2015.09.011. [DOI] [Google Scholar]
- Carrasco C. J.; Montilla F.; Galindo A. Molybdenum-Catalyzed Asymmetric Sulfoxidation with Hydrogen Peroxide and Subsequent Kinetic Resolution, Using an Imidazolium-Based Dicarboxylate Compound as Chiral Inductor. Catal. Commun. 2016, 84, 134–136. 10.1016/j.catcom.2016.06.021. [DOI] [Google Scholar]
- Carrasco C. J.; Montilla F.; Galindo A. Molybdenum-Catalyzed Enantioselective Sulfoxidation Controlled by a Nonclassical Hydrogen Bond between Coordinated Chiral Imidazolium-Based Dicarboxylate and Peroxido Ligands. Molecules 2018, 23, 1595. 10.3390/molecules23071595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer E. B. Chiral-at-Metal Complexes and Their Catalytic Applications in Organic Synthesis. Chem. Soc. Rev. 2012, 41, 3153–3167. 10.1039/c2cs15234g. [DOI] [PubMed] [Google Scholar]
- Cruchter T.; Larionov V. A. Asymmetric Catalysis with Octahedral Stereogenic-at-Metal Complexes Featuring Chiral Ligands. Coord. Chem. Rev. 2018, 376, 95–113. 10.1016/j.ccr.2018.08.002. [DOI] [Google Scholar]
- Li Z.-Z.; Yao S.-Y.; Wu J.-J.; Ye B.-H. In Situ Generation of Sulfoxides with Predetermined Chirality via a Structural Template with a Chiral-at-Metal Ruthenium Complex. Chem. Commun. 2014, 50, 5644–5647. 10.1039/c4cc01907e. [DOI] [PubMed] [Google Scholar]
- Li Z.-Z.; Wen A.-H.; Yao S.-Y.; Ye B.-H. Enantioselective Syntheses of Sulfoxides in Octahedral Ruthenium(II) Complexes via a Chiral-at-Metal Strategy. Inorg. Chem. 2015, 54, 2726–2733. 10.1021/ic502898e. [DOI] [PubMed] [Google Scholar]
- Li Z.-Z.; Yao S.-Y.; Wen A.-H.; Ye B.-H. Asymmetric Oxidation Synthesis of Modafinil Acid by Use of a Recyclable Chiral-at-Metal Complex. Eur. J. Inorg. Chem. 2015, 2015, 4335–4342. 10.1002/ejic.201500617. [DOI] [Google Scholar]
- Yao S.-Y.; Chen X.-Y.; Ou Y.-L.; Ye B.-H. Chiral Recognition and Dynamic Thermodynamic Resolution of Sulfoxides by Chiral Iridium(III) Complexes. Inorg. Chem. 2017, 56, 878–885. 10.1021/acs.inorgchem.6b02494. [DOI] [PubMed] [Google Scholar]
- Li L.-P.; Ye B.-H. Efficient Generation of Singlet Oxygen and Photooxidation of Sulfide into Sulfoxide via Tuning the Ancillary of Bicyclometalated Iridium(III) Complexes. Inorg. Chem. 2019, 58, 7775–7784. 10.1021/acs.inorgchem.9b00220. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Li H.; Qi W.; Yang Y.; Yan Y.; Li B.; Wu L. Supramolecular Assembly of Chiral Polyoxometalate Complexes for Asymmetric Catalytic Oxidation of Thioethers. J. Mater. Chem. 2012, 22, 9181–9188. 10.1039/c2jm16398e. [DOI] [Google Scholar]
- Xuan W.; Ye C.; Zhang M.; Chen Z.; Cui Y. A Chiral Porous Metallosalan-Organic Framework Containing Titanium-Oxo Clusters for Enantioselective Catalytic Sulfoxidation. Chem. Sci. 2013, 4, 3154–3159. 10.1039/c3sc50487e. [DOI] [Google Scholar]
- Yang Z.; Zhu C.; Li Z.; Liu Y.; Liu G.; Cui Y. Engineering Chiral Fe(Salen)-Based Metal–Organic Frameworks for Asymmetric Sulfide Oxidation. Chem. Commun. 2014, 50, 8775–8778. 10.1039/C4CC03308F. [DOI] [PubMed] [Google Scholar]
- Yadav M.; Bhunia A.; Jana S. K.; Roesky P. W. Manganese- and Lanthanide-Based 1D Chiral Coordination Polymers as an Enantioselective Catalyst for Sulfoxidation. Inorg. Chem. 2016, 55, 2701–2708. 10.1021/acs.inorgchem.5b02234. [DOI] [PubMed] [Google Scholar]
- Wei H.; Guo Z.; Liang X.; Chen P.; Liu H.; Xing H. Selective Photooxidation of Amines and Sulfides Triggered by a Superoxide Radical Using a Novel Visible-Light-Responsive Metal-Organic Framework. ACS Appl. Mater. Interfaces 2019, 11, 3016–3023. 10.1021/acsami.8b18206. [DOI] [PubMed] [Google Scholar]
- Russo A.; De Fusco C.; Lattanzi A. Organocatalytic Asymmetric Oxidations with Hydrogen Peroxide and Molecular Oxygen. ChemCatChem 2012, 4, 901–916. 10.1002/cctc.201200139. [DOI] [Google Scholar]
- Cibulka R.; Jurok R. Organokatalitycké Enantioselektivní Oxidace Sulfide na Sulfoxidy. Chem. Listy 2012, 106, 896–902. [Google Scholar]
- de Gonzalo G.; Fraaije M. W. Recent Developments in Flavin-Based Catalysis. ChemCatChem 2013, 5, 403–415. 10.1002/cctc.201200466. [DOI] [Google Scholar]
- Mojr V.; Budĕšínský M.; Cibulka R.; Kraus T. Alloxazine–Cyclodextrin Conjugates for Organocatalytic Enantioselective Sulfoxidations. Org. Biomol. Chem. 2011, 9, 7318–7326. 10.1039/c1ob05934c. [DOI] [PubMed] [Google Scholar]
- Hartman T.; Herzig V.; Budĕšínský M.; Jindřich J.; Cibulka R.; Kraus T. Flavin–Cyclodextrin Conjugates: Effect of the Structure on the Catalytic Activity in Enantioselective Sulfoxidations. Tetrahedron: Asymmetry 2012, 23, 1571–1583. 10.1016/j.tetasy.2012.10.017. [DOI] [Google Scholar]
- Liao S.; Čorić I.; Wang Q.; List B. Activation of H2O2 by Chiral Confined Brønsted Acids: A Highly Enantioselective Catalytic Sulfoxidation. J. Am. Chem. Soc. 2012, 134, 10765–10768. 10.1021/ja3035637. [DOI] [PubMed] [Google Scholar]
- Liu Z.-M.; Zhao H.; Li M.-Q.; Lan Y.-B.; Yao Q.-B.; Tao J.-C.; Wang X.-W. Chiral Phosphoric Acid-Catalyzed Asymmetric Oxidation of Aryl Alkyl Sulfides and Aldehyde-Derived 1,3-Dithianes: Using Aqueous Hydrogen Peroxide as the Terminal Oxidant. Adv. Synth. Catal. 2012, 354, 1012–1022. 10.1002/adsc.201100810. [DOI] [Google Scholar]
- Jindal G.; Sunoj R. B. Axially Chiral Imidodiphosphoric Acid Catalyst for Asymmetric Sulfoxidation Reaction: Insights on Asymmetric Induction. Angew. Chem., Int. Ed. 2014, 53, 4432–4436. 10.1002/anie.201309532. [DOI] [PubMed] [Google Scholar]
- Held F. E.; Stingl K. A.; Tsogoeva S. B. Synthesis of (R)-Modafinil via Organocatalyzed and Non-Heme Iron-Catalyzed Sulfoxidation Using H2O2 as an Environmentally Benign Oxidant. Symmetry 2017, 9, 88. 10.3390/sym9060088. [DOI] [Google Scholar]
- Olah G. A.; Nishimura J. Aromatic Substitution. XXXII. Aluminum Chloride Catalyzed Arenesulfinylation of Benzene and Toluene with Benzenesulfinyl and Substituted Benzenesulfinyl Chlorides in Nitromethane Solution. J. Org. Chem. 1974, 39, 1203–1205. 10.1021/jo00923a008. [DOI] [Google Scholar]
- Miao T.; Li P. H.; Zhang Y. C.; Wang L. A Sulfenylation Reaction: Direct Synthesis of 3-Arylsulfinylindoles from Arylsulfinic Acids and Indoles in Water. Org. Lett. 2015, 17, 832–835. 10.1021/ol503659t. [DOI] [PubMed] [Google Scholar]
- Nguyen N.-L. T.; Vo H.-T.; Duus F.; Luu T. X. T. Dramatic Influence of Ionic Liquid and Ultrasound Irradiation on the Electrophilic Sulfinylation of Aromatic Compounds by Sulfinic Esters. Molecules 2017, 22, 1458. 10.3390/molecules22091458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A. U.; Wimmer A.; König B. Visible-Light-Accelerated C-H Sulfinylation of Heteroarenes. Angew. Chem., Int. Ed. 2017, 56, 409–412. 10.1002/anie.201610210. [DOI] [PubMed] [Google Scholar]
- O’Donnell J. S.; Schwan A. L. Generation, Structure and Reactions of Sulfenic Acid Anions. J. Sulfur Chem. 2004, 25, 183–211. 10.1080/1741599042000220761. [DOI] [Google Scholar]
- Caupène C.; Boudou C.; Perrio S.; Metzner P. Remarkably Mild and Simple Preparation of Sulfenate Anions from β-Sulfinylesters: A New Route to Enantioenriched Sulfoxides. J. Org. Chem. 2005, 70, 2812–2815. 10.1021/jo0478003. [DOI] [PubMed] [Google Scholar]
- Maitro G.; Prestat G.; Madec D.; Poli G. Preparation of Allyl Sulfoxides by Palladium-Catalyzed Allylic Alkylation of Sulfenate Anions. J. Org. Chem. 2006, 71, 7449–7454. 10.1021/jo061359u. [DOI] [PubMed] [Google Scholar]
- Maitro G.; Vogel S.; Prestat G.; Madec D.; Poli G. Aryl Sulfoxides via Palladium-Catalyzed Arylation of Sulfenate Anions. Org. Lett. 2006, 8, 5951–5954. 10.1021/ol062315a. [DOI] [PubMed] [Google Scholar]
- Maitro G.; Vogel S.; Sadaoui M.; Prestat G.; Madec D.; Poli G. Enantioselective Synthesis of Aryl Sulfoxides via Palladium-Catalyzed Arylation of Sulfenate Anions. Org. Lett. 2007, 9, 5493–5496. 10.1021/ol702343g. [DOI] [PubMed] [Google Scholar]
- Bernoud E.; Le Duc G.; Bantreil X.; Prestat G.; Madec D.; Poli G. Aryl Sulfoxides from Allyl Sulfoxides via 2,3–Sigmatropic Rearrangement and Domino Pd-Catalyzed Generation/Arylation of Sulfenate Anions. Org. Lett. 2010, 12, 320–323. 10.1021/ol902620t. [DOI] [PubMed] [Google Scholar]
- Maitro G.; Prestat G.; Madec D.; Poli G. An Escapade in the World of Sulfenate Anions: Generation, Reactivity and Applications in Domino Processes. Tetrahedron: Asymmetry 2010, 21, 1075–1084. 10.1016/j.tetasy.2010.05.035. [DOI] [Google Scholar]
- Gelat F.; Jayashankaran J.; Lohier J.; Gaumont A.; Perrio S. Organocatalytic Asymmetric Synthesis of Sulfoxides from Sulfenic Acid Anions Mediated by a Cinchona-Derived Phase-Transfer Reagent. Org. Lett. 2011, 13, 3170–3173. 10.1021/ol2010962. [DOI] [PubMed] [Google Scholar]
- Gelat F.; Gaumont A.; Perrio S. Chiral Non-Racemic Sulfoxides by Asymmetric Alkylation of Alkanesulfenates in the Presence of a Chiral Ammonium Phase-Transfer Catalyst Derived from Cinchona Alkaloid. J. Sulfur Chem. 2013, 34, 596–605. 10.1080/17415993.2013.795226. [DOI] [Google Scholar]
- Zong L.; Ban X.; Kee C. W.; Tan C.-H. Catalytic Enantioselective Alkylation of Sulfenate Anions to Chiral Heterocyclic Sulfoxides Using Halogenated Pentanidium Salts. Angew. Chem., Int. Ed. 2014, 53, 11849–11853. 10.1002/anie.201407512. [DOI] [PubMed] [Google Scholar]
- Gelat F.; Lohier J. F.; Gaumont A. C.; Perrio S. tert-Butyl Sulfoxides: Key Precursors for Palladium-Catalyzed Arylation of Sulfenate Salts. Adv. Synth. Catal. 2015, 357, 2011–2016. 10.1002/adsc.201500368. [DOI] [Google Scholar]
- Schwan A. L.; Söderman S. C. Discoveries in Sulfenic Acid Anion Chemistry. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 275–286. 10.1080/10426507.2012.729116. [DOI] [Google Scholar]
- Schwan A. L.; Michalski M. M.; Findlay J. P. Origins and Applications of Stereoselective Sulfenate Anion Alkylation Reactions. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 692–697. 10.1080/10426507.2019.1603703. [DOI] [Google Scholar]
- Izquierdo F.; Chartoire A.; Nolan S. P. Direct S-Arylation of Unactivated Arylsulfoxides Using Pd(IPr*)(cin)Cl. ACS Catal. 2013, 3, 2190–2193. 10.1021/cs400533e. [DOI] [Google Scholar]
- Jia T. Z.; Bellomo A.; Montel S.; Zhang M. N.; El Baina K.; Zheng B.; Walsh P. J. Diaryl Sulfoxides from Aryl Benzyl Sulfoxides: A Single Palladium-Catalyzed Triple Relay Process. Angew. Chem., Int. Ed. 2014, 53, 260–264. 10.1002/anie.201307172. [DOI] [PubMed] [Google Scholar]
- Jia T. Z.; Zhang M. N.; Sagamanova I. K.; Wang C. Y.; Walsh P. J. Palladium Catalyzed Diaryl Sulfoxide Generation from Aryl Benzyl Sulfoxides and Aryl Chlorides. Org. Lett. 2015, 17, 1168–1171. 10.1021/acs.orglett.5b00092. [DOI] [PubMed] [Google Scholar]
- Jia T.; Zhang M.; Jiang H.; Wang C. Y.; Walsh P. J. Palladium-Catalyzed Arylation of Alkyl Sulfenate Anions. J. Am. Chem. Soc. 2015, 137, 13887–13893. 10.1021/jacs.5b08117. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Jia T.; Zhang M.; Walsh P. J. Palladium-Catalyzed Arylation of Aryl Sulfenate Anions with Aryl Bromides under Mild Conditions: Synthesis of Diaryl Sulfoxides. Org. Lett. 2016, 18, 972–975. 10.1021/acs.orglett.6b00073. [DOI] [PubMed] [Google Scholar]
- Jia T. Z.; Zhang M. N.; McCollom S. P.; Bellomo A.; Montel S.; Mao J. Y.; Dreher S. D.; Welch C. J.; Regalado E. L.; Williamson R. T.; et al. Palladium-Catalyzed Enantioselective Arylation of Aryl Sulfenate Anions: A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2017, 139, 8337–8345. 10.1021/jacs.7b03623. [DOI] [PubMed] [Google Scholar]
- Wu C.; Berritt S.; Liang X.; Walsh P. J. Palladium-Catalyzed Enantioselective Alkenylation of Sulfenate Anions. Org. Lett. 2019, 21, 960–964. 10.1021/acs.orglett.8b03943. [DOI] [PubMed] [Google Scholar]
- Wang L.; Chen M. J.; Zhang P. C.; Li W. B.; Zhang J. L. Palladium/PC-Phos-Catalyzed Enantioselective Arylation of General Sulfenate Anions: Scope and Synthetic Applications. J. Am. Chem. Soc. 2018, 140, 3467–3473. 10.1021/jacs.8b00178. [DOI] [PubMed] [Google Scholar]
- Yu H.; Li Z.; Bolm C. Nondirected Copper-Catalyzed Sulfoxidations of Benzylic C–H Bonds. Org. Lett. 2018, 20, 2076–2079. 10.1021/acs.orglett.8b00615. [DOI] [PubMed] [Google Scholar]
- Yu H.; Li Z.; Bolm C. Transition-Metal-Free Arylations of In-Situ Generated Sulfenates with Diaryliodonium Salts. Org. Lett. 2018, 20, 7104–7106. 10.1021/acs.orglett.8b03046. [DOI] [PubMed] [Google Scholar]
- Wang L.; Chen M.; Zhang J. Transition Metal-Free Base-Promoted Arylation of Sulfenate Anions with Diaryliodonium Salts. Org. Chem. Front. 2019, 6, 32–35. 10.1039/C8QO00914G. [DOI] [Google Scholar]
- Amos S. G. E.; Nicolai S.; Gagnebin A.; Le Vaillant F.; Waser J. Metal-Free Electrophilic Alkynylation of Sulfenate Anions with Ethynylbenziodoxolone Reagents. J. Org. Chem. 2019, 84, 3687–3701. 10.1021/acs.joc.9b00050. [DOI] [PubMed] [Google Scholar]
- Mączka W.; Wińska K.; Grabarczyk M. Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow. Catalysts 2018, 8, 624. 10.3390/catal8120624. [DOI] [Google Scholar]
- Li A.-T.; Zhang J.-D.; Yu H.-L.; Pan J.; Xu J.-H. Significantly Improved Asymmetric Oxidation of Sulfide with Resting Cells of Rhodococcus sp. in a Biphasic System. Process Biochem. 2011, 46, 689–694. 10.1016/j.procbio.2010.11.010. [DOI] [Google Scholar]
- Babiak P.; Kyslíková E.; Stěpánek V.; Valešová R.; Palyzová A.; Marešová H.; Hájíček J.; Kyslík P. Whole-Cell Oxidation of Omeprazole Sulfide to Enantiopure Esomeprazole with Lysinibacillus sp. B71. Bioresour. Technol. 2011, 102, 7621–7626. 10.1016/j.biortech.2011.05.052. [DOI] [PubMed] [Google Scholar]
- Mascotti M. L.; Orden A. A.; Bisogno F. R.; de Gonzalo G.; Kurina-Sanz M. Aspergillus Genus as a Source of New Catalysts for Sulfide Oxidation. J. Mol. Catal. B: Enzym. 2012, 82, 32–36. 10.1016/j.molcatb.2012.05.003. [DOI] [Google Scholar]
- Zhai X.-H.; Ma Y.-H.; Lai D.-Y.; Zhou S.; Chen Z.-M. Development of a Whole-Cell Biocatalyst with NADPH Regeneration System for Biosulfoxidation. J. Ind. Microbiol. Biotechnol. 2013, 40, 797–803. 10.1007/s10295-013-1288-0. [DOI] [PubMed] [Google Scholar]
- He Y.-C.; Ma C.-L.; Yang Z.-X.; Zhou M.; Xing Z.; Ma J.-T.; Yu H.-L. Highly Enantioselective Oxidation of Phenyl Methyl Sulfide and Its Derivatives into Optically Pure (S)-Sulfoxides with Rhodococcus sp. CCZU10–1 in an n-Octane–Water Biphasic System. Appl. Microbiol. Biotechnol. 2013, 97, 10329–10337. 10.1007/s00253-013-5258-2. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhuo J.; Zheng D.; Tian S.; Li Z. Stereoselective Oxidation of Sulfides to Optically Active Sulfoxides with Resting Cells of Pseudomonas monteilii CCTCC M2013683. J. Mol. Catal. B: Enzym. 2014, 106, 100–104. 10.1016/j.molcatb.2014.05.004. [DOI] [Google Scholar]
- Kylosova T. I.; Elkin A. A.; Grishko V. V.; Ivshina I. B. Biotransformation of Prochiral Sulfides into (R)-Sulfoxides Using Immobilized Gordonia terrae IEGM 136 Cells. J. Mol. Catal. B: Enzym. 2016, 123, 8–13. 10.1016/j.molcatb.2015.10.014. [DOI] [Google Scholar]
- Wu K.; Tang L.; Cui H.; Wan N.; Liu Z.; Wang Z.; Zhang S.; Cui B.; Han W.; Chen Y. Biocatalytical Asymmetric Sulfoxidation by Identifying Cytochrome P450 from Parvibaculum Lavamentivorans DS-1. ChemCatChem 2018, 10, 5410–5413. 10.1002/cctc.201801139. [DOI] [Google Scholar]
- Rioz-Martínez A.; de Gonzalo G.; Torres Pazmiño D. E.; Fraaije M. W.; Gotor V. Enzymatic Synthesis of Novel Chiral Sulfoxides Employing Baeyer–Villiger Monooxygenases. Eur. J. Org. Chem. 2010, 2010, 6409–6416. 10.1002/ejoc.201000890. [DOI] [PubMed] [Google Scholar]
- Rioz-Martínez A.; Kopacz M.; de Gonzalo G.; Torres Pazmiño D. E.; Gotor V.; Fraaije M. W. Exploring the Biocatalytic Scope of a Bacterial Flavin-Containing Monooxygenase. Org. Biomol. Chem. 2011, 9, 1337–1341. 10.1039/c0ob00988a. [DOI] [PubMed] [Google Scholar]
- Hibi M.; Kawashima T.; Yajima H.; Smirnov S. V.; Kodera T.; Sugiyama M.; Shimizu S.; Yokozeki K.; Ogawa J. Enzymatic Synthesis of Chiral Amino Acid Sulfoxides by Fe(II)/α-Ketoglutarate-Dependent Dioxygenase. Tetrahedron: Asymmetry 2013, 24, 990–994. 10.1016/j.tetasy.2013.07.017. [DOI] [Google Scholar]
- Bassanini I.; Ferrandi E. E.; Vanoni M.; Ottolina G.; Riva S.; Crotti M.; Brenna E.; Monti D. Peroxygenase-Catalyzed Enantioselective Sulfoxidations. Eur. J. Org. Chem. 2017, 2017, 7186–7189. 10.1002/ejoc.201701390. [DOI] [Google Scholar]
- Rondot L.; Girgenti E.; Oddon F.; Marchi-Delapierre C.; Jorge-Robin A.; Ménage S. Catalysis Without a Headache: Modification of Ibuprofen for the Design of Artificial Metalloenzyme for Sulfide Oxidation. J. Mol. Catal. A: Chem. 2016, 416, 20–28. 10.1016/j.molcata.2016.02.015. [DOI] [Google Scholar]
- Tang J.; Huang F.; Wei Y.; Bian H.; Zhang W.; Liang H. Bovine Serum Albumin–Cobalt(II) Schiff Base Complex Hybrid: An Efficient Artificial Metalloenzyme for Enantioselective Sulfoxidation Using Hydrogen Peroxide. Dalton Trans 2016, 45, 8061–8072. 10.1039/C5DT04507J. [DOI] [PubMed] [Google Scholar]
- Tang J.; Yao P.-F.; Xu X.-L.; Li H.-Y.; Huang F.-P.; Nie Q.-Q.; Luo M.-Y.; Yu Q.; Bian H.-D. Asymmetric Catalytic Sulfoxidation by a Novel VIV8 Cluster Catalyst in the Presence of Serum Albumin: a Simple and Green Oxidation System. RSC Adv. 2016, 6, 44154–44162. 10.1039/C6RA08153C. [DOI] [Google Scholar]
- Linde D.; Cañellas M.; Coscolín C.; Davó-Siguero I.; Romero A.; Lucas F.; Ruiz-Dueñas F. J.; Guallar V.; Martínez A. T. Asymmetric Sulfoxidation by Engineering the Heme Pocket of a Dye-Decolorizing Peroxidase. Catal. Sci. Technol. 2016, 6, 6277–6285. 10.1039/C6CY00539J. [DOI] [Google Scholar]
- Wang J.; Ilie A.; Reetz M. T. Chemo- and Stereoselective Cytochrome P450-BM3-Catalyzed Sulfoxidation of 1-Thiochroman-4-ones Enabled by Directed Evolution. Adv. Synth. Catal. 2017, 359, 2056–2060. 10.1002/adsc.201700414. [DOI] [Google Scholar]
- Chen K.-I.; Challinor V. L.; Kielmann L.; Sharpe P. C.; De Voss J. J.; Kappler U.; McEwan A. G.; Bernhardt P. V. Electrochemically Mediated Enantioselective Reduction of Chiral Sulfoxides. JBIC, J. Biol. Inorg. Chem. 2015, 20, 395–402. 10.1007/s00775-014-1215-5. [DOI] [PubMed] [Google Scholar]
- Yang J.; Yuan Z.; Zhou Y.; Zhao J.; Yang M.; Cheng X.; Ou G.; Chen Y. Asymmetric Reductive Resolution of Racemic Sulfoxides by Recombinant Methionine Sulfoxide Reductase from a Pseudomonas monteilii Strain. J. Mol. Catal. B: Enzym. 2016, 133, S588–S592. 10.1016/j.molcatb.2017.02.005. [DOI] [Google Scholar]
- Peng L.; Wen Y.; Chen Y.; Yuan Z.; Zhou Y.; Cheng X.; Chen Y.; Yang J. Biocatalytic Preparation of Chiral Sulfoxides through Asymmetric Reductive Resolution by Methionine Sulfoxide Reductase. ChemCatChem 2018, 10, 3284–3290. 10.1002/cctc.201800279. [DOI] [Google Scholar]
- Yang J.; Wen Y.; Peng L.; Chen Y.; Cheng X.; Chen Y. Identification of MsrA Homologues for the Preparation of (R)-Sulfoxides at High Substrate Concentrations. Org. Biomol. Chem. 2019, 17, 3381–3388. 10.1039/C9OB00384C. [DOI] [PubMed] [Google Scholar]
- Sagramora L.; Koch P.; Garbesi A.; Fava A. Asymmetric Oxidation of Bivalent Organic Sulphur Compounds. Absolute Configuration of Thiolsulphinate and Sulphinate Esters Obtained by Stereoselective Oxidation of Disulphides and Sulphenate Esters. Chem. Commun. 1967, 985–986. 10.1039/c19670000985. [DOI] [Google Scholar]
- Nemecek C.; Duñach E.; Kagan H. Asymmetric Oxidation of Some Sulfur Derivatives. New J. Chem. 1986, 10, 761–764. [Google Scholar]
- Hamel M.; Grach G.; Abrunhosa I.; Gulea M.; Masson S.; Vazeux M.; Drabowicz J.; Mikołajczyk M. Asymmetric Oxidation of Sulfenates to Sulfinates as a New Route to Optically Active ortho-Phosphorylated Phenyl Sulfoxides. Tetrahedron: Asymmetry 2005, 16, 3406–3415. 10.1016/j.tetasy.2005.09.005. [DOI] [Google Scholar]
- Phillips H. Investigations on the Dependence of Rotatory Power on Chemical Constitution. Part XXVII. The Optical Properties of N-Alkyl p-Toluenesulphinates. J. Chem. Soc., Trans. 1925, 127, 2552–2587. 10.1039/CT9252702552. [DOI] [Google Scholar]
- Evans J.W.; Fierman M. B.; Miller S. J.; Ellman J. A. Catalytic Enantioselective Synthesis of Sulfinate Esters Through the Dynamic Resolution of tert-Butanesulfinyl Chloride. J. Am. Chem. Soc. 2004, 126, 8134–8135. 10.1021/ja047845l. [DOI] [PubMed] [Google Scholar]
- Peltier H. M.; Evans J. W.; Ellman J. A. Catalytic Enantioselective Sulfinyl Transfer Using Cinchona Alkaloid Catalysts. Org. Lett. 2005, 7, 1733–1736. 10.1021/ol050275p. [DOI] [PubMed] [Google Scholar]
- Shibata N.; Matsunaga M.; Fukuzumi T.; Nakamura S.; Toru T. Cinchona Alkaloid-Sulfinyl Chloride Combinations: Catalytic Enantioselective Sulfinylation of Alcohols. Synlett 2005, 1699–1702. 10.1055/s-2005-871535. [DOI] [Google Scholar]
- Shibata N.; Matsunaga M.; Nakagawa M.; Fukuzumi T.; Nakamura S.; Toru T. Cinchona Alkaloid/Sulfinyl Chloride Combinations: Enantioselective Sulfinylating Agents of Alcohols. J. Am. Chem. Soc. 2005, 127, 1374–1375. correction J. Am. Chem. Soc. 2005, 127, 5271–5271. 10.1021/ja0430189. [DOI] [PubMed] [Google Scholar]
- Oertling H.; Reckziegel A.; Surburg H.; Bertram H.-J. Applications of Menthol in Synthetic Chemistry. Chem. Rev. 2007, 107, 2136–2164. 10.1021/cr068409f. [DOI] [PubMed] [Google Scholar]
- Andersen K. K. Synthesis of (+)-Ethyl p-Tolyl Sulfoxide from (−)-Menthyl (−)-p-Toluenesulfinate. Tetrahedron Lett. 1962, 3, 93–95. 10.1016/S0040-4039(00)71106-3. [DOI] [Google Scholar]
- Fernández I.; Khiar N.; Llera J. M.; Alcudia F. Asymmetric Synthesis of Alkane- and Arenesulfinates of Diacetone-d-glucose (DAG): an Improved and General Route to Both Enantiomerically Pure Sulfoxides. J. Org. Chem. 1992, 57, 6789–6796. 10.1021/jo00051a022. [DOI] [Google Scholar]
- Mikołajczyk M.; Drabowicz J.; Bujnicki B. Acid-catalysed Conversion of Sulphinamides into Sulphinates: A New Synthesis of Optically Active Sulphinates. J. Chem. Soc., Chem. Commun. 1976, 568–569. 10.1039/C39760000568. [DOI] [Google Scholar]
- Drabowicz J.; Ślebocka-Tilk H.; Mikołajczyk M. Stereochemistry and Kinetics of the Alkoxy-alkoxy Exchange in Sulphinates. Phosphorus Sulfur Relat. Elem. 1979, 6, 349–349. 10.1080/03086647908080448. [DOI] [Google Scholar]
- Mikołajczyk M.; Drabowicz J.; Ślebocka-Tilk H. Nucleophilic Substitution at Sulfur. Kinetic Evidence for Inversion of Configuration at Sulfinyl Sulfur in Acid-Catalyzed Transesterification of Sulfinates. J. Am. Chem. Soc. 1979, 101, 1302–1303. 10.1021/ja00499a053. [DOI] [Google Scholar]
- Mikołajczyk M.; Drabowicz J.; Bujnicki B. Nucleophilic Substitution at Sulfinyl Sulfur. Factors Affecting the Inversion to Retention Ratio in Acid-Catalyzed Alcoholysis of Chiral N,N-Diisopropyl p-Toluenesulfinamide. Tetrahedron Lett. 1985, 26, 5699–5702. 10.1016/S0040-4039(01)80924-2. [DOI] [Google Scholar]
- Bujnicki B.; Drabowicz J.; Mikołajczyk M. Acid Catalyzed Alcoholysis of Sulfinamides: Unusual Stereochemistry, Kinetics and a Question of Mechanism Involving Sulfurane Intermediates and Their Pseudorotation. Molecules 2015, 20, 2949–2972. 10.3390/molecules20022949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen E.; Chavda M. K.; Zikpi K. M.; Waggoner S. L.; Passini D. J.; Wolfe J. A.; Larson R.; Beckley C.; Hamaker C. G.; Hitchcock S. R. A Mixed Anhydride Approach to the Preparation of Sulfinate Esters and Allylic Sulfones: Trimethylacetic p-Toluenesulfinic Anhydride. Tetrahedron Lett. 2017, 58, 3073–3077. 10.1016/j.tetlet.2017.06.074. [DOI] [Google Scholar]
- Ji Y. Z.; Wang M.; Li H.-J.; Liu Y.; Wu Y.-C. Direct Sulfination of Nonactivated Alcohols with Arylsulfonylmethyl Isocyanides. Eur. J. Org. Chem. 2016, 2016, 4077–4083. 10.1002/ejoc.201600629. [DOI] [Google Scholar]
- Drabowicz J.; Legędź S.; Mikołajczyk M. Organosulphur Compounds-LXIX. Optically Active Sulphinates: a New Type of Enantioselective Asymmetric Synthesis and Kinetic Resolution. Tetrahedron 1988, 44, 5243–5251. 10.1016/S0040-4020(01)86031-2. [DOI] [Google Scholar]
- Rebiere F.; Samuel O.; Ricard L.; Kagan H. B. A General Route to Enantiomerically Pure Sulfoxides from a Chiral Sulfite. J. Org. Chem. 1991, 56, 5991–5999. 10.1021/jo00021a008. [DOI] [Google Scholar]
- Borsuk K.; Frelek J.; Łysek R.; Urbańczyk-Lipkowska Z.; Chmielewski M. Six-Membered Cyclic Sulfites Derived from Glucofuranose and 1,2,4-Butanetriol. Chirality 2001, 13, 533–540. 10.1002/chir.1173. [DOI] [PubMed] [Google Scholar]
- Coulomb J.; Certal V.; Larraufie M.-H.; Ollivier C.; Corbet J.-P.; Mignani G.; Fensterbank L.; Lacôte E.; Malacria M. Intramolecular Homolytic Substitution of Sulfinates and Sulfinamides. Chem. - Eur. J. 2009, 15, 10225–10232. 10.1002/chem.200900942. [DOI] [PubMed] [Google Scholar]
- Shyam P. K.; Kim Y. K.; Lee C.; Jang H.-Y. Copper-Catalyzed Aerobic Formation of Unstable Sulfinyl Radicals for the Synthesis of Sulfinates and Thiosulfonates. Adv. Synth. Catal. 2016, 358, 56–61. 10.1002/adsc.201500785. [DOI] [Google Scholar]
- Mikołajczyk M.; Drabowicz J. Optically Active O-Alkyl Alkylsulphinates. Tetrahedron Lett. 1972, 13, 2379–2382. 10.1016/S0040-4039(01)85308-9. [DOI] [Google Scholar]
- Cogan D. A.; Liu G.; Kim K.; Backes B. A.; Ellman J. A. Catalytic Asymmetric Oxidation of tert-Butyl Disulfide. Synthesis of tert-Butanesulfinamides, tert-Butyl Sulfoxide, and tert-Butanosulfinimines. J. Am. Chem. Soc. 1998, 120, 8011–8019. 10.1021/ja9809206. [DOI] [Google Scholar]
- Savige W. E.; Eager J.; Maclaren J. A.; Roxburgh C. M. The S-Monoxides of Cystine, Cystamine and Homocystine. Tetrahedron Lett. 1964, 5, 3289–3293. 10.1016/S0040-4039(01)89425-9. [DOI] [Google Scholar]
- Savige W. E.; Fava A. Asymmetric Synthesis and Racemization of Disulphide Monoxides. Chem. Commun. 1965, 417–418. 10.1039/c19650000417. [DOI] [Google Scholar]
- Kice J. L.; Large G. B. Synthesis of Optically Active Benzenethiolsulfinate by Asymmetric Oxidation of Phenyl Disulfide. Tetrahedron Lett. 1965, 6, 3537–3541. 10.1016/S0040-4039(01)99535-8. [DOI] [Google Scholar]
- Davis F. A.; Jenkins R. H. Jr.; Awad S. B.; Stringer O. D.; Watson W. H.; Galloy J. Chemistry of Oxaziridines. 3. Asymmetric Oxidation of Organosulfur Compounds Using Chiral 2-Sulfonyloxaziridines. J. Am. Chem. Soc. 1982, 104, 5412–5418. 10.1021/ja00384a028. [DOI] [Google Scholar]
- Bolm C.; Bienewald F. Asymmetric Sulfide Oxidation with Vanadium Catalysts and H2O2. Angew. Chem., Int. Ed. Engl. 1995, 34, 2640–2642. 10.1002/anie.199526401. [DOI] [Google Scholar]
- Liu G.; Cogan D. A.; Ellman J. A. Catalytic Asymmetric Synthesis of tert-Butanesulfinamide. Application to the Asymmetric Synthesis of Amines. J. Am. Chem. Soc. 1997, 119, 9913–9914. 10.1021/ja972012z. [DOI] [Google Scholar]
- Blum S. A.; Bergman R. G.; Ellman J. A. Enantioselective Oxidation of Di-tert-Butyl Disulfide with a Vanadium Catalyst: Progress toward Mechanism Elucidation. J. Org. Chem. 2003, 68, 150–155. 10.1021/jo0205560. [DOI] [PubMed] [Google Scholar]
- Weix D. J.; Ellman J. A. An Improved Synthesis of tert-Butanesulfinamide Suitable for Large Scale Production. Org. Lett. 2003, 5, 1317–1320. 10.1021/ol034254b. [DOI] [PubMed] [Google Scholar]
- Weix D. J.; Ellman J. A. (+)-2-Methyl-2-propanesulfinamide. Org. Synth. 2005, 82, 157–165. 10.1002/0471264229.os082.24. [DOI] [Google Scholar]
- Dragoli D. R.; Burdett M. T.; Ellman J. A. Design, Synthesis, and Utility of a Support-Bound tert-Butanesulfinamide. J. Am. Chem. Soc. 2001, 123, 10127–10128. 10.1021/ja016349j. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Wang X.; Wang Y.; Feng Y.; Zang Y. An Economic Approach to Chiral Thiosulfinates by Enantioselective Oxidation with Hydroperoxide. Synth. Commun. 2004, 34, 501–507. 10.1081/SCC-120027290. [DOI] [Google Scholar]
- Colonna S.; Gaggero N.; Carrea G.; Pasta P.; Alphand V.; Furstoss R. Enantioselective Synthesis of tert-Butyl tert-Butanethiosulfinate Catalyzed by Cyclohexanone Monooxygenase. Chirality 2001, 13, 40–42. 10.1002/1520-636X(2001)13:1<40::AID-CHIR8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Colonna C.; Pironti V.; Drabowicz J.; Brebion F.; Fensterbank L.; Malacria M. Enantioselective Synthesis of Thiosulfinates and of Acyclic Alkylidenemethylene Sulfide Sulfoxides. Eur. J. Org. Chem. 2005, 2005, 1727–1730. 10.1002/ejoc.200400892. [DOI] [Google Scholar]
- Boyd D. R.; Sharma N. D.; Kennedy M. A.; Shepherd S. D.; Malone J. F.; Alves-Areiás A.; Holt R.; Allenmark S. G.; Lemurell (née Andresson) M. A.; Dalton H.; Luckarift H. Enzyme-Catalysed Oxygenation and Deoxygenation Routes to Chiral Thiosulfinates. Chem. Commun. 2002, 2002, 1452–1453. 10.1039/b203139f. [DOI] [PubMed] [Google Scholar]
- Boyd D. R.; Sharma N. D.; King A. W. T.; Shepherd S. D.; Allen C. C. R.; Holt R.; Luckarift H.; Dalton H. Stereoselective Reductase-Catalysed Deoxygenation of Sulfoxides in Aerobic and Anaerobic Bacteria. Org. Biomol. Chem. 2004, 2, 554–561. 10.1039/b313714g. [DOI] [PubMed] [Google Scholar]
- Boyd D. R.; Sharma N. D.; Shepherd S. D.; Allenmark S. G.; Allen C. C. R. Enzyme-Catalysed Oxidation of 1,2-Disulfides to Yield Chiral Thiosulfinate, Sulfoxide and cis-Dihydrodiol Metabolites. RSC Adv. 2014, 4, 27607–27619. 10.1039/C4RA02923B. [DOI] [Google Scholar]
- Drabowicz J.; Łyżwa P.; Mikołajczyk M. A New Asymmetric Synthesis of Chiral t-Butyl t-Butanethiosulphinate. Phosphorus Sulfur Relat. Elem. 1983, 16, 267–270. 10.1080/03086648308080478. [DOI] [Google Scholar]
- Drabowicz J.; Pacholczyk M. A New Type of Asymmetric Synthesis of Sulphinic Acid Derivatives Using Chiral Carbodiimides. Phosphorus Sulfur Relat. Elem. 1987, 29, 257–263. 10.1080/03086648708080510. [DOI] [Google Scholar]
- Drabowicz J.; Mikołajczyk M. The First Stereoselective Synthesis of Optically Active Thiosulfinates. Tetrahedron Lett. 1985, 26, 5703–5706. 10.1016/S0040-4039(01)80925-4. [DOI] [Google Scholar]
- Liao J.; Sun X.; Cui X.; Yu K.; Zhu J.; Deng J. Facile Optical Resolution of tert-Butanethiosulfinate by Molecular Complexation with (R)-BINOL and Study of Chiral Discrimination of the Diastereomeric Complexes. Chem. - Eur. J. 2003, 9, 2611–2615. 10.1002/chem.200204653. [DOI] [PubMed] [Google Scholar]
- Ellman J. A. Applications of tert-Butanesulfinamide in the Asymmetric Synthesis of Amines. Pure Appl. Chem. 2003, 75, 39–46. 10.1351/pac200375010039. [DOI] [Google Scholar]
- Robak M. T.; Herbage M. A.; Ellman J. A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- Ellman J. A.; Owens T. D.; Tang T. P. N-tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines. Acc. Chem. Res. 2002, 35, 984–995. 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]
- Ferreira F.; Botuha C.; Chemla F.; Pérez-Luna A. t-Butanesulfinimines: Structure, Synthesis and Synthetic Applications. Chem. Soc. Rev. 2009, 38, 1162–1186. 10.1039/b809772k. [DOI] [PubMed] [Google Scholar]
- Craine M.; Raban M. The Chemistry of Sulfenamides. Chem. Rev. 1989, 89, 689–712. 10.1021/cr00094a001. [DOI] [Google Scholar]
- Datta M.; Buglass A. J. Fast and Efficient Synthesis of Sulfinamides by the Oxidation of Sulfenamides Using Potassium Fluoride and m-Chloroperoxybenzoic Acid. Synth. Commun. 2012, 42, 1760–1769. 10.1080/00397911.2010.543748. [DOI] [Google Scholar]
- Ma L.; Chen S.; Li G.; Zhu J.; Wang Q.; Tang Z. Chiral Brønsted-Acid-Catalyzed Asymmetric Oxidation of Sulfenamide by Using H2O2: A Versatile Access to Sulfinamide and Sulfoxide with High Enantioselectivity. ACS Catal. 2019, 9, 1525–1530. 10.1021/acscatal.8b04850. [DOI] [Google Scholar]
- Honraedt A.; Caillot G.; Gras E. N-t-Butanesulfinyl Amide: An Optimised and Versatile Access from Readily Available Starting Materials. C. R. Chim. 2013, 16, 350–357. 10.1016/j.crci.2013.03.001. [DOI] [Google Scholar]
- Taniguchi N. Copper-Catalyzed Oxidative Synthesis of Sulfinamides Using Thiols or Disulfides with Amines. Eur. J. Org. Chem. 2016, 2016, 2157–2162. 10.1002/ejoc.201600091. [DOI] [Google Scholar]
- Davis F. A.; Reddy R. E.; Szewczyk J. M.; Reddy G. V.; Portonovo P. S.; Zhang H.; Fanelli D.; Zhou P.; Carroll P. J. Asymmetric Synthesis and Properties of Sulfinimines (Thiooxime S-Oxides). J. Org. Chem. 1997, 62, 2555–2563. 10.1021/jo970077e. [DOI] [PubMed] [Google Scholar]
- Colonna S.; Giovini R.; Montanari F. Stereochemistry of Nucleophilic Substitutions at the Sulphur Atom. The Absolute Configuration of Sulphinamides. Chem. Commun. 1968, 865–866. 10.1039/c19680000865. [DOI] [Google Scholar]
- Davis F. A.; Reddy R. E.; Szewczyk J. M.; Portonovo P. S. Asymmetric Synthesis of Sulfinimines: Chiral Ammonia Imine Synthons. Tetrahedron Lett. 1993, 34, 6229–6232. 10.1016/S0040-4039(00)73717-8. [DOI] [Google Scholar]
- Davis F. A.; Zhang Y.; Andemichael Y.; Fang T.; Fanelli D. L.; Zhang H. Improved Synthesis of Enantiopure Sulfinimines (Thiooxime S-Oxide) from p-Toluenesulfinamide and Aldehydes and Ketones. J. Org. Chem. 1999, 64, 1403–1406. 10.1021/jo9820622. [DOI] [Google Scholar]
- Han Z.; Krishnamurthy D.; Grover P.; Fang Q. K.; Senanayake C. H. Properly Designed Modular Asymmetric Synthesis for Enantiopure Sulfinamide Auxiliaries from N-Sulfonyl-1,2,3-oxathiazolidine-2-oxide Agents. J. Am. Chem. Soc. 2002, 124, 7880–7881. 10.1021/ja0200692. [DOI] [PubMed] [Google Scholar]
- Han Z.; Krishnamurthy D.; Grover P.; Fang Q. K.; Su X.; Wilkinson H. S.; Lu Z.-H.; Magiera D.; Senanayake C. H. Practical and Highly Stereoselective Technology for Preparation of Enantiopure Sulfoxides and Sulfinamides Utilizing Activated and Functionally Differentiated N-Sulfonyl-1,2,3-Oxathiazolidine-2-Oxide Derivatives. Tetrahedron 2005, 61, 6386–6408. 10.1016/j.tet.2005.03.122. [DOI] [Google Scholar]
- Qin Y.; Wang C.; Huang Z.; Xiao X.; Jiang Y. Synthesis of Enantiopure tert-Butanesulfinamide from tert-Butanesulfinyloxazolidinone. J. Org. Chem. 2004, 69, 8533–8536. 10.1021/jo048576k. [DOI] [PubMed] [Google Scholar]
- Liu G.; Cogan D. A.; Owens T. D.; Tang T. P.; Ellman J. A. The Synthesis of Enantiomerically Pure N-tert-Butanesulfinyl Imines (tert-Butanesulfinimines) by the Direct Condensation of tert-Butanesulfinamide with Aldehydes and Ketones. J. Org. Chem. 1999, 64, 1278–1284. 10.1021/jo982059i. [DOI] [Google Scholar]
- Cogan D. A.; Ellman J. A. The Asymmetric Synthesis of α,α-Dibranched Amines by the Trimethylaluminum Mediated 1,2-Addition of Organolithiums to tert-Butanesulfinyl Ketimines. J. Am. Chem. Soc. 1999, 121, 268–269. 10.1021/ja983217q. [DOI] [Google Scholar]
- Datta G. K.; Ellman J. A. Racemization Free Protocol for the Synthesis of N-tert-Butanesulfinyl Ketimines. J. Org. Chem. 2010, 75, 6283–6285. 10.1021/jo1011625. [DOI] [PubMed] [Google Scholar]
- Wakayama M.; Ellman J. A. Recycling the tert-Butanesulfinyl Group in the Synthesis of Amines Using tert-Butanesulfinamide. J. Org. Chem. 2009, 74, 2646–2650. 10.1021/jo9001883. [DOI] [PubMed] [Google Scholar]
- Borg G.; Cogan D. A.; Ellman J. A. One-Pot Asymmetric Reductive Amination of Ketones to Prepare tert-Butanesulfinyl Protected Amines. Tetrahedron Lett. 1999, 40, 6709–6712. 10.1016/S0040-4039(99)01351-9. [DOI] [Google Scholar]
- Tanuwidjaja J.; Peltier H. M.; Ellman J. A. One-Pot Asymmetric Synthesis of Either Diastereomer of tert-Butanesulfinyl-protected Amines from Ketones. J. Org. Chem. 2007, 72, 626–629. 10.1021/jo0616512. [DOI] [PubMed] [Google Scholar]
- Cogan D. A.; Liu G.; Ellman J. A. Asymmetric Synthesis of Chiral Amines by Highly Diastereoselective 1,2-Additions of Organometallic Reagents to N-tert-Butanesulfinyl Imines. Tetrahedron 1999, 55, 8883–8904. 10.1016/S0040-4020(99)00451-2. [DOI] [Google Scholar]
- Tang T. P.; Ellman J. A. The tert-Butanesulfinyl Group: An Ideal Chiral Directing Group and Boc-Surrogate for Asymmetric β-Amino Acid Synthesis and Applications. J. Org. Chem. 1999, 64, 12–13. 10.1021/jo9820824. [DOI] [PubMed] [Google Scholar]
- Tang T. P.; Ellman J. A. Asymmetric Synthesis of β-Amino Acid Derivatives Incorporating a Broad Range of Substitution Patterns by Enolate Additions to tert-Butanesulfinyl Imines. J. Org. Chem. 2002, 67, 7819–7832. 10.1021/jo025957u. [DOI] [PubMed] [Google Scholar]
- Beenen M. A.; Weix D. J.; Ellman J. A. Asymmetric Synthesis of Protected Arylglycines by Rhodium-Catalyzed Addition of Arylboronic Acids to N-tert-Butanesulfinyl Imino Esters. J. Am. Chem. Soc. 2006, 128, 6304–6305. 10.1021/ja060529h. [DOI] [PubMed] [Google Scholar]
- Tang T. P.; Volkman S. K.; Ellman J. A. Asymmetric Synthesis of 1,2-Amino Alcohols Using tert-Butanesulfinyl Aldimines and Ketimines. J. Org. Chem. 2001, 66, 8772–8778. 10.1021/jo0156868. [DOI] [PubMed] [Google Scholar]
- Evans J. W.; Ellman J. A. Stereoselective Synthesis of 1,2-Disubstituted β-Amino Alcohols by Nucleophilic Addition to N-tert-Butanesulfinyl α-Alkoxyaldimines. J. Org. Chem. 2003, 68, 9948–9957. 10.1021/jo035224p. [DOI] [PubMed] [Google Scholar]
- Kochi T.; Tang T. P.; Ellman J. A. Development and Application of a New General Method for the Asymmetric Synthesis of syn- and anti-1,3-Amino Alcohols. J. Am. Chem. Soc. 2003, 125, 11276–11282. 10.1021/ja0363462. [DOI] [PubMed] [Google Scholar]
- Brinner K. M.; Ellman J. A. A Rapid and General Method for the Asymmetric Synthesis of 2-Substituted Pyrrolidines Using tert-Butanesulfinamide. Org. Biomol. Chem. 2005, 3, 2109–2113. 10.1039/b502080h. [DOI] [PubMed] [Google Scholar]
- Peltier H. M.; Ellman J. A. N-Sulfinyl Metalloenamine Conjugate Additions: Asymmetric Synthesis of Piperidines. J. Org. Chem. 2005, 70, 7342–7345. 10.1021/jo051020s. [DOI] [PubMed] [Google Scholar]
- Patterson A. W.; Ellman J. A. Asymmetric Synthesis of α,α-Dibranched Propargylamines by Acetylide Additions to N-tert-Butanesulfinyl Ketimines. J. Org. Chem. 2006, 71, 7110–7112. 10.1021/jo061160h. [DOI] [PubMed] [Google Scholar]
- Brak K.; Ellman J. A. Asymmetric Synthesis of α–Branched Allylic Amines by the Rh(I)-Catalyzed Addition of Alkenyltrifluoroborates to N-tert-Butanesulfinyl Aldimines. J. Am. Chem. Soc. 2009, 131, 3850–3851. 10.1021/ja9002603. [DOI] [PubMed] [Google Scholar]
- Tanuwidjaja J.; Peltier H. M.; Lewis J. C.; Schenkel L. B.; Ellman J. A. One-Pot Microwave-Promoted Synthesis of Nitriles from Aldehydes via tert-Butanesulfinyl Imines. Synthesis 2007, 2007, 3385–3389. 10.1055/s-2007-990805. [DOI] [Google Scholar]
- Khan H. A.; Ellman J. A. Asymmetric Synthesis of α-Aminophosphonate Esters by the Addition of Dialkyl Phosphites to tert-Butanesulfinyl Imines. Synthesis 2013, 45, 3147–3150. 10.1055/s-0033-1339712. [DOI] [Google Scholar]
- Beenen M. A.; An C.; Ellman J. A. Asymmetric Copper-Catalyzed Synthesis of alpha-Amino Boronate Esters from N-tert-Butanesulfinyl Aldimines. J. Am. Chem. Soc. 2008, 130, 6910–6911. 10.1021/ja800829y. [DOI] [PubMed] [Google Scholar]
- Kochi T.; Ellman J. A. Asymmetric α-Alkylation of N’-tert-Butanesulfinyl Amidines. Application to the Total Synthesis of (6R,7S)-7-Amino-7,8-dihydro-α-bisabolene. J. Am. Chem. Soc. 2004, 126, 15652–15653. 10.1021/ja044753n. [DOI] [PubMed] [Google Scholar]
- Brak K.; Ellman J. A. Total Synthesis of (−)-Aurantioclavine. Org. Lett. 2010, 12, 2004–2007. 10.1021/ol100470g. [DOI] [PubMed] [Google Scholar]
- Robak M. T.; Trincado M.; Ellman J. A. Enantioselective Aza-Henry Reaction with an N-Sulfinyl Urea Organocatalyst. J. Am. Chem. Soc. 2007, 129, 15110–15111. 10.1021/ja075653v. [DOI] [PubMed] [Google Scholar]
- Kimmel K. L.; Robak M. T.; Ellman J. A. Enantioselective Addition of Thioacetic Acid to Nitroalkenes via N-Sulfinyl Urea Organocatalysis. J. Am. Chem. Soc. 2009, 131, 8754–8755. 10.1021/ja903351a. [DOI] [PubMed] [Google Scholar]
- Kimmel K. L.; Robak M. T.; Thomas S.; Lee M.; Ellman J. A. Enantio- and Diastereoselective Addition of Thioacetic Acid to Nitroalkenes via N-Sulfinyl Urea Catalysis. Tetrahedron 2012, 68, 2704–2712. 10.1016/j.tet.2012.01.048. [DOI] [Google Scholar]
- Kimmel K. L.; Weaver J. D.; Ellman J. A. Enantio- and Diastereoselective Addition of Cyclohexyl Meldrum’s Acid to β- and α,β-Disubstituted Nitroalkenes via N-Sulfinyl Urea Catalysis. Chem. Sci. 2012, 3, 121–125. 10.1039/C1SC00441G. [DOI] [Google Scholar]
- Kimmel K. L.; Weaver J. D.; Lee M.; Ellman J. A. Catalytic Enantioselective Protonation of Nitronates Utilizing an Organocatalyst Chiral Only at Sulfur. J. Am. Chem. Soc. 2012, 134, 9058–9061. corrigendum J. Am. Chem. Soc. 2012, 134, 11828. 10.1021/ja3026196. [DOI] [PubMed] [Google Scholar]
- Phelan J. P.; Ellman J. A. Catalytic Enantioselective Addition of Pyrazol-5-ones to Trisubstituted Nitroalkenes with an N-Sulfinylurea Organocatalyst. Adv. Synth. Catal. 2016, 358, 1713–1718. 10.1002/adsc.201600110. [DOI] [Google Scholar]
- Robak M. T.; Herbage M. A.; Ellman J. A. Development of an N-Sulfinyl Prolinamide for the Asymmetric Aldol Reaction. Tetrahedron 2011, 67, 4412–4416. 10.1016/j.tet.2011.03.030. [DOI] [Google Scholar]
- Owens T. D.; Souers A. J.; Ellman J. A. The Preparation and Utility of Bis(sulfinyl)imidoamidine Ligands for the Copper-Catalyzed Diels Alder Reaction. J. Org. Chem. 2003, 68, 3–10. 10.1021/jo020524c. [DOI] [PubMed] [Google Scholar]
- Schenkel L. B.; Ellman J. A. Novel Sulfinyl Imine Ligands for Asymmetric Catalysis. Org. Lett. 2003, 5, 545–548. 10.1021/ol027468m. [DOI] [PubMed] [Google Scholar]
- Schenkel L. B.; Ellman J. A. Application of P,N-Sulfinyl Imine Ligands to Iridium-Catalyzed Asymmetric Hydrogenation of Olefins. J. Org. Chem. 2004, 69, 1800–1802. 10.1021/jo035675+. [DOI] [PubMed] [Google Scholar]
- Wangweerawong A.; Hummel J. R.; Bergman R. G.; Ellman J. A. Preparation of Enantiomerically Pure Perfluorobutanesulfinamide and Its Application to the Asymmetric Synthesis of α-Amino Acids. J. Org. Chem. 2016, 81, 1547–1557. 10.1021/acs.joc.5b02700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangweerawong A.; Kolmar S.; Ellman J. A. Preparation of (S)-Nonafluorobutanesulfinamide. Org. Synth. 2016, 93, 319–330. 10.15227/orgsyn.093.0319. [DOI] [Google Scholar]
- Wangweerawong A.; Bergman R. G.; Ellman J. A. Asymmetric Synthesis of α-Branched Amines via Rh(III)-Catalyzed C–H Bond Functionalization. J. Am. Chem. Soc. 2014, 136, 8520–8523. 10.1021/ja5033452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pindi S.; Wu J.; Li G. Design, Synthesis, and Applications of Chiral N-2-Phenyl-2-propyl Sulfinyl Imines for Group-Assisted Purification (GAP) Asymmetric Synthesis. J. Org. Chem. 2013, 78, 4006–4012. 10.1021/jo400354r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R.-H.; Shi X.-X. Practical and Highly Stereoselective Method for the Preparation of Several Chiral Arylsulfinamides and Arylsulfinates Based on the Spontaneous Crystallization of Diastereomerically Pure N-Benzyl-N-(1-phenylethyl)-arylsulfinamides. Tetrahedron: Asymmetry 2011, 22, 387–393. 10.1016/j.tetasy.2011.01.028. [DOI] [Google Scholar]
- Harmata M.; Zheng P.; Huang C.; Gomes M. G.; Ying W.; Ranyanil K.-O.; Balan G.; Calkins N. L. An Expedient Synthesis of Sulfinamides from Sulfonyl Chlorides. J. Org. Chem. 2007, 72, 683–685. corrigendum: J. Org. Chem. 2007, 72, 4586. 10.1021/jo062296i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamińska K.; Wojaczyńska E.; Skarżewski J.; Kochel A.; Wojaczyński J. Application of Sulfonyl Chlorides and Chiral Amines in the Efficient Synthesis of Nonracemic Sulfinamides. Tetrahedron: Asymmetry 2017, 28, 561–566. 10.1016/j.tetasy.2017.03.004. [DOI] [Google Scholar]
- Yu H.; Li Z.; Bolm C. Copper-Catalyzed Transsulfinamidation of Sulfinamides as a Key Step in the Preparation of Sulfonamides and Sulfonimidamides. Angew. Chem., Int. Ed. 2018, 57, 15602–15605. 10.1002/anie.201810548. [DOI] [PubMed] [Google Scholar]
- Wei J.; Sun Z. tert-Butyl Sulfoxide as a Starting Point for the Synthesis of Sulfinyl Containing Compounds. Org. Lett. 2015, 17, 5396–5399. 10.1021/acs.orglett.5b02743. [DOI] [PubMed] [Google Scholar]
- Schroeck C. W.; Johnson C. R. Chemistry of Sulfoxides and Related Compounds. XXXI. Aluminum Amalgam Reduction of Aryl Sulfoximines and Related Compounds. J. Am. Chem. Soc. 1971, 93, 5305–5306. 10.1021/ja00749a083. [DOI] [Google Scholar]
- Tsujihara K.; Furukawa N.; Oae S. Pyrolysis of N-p-Toluenesulphonylsulphilimines. Tetrahedron 1971, 27, 4921–4930. 10.1016/S0040-4020(01)98197-9. [DOI] [Google Scholar]
- Williams T. R.; Booms R. E.; Cram D. J. Three New Organosulfur Reactions and the First Example of a Monoligostatic Stereochemical Cycle. J. Am. Chem. Soc. 1971, 93, 7338–7340. 10.1021/ja00755a048. [DOI] [Google Scholar]
- Savile C. K.; Magloire V. P.; Kazlauskas R. J. Subtilisin-Catalyzed Resolution of N-Acyl Arylsulfinamides. J. Am. Chem. Soc. 2005, 127, 2104–2113. 10.1021/ja045397b. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wang Z.; Guo B.; Cai Q. Asymmetric Synthesis of N-Aryl Sulfinamides: Copper(I)-Catalyzed Coupling of Sulfinamides with Aryl Iodides via Kinetic Resolution. Tetrahedron Lett. 2016, 57, 2379–2381. 10.1016/j.tetlet.2016.04.049. [DOI] [Google Scholar]
- Dai Q.; Zhang J. Direct Synthesis of Sulfinamides by the Copper-Catalyzed Electrophilic Amidation of Sulfenate Anions. Adv. Synth. Catal. 2018, 360, 1123–1127. 10.1002/adsc.201701510. [DOI] [Google Scholar]
- Wang Q.; Tang X.-Y.; Shi M. Metal-Free Cross-Coupling of Arylboronic Acids and Derivatives with DAST-Type Reagents for Direct Access to Diverse Aromatic Sulfinamides and Sulfonamides. Angew. Chem., Int. Ed. 2016, 55, 10811–10815. 10.1002/anie.201605066. [DOI] [PubMed] [Google Scholar]
- Bentley H. R.; Booth R. G.; Greer E. N.; Heathcote J. G.; Hutchinson J. B.; Mohan T. Action of Nitrogen Trichloride on Proteins: Production of Toxic Derivative. Nature 1948, 161, 126–127. 10.1038/161126b0. [DOI] [PubMed] [Google Scholar]
- Bentley H. R.; McDermott E. E.; Pace J.; Whitehead J. K.; Moran T. Action of Nitrogen Trichloride (’Agene’) on Proteins: Isolation of Crystalline Toxic Factor. Nature 1949, 164, 438–439. 10.1038/164438a0. [DOI] [PubMed] [Google Scholar]
- Bentley H. R.; McDermott E. E.; Pace J.; Whitehead J. K.; Moran T. Action of Nitrogen Trichloride on Proteins: Progress in the Isolation of the Toxic Factor. Nature 1949, 163, 675–676. 10.1038/163675a0. [DOI] [PubMed] [Google Scholar]
- Bentley H. R.; McDermott E. E.; Pace J.; Whitehead J. K.; Moran T. Toxic Factor form “Agenized” Proteins: Methionine as the Essential Reactant. Nature 1950, 165, 150–151. 10.1038/165150a0. [DOI] [PubMed] [Google Scholar]
- Bentley H. R.; McDermott E. E.; Whitehead J. K. Action of Nitrogen Trichloride on Proteins: a Synthesis of the Toxic Factor from Methionine. Nature 1950, 165, 735–735. 10.1038/165735b0. [DOI] [PubMed] [Google Scholar]
- Bentley H. R.; Whitehead J. K. Dimethylsulphoximine. J. Chem. Soc. 1950, 2081–2082. 10.1039/jr9500002081. [DOI] [Google Scholar]
- Bizet V.; Kowalczyk R.; Bolm C. Fluorinated Sulfoximines: Syntheses, Properties and Applications. Chem. Soc. Rev. 2014, 43, 2426–2438. 10.1039/c3cs60427f. [DOI] [PubMed] [Google Scholar]
- Bizet V.; Hendriks C. M. M.; Bolm C. Sulfur Imidations: Access to Sulfimides and Sulfoximines. Chem. Soc. Rev. 2015, 44, 3378–3390. 10.1039/C5CS00208G. [DOI] [PubMed] [Google Scholar]
- Frings M.; Bolm C.; Blum A.; Gnamm C. Sulfoximines from a Medicinal Chemist’s Perspective: Physicochemical and in vitro Parameters Relevant for Drug Discovery. Eur. J. Med. Chem. 2017, 126, 225–245. 10.1016/j.ejmech.2016.09.091. [DOI] [PubMed] [Google Scholar]
- Lücking U. Sulfoximines: A Neglected Opportunity in Medicinal Chemistry. Angew. Chem., Int. Ed. 2013, 52, 9399–9408. 10.1002/anie.201302209. [DOI] [PubMed] [Google Scholar]
- Sirvent J. A.; Lücking U. Novel Pieces for the Emerging Picture of Sulfoximines in Drug Discovery: Synthesis and Evaluation of Sulfoximine Analogues of Marketed Drugs and Advanced Clinical Candidates. ChemMedChem 2017, 12, 487–501. 10.1002/cmdc.201700044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmata M.; Hong X. Recent Progress in the Chemistry of 2,1-Benzothiazines. Prog. Heterocycl. Chem. 2008, 19, 1–43. 10.1016/S0959-6380(08)80003-7. [DOI] [Google Scholar]
- Bull J. A.; Degennaro L.; Luisi R. Straightforward Strategies for the Preparation of NH-Sulfoximines: A Serendipitous Story. Synlett 2017, 28, 2525–2538. 10.1055/s-0036-1590874. [DOI] [Google Scholar]
- Wang Y.; Hong X.; Deng Z. Progress in the Synthesis and Application of Sulfoximines. Youji Huaxue 2012, 32, 825–833. 10.6023/cjoc1110053. [DOI] [Google Scholar]
- Zhou H.; Chen Z. Recent Achievements in the Synthesis of Sulfoximine Derivatives. Youji Huaxue 2018, 38, 719–737. 10.6023/cjoc201710002. [DOI] [Google Scholar]
- Reggelin M.; Zur C. Sulfoximines: Structures, Properties and Synthetic Applications. Synthesis 2000, 2000, 1–64. 10.1055/s-2000-6217. [DOI] [Google Scholar]
- Brandt J.; Gais H.-J. An efficient resolution of (±)-S-Methyl-S-phenylsulfoximine with (+)-10-Camphorsulfonic Acid by the Method of Half-Quantities. Tetrahedron: Asymmetry 1997, 8, 909–912. 10.1016/S0957-4166(97)00046-3. [DOI] [Google Scholar]
- Gilchrist T. L.; Moody C. J. The Chemistry of Sulfilimines. Chem. Rev. 1977, 77, 409–435. 10.1021/cr60307a005. [DOI] [Google Scholar]
- Nishikori H.; Ohta C.; Oberlin E.; Irie R.; Katsuki T. Mn-Salen Catalyzed Nitrene Transfer Reaction: Enantioselective Imidation of Alkyl Aryl Sulfides. Tetrahedron 1999, 55, 13937–13946. 10.1016/S0040-4020(99)00853-4. [DOI] [Google Scholar]
- Kresze G.; Wustrow B. Stereochemie der Oxydation am S-Atom von Sulfiminen. Ein Optisch-Aktives Derivat des Vierbindigen Schwefels. Chem. Ber. 1962, 95, 2652–2659. 10.1002/cber.19620951110. [DOI] [Google Scholar]
- Johnson C. R.; Rigau J. J. Sulfilimines and Sulfoximines Derived from 4-t-Butylthiane. J. Org. Chem. 1968, 33, 4340–4343. 10.1021/jo01276a007. [DOI] [Google Scholar]
- Macé Y.; Urban C.; Pradet C.; Marrot J.; Blazejewski J.-C.; Magnier E. Sulfilimines and Sulfoximines by Reaction of Nitriles with Perfluoroalkyl Sulfoxides. Eur. J. Org. Chem. 2009, 2009, 3150–3153. 10.1002/ejoc.200900410. [DOI] [Google Scholar]
- Arndt K. E.; Bland D. C.; Irvine N. M.; Powers S. L.; Martin T. P.; McConnell J. R.; Podhorez D. E.; Renga J. M.; Ross R.; Roth G. A.; et al. Development of a Scalable Process for the Crop Protection Agent Isoclast. Org. Process Res. Dev. 2015, 19, 454–462. 10.1021/acs.oprd.5b00007. [DOI] [Google Scholar]
- Veale H. S.; Levin J.; Swern D. New Method of Preparation of Acyl- and Sulfonylsulfoximines. Ruthenium Tetroxide Oxidation of Sulfilimines. Tetrahedron Lett. 1978, 19, 503–506. 10.1016/S0040-4039(01)85317-X. [DOI] [Google Scholar]
- Swern D.; Veale H. S.; Huang S. L.; Heintzelman R. W. Novel Methods for the Preparation of Sulfoximines. Phosphorus Sulfur Relat. Elem. 1979, 6, 295–296. 10.1080/03086647908080421. [DOI] [Google Scholar]
- Garcia Mancheño O.; Bistri O.; Bolm C. Iodinane- and Metal-Free Synthesis of N-Cyano Sulfilimines: Novel and Easy Access of NH-Sulfoximines. Org. Lett. 2007, 9, 3809–3811. 10.1021/ol7016577. [DOI] [PubMed] [Google Scholar]
- Wang J.; Frings M.; Bolm C. Enantioselective Nitrene Transfer to Sulfides Catalyzed by a Chiral Iron Complex. Angew. Chem., Int. Ed. 2013, 52, 8661–8665. 10.1002/anie.201304451. [DOI] [PubMed] [Google Scholar]
- Lebel H.; Piras H.; Bartholoméüs J. Rhodium-Catalyzed Stereoselective Amination of Thioethers with N-Mesyloxycarbamates: DMAP and Bis(DMAP)CH2Cl2 as Key Additives. Angew. Chem., Int. Ed. 2014, 53, 7300–7304. 10.1002/anie.201402961. [DOI] [PubMed] [Google Scholar]
- Lebel H.; Piras H. Stereoselective Synthesis of Chiral Sulfilimines from N-Mesyloxycarbamates: Metal-Nitrenes versus Metal-Nitrenoids Species. J. Org. Chem. 2015, 80, 3572–3585. 10.1021/acs.joc.5b00256. [DOI] [PubMed] [Google Scholar]
- Lai C.; Mathieu G.; Gabrielli Tabarez L. P.; Lebel H. Batch and Continuous-Flow Iron(II)-Catalyzed Synthesis of Sulfilimines and Sulfoximines using N-Mesyloxycarbamates. Chem. - Eur. J. 2019, 25, 9423–9426. 10.1002/chem.201900953. [DOI] [PubMed] [Google Scholar]
- Akasaka T.; Yoshimura T.; Furukawa N.; Oae S. Stereochemistry of the Alkaline Hydrolysis of N-Chlorosulfimides to Sulfoximides. Chem. Lett. 1978, 7, 417–420. 10.1246/cl.1978.417. [DOI] [Google Scholar]
- Furukawa N.; Akutagawa K.; Oae S. Convenient Preparation and Spectroscopic Studies of Sulfoximines and Sulfonediimines: Key Intermediate N-Chlorosulfilimine. Phosphorus Sulfur Relat. Elem. 1984, 20, 1–14. 10.1080/03086648408077606. [DOI] [Google Scholar]
- Wang J.; Frings M.; Bolm C. Iron-Catalyzed Imidative Kinetic Resolutions of Racemic Sulfoxides. Chem. - Eur. J. 2014, 20, 966–969. 10.1002/chem.201303850. [DOI] [PubMed] [Google Scholar]
- Dannenberg C. A.; Fritze L.; Krauskopf F.; Bolm C. Access to N-Cyanosulfoximines by Transition Metal-Free Iminations of Sulfoxides. Org. Biomol. Chem. 2017, 15, 1086–1090. 10.1039/C6OB02691E. [DOI] [PubMed] [Google Scholar]
- Yu H.; Li Z.; Bolm C. Iron(II)-Catalyzed Direct Synthesis of NH Sulfoximines from Sulfoxides. Angew. Chem., Int. Ed. 2018, 57, 324–327. 10.1002/anie.201710498. [DOI] [PubMed] [Google Scholar]
- Gutmann B.; Elsner P.; O’Kearney-McMullan A.; Goundry W.; Roberge D. M.; Kappe C. O. Development of a Continuous Flow Sulfoxide Imidation Protocol Using Azide Sources under Superacidic Conditions. Org. Process Res. Dev. 2015, 19, 1062–1067. 10.1021/acs.oprd.5b00217. [DOI] [Google Scholar]
- Zenzola M.; Doran R.; Luisi R.; Bull J. A. Synthesis of Sulfoximine Carbamates by Rhodium-Catalyzed Nitrene Transfer of Carbamates to Sulfoxides. J. Org. Chem. 2015, 80, 6391–6399. 10.1021/acs.joc.5b00844. [DOI] [PubMed] [Google Scholar]
- Zenzola M.; Doran R.; Degennaro L.; Luisi R.; Bull J. A. Transfer of Electrophilic NH Using Convenient Sources of Ammonia: Direct Synthesis of Sulfoximines from Sulfoxides. Angew. Chem., Int. Ed. 2016, 55, 7203–7207. 10.1002/anie.201602320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tota A.; Zenzola M.; Chawner S. J.; St John-Campbell S.; Carlucci C.; Romanazzi G.; Degennaro L.; Bull J. A.; Luisi R. Synthesis of NH-Sulfoximines from Sulfides by Chemoselective One-Pot N- and O-Transfers. Chem. Commun. 2017, 53, 348–351. 10.1039/C6CC08891K. [DOI] [PubMed] [Google Scholar]
- Tota A.; St John-Campbell S.; Briggs E. L.; Estévez G. O.; Afonso M.; Degennaro L.; Luisi R.; Bull J. A. Highly Chemoselective NH- and O-Transfer to Thiols Using Hypervalent Iodine Reagents: Synthesis of Sulfonimidates and Sulfonamides. Org. Lett. 2018, 20, 2599–2602. 10.1021/acs.orglett.8b00788. [DOI] [PubMed] [Google Scholar]
- Degennaro L.; Tota A.; De Angelis S.; Andresini M.; Cardellicchio C.; Capozzi M. A.; Romanazzi G.; Luisi R. A Convenient, Mild, and Green Synthesis of NH-Sulfoximines in Flow Reactors. Eur. J. Org. Chem. 2017, 2017, 6486–6490. 10.1002/ejoc.201700850. [DOI] [Google Scholar]
- Reggelin M.; Weinberger H. One-Pot Synthesis of (S)-4-Isopropyl-2-p-toluene-4,5-dihydro-[1.2λ6,3]oxathiazole 2-Oxides: Efficient Precursors of Optically Active Sulfoximines. Tetrahedron Lett. 1992, 33, 6959–6962. 10.1016/S0040-4039(00)60906-1. [DOI] [Google Scholar]
- Reggelin M.; Mehler C.; Kaiser J. P. Synthesis of Bis(4-methylphenylsulfonimidoyl)methane – The First ‘Free’ Geminal Bis(sulfoximine). Synlett 2012, 23, 1095–1098. 10.1055/s-0031-1290759. [DOI] [Google Scholar]
- Kühl J.; Reggelin M. 3-Oxo-1,3λ6,4-oxathiazines: A Novel Class of Heterocyclic S,O-Acetals. Synthesis 2017, 49, 403–408. 10.1055/s-0036-1588679. [DOI] [Google Scholar]
- Matos P. M.; Lewis W.; Moore J. C.; Stockman R. A. Sulfonimidates: Useful Synthetic Intermediates for Sulfoximine Synthesis via C–S Bond Formation. Org. Lett. 2018, 20, 3674–3677. 10.1021/acs.orglett.8b01473. [DOI] [PubMed] [Google Scholar]
- Harmata M.; Schlemper E. O. Lewis Acid Mediated Reactions of N-Phenyl-(4-Methylphenyl)-Sulfoximidoyl Chloride with Alkynes. A Novel Route to Benzothiazines. Tetrahedron Lett. 1987, 28, 5997–6000. 10.1016/S0040-4039(00)96846-1. [DOI] [Google Scholar]
- Harmata M.; Claassen R. J. II; Barnes C. L. Lewis Acid Mediated Reactions of N-Phenyl-(4-Methylphenyl)-Sulfoximidoyl Chloride with Alkenes. J. Org. Chem. 1991, 56, 5059–5062. 10.1021/jo00017a015. [DOI] [Google Scholar]
- Harmata M.; Kahraman M. Lewis Acid Mediated Reaction of N-Aryl Sulfonimidoyl Chlorides with Alkenes. Some Steric Effects of Alkene Substitution. J. Org. Chem. 1998, 63, 6845–6851. 10.1021/jo980503b. [DOI] [PubMed] [Google Scholar]
- Kowalczyk R.; Edmunds A. J. F.; Hall R. G.; Bolm C. Synthesis of CF3-Substituted Sulfoximines from Sulfonimidoyl Fluorides. Org. Lett. 2011, 13, 768–771. 10.1021/ol103030w. [DOI] [PubMed] [Google Scholar]
- Furusho Y.; Okada Y.; Takata T. Synthesis of Diarylsulfoximines by the Friedel-Crafts Reaction of Sulfonimidoyl Chlorides. Bull. Chem. Soc. Jpn. 2000, 73, 2827–2828. 10.1246/bcsj.73.2827. [DOI] [Google Scholar]
- Ye W.; Zhang L.; Ni C.; Rong J.; Hu J. Stereoselective [3 + 2] Cycloaddition of N-tert-Butanesulfinyl Imines to Arynes Facilitated by a Removable PhSO2CF2 Group: Synthesis and Transformation of Cyclic Sulfoximines. Chem. Commun. 2014, 50, 10596–10599. 10.1039/C4CC05042H. [DOI] [PubMed] [Google Scholar]
- Moragas T.; Liffey R. M.; Regentová D.; Ward J.-P. S.; Dutton J.; Lewis W.; Churcher I.; Walton L.; Souto J. A.; Stockman R. A. Sigmatropic Rearrangement of Vinyl Aziridines: Expedient Synthesis of Cyclic Sulfoximines from Chiral Sulfinimines. Angew. Chem., Int. Ed. 2016, 55, 10047–10051. 10.1002/anie.201604188. [DOI] [PubMed] [Google Scholar]
- Aota Y.; Kano T.; Maruoka K. Asymmetric Synthesis of Chiral Sulfoximines through the S-Alkylation of Sulfinamides. Angew. Chem., Int. Ed. 2019, 58, 17661–17665. 10.1002/anie.201911021. [DOI] [PubMed] [Google Scholar]
- Aota Y.; Kano T.; Maruoka K. Asymmetric Synthesis of Chiral Sulfoximines via the S-Arylation of Sulfinamides. J. Am. Chem. Soc. 2019, 141, 19263–19268. 10.1021/jacs.9b11298. [DOI] [PubMed] [Google Scholar]
- Aota Y.; Maeda Y.; Kano T.; Maruoka K. Efficient Synthesis of Cyclic Sulfoximines from N-Propargylsulfinamides through Sulfur–Carbon Bond Formation. Chem. - Eur. J. 2019, 25, 15755–15758. 10.1002/chem.201904501. [DOI] [PubMed] [Google Scholar]
- Lohier J.-F.; Glachet T.; Marzag H.; Gaumont A.-C.; Reboul V. Mechanistic Investigation of the NH-Sulfoximination of Sulfide. Evidence for λ6-Sulfanenitrile Intermediates. Chem. Commun. 2017, 53, 2064–2067. 10.1039/C6CC09940H. [DOI] [PubMed] [Google Scholar]
- Chaabouni S.; Lohier J.-F.; Barthelemy A. L.; Glachet T.; Anselmi E.; Dagousset G.; Diter P.; Pégot B.; Magnier E.; Reboul V. One-Pot Synthesis of Aryl- and Alkyl S-Perfluoroalkylated NH-Sulfoximines from Sulfides. Chem. - Eur. J. 2018, 24, 17006–17010. 10.1002/chem.201805055. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Zhou B.; Zhou S.; Zhou S.; Wei W.; Liu J.; Zhan Y.; Cheng D.; Chen M.; Li Y.; et al. Sulfimine-Promoted Fast O Transfer: One–step Synthesis of Sulfoximine from Sulfide. ChemistrySelect 2017, 2, 1620–1624. 10.1002/slct.201700132. [DOI] [Google Scholar]
- Glachet T.; Franck X.; Reboul V. Late-Stage Sulfoximination: Improved Synthesis of the Anticancer Drug Candidate Atuveciclib. Synthesis 2019, 51, 971–975. 10.1055/s-0037-1610316. [DOI] [Google Scholar]
- McGrath M. J.; Bolm C. The Use of Chiral Lithium Amides in the Desymmetrisation of N-Trialkylsilyl Dimethyl Sulfoximines. Beilstein J. Org. Chem. 2007, 3, 33. 10.1186/1860-5397-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A. G.; McGrath M. J.; García Mancheño O.; Bolm C. Asymmetric Synthesis of S,S-Dialkyl-Substituted Sulfoximines and Related Heterocycles. Synthesis 2011, 2011, 3827–3828. 10.1055/s-0030-1260260. [DOI] [Google Scholar]
- Dong S.; Frings M.; Cheng H.; Wen J.; Zhang D.; Raabe G.; Bolm C. Organocatalytic Kinetic Resolution of Sulfoximines. J. Am. Chem. Soc. 2016, 138, 2166–2169. 10.1021/jacs.6b00143. [DOI] [PubMed] [Google Scholar]
- Shen B.; Wan B.; Li X. Enantiodivergent Desymmetrization in the Rhodium(III)-Catalyzed Annulation of Sulfoximines with Diazo Compounds. Angew. Chem., Int. Ed. 2018, 57, 15534–15538. 10.1002/anie.201810472. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Cramer N. Enantioselective Synthesis of Chiral-at-Sulfur 1,2-Benzothiazines by CpxRhIII-Catalyzed C-H Functionalization of Sulfoximines. Angew. Chem., Int. Ed. 2018, 57, 15539–15543. 10.1002/anie.201810887. [DOI] [PubMed] [Google Scholar]
- Brauns M.; Cramer N. Efficient Kinetic Resolution of Sulfur-Stereogenic Sulfoximines by Exploiting CpXRhIII-Catalyzed C-H Functionalization. Angew. Chem., Int. Ed. 2019, 58, 8902–8906. 10.1002/anie.201904543. [DOI] [PubMed] [Google Scholar]
- Bolm C.; Kahmann J. D.; Moll G. Sulfoximines in Pseudopeptides. Tetrahedron Lett. 1997, 38, 1169–1172. 10.1016/S0040-4039(97)00001-4. [DOI] [Google Scholar]
- Bolm C.; Moll G.; Kahmann J. D. Synthesis of Pseudopeptides with Sulfoximines as Chiral Backbone Modifying Elements. Chem. - Eur. J. 2001, 7, 1118–1128. 10.1002/1521-3765(20010302)7:5<1118::AID-CHEM1118>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Bolm C.; Müller D.; Dalhoff C.; Hackenberger C. P. R.; Weinhold E. The Stability of Pseudopeptides Bearing Sulfoximines as Chiral Backbone Modifying Element towards Proteinase K. Bioorg. Med. Chem. Lett. 2003, 13, 3207–3211. 10.1016/S0960-894X(03)00697-8. [DOI] [PubMed] [Google Scholar]
- Bolm C.; Hildebrand J. P. Palladium-Catalyzed Carbon-Nitrogen Bond Formation: A Novel, Catalytic Approach towards N-Arylated Sulfoximines. Tetrahedron Lett. 1998, 39, 5731–5734. 10.1016/S0040-4039(98)01199-X. [DOI] [Google Scholar]
- Bolm C.; Hildebrand J. P. Palladium-Catalyzed N-Arylation of Sulfoximines with Aryl Bromides and Aryl Iodides. J. Org. Chem. 2000, 65, 169–175. 10.1021/jo991342u. [DOI] [PubMed] [Google Scholar]
- Langner M.; Rémy P.; Bolm C. Highly Modular Synthesis of C1-Symmetric Aminosulfoximines and their Use as Ligands in Copper-Catalyzed Asymmetric Mukaiyama-Aldol Reactions. Chem. - Eur. J. 2005, 11, 6254–6265. 10.1002/chem.200500497. [DOI] [PubMed] [Google Scholar]
- Benetskiy E. B.; Bolm C. Synthesis of Phosphorylated Sulfoximines and Sulfinamides and Their Application as Ligands in Asymmetric Metal Catalysis. Tetrahedron: Asymmetry 2011, 22, 373–378. 10.1016/j.tetasy.2011.02.005. [DOI] [Google Scholar]
- Frings M.; Thomé I.; Bolm C. Synthesis of Chiral Sulfoximine-based Thioureas and their Application in Asymmetric Organocatalysis. Beilstein J. Org. Chem. 2012, 8, 1443–1451. 10.3762/bjoc.8.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolm C.; Hackenberger C. P. R.; Simić O.; Verrucci M.; Müller D.; Bienewald F. A Mild Synthetic Procedure for the Preparation of N-Alkylated Sulfoximines. Synthesis 2002, 2002, 879–887. 10.1055/s-2002-28514. [DOI] [Google Scholar]
- Wang H.; Zhang D.; Sheng H.; Bolm C. Sulfoximidoyl-Containing Hypervalent Iodine(III) Reagents: 1-Sulfoximidoyl-1,2-benziodoxoles. J. Org. Chem. 2017, 82, 11854–11858. 10.1021/acs.joc.7b01535. [DOI] [PubMed] [Google Scholar]
- Wang H.; Cheng Y.; Becker P.; Raabe G.; Bolm C. Synthesis of Sulfoximidoyl-Containing Hypervalent Iodine(III) Reagents and Their Use in Transition-Metal-Free Sulfoximidations of Alkynes. Angew. Chem., Int. Ed. 2016, 55, 12655–12658. 10.1002/anie.201605743. [DOI] [PubMed] [Google Scholar]
- Harmata M.; Pavri N. A One-Pot, One-Operation 3 + 3 Annulation Approach to Benzothiazines. Angew. Chem., Int. Ed. 1999, 38, 2419–2421. 10.1002/(SICI)1521-3773(19990816)38:16<2419::AID-ANIE2419>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Harmata M.; Hong X. The Intramolecular, Stereoselective Addition of Sulfoximine Carbanions to α,β- Unsaturated Esters. J. Am. Chem. Soc. 2003, 125, 5754–5756. 10.1021/ja034744z. [DOI] [PubMed] [Google Scholar]
- Harmata M.; Rayanil K.; Espejo V.; Barnes C. L. Benzothiazines in Synthesis. Further Studies of the Intramolecular, Stereoselective Addition of Sulfonimidoyl Carbanions to α,β-Unsaturated Functional Groups. J. Org. Chem. 2009, 74, 3214–3216. 10.1021/jo900151d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmata M.; Hong X.; Ghosh S. K. Microwave-Assisted N-Arylation of a Sulfoximine with Aryl Chlorides. Tetrahedron Lett. 2004, 45, 5233–5236. 10.1016/j.tetlet.2004.05.027. [DOI] [Google Scholar]
- Harmata M.; Hong X. Palladium-Catalyzed Cross-Coupling Reaction of a Sulfoximine with Aryl Dichlorides under Microwave Irradiation. Synlett 2007, 2007, 0969–0973. 10.1055/s-2007-973872. [DOI] [Google Scholar]
- Levchenko E. S.; Markovskii L. N.; Shermolovich Y. G. Chemistry of Sulfonimide Acids Derivatives. Russ. J. Org. Chem. 2000, 36, 143–157. [Google Scholar]
- Levchenko E. S.; Kirsanov A. V. Khlorangidridy N-Dikhlorfosfinilareniminosul’fokislot. Zh. Obshch. Khim. 1960, 30, 1553–1561. [Google Scholar]
- Jonsson E. U.; Bacon C. C.; Johnson C. R. Preparation of Sulfonimidoyl Chlorides by Chlorination of Sulfinamides. J. Am. Chem. Soc. 1971, 93, 5306–5308. 10.1021/ja00749a084. [DOI] [Google Scholar]
- Johnson C. R.; Jonsson E. U.; Bacon C. C. Preparation and Reactions of Sulfonimidoyl Chlorides. J. Org. Chem. 1979, 44, 2055–2061. 10.1021/jo01327a001. [DOI] [Google Scholar]
- Johnson C. R.; Wambsgans A. Sulfonimidoyl Chlorides by Oxidation of Sulfinamides with tert-Butyl Hypochlorite. J. Org. Chem. 1979, 44, 2278–2280. 10.1021/jo01327a051. [DOI] [Google Scholar]
- Jonsson E. U.; Johnson C. R. The Stereochemistry of Substitution at Tetracoordinate Hexavalent Sulfur. Nucleophilic Reactions at Sulfur in Sulfonimidoyl Compounds. J. Am. Chem. Soc. 1971, 93, 5308–5309. 10.1021/ja00749a085. [DOI] [Google Scholar]
- Roy A. Novel Synthesis of Sulfonimidoyl Halides and Sulfonimidates from N-Silylated Sulfonamides and Dihalophosphoranes. J. Am. Chem. Soc. 1993, 115, 2598–2603. 10.1021/ja00060a008. [DOI] [Google Scholar]
- Johnson C. R.; Bis K. G.; Cantillo J. H.; Meanwell N. A.; Reinhard M. F. D.; Zeller J. R.; Vonk G. P. Preparation and Reactions of Sulfonimidoyl Fluorides. J. Org. Chem. 1983, 48, 1–3. 10.1021/jo00149a001. [DOI] [Google Scholar]
- van Leusen D.; van Leusen A. M. Synthesis of Arylsulfonimidoyl Fluorides and Their Use in the Preparation of (Arylsulfonimidoyl)Methyl Isocyanides. Partial Resolution of Optically Active S-Phenyl- N-Tosylsulfonimidoyl Fluoride. Recl. Trav. Chim. Pays-Bas 1984, 103, 41–45. 10.1002/recl.19841030201. [DOI] [Google Scholar]
- Kolesnik N. P.; Rozhenko A. B.; Kinzhybalo V.; Lis T.; Shermolovich Y. G. 1-Oxo-1-fluoro-1,2,4-benzothiadiazines - A New Type of Cyclic Sulfonimidoyl Fluorides. J. Fluorine Chem. 2014, 160, 16–19. 10.1016/j.jfluchem.2014.01.008. [DOI] [Google Scholar]
- Gao B.; Li S.; Wu P.; Moses J. E.; Sharpless K. B. SuFEx Chemistry of Thionyl Tetrafluoride (SOF4) with Organolithium Nucleophiles: Synthesis of Sulfonimidoyl Fluorides, Sulfoximines, Sulfonimidamides, and Sulfonimidates. Angew. Chem., Int. Ed. 2018, 57, 1939–1943. 10.1002/anie.201712145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis B. C.; Iley J. N. Pseudomolecular Rearrangement of O-Ethyl N-Methyl Toluene-4-sulphonimidate to N-Ethyl-N-methyltoluene-4-sulfonamide and its Relevance to the Nucleophilic Properties of Neutral Sulphonamides. J. Chem. Soc., Perkin Trans. 2 1985, 2, 699–703. 10.1039/p29850000699. [DOI] [Google Scholar]
- Roy A. K.; Burns G. T.; Lie G. C.; Grigoras S. Poly(alkyl/aryloxothiazenes): Inorganic Polymers with a Sulfur(VI)-Nitrogen Backbone. Synthesis, Characterization, and Theoretical Calculations. J. Am. Chem. Soc. 1993, 115, 2604–2612. 10.1021/ja00060a009. [DOI] [Google Scholar]
- Reggelin M.; Welcker R. New Stereocontrolled Synthesis of Cyclic Sulfonimidates. Tetrahedron Lett. 1995, 36, 5885–5886. 10.1016/00404-0399(50)1195N-. [DOI] [Google Scholar]
- Reggelin M.; Junker B. Cyclic Sulfonimidates by Dynamic Diastereomer-Differentiating Cyclisation: Large-Scale Synthesis and Mechanistic Studies. Chem. - Eur. J. 2001, 7, 1232–1239. 10.1002/1521-3765(20010316)7:6<1232::AID-CHEM1232>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Leca D.; Fensterbank L.; Lacôte E.; Malacria M. A New Practical One-Pot Access to Sulfonimidates. Org. Lett. 2002, 4, 4093–4095. 10.1021/ol026837b. [DOI] [PubMed] [Google Scholar]
- Leca D.; Song K.; Amatore M.; Fensterbank L.; Lacôte E.; Malacria M. Iodine(III)-Mediated Preparations of Nitrogen-Containing Sulfur Derivatives: Dramatic Influence of the Sulfur Oxidation State. Chem. - Eur. J. 2004, 10, 906–916. 10.1002/chem.200305525. [DOI] [PubMed] [Google Scholar]
- Felim A.; Toussaint A.; Phillips C. R.; Leca D.; Vagstad A.; Fensterbank L.; Lacôte E.; Malacria M. Improved Method for the Iodine(III)-Mediated Preparation of Aryl Sulfonimidates. Org. Lett. 2006, 8, 337–339. 10.1021/ol052790t. [DOI] [PubMed] [Google Scholar]
- Nandi G. C.; Arvidsson P. I. Sulfonimidamides: Synthesis and Applications in Preparative Organic Chemistry. Adv. Synth. Catal. 2018, 360, 2976–3001. 10.1002/adsc.201800273. [DOI] [Google Scholar]
- Chinthakindi P. K.; Naicker T.; Thota N.; Govender T.; Kruger H. G.; Arvidsson P. I. Sulfonimidamides in Medicinal and Agricultural Chemistry. Angew. Chem., Int. Ed. 2017, 56, 4100–4109. 10.1002/anie.201610456. [DOI] [PubMed] [Google Scholar]
- García Mancheño O.; Bolm C. Synthesis of Sulfonimidamides from Sulfinamides by Oxidation with N-Chlorosuccinimide. Beilstein J. Org. Chem. 2007, 3, 25. 10.1186/1860-5397-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worch C.; Atodiresei I.; Raabe G.; Bolm C. Synthesis of Enantiopure Sulfonimidamides and Elucidation of Their Absolute Configuration by Comparison of Measured and Calculated CD Spectra and X-Ray Crystal Structure Determination. Chem. - Eur. J. 2010, 16, 677–683. 10.1002/chem.200901715. [DOI] [PubMed] [Google Scholar]
- Steurer M.; Bolm C. Synthesis of Amino-Functionalized Sulfonimidamides and Their Application in the Enantioselective Henry Reaction. J. Org. Chem. 2010, 75, 3301–3310. 10.1021/jo100326x. [DOI] [PubMed] [Google Scholar]
- Richards-Taylor C. S.; Martínez-Lamenca C.; Leenaerts J. E.; Trabanco A. A.; Oehlrich D. The Synthesis of Trifluoromethyl-sulfonimidamides from Sulfinamides. J. Org. Chem. 2017, 82, 9898–9904. 10.1021/acs.joc.7b01628. [DOI] [PubMed] [Google Scholar]
- Wright M.; Martínez-Lamenca C.; Leenaerts J. E.; Brennan P. E.; Trabanco A. A.; Oehlrich D. Bench-Stable Transfer Reagent Facilitates the Generation of Trifluoromethyl-sulfonimidamides. J. Org. Chem. 2018, 83, 9510–9516. 10.1021/acs.joc.8b01244. [DOI] [PubMed] [Google Scholar]
- Cathers B. E.; Schloss J. V. The Sulfonimidamide as a Novel Transition State Analog for Aspartic Acid and Metallo Proteases. Bioorg. Med. Chem. Lett. 1999, 9, 1527–1532. 10.1016/S0960-894X(99)00241-3. [DOI] [PubMed] [Google Scholar]
- Izzo F.; Schäfer M.; Stockman R.; Lücking U. A New, Practical One-Pot Synthesis of Unprotected Sulfonimidamides by Transfer of Electrophilic NH to Sulfinamides. Chem. - Eur. J. 2017, 23, 15189–15193. 10.1002/chem.201703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Gibson J. A Convenient Synthetic Route to Sulfonimidamides from Sulfonamides. RSC Adv. 2015, 5, 4171–4174. 10.1039/C4RA14056G. [DOI] [Google Scholar]
- Chinthakindi P. K.; Benediktsdottir A.; Ibrahim A.; Wared A.; Aurell C.-J.; Pettersen A.; Zamaratski E.; Arvidsson P. I.; Chen Y.; Sandström A. Synthesis of Sulfonimidamide-Based Amino Acid Building Blocks with Orthogonal Protecting Groups. Eur. J. Org. Chem. 2019, 2019, 1045–1057. 10.1002/ejoc.201801541. [DOI] [Google Scholar]
- Zasukha S. V.; Timoshenko V. M.; Tolmachev A. A.; Pivnytska V. O.; Gavrylenko O.; Zhersh S.; Shermolovich Y.; Grygorenko O. O. Sulfonimidamides and Imidosulfuric Diamides: Compounds from an Underexplored Part of Biologically Relevant Chemical Space. Chem. - Eur. J. 2019, 25, 6928–6940. 10.1002/chem.201900440. [DOI] [PubMed] [Google Scholar]
- Toth J. E.; Grindey G. B.; Ehlhardt W. J.; Ray J. E.; Boder G. B.; Bewley J. R.; Klingerman K. K.; Gates S. B.; Rinzel S. M.; Schultz R. M.; et al. Sulfonimidamide Analogs of Oncolytic Sulfonylureas. J. Med. Chem. 1997, 40, 1018–1025. 10.1021/jm960673l. [DOI] [PubMed] [Google Scholar]
- Wen J.; Cheng H.; Dong S.; Bolm C. Copper-Catalyzed S-C/S-N Bond Interconversions. Chem. - Eur. J. 2016, 22, 5547–5550. 10.1002/chem.201600661. [DOI] [PubMed] [Google Scholar]
- Davies T. Q.; Hall A.; Willis M. C. One-Pot, Three-Component Sulfonimidamide Synthesis Exploiting the Sulfinylamine Reagent N-Sulfinyltritylamine, TrNSO. Angew. Chem., Int. Ed. 2017, 56, 14937–14941. 10.1002/anie.201708590. [DOI] [PubMed] [Google Scholar]
- Kluge R.; Hocke H.; Schulz M.; Schilke F. Preparation of Racemic and Enantiopure Arenesulfonimidoyl on Sulfur Azoles - New Compounds Chiral on Sulfur. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 149, 179–206. 10.1080/10426509908037031. [DOI] [Google Scholar]
- Nandi G. C.; Jesin I. Direct Synthesis of N-Acyl Sulfonimidamides and N-Sulfonimidoyl Amidines from Sulfonimidoyl Azides. Adv. Synth. Catal. 2018, 360, 2465–2469. 10.1002/adsc.201800215. [DOI] [Google Scholar]
- Bremerich M.; Conrads C. M.; Langletz T.; Bolm C. Additions to N-Sulfinylamines as an Approach for the Metal-free Synthesis of Sulfonimidamides: O-Benzotriazolyl Sulfonimidates as Activated Intermediates. Angew. Chem., Int. Ed. 2019, 58, 19014–19020. 10.1002/anie.201911075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs E. L.; Tota A.; Colella M.; Degennaro L.; Luisi R.; Bull J. A. Synthesis of Sulfonimidamides from Sulfenamides via an Alkoxyamino-λ6-sulfanenitrile Intermediate. Angew. Chem., Int. Ed. 2019, 58, 14303–14310. 10.1002/anie.201906001. [DOI] [PubMed] [Google Scholar]
- Funes Maldonado M.; Sehgelmeble F.; Bjarnemark F.; Svensson M.; Ahman J.; Arvidsson P. I. Synthesis and Arylation of Unprotected Sulfonimidamides. Tetrahedron 2012, 68, 7456–7462. 10.1016/j.tet.2012.06.072. [DOI] [Google Scholar]
- Nandi G. C.; Kota S. R.; Govender T.; Kruger H. G.; Arvidsson P. I. Cu(OAc)2 Promoted Chan–Evans–Lam C–N Cross Coupling Reactions on the N- and N’-Nitrogen Atoms of Sulfonimidamides with Aryl Boronic Acids. Tetrahedron 2014, 70, 5428–5433. 10.1016/j.tet.2014.06.122. [DOI] [Google Scholar]
- Battula S. R. K.; Subbareddy G. V.; Chakravarthy I. E. A Mild and Efficient Copper-Catalyzed N-Arylation of Unprotected Sulfonimidamides Using Boronic Acids. Tetrahedron Lett. 2014, 55, 517–520. 10.1016/j.tetlet.2013.11.084. [DOI] [Google Scholar]
- Borhade S. R.; Sandstrom A.; Arvidsson P. I. Synthesis of Novel Aryl and Heteroaryl Acyl Sulfonimidamides via Pd-Catalyzed Carbonylation Using a Nongaseous Precursor. Org. Lett. 2013, 15, 1056–1059. 10.1021/ol400049m. [DOI] [PubMed] [Google Scholar]
- Thota N.; Makam P.; Rajbongshi K. K.; Nagiah S.; Abdul N. S.; Chuturgoon A. A.; Kaushik A.; Lamichhane G.; Somboro A. M.; Kruger H. G.; et al. N-Trifluoromethylthiolated Sulfonimidamides and Sulfoximines: Anti-microbial, Anti-mycobacterial, and Cytotoxic Activity. ACS Med. Chem. Lett. 2019, 10, 1457–1461. 10.1021/acsmedchemlett.9b00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzaro S.; Desage-El Murr M.; Fensterbank L.; Lacôte E.; Malacria M. Copper-Catalyzed N-Arylation of Sulfonimidamides. Synlett 2011, 2011, 849–851. 10.1055/s-0030-1259682. [DOI] [Google Scholar]
- Leca D.; Toussaint A.; Mareau C.; Fensterbank L.; Lacôte E.; Malacria M. Efficient Copper-Mediated Reactions of Nitrenes Derived from Sulfonimidamides. Org. Lett. 2004, 6, 3573–3575. 10.1021/ol0485520. [DOI] [PubMed] [Google Scholar]
- Izzo F.; Schäfer M.; Lienau P.; Ganzer U.; Stockman R.; Lücking U. Exploration of Novel Chemical Space: Synthesis and in vitro Evaluation of N-Functionalized Tertiary Sulfonimidamides. Chem. - Eur. J. 2018, 24, 9295–9304. 10.1002/chem.201801557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worch C.; Bolm C. Copper-mediated Cross-Coupling Reactions of N-Protected Sulfonimidamides and Aryl Halides. Synthesis 2008, 2008, 739–742. 10.1055/s-2008-1032139. [DOI] [Google Scholar]
- Bachon A.-K.; Hermann A.; Bolm C. 3D Heterocycles from Sulfonimidamides by Sequential C–H Bond Alkenylation/Aza-Michael Cyclization. Chem. - Eur. J. 2019, 25, 5889–5892. 10.1002/chem.201900920. [DOI] [PubMed] [Google Scholar]
- Worch C.; Bolm C. Use of Prolyl Sulfonimidamides in Solvent-Free Organocatalytic Asymmetric Aldol Reactions. Synlett 2009, 2009, 2425–2428. 10.1055/s-0029-1217727. [DOI] [Google Scholar]
- Patureau F. W.; Worch C.; Siegler M. A.; Spek A. L.; Bolm C.; Reek J. N. H. SIAPhos: Phosphorylated Sulfonimidamides and their Use in Iridium-Catalyzed Asymmetric Hydrogenations of Sterically Hindered Cyclic Enamides. Adv. Synth. Catal. 2012, 354, 59–64. 10.1002/adsc.201100692. [DOI] [Google Scholar]
- Liang C.; Collet F.; Robert-Peillard F.; Müller P.; Dodd R. H.; Dauban P. Toward a Synthetically Useful Stereoselective C-H Amination of Hydrocarbons. J. Am. Chem. Soc. 2008, 130, 343–350. 10.1021/ja076519d. [DOI] [PubMed] [Google Scholar]
- Lenstra D. C.; Vedovato V.; Ferrer Flegeau E.; Maydom J.; Willis M. C. One-Pot Sulfoxide Synthesis Exploiting a Sulfinyl-Dication Equivalent Generated from a DABSO/Trimethylsilyl Chloride Sequence. Org. Lett. 2016, 18, 2086–2089. 10.1021/acs.orglett.6b00712. [DOI] [PubMed] [Google Scholar]